

Summary
Histone deacetylases (HDACs) are ubiquitous enzymes that cleave post-translational ε-N-acyllysine modifications. The continued identification of diverse acyl modifications at lysine residues in proteins has resulted in discovery of new insight into the biological roles of these enzymes. Here, we describe a fluorogenic high-throughput screening protocol to identify deacylase activities. We describe the careful optimization of continuous, coupled enzyme assays, which provide efficient determination of kinetic parameters. These techniques can facilitate inhibitor assay design and provide fundamental understanding of HDAC biochemistry.
For complete details on the use and execution of this protocol, please refer to Moreno-Yruela et al. (2018).
Subject areas: Molecular/chemical probes, Protein biochemistry
Graphical abstract
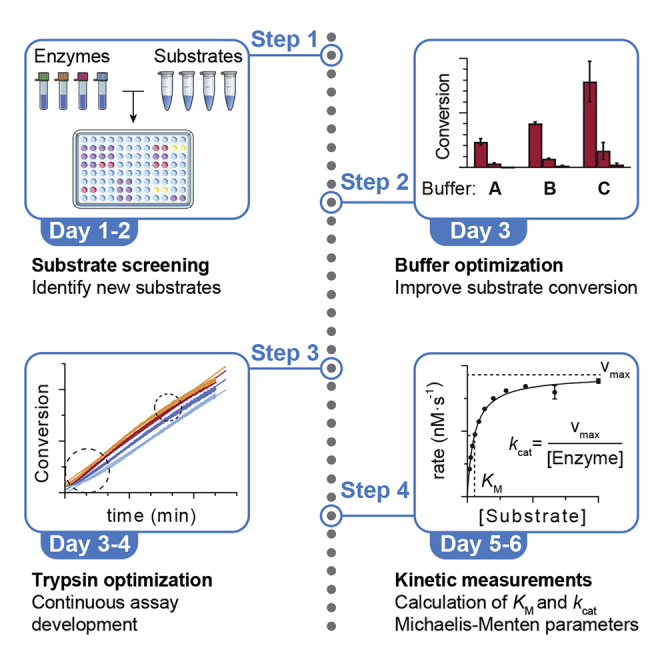
Highlights
-
•
Histone deacetylases hydrolyze multiple different acyl modifications of lysine
-
•
Fluorogenic substrates enable high-throughput screening of deacylase activities
-
•
We develop continuous, coupled enzyme assays by careful optimization
-
•
Continuous assays allow accurate determination of enzyme kinetic parameters
Histone deacetylases (HDACs) are ubiquitous enzymes that cleave post-translational ε-N-acyllysine modifications. The continued identification of diverse acyl modifications at lysine residues in proteins has resulted in discovery of new insight into the biological roles of these enzymes. Here, we describe a fluorogenic high-throughput screening protocol to identify deacylase activities. We describe the careful optimization of continuous, coupled enzyme assays, which provide efficient determination of kinetic parameters. These techniques can facilitate inhibitor assay design and provide fundamental understanding of HDAC biochemistry.
Before you begin
Preparation of reagents and workspace
Timing: 1–2 h
-
1.Prepare Tris or HEPES assay buffer (Tris buffer was used for broad deacylase activity screening, HEPES buffer was used for HDAC11 deacylation assays)(Moreno-Yruela et al., 2018).
-
a.Titrate the corresponding solution B (∼350 mL) into solution A (600 mL) until the desired pH is reached (Tris: pH 8.0, HEPES: pH 7.4). Use leftover solutions A and B to further adjust the pH (see Materials and equipment for composition of each solution).
-
a.
-
2.
Prepare substrate stocks in DMSO at 20 mM concentration, or thaw existing frozen stocks at 25°C or by shaking at 37°C.
Note: Due to its potential HDAC inhibition effect (Marks and Breslow, 2007), final assay DMSO content should not be above 2%‒3%. Therefore, the desirable substrate DMSO stock concentration is >100 times the final assay concentration.
-
3.
Verify that the amount needed of each substrate and enzyme stock is available.
-
4.
Weigh 20‒25 mg BSA in a 50 mL centrifuge tube and dissolve in assay buffer to reach a concentration of 0.5 mg/mL.
Note: BSA concentration may vary in follow-up screenings and time-course experiments.
Note: BSA-containing assay buffers can be stored at 4°C and reused for 7 days.
-
5.
Weigh 30‒40 mg trypsin in a 15 mL centrifuge tube and dissolve in assay buffer containing BSA, to reach a concentration of 5.0 mg/mL of trypsin. Keep this solution on ice until use.
Note: Trypsin concentration may vary in follow-up screenings and time-course experiments.
Note: The trypsin solution is made fresh every day and with the same batch of buffer used for the assay.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Bovine serum albumin (BSA, heat shock fraction, protease free, essentially globulin free, pH 7, ≥98%) | Sigma-Aldrich | Cat#A3059 |
| HDAC1 (full length with C-terminal His-tag, C-terminal FLAG-tag) | BPS Bioscience | Cat#50051 |
| HDAC2 (full length with C-terminal His-tag) | BPS Bioscience | Cat#50002 |
| HDAC3/NcoR2 (full length with C-terminal His-tag) | BPS Bioscience | Cat#50003 |
| HDAC4 (aa 627–1,084 with N-terminal GST-tag, C-terminal His-tag) | BPS Bioscience | Cat#50004 |
| HDAC5 (aa 656–1,122 with C-terminal His-tag) | BPS Bioscience | Cat#50005 |
| HDAC6 (full length with C-terminal FLAG-tag) | BPS Bioscience | Cat#50056 |
| HDAC7 (aa 383–end with N-terminal His-tag) | Millipore | Cat#14-832 |
| HDAC8 (full length with C-terminal His-tag) | BPS Bioscience | Cat#50008 |
| HDAC9 (aa 604–1066 with C-terminal His-tag) | BPS Bioscience | Cat#50009 |
| HDAC10 (aa 2–631 with N-terminal FLAG-tag) | BPS Bioscience | Cat#50060 |
| HDAC11 (full length untagged) | BPS Bioscience | Cat#50021 |
| HEPES | Sigma-Aldrich | Cat#H4034 |
| HEPES sodium salt | Sigma-Aldrich | Cat#H3784 |
| KCl | Sigma-Aldrich | Cat#P9541 |
| MgCl2 | Sigma-Aldrich | Cat#M4880 |
| NaCl | Sigma-Aldrich | Cat#71380 |
| Tris(2-carboxyethyl)phosphine (TCEP) HCl salt | Sigma-Aldrich | Cat#C4706 |
| Tris(hydroxymethyl)aminomethane (Tris) | Sigma-Aldrich | Cat#T1503 |
| Tris(hydroxymethyl)aminomethane (Tris) HCl salt | Sigma-Aldrich | Cat#T5941 |
| Trypsin (TPCK treated, essentially salt-free, lyophilized powder, ≥10,000 BAEE units/mg protein) | Sigma-Aldrich | Cat#T1426 |
| Tween 20 | Fisher Scientific | Cat#BP337 |
| 1a) Ac-Leu-Gly-Lys(Ac)-AMC | (Wegener et al., 2003, Madsen and Olsen, 2012b) | n/a |
| 1b) Ac-Leu-Gly-Lys(Tfa)-AMC | (Bradner et al., 2010, Madsen and Olsen, 2012b) | n/a |
| 1f) Ac-Leu-Gly-Lys(i-But)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 1g) Ac-Leu-Gly-Lys(i-Val)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 1i) Ac-Leu-Gly-Lys(Cr)-AMC | (Madsen and Olsen, 2012b) | n/a |
| 1r) Ac-Leu-Gly-Lys(Glu)-AMC | (Anderson et al., 2017) | n/a |
| 1s) Ac-Leu-Gly-Lys(Hmg)-AMC | (Anderson et al., 2017) | n/a |
| 1t) Ac-Leu-Gly-Lys(Bio)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 2a) Ac-Arg-His-Lys-Lys(Ac)-AMC | Fluor de lys-SIRT1 substrate (Enzo Life Sciences) | Cat#KI177 |
| 3a) Ac-Arg-His-Lys(Ac)-Lys(Ac)-AMC | Fluor de lys-HDAC8 substrate (Enzo Life Sciences) | Cat#KI178 |
| 4a) Ac-Thr-Ala-Arg-Lys(Ac)-AMC | (Madsen and Olsen, 2012b) | n/a |
| 4d) Ac-Thr-Ala-Arg-Lys(Dec)-AMC | (Madsen et al., 2016) | n/a |
| 4e) Ac-Thr-Ala-Arg-Lys(Myr)-AMC | (Madsen et al., 2016) | n/a |
| 4x) Ac-Thr-Ala-Arg-Lys(Lau)-AMC | (Madsen et al., 2016) | n/a |
| 5a) Ac-Gln-Pro-Lys-Lys(Ac)-AMC | Fluor de lys-SIRT2 substrate (Enzo Life Sciences) and (Madsen et al., 2016) | Cat#KI179 |
| 5c) Ac-Gln-Pro-Lys-Lys(Hex)-AMC | (Madsen et al., 2016) | n/a |
| 5d) Ac-Gln-Pro-Lys-Lys(Dec)-AMC | (Madsen et al., 2016) | n/a |
| 5e) Ac-Gln-Pro-Lys-Lys(Myr)-AMC | (Madsen et al., 2016) | n/a |
| 5f) Ac-Gln-Pro-Lys-Lys(i-But)-AMC | (Madsen et al., 2016) | n/a |
| 5g) Ac-Gln-Pro-Lys-Lys(i-Val)-AMC | (Madsen et al., 2016) | n/a |
| 5i) Ac-Gln-Pro-Lys-Lys(Cr)-AMC | (Madsen et al., 2016) | n/a |
| 5u) Ac-Gln-Pro-Lys-Lys(But)-AMC | (Madsen et al., 2016) | n/a |
| 5v) Ac-Gln-Pro-Lys-Lys(Oct)-AMC | (Madsen et al., 2016) | n/a |
| 5x) Ac-Gln-Pro-Lys-Lys(Lau)-AMC | (Madsen et al., 2016) | n/a |
| 5y) Ac-Gln-Pro-Lys-Lys(Pal)-AMC | (Madsen et al., 2016) | n/a |
| 6a) Ac-Glu-Thr-Asp-Lys(Ac)-AMC | (Galleano et al., 2016) | n/a |
| 6d) Ac-Glu-Thr-Asp-Lys(Dec)-AMC | (Galleano et al., 2016) | n/a |
| 6e) Ac-Glu-Thr-Asp-Lys(Myr)-AMC | (Galleano et al., 2016) | n/a |
| 6f) Ac-Glu-Thr-Asp-Lys(i-But)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 6g) Ac-Glu-Thr-Asp-Lys(i-Val)-AMC | (Galleano et al., 2016) | n/a |
| 6h) Ac-Glu-Thr-Asp-Lys(2-MeBut)-AMC | (Galleano et al., 2016) | n/a |
| 6i) Ac-Glu-Thr-Asp-Lys(Cr)-AMC | (Galleano et al., 2016) | n/a |
| 6j) Ac-Glu-Thr-Asp-Lys(MeAcr)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 6k) Ac-Glu-Thr-Asp-Lys(2-MeCr)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 6l) Ac-Glu-Thr-Asp-Lys(3-MeCr)-AMC | (Moreno-Yruela et al., 2018) | n/a |
| 6m) Ac-Glu-Thr-Asp-Lys(2E-Dece)-AMC | (Galleano et al., 2016) | n/a |
| 6n) Ac-Glu-Thr-Asp-Lys(3-HydroxyBut)-AMC | (Galleano et al., 2016) | n/a |
| 6o) Ac-Glu-Thr-Asp-Lys(3-HydroxyDec)-AMC | (Galleano et al., 2016) | n/a |
| 6p) Ac-Glu-Thr-Asp-Lys(AcAc)-AMC | (Galleano et al., 2016) | n/a |
| 6q) Ac-Glu-Thr-Asp-Lys(3-OxoDec)-AMC | (Galleano et al., 2016) | n/a |
| 6x) Ac-Glu-Thr-Asp-Lys(Lau)-AMC | (Galleano et al., 2016) | n/a |
| 6z) Ac-Glu-Thr-Asp-Lys(2E-Laue)-AMC | (Galleano et al., 2016) | n/a |
| 6æ) Ac-Glu-Thr-Asp-Lys(3-HydroxyLau)-AMC | (Galleano et al., 2016) | n/a |
| 6ø) Ac-Glu-Thr-Asp-Lys(3-OxoLau)-AMC | (Galleano et al., 2016) | n/a |
| Software and algorithms | ||
| Prism 8 or 9 (for Mac and Windows) | GraphPad | n/a |
| Other | ||
| Corning 96-well half-area plates, black, polystyrene | Fisher Scientific | Cat#3686 |
| Microcentrifuge tubes (1.5 mL Eppendorfs) | Hounisen Lab Equipment | Cat#83.46.00 |
| V-bottom 96-well plates, clear, polystyrene | Greiner Bio-One | Cat#651161 |
| FLUOstar Omega microplate reader | BMG Labtech | n/a |
Materials and equipment
Tris assay buffer – solution A
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris HCl salt | 50 mM | 5.516 g |
| NaCl | 137 mM | 5.604 g |
| KCl | 2.7 mM | 141 mg |
| MgCl2 | 1.0 mM | 67 mg |
| ddH2O | n/a | 700 mL |
| Total | n/a | 700 mL |
Tris assay buffer – solution B
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris | 50 mM | 2.423 g |
| NaCl | 137 mM | 3.203 g |
| KCl | 2.7 mM | 81 mg |
| MgCl2 | 1.0 mM | 38 mg |
| ddH2O | n/a | 400 mL |
| Total | n/a | 400 mL |
HEPES assay buffer – solution A
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES | 50 mM | 8.339 g |
| KCl | 100 mM | 5.217 g |
| TCEP HCl salt | 0.2 mM | 40 mg |
| Tween 20 | 0.001% | 7.0 μL |
| ddH2O | n/a | 700 mL |
| Total | n/a | 700 mL |
HEPES assay buffer – solution B
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES sodium salt | 50 mM | 7.808 g |
| KCl | 100 mM | 2.984 g |
| TCEP HCl salt | 0.2 mM | 23 mg |
| Tween 20 | 0.001% | 4.0 μL |
| ddH2O | n/a | 400 mL |
| Total | n/a | 400 mL |
Note: Assay buffers without BSA can be stored at 4°C for several months.
Alternatives: Substrate and trypsin dilutions are performed either in Eppendorf tubes or in clear polystyrene 96-well plates.
Alternatives: Here, fluorescence is recorded in a FLUOstar Omega microplate reader (excitation: 355–20 nm, emission: 450–10 nM), where the plate is shaken once for 10 s at 300 rpm and then read with 5 flashes per well either a single time or every 30 s. Similar plate readers with excitation at ∼365 nm and emission at ∼440 nm are also suitable. Shaking of the plate before fluorescence recording is highly encouraged to achieve homogeneous distribution of the fluorophore in the solutions.
Step-by-step method details
ε-N-Acyllysine substrate screening
Recombinant HDACs are incubated with a library of ε-N-acyllysine-containing peptide substrates, and final addition of trypsin and fluorophore release is used to measure conversion.
Note: Assay plates contain two internal replicates of every condition and of the background controls for each substrate (no enzyme added, Table 1). Data from these replicates is averaged to afford a single assay result. Then, the entire screening is repeated twice using freshly made substrate and enzyme dilutions, and data are reported as the average of both results (n = 2 independent experiments).
Table 1.
Example of plate layout (half plate) for substrate screening against HDACs 1‒3
| (HDAC) (Substrate) |
1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| A | NE 1a |
NE 1a |
NE 1b |
NE 1b |
NE 2a |
NE 2a |
| B | HDAC1 1a |
HDAC1 1a |
HDAC1 1b |
HDAC1 1b |
HDAC1 2a |
HDAC1 2a |
| C | HDAC2 1a |
HDAC2 1a |
HDAC2 1b |
HDAC2 1b |
HDAC2 2a |
HDAC2 2a |
| D | HDAC3 1a |
HDAC3 1a |
HDAC3 1b |
HDAC3 1b |
HDAC3 2a |
HDAC3 2a |
| E | NE 3a |
NE 3a |
NE 4a |
NE 4a |
NE 4d |
NE 4d |
| F | HDAC1 3a |
HDAC1 3a |
HDAC1 4a |
HDAC1 4a |
HDAC1 4d |
HDAC1 4d |
| G | HDAC2 3a |
HDAC2 3a |
HDAC2 4a |
HDAC2 4a |
HDAC2 4d |
HDAC2 4d |
| H | HDAC3 3a |
HDAC3 3a |
HDAC3 4a |
HDAC3 4a |
HDAC3 4d |
HDAC3 4d |
NE, no enzyme added.
Note: Stock solution concentrations are provided with a number in parenthesis (e.g., 2×) that refers to how many times it will be diluted to achieve the final assay concentration.
Note: Experiments are performed in a volume of 25 μL per well, followed by addition of 25 μL of developer solution.
Optional: Include known enzyme‒substrate pairs in the screening as positive controls. For example: LGKac (1a) for HDACs 1‒3 and 6 (Wegener et al., 2003), LGKtfa (1b) for HDACs 4, 5 and 7‒9 (Bradner et al., 2010), or QPKKmyr (5e) for SIRTs 1‒3 (Chiang and Lin, 2016).
-
1.
Prepare substrate stocks in BSA-containing Tris assay buffer at 83.3 μM concentration (1.78×).
-
2.
Prepare enzyme stocks in BSA-containing Tris assay buffer at 125 nM concentration (2.5×).
Optional: Prepare 200 μL extra of enzyme stocks and use a liquid reservoir and a multichannel pipette for addition of the enzyme (point 5).
Note: Keep enzyme solutions on ice as much as possible.
-
3.
Add 15 μL of 1.78× substrate stocks to the corresponding wells of half-area black 96-well plates (see Table 1 for a plate layout example).
-
4.
Add 10 μL of BSA-containing assay buffer to the control wells (where no enzyme will be added).
-
5.
Add 10 μL of 2.5× enzyme stocks to the corresponding wells.
-
6.
Cover the plates with a transparent sticker and incubate at 37°C for 60 min with gentle shaking (e.g., 750 rpm).
-
7.
Take out the plates from the incubator, remove the sticker, and add 25 μL of trypsin solution (5.0 mg/mL) to every well.
-
8.
Cover the plates again and let them stand at 25°C for 90 min for development.
-
9.
Read the fluorescence from each well using a plate reader.
-
10.
Calculate the average fluorescence of each replicate pair.
-
11.
For each experiment, subtract background fluorescence from the corresponding control wells without enzyme added (always subtract the background from the same substrate, because background fluorescence may change with chemical structure).
-
12.
Repeat steps 1–11.
-
13.
Represent data as average ± SEM of both screenings (n = 2 independent experiments) using a data analysis software (e.g., GraphPad Prism), as bar graphs for individual enzymes or as a heatmap (Figure 1).
Optional: A 7-amino-4-methylcoumarin (AMC) fluorescence standard curve can be obtained by preparing dilutions of free AMC fluorophore in BSA-containing assay buffer and measuring the fluorescence with the plate reader. This standard curve can be added to the data-processing software and used to convert fluorescence units into AMC concentration for each experiment, which corresponds to amount of substrate converted. In GraphPad Prism, this can be done using the function “Transform ‒ User-defined Y function.”
Optional: Represent screening data as a heatmap. In order to highlight differences in conversion, manually select range intervals that would differentiate most of the values in the heatmap (in GraphPad Prism, select “Colormap: categorical”). Change the color of each interval so that differences are clearly visible in web and in print versions (color advice sites such as colorbrewer2.org can be used as inspiration). Here, Adobe Illustrator was used to add frames, adjust the legend, and organize the labels (Figure 1).
Figure 1.

Representation of screening data as a heatmap
Since only mean values are included, supplementary raw data or bar graphs should be reported for such representations. Heatmap reprinted with permission from Moreno-Yruela et al. (2018).
Buffer optimization
The selected HDAC(s) and substrate(s) are incubated in Tris of HEPES buffer with varying amounts of BSA to find optimal assay conditions (e.g., 0.00‒0.50 mg/mL). These conditions can then be applied for testing of future enzyme batches or for inhibition/activation experiments.
Note: Experiments are performed in a volume of 25 μL per well, followed by addition of 25 μL of developer solution.
-
14.
Prepare Tris and HEPES buffers with different concentrations of BSA (for example, 0.5 mg/mL and 0.05 mg/mL) (Troubleshooting 1).
-
15.
Prepare corresponding trypsin solutions in each assay buffer.
Optional: Depending on the peptide sequence of the selected substrate(s), the concentration and development time with trypsin may vary. This can be tested by measuring fluorescence at several time points during the development phase and checking when the maximum signal stays constant. For example, development of Ac-Gln-Pro-Lys-Lys-AMC (QPKK) requires 90 min incubation with a final concentration of 2.5 mg/mL of trypsin (5.0 mg/mL trypsin stock), whereas Ac-Leu-Gly-Lys-AMC (LGK) and Ac-Glu-Thr-Asp-Lys-AMC (ETDK) can be developed in 15 min with a final concentration of 0.2 mg/mL of trypsin (0.4 mg/mL tryspin stock) (Galleano et al., 2016).
-
16.
Repeat steps 1‒13 for the selected enzymes and substrates, with the following modifications: prepare substrate and enzyme stocks in each of the BSA-containing assay buffers (steps 1 and 2), and develop each experiment with the matching trypsin solution (step 7).
Optimization of trypsin concentration for time-course experiments
Small amounts of trypsin are added from the beginning of the experiment in order to record substrate conversion in real time. Trypsin concentration requires optimization, so that the recorded rate corresponds to that of the deacylation reaction (too low trypsin concentration limits the reaction rate to that of trypsin) and to be able to measure the steady state for the longest time possible (too high trypsin concentration can compromise enzyme stability).
Note: Experiments are performed in a volume of 50 μL per well.
-
17.
Prepare trypsin stocks (5×) in the selected BSA-containing assay buffer at decreasing concentrations (for example: 1,000, 500, 250 and 125 μg/mL).
-
18.
Prepare a 333 μM or 666 μM substrate stock in BSA-containing assay buffer (3.33×).
-
19.
Prepare enzyme stocks (2×) in the selected BSA-containing assay buffer at decreasing concentrations (for example: 200, 20, and 2 nM).
-
20.
Add 15 μL of 3.33× substrate stock to every well (see Table 2 for a plate layout example).
-
21.
Add 10 μL of the corresponding 5× trypsin stocks to the wells.
-
22.
Add 25 μL of BSA-containing assay buffer to the negative control wells.
-
23.
Make sure the plate reader is set up with the time-course experiment ready to be launched (in order to measure fluorescence as soon as possible after enzyme addition).
-
24.
Add 25 μL of the corresponding 2× enzyme stocks to the wells, place the plate in the plate reader, and start recording fluorescence of each well every 30 s.
CRITICAL: Fast addition of enzyme is advised to avoid different starting times between experiments. A repetitive or multichannel pipette can be used for this purpose.
-
25.
Record fluorescence for 30‒60 min, or until progression curves are not linear (Figure 2).
-
26.Plot fluorescence measurements vs. time and calculate initial rates for each experiment (in GraphPad Prism, this can be done with “Analyze” – “Nonlinear regression” – “Straight line”).
-
a.Exclude data points at the start and end of the assay whenever they do not fit a linear trend, because lack of linearity indicates that the system is not in steady state (Stein, 2011a).
-
a.
Note: At low concentrations of trypsin, it is common to observe a long “lag phase” until the deacylated substrate is accumulated and steady-state fluorophore release is reached (Figure 2). Avoid choosing such conditions for future experiments, if possible.
-
27.
Find a concentration of enzyme at which assay progression curves are obtained with appropriate signal-to-noise ratio, but not as high as to consume all substrate within the assay time frame. Ideally, fluorescence units should increase to at least 2-fold higher values than the background signal during the first 15 min of reaction, and the error obtained between internal replicates should be smaller than the fluorescence increase during a period of 2‒3 min.
-
28.
For the set concentration of enzyme, find the lowest trypsin concentration at which the conversion rate remains unchanged. This should also be the concentration at which linearity is kept for the longest period of time (Figure 2).
Note: Lower conversion rates indicate that the fluorophore release by trypsin, and not the deacylation event, is the rate-limiting step of the experiment.
Optional: Fine-tune enzyme and trypsin concentrations in order to extend assay linearity and/or spare material (Troubleshooting 2).
Table 2.
Example of plate layout for trypsin optimization
| [HDAC] [Trypsin] |
1 | 2 | 3 | 4 |
|---|---|---|---|---|
| A | NE 200 μg/mL |
NE 100 μg/mL |
NE 50 μg/mL |
NE 25 μg/mL |
| B | NE 200 μg/mL |
NE 100 μg/mL |
NE 50 μg/mL |
NE 25 μg/mL |
| C | 100 nM 200 μg/mL |
100 nM 100 μg/mL |
100 nM 50 μg/mL |
100 nM 25 μg/mL |
| D | 100 nM 200 μg/mL |
100 nM 100 μg/mL |
100 nM 50 μg/mL |
100 nM 25 μg/mL |
| E | 10 nM 200 μg/mL |
10 nM 100 μg/mL |
10 nM 50 μg/mL |
10 nM 25 μg/mL |
| F | 10 nM 200 μg/mL |
10 nM 100 μg/mL |
10 nM 50 μg/mL |
10 nM 25 μg/mL |
| G | 1 nM 200 μg/mL |
1 nM 100 μg/mL |
1 nM 50 μg/mL |
1 nM 25 μg/mL |
| H | 1 nM 200 μg/mL |
1 nM 100 μg/mL |
1 nM 50 μg/mL |
1 nM 25 μg/mL |
NE, no enzyme added.
Figure 2.

Example of data obtained for the optimization of trypsin concentration, with associated linear regression curves
Here, enzyme concentrations of 4, 2, and 1 nM were incubated with substrate 6e (ETDKmyr, 60 μM) and trypsin at 200, 100, 50, or 25 μg/mL concentration. Sample data corresponding to 4 nM enzyme concentration is used to highlight examples of long lag phase and of loss of linearity. Data points not fitting to a linear regression should be excluded in order to obtain an accurate measure of the steady state. Data represent mean ± SD of 2 internal replicates.
Determination of Michaelis-Menten kinetic parameters
With the optimized time-course assay conditions, the selected HDAC is incubated with substrate at different concentrations, and the initial conversion rates are used to calculate kinetic parameters. These are critical for the design of continuous or discontinuous inhibition assays (Moreno-Yruela et al., 2019b), and for the accurate comparison of substrates.
Note: Experiments are performed in a volume of 50 μL per well.
-
29.
Prepare a trypsin stock (3.33×) in BSA-containing assay buffer at the chosen concentration.
-
30.
Prepare a 1 mM substrate stock (5×) and 10 additional dilutions (1.5-fold, 1,000‒17.3 μM) in BSA-containing assay buffer.
Optional: Use a clear V-bottom 96-well plate for the dilution series. This will allow dilution of several substrates at a time, detection of substrate precipitation, and facilitate the transfer to the assay plate using a multichannel pipette.
-
31.
Prepare an enzyme stock (2×) in BSA-containing assay buffer at the chosen concentration.
-
32.
Add 10 μL of the corresponding 5× substrate stocks to each well (see Table 3 for a plate layout example).
-
33.
Add 15 μL of 3.33× trypsin stock to every well.
-
34.
Add 25 μL of BSA-containing assay buffer to the negative control wells.
-
35.
Make sure the plate reader is set up with the time-course experiment ready to be launched (in order to measure fluorescence as soon as possible after enzyme addition).
-
36.
Add 25 μL of the 2× enzyme stock to the corresponding wells, place the plate in the plate reader, and start recording fluorescence of each well every 30 s.
-
37.
Record fluorescence for 30‒60 min, or until progression curves are not linear (Figure 3).
-
38.
Plot fluorescence measurement replicates (Y) vs. time (X). In GraphPad Prism, label each Y column with the corresponding substrate concentration, as these labels will be used for later calculations.
Optional: In case of having obtained a standard curve, transform fluorescence units into [AMC] with it. In GraphPad Prism, use “Transform” – “User-defined Y functions” and the custom function.
-
39.Calculate initial rates for each experiment by fitting data to a linear regression. In GraphPad Prism, use “Analyze” – “Nonlinear regression” ‒ “Lines” – “Straight line.”
-
a.Exclude data points at the start and end of the assay whenever they do not fit a linear trend. This ensures only the steady state is taken into account for the calculation of kinetic parameters (Stein, 2011a).
-
b.Create a summary table and associated graph with the slope ± SEM of each experiment (conversion rate, Y) and the substrate concentration (X). In GraphPad Prism, do this by checking the box “Create a summary table and graph” in the “Output” tab, select only “Slope,” and mark “Create a: XY graph with X values coming from column titles.”
-
a.
-
40.
Check that negative control experiments have slopes close to 0.
-
41.
Check that experiment replicates follow the same trend (also indicated by low slope SEM).
-
42.
Repeat steps 29 to 41.
-
43.
Prepare a new data sheet with the slopes of each experiment (Y) and the concentration of substrate (X) (Figure 3).
Optional: Divide conversion rate data (slopes) by the concentration of enzyme employed, in order to normalize data across experiments. Then, the calculated vmax will be equal to the turnover number of the enzyme (kcat). In GraphPad Prism, use “Transform Y values using Y=Y/K,” and use the concentration of enzyme as “K.”
-
44.Fit data to the Michaelis-Menten equation (Figure 3). In GraphPad Prism, use “Analyze” – “Nonlinear regression” – “Enzyme kinetics” ‒ “Michaelis-Menten.”
-
a.Create a summary table and graph with vmax and KM values. Troubleshooting 1, Troubleshooting 3
-
a.
Optional: Repeat steps 29 to 41 and plot additional data sets until the kinetic values are obtained with sufficient certainty (ideally, SEM <10% for both vmax and KM values). Alternative substrate concentration ranges might be needed in order to define the Michaelis-Menten curve accurately.
-
45.
If not done in the optional step before, calculate the enzyme kcat by dividing the vmax by the concentration of enzyme employed.
Table 3.
Example of plate layout (half plate) for determination of kinetic parameters
| (HDAC) [Substrate] | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| A | NE 200 μM |
NE 133.3 μM |
NE 88.9 μM |
NE 59.3 μM |
NE 39.5 μM |
NE 26.3 μM |
| B | NE 200 μM |
NE 133.3 μM |
NE 88.9 μM |
NE 59.3 μM |
NE 39.5 μM |
NE 26.3 μM |
| C | HDAC11 200 μM |
HDAC11 133.3 μM |
HDAC11 88.9 μM |
HDAC11 59.3 μM |
HDAC11 39.5 μM |
HDAC11 26.3 μM |
| D | HDAC11 200 μM |
HDAC11 133.3 μM |
HDAC11 88.9 μM |
HDAC11 59.3 μM |
HDAC11 39.5 μM |
HDAC11 26.3 μM |
NE, no enzyme added.
Figure 3.

Example of kinetic data and fitting to the Michaelis-Menten equation
Kinetic evaluation of (A) substrate ETDKmyr (6e) for HDAC11 (Moreno-Yruela et al., 2018) and (B) substrate LGKac (1a) for HDAC3. HDAC11 experiments were linear for up to 65 min, whereas HDAC3 only retained linearity for 25 min (progression data represent mean ± SD of 2 internal replicates). XY data sheets are created with substrate concentrations and corresponding slopes from at least 2 experiments, and data are fitted to the Michaelis-Menten equation to generate the represented curves (steady-state data represent mean ± SEM of 2 individual experiments). 1.5-fold dilutions of substrate concentration are preferred, as in B, for better definition of the curve and calculation of KM and kcat parameters. HDAC11 plot reprinted with permission from Moreno-Yruela et al. (2018).
Expected outcomes
ε-N-Acyllysine substrate screening
Enzyme‒substrate pairs included as positive controls should provide fluorescence units higher than 3 times the background (wells without enzyme). Additional hits could be converted to different extents depending on the peptide sequence (Figure 1).
Buffer optimization
In our experience, both Tris and HEPES buffers are appropriate for studying HDACs and sirtuins, and only minor differences in substrate conversion are observed between both. BSA content, however, can have a more substantial effect, and it especially interferes with long-chain acyl-lysine substrates at concentrations below 10 μM (Troubleshooting 1).
Optimization of trypsin concentration for time-course experiments
Assay progression curves should appear at a sufficient signal-to-noise ratio, as judged by the signal compared to the background and the error obtained between replicates. Ideally, fluorescence units should be at least 2-fold higher than the background after 15 min of reaction and the difference between internal replicate pairs should be smaller than the increase in fluorescence within a period of 2‒3 min. Lag times of 3‒5 min are expected at most trypsin concentrations and higher trypsin concentrations likely lead to shorter periods of linearity due to proteolysis of the HDAC enzyme (Figure 2). For a set concentration of HDAC, all assay progression curves should provide similar slopes, and any lower values would indicate non-sufficient trypsin concentration.
Determination of Michaelis-Menten kinetic parameters
The obtained conversion rates should adjust to the Michaelis-Menten equation and accurately define the turning point of the curve, in order to enable precise KM and kcat calculation. Efficiencies (kcat/KM) between 102 and 105 s−1·M−1 can be expected (Madsen and Olsen, 2012a, Moreno-Yruela et al., 2018).
Limitations
This protocol relies on trypsin for continuous release of the fluorescent probe. Therefore, any deacylase enzymes degraded rapidly by trypsin cannot be studied in this fashion. These enzymes would require discontinuous determination of substrate conversion at >2 time points, by end-point addition of trypsin (Madsen and Olsen, 2012a). In other cases, trypsin can cleave just the unstructured N- and/or C-termini of the enzyme, as it happens to HDACs 1‒3 (Moreno-Yruela et al., 2019a). This cleavage should be assessed by gel electrophoresis, and whether it affects enzyme activity can be determined by comparing the kinetic parameters obtained from this protocol to those obtained from discontinuous experiments or alternative methods without trypsin (Stein, 2011a, Moreno-Yruela et al., 2019a).
Calculation of the kcat parameter requires knowing the concentration of active enzyme in the solution, which is difficult to define, as enzyme preparations can contain some inactive or unfolded fraction of the protein. Therefore, kcat values should always be considered approximate and be used for relative comparison of substrates for a single enzyme.
Troubleshooting
Problem 1
Initial velocity data fit a sigmoidal regression rather than the Michaelis-Menten equation (Figure 4A).
Figure 4.

Deviations from Michaelis-Menten kinetics
(A) Sigmoidal dependence on substrate concentration, as obtained when BSA sequesters substrate molecules.
(B) Conversion rate drop at high substrate concentration, which indicates substrate inhibition.
Potential solution
Sigmoidal dependence of the initial rates on substrate concentration can indicate positive cooperativity, where binding of one substrate molecule to the enzyme promotes binding of an additional molecule (Stein, 2011b). This is common for homodimeric enzymes, but unlikely for HDACs and sirtuins. In our case, we found this effect to appear with substrates featuring a long-chain acyl modification when a concentration of 0.5 mg/mL of BSA was present in the mixture. Since BSA can bind fatty acids, we consider BSA could sequester substrate molecules from the solution and lower the effective substrate concentration. This effect can be detected when substrate concentrations lower than 10 μM are tested, since the apparent conversion rates are lower than those expected from Michaelis-Menten behavior (Figure 4A). Thus, the problem could be solved using a lower concentration of BSA (for example, 0.05 mg/mL) or even removing BSA completely from the mixture (Galleano et al., 2016, Moreno-Yruela et al., 2018). However, removing BSA could lead to alternative problems such as a higher uncertainty in the concentration of enzyme, because BSA is used to block unspecific protein binding to surfaces in assay tubes and plates.
Problem 2
Assay progression curves are linear for less than 15 min, even at low concentrations of trypsin.
Potential solution
Loss of linearity during the assay could be due to excessive substrate consumption, product inhibition, or to lack of enzyme stability. Each point should be addressed by different means. First, verification that substrate conversion has not exceeded 10% of the initial concentration and testing the enzyme at a lower concentration would exclude a decrease in conversion rate due to a decrease in substrate concentration. If product inhibition is suspected, perform inhibition experiments using the carboxylic acid formed as product of the enzymatic reaction. Alternatively, some deacylase enzymes have limited stability in buffer. In those cases, perform a more extensive buffer screen including, for example, polyethylene glycol (PEG), which has been found to stabilize HDAC8 and provide extended activity over time (10% PEG8kDa). Additionally, the temperature of the experiment could be lowered. Trypsin digestion of the deacylase enzyme could also be responsible for the limited stability and this could be checked by gel electrophoresis of the enzyme before and after the time-course experiment.
Problem 3
Initial velocity data drop at high concentrations of substrate (Figure 4B).
Potential solution
Certain enzyme‒substrate complexes can be stabilized by an additional molecule of substrate, leading to enzyme inhibition (Stein, 2011b). In this case, fitting the data to the appropriate equation permits the calculation of vmax, KM and the dissociation constant of the two molecules of substrate to inhibit the enzyme (Ki). In GraphPad Prism, find this equation using “Analyze” – “Nonlinear regression” – “Enzyme kinetics” ‒ “Substrate inhibition.”
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christian A. Olsen (cao@sund.ku.dk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Original data for screening results and HDAC11 kinetic parameters in the paper are available in Moreno-Yruela et al., 2018.
Acknowledgments
The authors thank Dr. Andreas S. Madsen, Dr. Iacopo Galleano, and other members of the Olsen lab for helping develop and optimize the current protocol. This work was supported by the European Research Council (ERC-CoG-725172 – SIRFUNCT; C.A.O.) and the Lundbeck Foundation (running cost grant R289-2018-2074; C.A.O.).
Author contributions
Conceptualization, C.M.-Y. and C.A.O.; methodology, C.M.-Y.; investigation, C.M.-Y.; writing ‒ original draft, C.M.Y.; writing ‒ review & editing, C.M.-Y. and C.A.O.; visualization, C.M.-Y.; supervision, C.A.O.; project administration, C.A.O.; funding acquisition, C.A.O.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Carlos Moreno-Yruela, Email: carlos.myruela@sund.ku.dk.
Christian A. Olsen, Email: cao@sund.ku.dk.
References
- Anderson K.A., Huynh F.K., Fisher-Wellman K., Stuart J.D., Peterson B.S., Douros J.D., Wagner G.R., Thompson J.W., Madsen A.S., Green M.F. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 2017;25:838–855. doi: 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner J.E., West N., Grachan M.L., Greenberg E.F., Haggarty S.J., Warnow T., Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010;6:238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang Y.L., Lin H. An improved fluorogenic assay for SIRT1, SIRT2, and SIRT3. Org. Biomol. Chem. 2016;14:2186–2190. doi: 10.1039/c5ob02609a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galleano I., Schiedel M., Jung M., Madsen A.S., Olsen C.A. A continuous, fluorogenic sirtuin 2 deacylase assay: substrate screening and inhibitor evaluation. J. Med. Chem. 2016;59:1021–1031. doi: 10.1021/acs.jmedchem.5b01532. [DOI] [PubMed] [Google Scholar]
- Madsen A.S., Andersen C., Daoud M., Anderson K.A., Laursen J.S., Chakladar S., Huynh F.K., Colaço A.R., Backos D.S., Fristrup P. Investigating the sensitivity of NAD+-dependent sirtuin deacylation activities to NADH. J. Biol. Chem. 2016;291:7128–7141. doi: 10.1074/jbc.M115.668699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen A.S., Olsen C.A. Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angew. Chem. Int. Ed. Engl. 2012;51:9083–9087. doi: 10.1002/anie.201203754. [DOI] [PubMed] [Google Scholar]
- Madsen A.S., Olsen C.A. Substrates for efficient fluorometric screening employing the NAD-dependent sirtuin 5 lysine deacylase (KDAC) enzyme. J. Med. Chem. 2012;55:5582–5590. doi: 10.1021/jm300526r. [DOI] [PubMed] [Google Scholar]
- Marks P.A., Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- Moreno-Yruela C., Fass D.M., Cheng C., Herz J., Olsen C.A., Haggarty S.J. Kinetic tuning of HDAC inhibitors affords potent inducers of progranulin expression. ACS Chem. Neurosci. 2019;10:3769–3777. doi: 10.1021/acschemneuro.9b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Yruela C., Galleano I., Madsen A.S., Olsen C.A. Histone deacetylase 11 is an ε-N-myristoyllysine hydrolase. Cell Chem. Biol. 2018;25:849–856. doi: 10.1016/j.chembiol.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Moreno-Yruela C., Madsen A.S., Olsen C.A. Kinetic characterization of inhibitors of histone deacetylases (HDACs) and sirtuins. Protocol Exchange. 2019 doi: 10.21203/rs.2.13042/v1. [DOI] [Google Scholar]
- Stein R.L. First Edition. John Wiley & Sons; 2011. Kinetics of single-substrate enzymatic reactions. Kinetics of Enzyme Action: Essential Principles for Drug Hunters. [Google Scholar]
- Stein R.L. First Edition. John Wiley & Sons; 2011. Kinetics of single-substrate enzyme reactions: special topics. Kinetics of Enzyme Action: Essential Principles for Drug Hunters. [Google Scholar]
- Wegener D., Wirsching F., Riester D., Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol. 2003;10:61–68. doi: 10.1016/s1074-5521(02)00305-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data for screening results and HDAC11 kinetic parameters in the paper are available in Moreno-Yruela et al., 2018.