

Abstract
Background
Chemotherapy is the first line of treatment for cancer patients. However, the side effects cause severe muscle atrophy or chemotherapy‐induced cachexia. Previously, the NF‐κB/MuRF1‐dependent pathway was shown to induce chemotherapy‐induced cachexia. We hypothesized that acute collateral toxic effects of chemotherapy on muscles might involve other unknown pathways promoting chemotherapy‐induced muscle atrophy. In this study, we investigated differential effects of chemotherapeutic drugs and probed whether alternative molecular mechanisms lead to cachexia.
Methods
We employed mouse satellite stem cell‐derived primary muscle cells and mouse C2C12 progenitor cell‐derived differentiated myotubes as model systems to test the effect of drugs. The widely used chemotherapeutic drugs, such as daunorubicin (Daun), etoposide (Etop), and cytarabine (Ara‐C), were tested. Molecular mechanisms by which drug affects the muscle cell organization at epigenetic, transcriptional, and protein levels were measured by employing chromatin immunoprecipitations, endogenous gene expression profiling, co‐immunoprecipitation, complementation assays, and confocal microscopy. Myotube function was examined using the electrical stimulation of myotubes to monitor contractile ability (excitation–contraction coupling) post drug treatment.
Results
Here, we demonstrate that chemotherapeutic drugs disrupt sarcomere organization and thereby the contractile ability of skeletal muscle cells. The sarcomere disorganization results from severe loss of molecular motor protein MyHC‐II upon drug treatment. We identified that drugs impede chromatin targeting of SETD7 histone methyltransferase and disrupt association and synergetic function of SETD7 with p300 histone acetyltransferase. The compromised transcriptional activity of histone methyltransferase and acetyltransferase causes reduced histone acetylation and low occupancy of active RNA polymerase II on MyHC‐II, promoting drastic down‐regulation of MyHC‐II expression (~3.6‐fold and ~4.5‐fold reduction of MyHC‐IId mRNA levels in Daun and Etop treatment, respectively. P < 0.0001). For MyHC‐IIa, gene expression was down‐regulated by ~2.6‐fold and ~4.5‐fold in Daun and Etop treatment, respectively (P < 0.0001). Very interestingly, the drugs destabilize SUMO deconjugase SENP3. Reduction in SENP3 protein level leads to deregulation of SETD7–p300 function. Importantly, we identified that SUMO deconjugation independent role of SENP3 regulates SETD7–p300 functional axis.
Conclusions
The results show that the drugs critically alter SENP3‐dependent synergistic action of histone‐modifying enzymes in muscle cells. Collectively, we defined a unique epigenetic mechanism targeted by distinct chemotherapeutic drugs, triggering chemotherapy‐induced cachexia.
Keywords: Chemotherapy‐induced cachexia, Epigenetics, Muscle atrophy, p300, Sarcomere organization, SENP3, SETD7
Introduction
Mobility is one of the defined features of the animals, indispensable for survivability and biological success. Skeletal muscles—constitute ~40% 1 of human body mass—generate force that powers movements through interactions between thick (actin) and thin (myosin) filaments. The individual muscle cells are comprising of several myofibrils, which house linear arrays of the micrometre‐sized multimolecular structures called ‘sarcomere’—the fundamental contractile unit of striated skeletal and cardiac muscles. 2 , 3 The ATP‐driven cyclical interaction between the actin and myosin filaments within the sarcomeres causes muscle shortening, generating the force to drive movements at the molecular as well as organismic level. Precise assembly and accurate functioning of the sarcomeric proteins is prerequisite for the correct contractile function of the muscle. Deregulation of precise assembly or dysfunction of the contractile proteins can lead to altered force generation and consequently defective contractile function due to sarcomere disarray—a feature associated with myopathies and muscle wasting. 4 , 5 One striking example of sarcomere disarray and muscle atrophy is associated with cachexia prevalent in cancer patients. The prominent feature of cancer cachexia is severe loss of body mass due to muscle atrophy. Cancer cachexia is a debilitating trait prevalent in more than 50% of cancer patients and a leading cause of death in nearly 30% of patients. 6 , 7 , 8 , 9 , 10
For the past few decades, standard chemotherapy—including combinations of nucleoside analogue cytarabine (Ara‐C), anthracycline daunorubicin (Daun; variant of doxorubicin), and VP16 etoposide (Etop)—has remained the widely used and effective therapies available for treating various human cancers such as leukaemia and breast cancers. 11 , 12 , 13 Interestingly, few recent reports revealed that apart from targeting cancers, chemotherapeutic drugs induce cachexia. 7 , 14 , 15 Thus, in addition to cancer‐induced cachexia, chemotherapy‐induced cachexia exacerbates the condition, leading to profound loss of muscle mass and function. 16 Chemotherapeutic agents, including Ara‐C, Daun, and Etop‐induced oxidative stress, have been shown as a possible cause of various side effects, including muscle wasting. 17 , 18 Moreover, chemotherapeutic drug Daun was also reported to cause muscle atrophy by activating Atrogin/MuRF1‐dependent ubiquitin–proteasome pathway. 19 On the basis of these studies, we hypothesize that the intense collateral toxic effect, particularly on muscles, may involve other unknown mechanisms promoting chemotherapy‐induced muscle atrophy.
In this study, we showed chemotherapeutic drugs Daun and Etop target small ubiquitin‐like modifier (SUMO) isopeptidase SENP3 and perturb contractile function of muscle. Post‐translational modification of proteins by the evolutionary conserved SUMO proteins through SUMOylation pathway has emerged as a key regulatory switch controlling ~18% of total human proteome. 20 , 21 , 22 , 23 The regulation by SUMOylation mediates almost every aspects of cell function including early mammalian developmental stages, adult life, and cellular stress response. 24 , 25 , 26 , 27 , 28 , 29 , 30 Three major SUMO isoforms (SUMO1 and highly related SUMO2/3) express in mammalian cell. Covalent addition of the C terminus of SUMO moiety to ε‐acceptor lysine of target proteins is important to alter protein assemblies, localization, stability, and their function in a dynamic and reversible manner. Deconjugation/deSUMOylation of SUMO moiety from target proteins by the SUMO‐specific isopeptidases (SENP) contributes to the reversible and dynamic features of this pathway. The highly dynamic and reversible nature of SUMOylation is partly due to the rapid deSUMOylation role of SENPs. Hence, the SENPs serve as the important determinants of SUMOylation process. 31 , 32 , 33 The most studied class, i.e. ULP/SENP class (SENP1, 2, 3, 5, 6, and 7), a group of cysteine proteases, contains conserved catalytic domain and display distinct subcellular localizations, substrate specificities, and functions. For instance, SENP3, which is predominantly localized to the nucleolus, preferentially cleaves SUMO2/3‐conjugated substrate proteins. Earlier studies established a major role of SENP3 in ribosome maturation. 34 , 35 , 36 In addition, nuclear SENP3 function was also reported to be involved in the regulation of various classes of genes by regulating distinct histone methyltransferase complex such as MLL1/2 and SETD7 complexes. 37 , 38 One common theme that emerged from all these studies is the importance of catalytic site‐dependent function of SENP3, controlling SUMOylation status of SENP3's interaction partners. Whether SENP3 plays additional roles apart from its well‐characterized SUMO deconjugation activities is not clear. Recently, SENP3 was reported as a key regulator of SETD7‐driven transcriptional programme crucial for correct sarcomere assembly and contractile function of skeletal muscle cells. The SENP3–SETD7 transcriptional regulation was disrupted when muscle atrophy was induced by pro‐inflammatory cytokines. 38 A potential connection of SENP3‐regulated mechanisms with chemotherapy‐induced cachexia however is unknown.
Materials and methods
Satellite stem cell (satellite cells) extraction, culture, and differentiation
Thirty‐day‐old male and female wild‐type C57BL/6J mice were used for the experiments. All the experimental procedures were performed under the ethical approval of the Italian Ministry of Health and the Institutional Animal Care and Use Committee (authorization no. 83/2019‐PR). The animals were maintained in an authorized facility at San Raffaele Hospital, Milan (authorization no. N. 127/2012‐A). Hindlimb muscles were isolated from sacrificed mice and digested 90 min in 2.4 U/mL of Dispase II (Roche, S.p.A., Monza, Italy, 04942078001), 2 ug/mL of Collagenase A (Roche, 1013586001), 0.2 mM CaCl2 (Sigma, Merck Life Science S.r.L, Milan, Italy, C5670), 4 mM MgCl2 (Sigma, M8266), and 10 ng/mL DNase I (Roche, 1014159001) in PBS1X (Euroclone, S.p.A., Pero, Italy, ECB4004L) at 37°C in a moving water bath. The sample were resuspended in Hank's balanced salt solution (Gibco, Thermo Fisher Scientific, Monza, Italy, 14025‐050) supplemented with 0.1% bovine serum albumin (Sigma, A7030). Cell suspension was serially filtered with 70 (Falcon, VWR International S.r.L., Milan, Italy, 352350) and 40 μm filters (Falcon, 352340) and stained with antibodies: PB‐CD45 1:50 (eBioscience, Thermo Fisher Scientific, Monza, Italy, 48‐0451), PB‐CD31 1:50 (eBioscience, 48‐0311), PB‐Ter119 1:50 (eBioscience, 48‐5921), FITC‐Sca1 1:50 (eBioscience, 115981), and APC‐α7integrin 1:200 (AbLab, UBC antibody lab, Vancouver, Canada, 67‐001‐05) and sorted with BD FACS ARIA III SORP for PB‐CD45−/PB‐CD31−/PB‐Ter119−/FITC‐Sca1−/APC‐α7integrin+. The satellite cells obtained in this way were plated on precoated 6 well dishes (Matrigel −1 mg/mL, Corning, Merck Life Science S.r.L., Milan, Italy, cat no. 354234) and allowed to grow in Dulbecco's modified Eagle's medium (DMEM) (Gibco, 10569‐010) supplemented with 20% fetal bovine serum (Corning, 35‐015‐CV), 10% horse serum (HS) (Gibco, 26050‐080), 1% chicken embryo extract (Seralab, CE650‐DL), 1% penicillin/streptomycin (Euroclone, ECB 3001), and 2.5 ng/mL bFGF (Gibco, 13256029) for 4 days, changing medium every 2 days. After the cells started differentiating, they were grown further for 2 more days in DMEM supplemented with 5% HS and 1% penicillin/streptomycin until complete differentiation into myotubes.
Satellite cells treatment
Differentiated myotubes were treated for 10–12 h with Daunorubicin hydrochloride (Sigma‐Merck, Merck Life Science S.r.L., Milan, Italy, 30450‐5MG) to a final concentration of 1 μM in water, always cultured in DMEM supplemented with 5% HS and 1% penicillin/streptomycin. At the end of the treatment, all the myotubes were collected for further applications.
Cell culture and drug treatment
Undifferentiated C2C12 myoblasts were cultured using growth media (DMEM with 20% fetal bovine serum). At nearly 90% confluency, cells were differentiated with media containing 2% HS to generate mature myotubes. The myotubes (after 6 days of differentiation) were treated with antineoplastic drugs Ara‐C (C1768, Sigma‐Aldrich, Taufkirchen, Germany), Daun (23541‐50‐6, Sigma‐Aldrich), and Etop (E1383, Sigma‐Aldrich), respectively. The details of final doses and incubation times are indicated in the figure legends.
Immunofluorescence and confocal microscopy
The myotubes grown and treated on the coverslips were fixed with paraformaldehyde for 15 min at room temperature in dark condition followed by cell permeabilization with 0.5% Triton X‐100 in phosphate‐buffered saline (PBS). The coverslips were then processed using standard fluorescence staining protocols. Slides were scanned, and images were captured using Olympus FV1000 (Olympus Deutschland GmbH, Hamburg, Germany) confocal microscope. Details of antibodies used in immunofluorescence experiments are indicated in the figure legends.
Contractility assay of C2C12 myotubes
Myoblasts grown on the laminin‐coated coverslips were differentiated for 6 days. On the 6th day, antineoplastic drugs and respective drug controls were added; 10–12 h after drug treatment, coverslips were placed in a custom‐made chamber (~200 μL volume) consisting of a plastic ring placed on a glass for electrical stimulation. This set‐up was connected to a (i) thermistor probe (Green Leaf Scientific, Dublin, Ireland), temperature controller (TC‐II, micro‐temperature controller, Cell MicroControls, Norfolk, VA 23517, USA), to maintain the temperature at 37°C and to (ii) an electric field stimulator (MyoPacer Field Stimulator, IonOptix Corp, Amsterdam, Netherlands) for delivering transistor–transistor logic impulses (4 ms duration, 1 Hz frequency). Bright‐field microscope (Olympus IX51, Olympus Deutschland GmbH, Hamburg, Germany) fitted with a high‐speed camera (optiMOS, QImaging) was used to record cell's response to electrical stimulation. Videos were processed and analysed using ImageJ software 39 using standard analysis functions.
Quantitative reverse transcription polymerase chain reaction
Total RNA was isolated by using high Pure RNA isolation kit (Roche), followed by the cDNA synthesis (using 700 ng total RNA) using Transcriptor 1st strand cDNA synthesis kit (Roche). The quantitative PCR (qPCR) assays were performed with SYBR green PCR master mix kit (Cat no. 4309155; Thermo Fisher Scientific GmbH, Schwerte, Germany) and qPCR machine (Quant Studio 6 Flex Real‐Time PCR System, Thermo Fisher Scientific). The expression of housekeeping gene GAPDH was used. Quantification of mRNA expression was performed using ΔΔCT method. To check the effect of drug on sarcomere‐related gene expression, we employed the RT2 Profiler PCR array system (cat no. PAMM‐099Z, Qiagen, Hilden, Germany) according to the manufacturer's instruction. The primer pairs were used for quantitative reverse transcription polymerase chain reaction (RT‐qPCR) to amplify cDNA, which are mentioned in the Supporting Information.
Immunoprecipitation and chromatin immunoprecipitation
Approximately 10 million cells (myotubes) were lysed in a buffer containing 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% TritonX‐100 with freshly added protease and phosphatase inhibitors (Complete/PhosphoSTOP, Roche), and 10 mM NEM (SENP inhibitor). Immunoprecipitation (IP) was performed by incubating the cell lysate with indicated antibodies (detail in figure legends) for overnight (~12 h); protein–antibody complexes were captured by protein G dynabeads (Invitrogen) for ~1 h at cold room. This was followed by 3× wash with 1:4 diluted lysis buffers. The immunoprecipitates (IPs) were eluted by boiling the beads with SDS‐PAGE loading buffer. Elute was loaded on SDS‐PAGE and analysed by western blots. Chromatin immunoprecipitation (ChIP) assays were performed according to the published method38. Formaldehyde cross‐linking of myotubes at a final concentration of 1.47% was performed for 10 min at room temperature. To quench cross‐linking, 125 mM glycine was added and incubated for 5 min at room temperature. This was followed by washing the cells twice with PBS and lysed in 1 mL of ChIP lysis buffer containing 150 mM NaCl, 50 mM Tris‐HCl (pH 7.5), 5 mM EDTA, NP‐40 (0.5% vol/vol), Triton X‐100 (1.0% vol/vol), and freshly added protease inhibitor, phosphatase inhibitor cocktail (Roche). Sonification (15 pulses—1 s ON and 1 s with 45% amplitude and 6 rounds of pulses) was performed with VCX 130 Vibra Cell sonoficator (Sonics & Materials, Inc., Newtown, USA). Chromatin was then isolated by centrifugation at 1647 g for 15 min at 4°C; 2% chromatin was kept as an input control, and overnight incubation of 4 μg of specific antibodies was performed with the remaining sample. Protein G dynabead was added and incubated at cold for 1 h to capture the immunoprecipitated chromatin complex. Beads were then washed twice with normal ChIP lysis buffer, thrice with ChIP lysis buffer with 500 mM NaCl, twice washes again with normal ChIP lysis, and the final wash was with cold PBS. Reverse‐cross‐linking and DNA purification was done by adding 100 μL of 10% (wt/vol) Chelex‐100 slurry to the washed beads followed by vigorous vortexing and boiling at 95°C for 10 min and thereafter centrifugation at 1647 g for 1 min of 4°C. After real‐time PCR of ChIPed DNA, data calculation was performed by regularly used percentage of input chromatin method 38 to analyse ChIP‐qPCR data. Typically, the average of at least three technical replicate of raw percentage of input values was considered for a particular experiment. Data were plotted as an average of three biological replicates (unless mentioned otherwise in the corresponding legend) with ± SEM. The primer pairs were used for ChIP‐qPCR to amplify ChIPed DNA, which are mentioned in the Supporting Information.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism 5 (GraphPad software, Inc., USA). Data are plotted from the average (± SEM) of at least three independent experiments with triplicates (unless stated otherwise). Stars indicate statistically significant difference calculated with unpaired t‐test. Detailed descriptions of statistical analysis and P‐values are described in the corresponding figure legends (wherever applicable).
Results
Chemotherapeutic drugs disrupt contractile ability of sarcomere
We set out to investigate the effect of chemotherapeutic drugs (Ara‐C, Daun, and Etop) on the primary function of muscle, i.e. the ability of contraction. For this purpose, the progenitor C2C12 myoblast cells were differentiated for 6 days into mature muscle cells with myofibrils. The mature myotubes were placed under electric stimulation to monitor their contractile ability. With the MyoPacer® EP Cell Stimulator (IonOptix Corp.) device, 4 ms electric pulse of 1 Hz (1 pulse/s) was applied. In this set‐up, we observed that control cells responded to the electric stimulation by exhibiting synchronized contraction (Supporting Information, Movie S1 and Supporting Information, Figure S1A), which is the functional read‐out of actin–myosin cross‐bridge cycling through the excitation–contraction coupling process. 40 The myotubes with Daun and Etop treatment resulted in compromised contraction ability (Supporting Information, Movies S3–S4 and Figure S1A). The Ara‐C treated cells however displayed comparable contractile ability to the control cells ( Movies S1–S2 and Figure S1A). These results suggested drug‐specific effects on muscle function. Noteworthy, the concentration of drugs used in our experiments is within the recommended dose established earlier. We have also tested lower doses in our study; for instance, we used 1 μM Daun instead of 10 μM Daun used elsewhere. 12 , 41 , 42 , 43 The observed defective contraction presented here is with a lower Daun concentration ( Movies S3–S4 and Figure S1A).
The contractile ability of muscle cells is a direct consequence of sarcomere protein assembly and function; thus, we argued that the chemotherapeutic drugs might interfere with the assembly of sarcomere proteins. alpha‐Actinin, a major component and a prominent marker of the Z‐disc of sarcomere, organizes large macromolecular protein complexes and provides structural stability necessary for correct sarcomere organization. 44 Because chemotherapeutic agents earlier have shown to induce protein degradation, we probed if the drug treatment led to degradation of alpha‐actinin. Figure 1A shows no significant changes in alpha‐actinin protein levels after Ara‐C, Daun, and Etop treatment. We furthermore checked the sarcomere organization in drug‐treated muscle cells by immunostaining the cells with an antibody specific to the alpha‐actinin to mark the Z‐discs. As shown in Figures 1B and S1B, control cells showed well‐resolved typical striated pattern of sarcomeres. Interestingly, both Daun and Etop treatment resulted insevere disorganization of sarcomereic pattern. Ara‐C treatment—which did not significantly change contractile property of myotubes—did not have major defect in sarcomere organization. These observations indicated that (i) chemotherapy‐induced cachexia severely compromised sarcomere structures and consequently muscle cell contractile function and (ii) the effect of chemotherapy on muscle function is drug specific.
Figure 1.
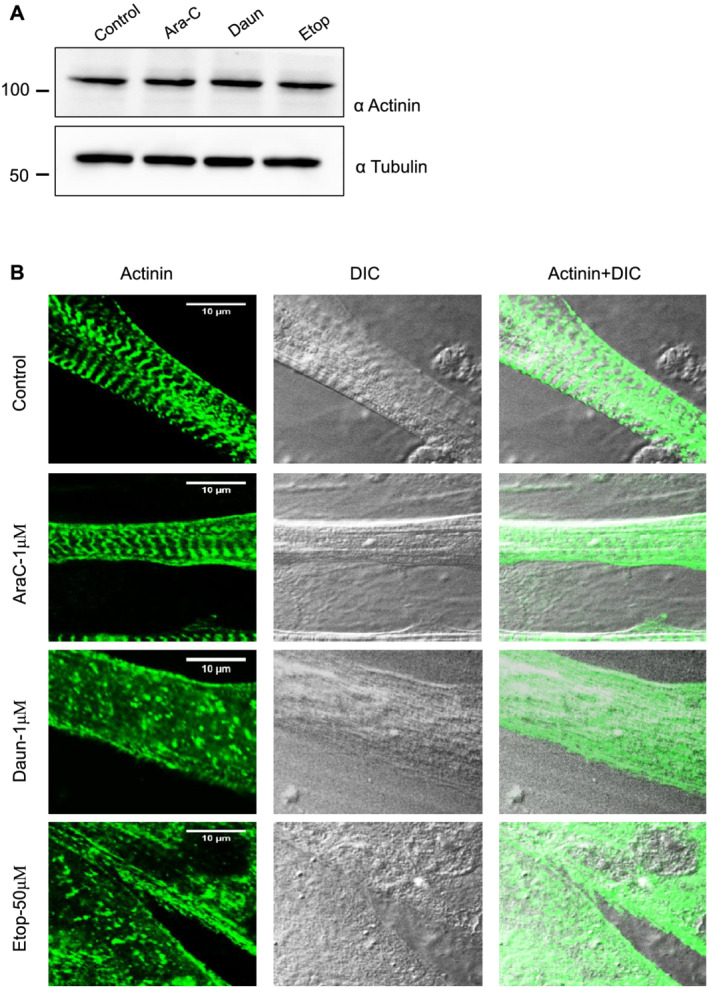
Chemotherapeutic drugs daunorubicin (Daun) and etoposide (Etop) disrupt sarcomere organization. (A) Progenitor myoblasts were differentiated for 6 days and followed by overnight treatment of myotubes with indicated chemotherapeutic drugs [1 μM cytarabine (Ara‐C), 1 μM Daun, and 50 μM Etop, respectively] overnight. Western blot shows status of actinin expression status after drug treatment. A representative of three biological experiments. (B) Confocal images showing differentiated myotubes with and without drug treatment. Myotubes were probed with an antibody against alpha‐actinin indicating sarcomeric Z‐disc striation of a fully matured myotubes in control cells. Scale bar—10 μm. DIC, differential interference contrast microscopy.
Chemotherapeutic drugs alter transcription status of sarcomeric genes
Previous studies have reported the direct association of the drugs Daun and Etop with DNA. 11 We therefore asked whether these agents may influence the transcriptional process. Because the drugs affected sarcomere structure and thereby function, we employed a skeletal muscle‐specific gene expression array (RT2 Profiler PCR array system, cat no. PAMM‐099Z, Qiagen) comprising 84 different genes important for skeletal muscle function. Next, we differentiated myoblasts for 6 days into matured myotubes and performed overnight Daun (1 μM) treatment. Thorough analysis of the gene expression array showed that compared with the control treatment, addition of Daun resulted in no significant changes in 71 genes. Out of the 84 genes, expressions of 13 genes were deregulated (Figure 2A and Supporting Information, Figure S2). The deregulated genes could be attributed to two main categories, namely, the genes positively responsible for sarcomere functions and the genes known to be responsible for inducing muscle atrophy in cachexia. Very interestingly, there was an inverse relationship of expression pattern between these two groups (Figure 2B). Daun treatment up‐regulated genes, i.e. Nos2, Pdk4, Igbp3, Mapk14, Trim63, Mstn, and Mmp3, that are responsible for muscle atrophy and down‐regulated genes Myh1, Myh2, Myod1, Myog, Pax7, and Igf1 that are necessary for proper muscle growth and function. The expression of other sarcomeric contractile genes such as Actinin (Actn3), Titin (Ttn), Myotilin (Myot), Nebulin (Neb), and Troponin (Tnnc1, Tnni2, Tnnt1, and Tnnt3) remained unchanged. In our screen, we found myosin heavy chain gene Myh1 (MyHC‐IId) as the strongest down‐regulated (~17.32‐fold) gene (Figure S2A). MyHC‐IId encodes for the force‐generating molecular motor protein myosin II (MyHC‐IId). As Daun most strongly affected MyHC‐IId, we focused our further studies to investigate in detail how cancer chemotherapeutics influence muscle function by specifically regulating MyHC‐IId. The other strongly down‐regulated gene was Myh2 or MyHC‐IIa (approximately seven‐fold). Therefore, we also monitored MyHC‐IIa gene.
Figure 2.
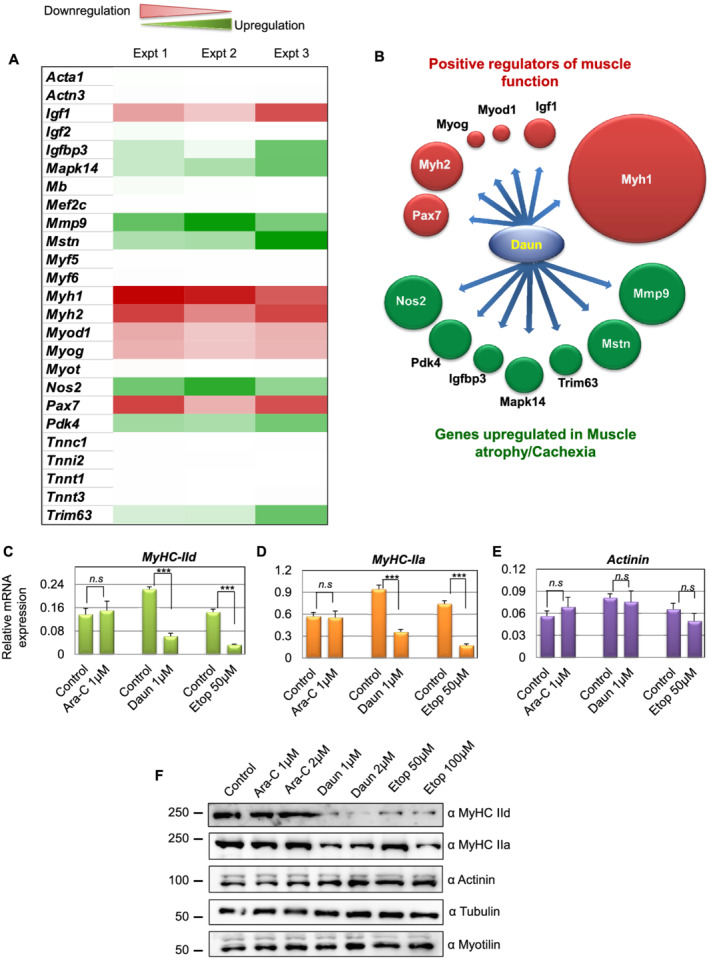
Chemotherapeutic drugs daunorubicin (Daun) and etoposide (Etop) alter gene expression of key muscle‐specific gene, specifically the Myh1 (MyHC‐IId). (A) Differentiated myotubes were treated overnight with either control or 1 μM Daun, followed by isolation of total RNA, the cDNA preparation, and RT‐qPCR to quantify relative changes of gene expression (as indicated in the figure) in the skeletal muscle‐specific gene expression arrays. ΔΔCT method was used for data analysis. Data normalization was performed against five different housekeeping genes. The heat map illustrates extent of changes in gene expressions compared with housekeeping genes. Data from three independent experiments are presented. Red colour represents down‐regulation, and green colour shows up‐regulation of corresponding genes. (B) Schematic representation of two distinct functionally opposite clusters of regulators. The size of the spheres corresponds to the level of up‐regulation and down‐regulation relative to the highest affected gene expression, i.e. Myh1. After Daun treatment, genes necessary for muscle cell growth and function were down‐regulated (green spheres), and the genes promoting muscle atrophy were up‐regulated (red spheres). (C–E) Quantitative reverse transcription polymerase chain reaction results indicating mRNA expression of (C) Myh1, (D) Myh2, and (E) Actinin tested with independent primers. The cells were treated overnight with indicated drugs, followed by mRNA isolation and cDNA synthesis. The data represent average (± SEM) from three biological experiments with technical duplicates in each experiment. The stars indicate statistically significant changes in the expression levels, unpaired t‐test, P < 0.0001. n.s, not significant. (F) Representative western blot showing changes in expression of indicated protein levels upon cytarabine (Ara‐C), Daun, and Etop treatment. Number of experiments = 3.
Next, we validated the gene expression screening by performing independent RT‐qPCRs with different qPCR primer. We observed ~3.6‐fold and ~4.5‐fold down‐regulation of MyHC‐IId mRNA in Daun and Etop treatment, respectively (Figure 2C–E). The MyHC‐IIa (Myh2) was down‐regulated by ~2.6‐fold and ~4.4‐fold, respectively. The drug treatment had no significant effect on actinin mRNA expression. Interestingly, the Ara‐C treatment barely had any effect on MyHC‐IIa, MyHC‐IId, and actinin expression. We additionally examined the affected gene products at protein level. As shown in Figure 2F, consistent with our observations at mRNA level, we observed strong reduction in MyHC‐IId levels but not other sarcomeric proteins—such as myotilin and actinin (Figure 2F) as a result of drug treatment. A dose dependent effect of drug on MyHC IIa protein level is also seen following Etop treatment (Figures 2F and Figure S2C). Again, the Ara‐C treatment showed no noticeable effect on the expression of MyHC‐IId and MyHC‐IIa. Taken together, chemotherapeutic drugs Daun and Etop specifically targeted major molecular motor protein, Myosin II. Furthermore, these observations strongly indicate drug‐specific influence on muscle function, conceivably through different modulatory mechanisms.
Chromatin targeting of SETD7 is perturbed by daunorubicin and etoposide
To uncover the mechanism whereby the chemotherapeutic drugs regulate transcriptional output of MyHC‐IId, we mainly focused our attention to the Daun and Etop‐driven effects, as our earlier results showed no significant influence of Ara‐C in this context.
Histone monomethyltransferase, SETD7, is an important determinant for skeletal myofibril assembly, which critically regulates MyHC‐IId gene expression. 38 , 45 We reasoned whether down‐regulation of MyHC‐IId transcription was due to degradation of SETD7 protein upon drug treatment. As shown in Figure 3A, SETD7 protein level remained nearly unchanged after Daun and Etop treatment.
Figure 3.
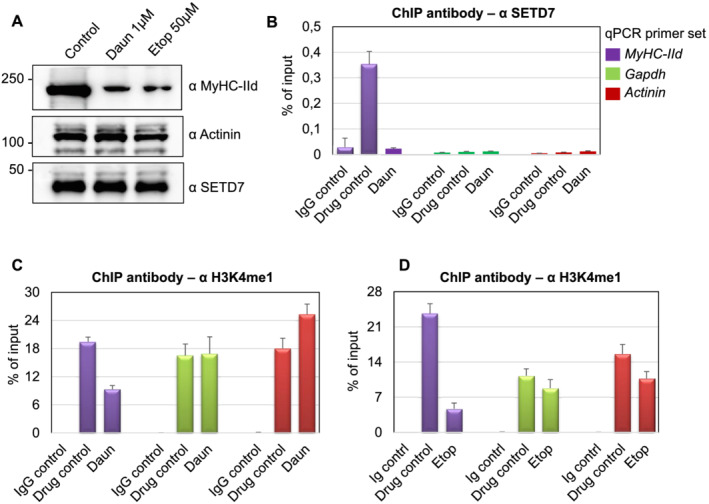
Daunorubicin (Daun) and etoposide (Etop) treatment perturbed SETD7 recruitment and H3K4me1 level on MyHC‐IId gene. (A) Western blot showing indicated changes in protein expression status upon Daun and Etop treatment. (B) Chromatin immunoprecipitation (ChIP) assays were performed with the specified antibodies. Coloured boxes in the inset represent primer pairs of the corresponding gene promoter used to amplify ChIPed DNA template by quantitative polymerase chain reaction (qPCR). Individual bars show the status of SETD7 chromatin occupancy on indicated genes after Daun treatment. Data represented as an average of at least three independent experiments with triplicates used in each experiment. (C) Same as in (B), except anti‐rabbit H3K4me1 antibody was used for ChIP to measure H3K4me1 marks on the indicated gene after Daun treatment. (D) Same as in (C), except ChIP assays were performed with Etop‐treated myotubes.
Because correct recruitment of epigenetic regulators such as SETD7 to its target genes is crucial mechanisms that govern histone methyltransferase function and gene expression programme, we, therefore, checked the presence of SETD7 on chromatin. We optimized ChIP assays and designed primers to measure chromatin associated events on MyHC‐IId promoter as the drugs specifically targeted MyHC‐IId gene expression. We used Actinin and gapdh as control genes as their expression remained unperturbed following the drug treatment. Myotubes differentiated for 6 days were treated with Daun and Etop for 10–12 h. The treated cells, fixed in formaldehyde, were used for chromatin isolation. ChIPs were performed with an antibody directed against SETD7 or a non‐specific IgG as a control. The real‐time qPCR experiments using ChIP‐enriched DNA templates showed that in comparison with the IgG control, SETD7 was specifically enriched (~12‐fold) on MyHC‐IId promoter. Daun treatment strongly reduced (~11‐fold) the association of SETD7 on chromatin (Figure 3B), whereas the control genes did not show any detectable alterations. Similar to Daun treatment, Etop treatment also showed significant reduction of SETD7 recruitment on MyHC‐IId promoter (Supporting Information, Figure S3).
SETD7 adds a monomethyl group on H3K4 (H3K4me1) and is reported to be important for myogenic differentiation and proper myofibrils formation through up‐regulating transcription of mainly MyHC‐II genes. 45 , 46 Because the drugs disrupted targeting of SETD7 on Myosin II genes, we investigated the downstream signalling (i.e. histone modifications) of this event in details. We examined if drug treatment‐mediated reduction of SETD7 from chromatin results into spatial reduction of SETD7 enzymatic activities. By using anti‐H3K4me1 antibody in ChIP assays, we detected strong presence of H3K4me1 epigenetic marks (~20–24‐fold) on MyHC‐IId in control cells. Treatment of myotubes with Daun and Etop however reduced H3K4me1 marks from chromatin by approximately two‐fold to five‐fold, respectively (Figure 3C,D). Actinin and Gapdh genes also showed a strong enrichment of H3K4me1 marks; however, upon drug treatment, no significant alteration of H3K4me1 was detected on these genes.
Together, these ChIP results suggested that Daun and Etop treatment resulted in alteration of epigenetic changes (H3K4me1 marks) through reduction of SETD7 chromatin association specifically on MyHC‐IId.
Daunorubicin and etoposide alter histone acetylation level on MyHC‐IId
Histone acetylation plays a key role in positively regulating gene expression during myofibril formation. Besides, SETD7‐mediated H3K4me1‐modified chromatin is primed for histone acetylation. 47 Therefore, we wondered if Daun and Etop could further interfere with histone acetylation. Very interestingly, our western blots revealed a temporal expression pattern of the major histone acetyltransferase p300 during myogenic differentiation. The p300 level increased during the course of differentiation, similar to that of SETD7 histone methyltransferase and other well‐established myogenic differentiation markers such as MyHC‐IId and actinin proteins (Figure 4A). These results suggested that (i) together with SETD7, p300 might also have a predominant role in MyHC‐IId gene expression and (ii) the chemotherapeutic drugs might target possible synergetic action of SETD7 and p300, presumably required for proper MyHC‐IId gene expression.
Figure 4.
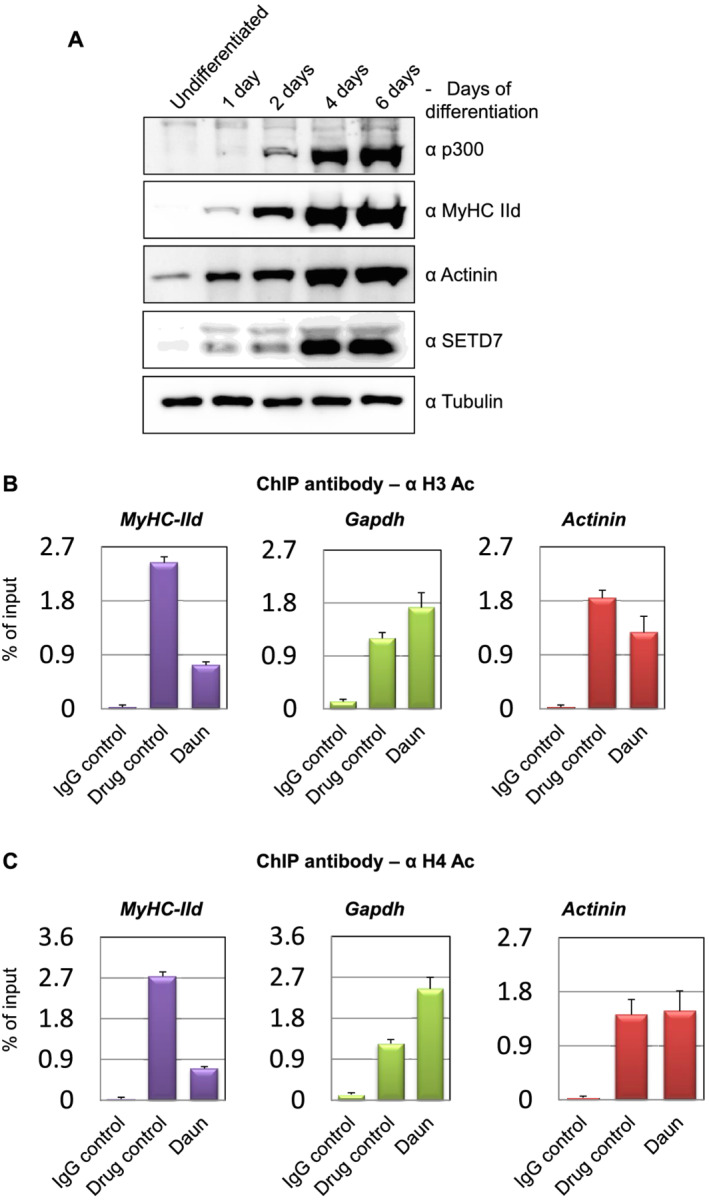
p300, a major histone acetyltransferase up‐regulated in differentiated myotube. Chemotherapy perturbed histone acetylation on MyHC‐IId. (A) Western blot showing indicated changes in temporal protein expression status of p300 histone acetyltransferase and the expression of other indicated proteins during the course of myogenic differentiation. (B) Chromatin immunoprecipation (ChIP) using a rabbit polyclonal antibody that detects acetylated H3 (typically acetylates H3 on lysine 9 + 14 + 18 + 23 + 27). The acetylation levels on different promoters (MyHC‐IId, Gapdh, and Actinin) after daunorubicin (Daun) treatment are presented in the bar diagram. (C) Same as in (B), except ChIP performed with an antibody that detects acetylated H4 (typically acetylates H4 on lysine 5 + 8 + 12 + 16). Upon Daun treatment of myotubes, the changes in the acetylation level are indicated.
To test these possibilities, we used two different rabbit polyclonal antibodies in ChIP assays that detected acetylated residues on H3 and H4, respectively. Compared with the control treatment, Daun‐treated myotubes resulted in ~3.3‐fold reduction in H3 acetylation and ~3.8‐fold reduction in H4 acetylation marks on MyHC‐IId (Figure 4B,C). We observed similar reduction of acetylation marks of ~3.7‐fold and ~2.9‐fold in Etop treated cells for H3 and H4, respectively (Supporting Information, Figure S4A,B). Again, actinin and gapdh genes exhibited no similar reduction in acetylation marks in response to drug treatments.
Chemotherapeutic drugs disrupt association of SETD7–p300 and chromatin recruitment of p300
As the histone acetylation levels were reduced in response to drug treatment, we further examined if this was a result of degradation of the histone acetyltransferase p300 at protein level. The drug treatment caused no significant change in the p300 protein levels (Figure 5A). This observation prompted us to test if the SETD7–p300 association and subsequently p300 chromatin targeting was compromised upon drug treatment. To test this hypothesis, we perform endogenous SETD7 IP assays from either control or drug‐treated myotubes. Interestingly, compared with the control cells, p300 protein signal in SETD7 IPs was drastically reduced in Daun‐treated myotubes (Figure 5B). More interestingly, chromatin occupancy of p300 on MyHC‐IId promoter was reduced ~15‐fold and ~2.9‐fold, respectively, upon Daun and Etop treatment (Figure 5C,D). On the contrary, the chromatin targeting of p300 on Actinin and Gapdh genes again remained largely unaffected upon drug treatments.
Figure 5.
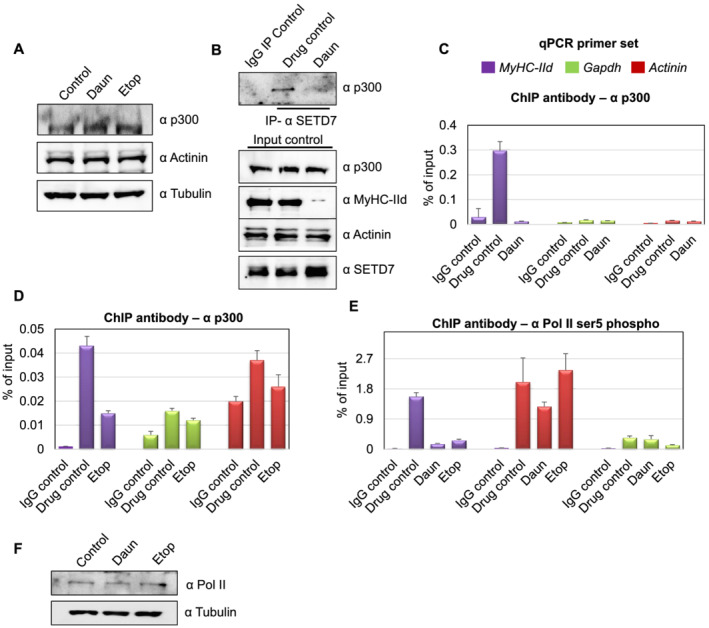
Chemotherapeutic drugs impede SETD7–p300 association and p300 chromatin targeting. (A) Western blot shows p300 and other indicated protein level after daunorubicin (Daun) and etoposide (Etop) treatment. (B) Upper panel shows SETD7–p300 interaction. The endogenous SETD7 was immunoprecipitated (anti‐mouse SETD7 antibody) from control vs. Daun‐treated myotubes and probed with p300 antibody. The lower panel shows the input control probed with indicated antibodies. (C) Chromatin immunoprecipitation (ChIP) assays were performed from Daun‐treated myotubes (nearly 10 million) extract using p300 antibody. Coloured boxes represent primer pairs for corresponding genes. (D) Same as in (C), except ChIP assays were performed from Etop‐treated and roughly 2 million myotubes. (E) Similar to (C), except ChIP assays were performed from with anti‐RNA pol II (phosphorylated at ser5) antibody with chromatin originated from Daun and Etop‐treated myotubes, respectively. (F) Western blot showing RNA pol II level after Daun and Etop treatment. qPCR, quantitative polymerase chain reaction.
The association of phosphorylated RNA polymerase II (pol II) on chromatin ultimately delineate the active transcriptional state of a gene. To investigate whether drug‐induced epigenetic changes have an effect on pol II association, we probed the status of transcriptionally active RNA polymerase on MyHC‐IId gene. We introduced anti‐phosphorylated pol II (phosphorylated ser5) antibody in our ChIP experiments. We detected ~10‐fold and ~5.8‐fold reduction in pol II association with MyHC‐IId upon Daun and Etop treatment, respectively (Figure 5E). Notably, analysis of protein lysate revealed that the reduction of pol II chromatin association was not due to reduction of overall pol II protein level (Figure 5F).
Collectively, these observations suggested a chemotherapeutic drug‐triggered pathway that (i) affected SETD7 chromatin targeting and (ii) disrupted SETD7–p300 association, thus altering epigenetic mechanisms specifically disrupting the active transcription of major molecular motor protein coding gene MyHC‐IId.
Degradation of SENP3 in drug‐treated myotubes alters MyHC‐IId gene expression
On the basis of these results, we wondered if the abnormal chromatin association of SETD7 was the first affected step in the transcriptional events, or the chemotherapeutic drugs may affect steps prior to the SETD7 targeting. To gain further molecular insights, we investigated how exactly the chemotherapeutic drugs regulate SETD7 targeting, which had further cascading effect on p300 activity specifically on MyHC‐IId.
Earlier studies established a role of the SUMO isopeptidase SENP3 in proper targeting of SETD7 to its target gene, MyHC‐IId in particular. 38 We therefore examined the possible connection of SENP3 with the chemotherapeutic drugs. The expression and localization of SENP3 were reported as deterministic steps towards its function, such as the transcriptional regulator of the MyHC‐IId gene. We measured SENP3 mRNA level, which was nearly unchanged after drug treatment (Figure 6A). Surprisingly, treatment of Daun and Etop resulted in strong down‐regulation of SENP3 protein level (Figure 6B), similar to reduction of MyHC‐IId level (Figure 6B). The expression of the other sarcomere contractile protein actinin showed no significant change (Figure 6B). Interestingly, Ara‐C, which did not have strong effect on MyHC‐IId expression, also did not show strong effect on SENP3 protein level (Figure 6B). These results indicated that Daun and Etop‐targeted SENP3 degradation, which translated into the deregulated transcription of MyHC‐IId, results in considerably decreased MyHC‐IId protein and perturbed sarcomere organization.
Figure 6.
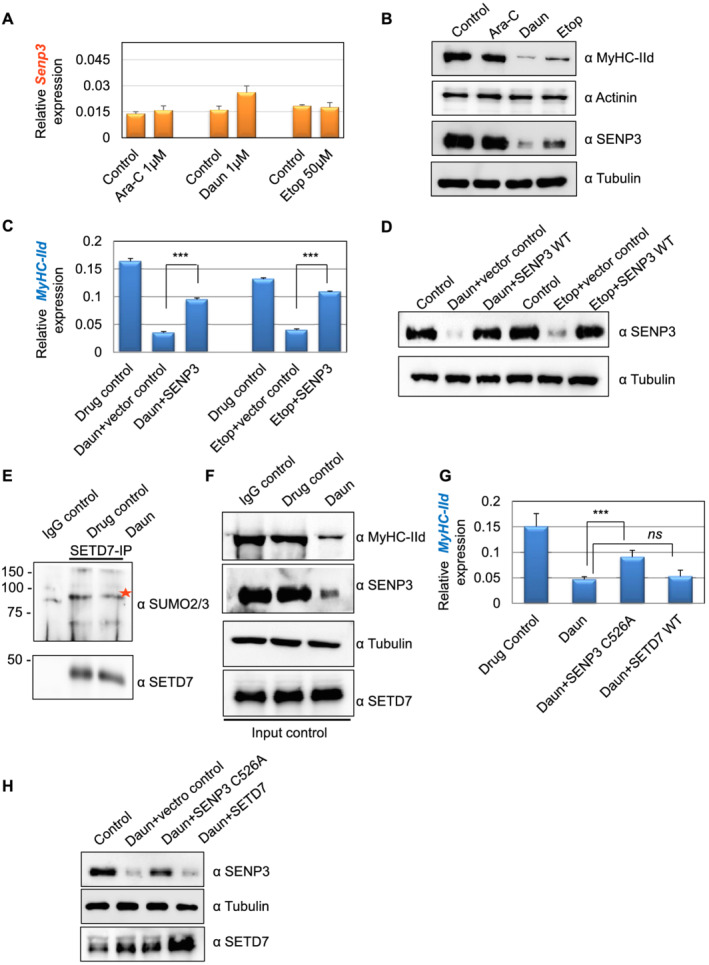
Chemotherapeutic drugs target SENP3‐specific transcriptional programme of MyHC‐IId. (A) SENP3 mRNA expression levels in drug‐treated cells were measured using quantitative reverse transcription polymerase chain reaction assays. (B) The cell lysates corresponding to (A) were analysed in western blots. (C) The Myh1 expression monitored under different conditions using quantitative reverse transcription polymerase chain reaction assays. At 3 days of differentiation, 0.5 μg of SENP3 plasmid was transfected by using lipfectamine LTX reagent. After 48 h of transfection, the cells were treated overnight with indicated drugs to trigger chemotherapy‐induced cachexia. Individual bars are the average (± SEM) of at least three independent experiments with technical duplicates in each experiment. The stars indicate statistically significant changes in the expression levels, unpaired t‐test, P < 0.0001. (D) Western blots probed for indicated protein expression corresponding to (C). (E) To check SETD7 SUMOylation, endogenous SETD7 was immunoprecipitated by using an anti‐mouse SETD7 antibody from control vs. daunorubicin (Daun)‐treated myotubes and followed by SDS‐PAGE analysis of the immnoprecipitates by SUMO2/3 antibody. Red star shows major SETD7 SUMOylated species. (F) Western blots show indicated protein expression level in the cell lysate corresponding to (E). (G) Similar to (C), except SENP3 C526A was transiently expressed in this assay. Individual bars are the average (± SEM) of two independent experiments with technical triplicates in each experiment. Stars indicate statistically significant difference, unpaired t‐test, P = 0.028. ns, not significant. (H) Western blots show indicated protein expression level corresponding to (G). Ara‐C, cytarabine; Etop, etoposide.
Next, we set up complementation assays to check if the drug‐mediated transcriptional defect was specific and directed towards SENP3. To this end, we transiently expressed low levels of SENP3 before treating the differentiated myotubes with Daun, Etop, or drug control. Similar to our previous observations (Figure 2C), MyHC‐IId mRNA was down‐regulated by ~4.7‐fold and ~3.2‐fold, respectively, upon Daun and Etop treatment (Figure 6C,D). Interestingly, transient expression of SENP3 could partially but significantly restore (~60%) defective MyHC‐IId expression caused by Daun treatment (Figure 6C). Similarly, defective MyHC‐IId expression due to Etop treatment was also restored up to ~82% upon transiently improving SENP3 level (Figure 6C).
SENP3's catalytic site‐independent function is important in chemotherapy‐induced cachexia
SENP3 preferentially cleaves SUMO2/3 from SUMOylated substrates. Earlier studies revealed SENP3 deSUMOylates SETD7, as signal intensity of SUMOylated SETD7 species was increased in SENP3‐depleted myotubes. 38 The deSUMOylation of SETD7 was important for its association with RNA pol II. Therefore, we asked (i) whether Daun and Etop‐mediated degradation of SENP3 could cause increased SETD7 SUMOylation and (ii) whether increased SETD7 SUMOylation perturbed downstream events, i.e. disruption of SETD7–p300 synergetic activity. To dissect out these possibilities, we immunoprecipitated endogenous SETD7 and probed with SUMO2/3 antibody to detect SUMOylated SETD7 status. Compared with the control IgG antibody IPs, the SETD7 antibody IPs showed characteristic higher molecular weight SUMO2/3‐conjugate signals of SETD7 in control myotubes. Interestingly, we consistently observed that SETD7 SUMOylation level was not increased upon drug treatment (Figure 6E). Instead, we observed reduction in the SETD7 SUMOylation level. The effect of drug treatment was nonetheless evident as SENP3 and MyHC‐IId protein levels were reduced (Figure 6F). Notably, the drug treatment also reduced the global SUMO conjugation level (Supporting Information, Figure S5). This suggested that the drug—by degrading SENP3 level—may interfere with SETD7 chromatin targeting.
To test if the chromatin targeting of SETD7 by SENP3 is dependent on SENP3's catalytic role, we performed further complementary assays using catalytic site mutant SENP3 C526A, incapable of deconjugating SUMO. Daun treatment led to ~3.2‐fold down‐regulation of MyHC‐IId mRNA. Interestingly, like the catalytic site active SENP3 (Figure 6C), transiently restoring SENP3 C526A level also significantly rescued (~60%) defective MyHC‐IId expression caused by Daun treatment (Figure 6G,H). Notably, increasing the level of SETD7 protein by transient expression in the same set‐up could not rescue defective MyHC‐IId expression programme (Figure 6G,H), indicating that an upstream regulator or binding partner may be required for its function. In the same figure, we have provided evidence that this upstream regulator is indeed the SENP3, and Daun treatment led to destabilization of SENP3 level (Figure 6B), resulting in defective targeting of SETD7 to Myosin II gene (Figure 3B). Improving SENP3 level—but not just SETD7 level—could partially but significantly rescue defective MyHC‐IId gene expression caused by Daun treatment (Figure 6C, D, G, and H).
In order to gain critical insights into the specificity of SETD7‐mediated MyHC‐IId expression, we performed SETD7 knockdown by two different siRNA. The Supporting Information, Figure S7A shows that SETD7 siRNA leads to down‐regulation of MyHC‐IId gene expression (3.5‐fold down‐regulation in SETD7.1 siRNA and 6.3‐fold in SETD7.2 siRNA‐mediated knockdown assays) in differentiated C2C12 myotubes. Moreover, chromatin IP experiments (Figure S7C,D) shows that SETD7 siRNA also results in statistically significant reduction of H3K4 mono‐methylation marks by nearly 2.2‐fold and 1.8‐fold from Myh1 (MyHC‐IId) as well as Myh2 (MyHC‐IIa) gene, respectively.
In summary, these outcomes suggested that the effect of chemotherapeutic drugs on MyHC‐IId transcriptional pathway was critically affected via SENP3‐mediated chromatin targeting of SETD7, irrespective of SENP3's catalytic activity. It appears that as drug treatment already reduced SETD7 SUMOylation levels, the catalytic dead SENP3 C526A mutant could still recruit SETD7 to MyHC‐IId and partially rescue defective MyHC‐IId expression.
Chemotherapeutic drugs induced cachexia in primary muscle cells
We further tested our observations in mouse primary muscle cells. We harvested satellite stem cells from 30‐day‐old adult mice. The satellite stem cells were then further differentiated into primary myotubes and followed by treatment with Daun. In agreement with our cell culture model, Daun treatment led to severely disorganized sarcomeres in primary myotubes (Figure 7A and Supporting Information, Figure S6). Moreover, Daun treatment also exhibited strong down‐regulation of SENP3 protein, and subsequently, the MyHC‐IId mRNA was down‐regulated by approximately five‐fold (Figure 7B,C). Again, the drug treatment had specific effect only on MyHC‐IId gene as no distinguishable effect of drug treatment was observed in SENP3 mRNA or other contractile sarcomere gene, such as actinin (Figure 7D,E). Moreover, Etop treatment also reduced MyHC‐IId (~6.3‐fold) and MyHC‐IIa (approximately four‐fold) gene expression, without any significant changes in Senp3 gene expression. To further consolidate SETD7‐mediated mechanism, we treated mouse satellite stem cell‐derived primary myotubes with increasing dose of SETD7 inhibitor, sinefungin. The MyHC‐IId expression was found down‐regulated by 2.1‐fold and 4.3‐fold, respectively, at 1 and 5 μg/mL of sinefungin treatment. Similarly, MyHC‐IIa mRNA levels were reduced by nearly 2.3‐fold and 3.1‐fold, respectively, following the sinefungin treatment (Figure S7E,F).
Figure 7.
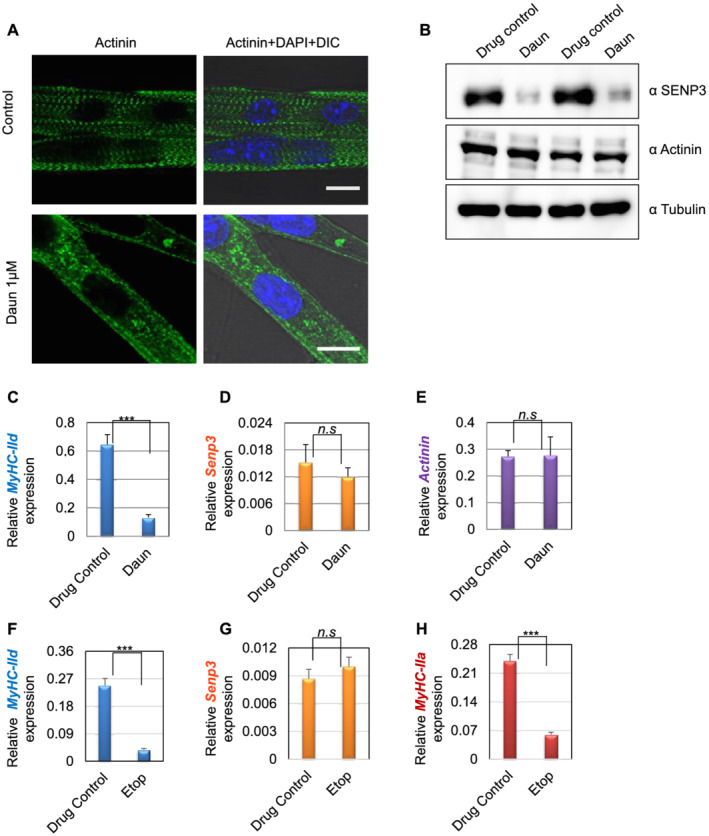
Effect of drug treatment on stem cell‐derived muscle cells. (A) Satellite stem cells were isolated from wild‐type C57BL/6J adult mice (30 days old) followed by differentiation into matured myotubes. After overnight daunorubicin (Daun) treatment, immunofluorescence staining followed by confocal microscopy was performed with an antibody against alpha‐actinin to probe for Z‐disc striation of a fully matured primary myotubes in control cells as well as Daun‐treated cells. The size of both the scale bars—10 μm each. DIC, differential interference contrast microscopy. (B) Western blots show status of indicated protein expression in response to drug treatment in primary myotubes. (C–E) Expression of indicated mRNAs were monitored in quantitative reverse transcription polymerase chain reaction assay. Data represent average (± SEM) of stem cells isolated from two different mice, with technical duplicates for each experiment. Stars indicate statistically significant difference, unpaired t‐test, P = 0.0004. n.s, not significant. (D–H) Similar to (C), except the primary myotubes were treated overnight with etoposide (Etop). Data represent average (± SEM) of primary myotubes from two different mice, with technical quadruplicates for each experiment. Stars indicate statistically significant difference, unpaired t‐test, P < 0.0001. n.s, not significant.
Taken together, these outcomes indicate that Daun and Etop target SENP3‐SETD7‐dependent transcriptional mechanism specific for MyHC‐II transcription in skeletal muscle cells.
Discussion
We identified a unique molecular mechanism responsible for chemotherapy‐induced cachexia. Our findings demonstrated a remarkable link of chemotherapeutic drugs modulating epigenetics processes by targeting SENP3‐regulated SETD7–p300 transcriptional mechanisms. The altered epigenetic marks exhibited a primary impact on the transcriptional status of the molecular motor coding gene of skeletal muscle, the Myosin II. The defective myosin II gene expression induced by Daun and Etop ultimately disrupted sarcomere organization and contractile propensity of myotubes. Importantly, the chemotherapy‐induced defects in muscle cells were reversible; i.e. the MyHC‐IId expression could be partially restored by improving SENP3 levels in these cells. Unlike Daun and Etop, Ara‐C showed no significant defect in muscle cells organization in this study. Thus, our findings identified distinct drug‐specific regulatory mechanisms as a basis of chemotherapy‐induced muscle atrophy.
Standard chemotherapy has continued as the major therapy available for treating human cancers. In spite of the clinical success, chemotherapy treatment suffers from severe side effects including muscle atrophy, greatly enhancing the burden of the disease. Doxorubicin (a variant of Daun) induces MuRF1‐regulated protein degradation towards promoting muscle atrophy. 19 The other classes of drugs like cisplatin and adriamycin also use NF‐κB pathway but independent of MuRF1‐dependent protein degradation. 48 These reports argued in favour of other uncharacterized pathways associated with chemotherapy‐induced cachexia, specific for a particular class of drug. Our data strengthened this rationale because we found a distinct pathway involving SENP3–SETD7–p300, as a prime target of Daun and Etop treatment, causing muscle atrophy. This mechanism was restricted to Daun and Etop, as Ara‐C did not have any significant impact on sarcomere organization and function in our study.
An interesting observation that Daun targeted two different categories of gene group having opposite roles in muscle function was noteworthy. In our gene expression array, we observed down‐regulation—albeit not as strongly as MyHC‐IId—of four other genes important for myogenic differentiation and muscle function (Figure 2B). None of these genes, except MyHC‐IIa, are known SENP3 targets. This indicates that apart from targeting SENP3–SETD7–p300 axis towards the regulation of MyHC‐II, it is possible that Daun and Etop also affect other pathways, specific for other genes. For example, insulin‐like growth factor 1 (IGF‐1) was shown to mediate muscle growth by stimulating Akt, mTOR‐mediated protein synthesis. 49 IGF‐1 was also down‐regulated in our study upon Daun treatment. On the contrary, the up‐regulated set of genes are known to be involved in muscle function inhibition and the promotion of muscle atrophy. For instance, the expression of myostatin (Mstn), Pdk4 genes—negative regulators of muscle growth—is up‐regulated in various cancers and causes muscle atrophy. 10 , 50 , 51 Thus, it is conceivable that Daun and Etop may target other unknown regulators or SENP3‐independent pathway/s as mechanisms leading to muscle atrophy.
Here, we demonstrated that in muscle cells, Daun and Etop targeted epigenetic processes mainly through modulating SENP3‐SETD7–p300 transcriptional axis specific for Myosin II. The expression of other sarcomeric proteins such as actinin and myotilin remained largely unaltered. Degradation of SENP3 at protein level appears as a nucleation point and rate‐limiting step in this process leading to defective SETD7 chromatin recruitment. The alteration in downstream events included the subsequent reduction in H3K4me1, decreased chromatin association of p300 and histone acetylation. We found that the p300 expression itself was temporally regulated during myogenic differentiation corresponding to SENP3 and SETD7 expression. The up‐regulation of p300—together with SENP3 and SETD7—indicated that the trio most likely operated in a functional protein complex in differentiated myotubes. This notion was further supported by reduced SETD7–p300 interaction after drug treatment (Figure 5B).
Another important aspect of this study was the role of SETD7 SUMOylation in this context. Earlier, SENP3‐dependent SETD7 deSUMOylation was reported, and the reduction in SENP3 levels correlated with increased SUMO‐modified species of SETD7. 38 In this study, chemotherapeutic drug treatment degraded SENP3; we however did not observe increased SUMO‐modified species of SETD7. On the contrary, Daun treatment decreased SETD7 SUMOylation level (Figure 6E). We also observed a decrease in global SUMO conjugation as a result of Daun and Etop treatment (Figure S5). Recently, we showed that SENP3 deSUMOylates SETD7 and delivers it to the chromatin. 38 On the basis of our earlier and current studies, we propose that, in this context, SENP3 plays a dual role under physiological condition, i.e. (i) a deSUMOylase and (ii) a recruiter of deSUMOylated interaction partner (SETD7) to chromatin. We reason that the SENP3 mutant devoid of its deSUMOylase activity nevertheless has its recruitment ability intact and recruits SETD7—which was already deSUMOylated by the action of drugs. Our rescue assays using catalytic inactive mutant of SENP3 indeed supported the aforementioned assumption. Transient expression of both catalytic active and catalytic dead mutant of SENP3 led to partial but significant rescue of defective MyHC‐IId gene expression elicited by the drug treatment (Figure 6C and G). The presence of SENP3 is critical for this pathway. Therefore, we propose that the impact of drug treatment was a consequence of compromised chromatin targeting of downstream transcriptional elements (SETD7) via SENP3 and perhaps to the lower extent a result of its deSUMOylation activity in this context. At present, it is unclear how Daun and Etop specifically promoted SENP3 degradation.
Our main focus in this study was to unravel the mechanism whereby the drug treatment influenced sarcomere contractile gene regulation. We illustrated SENP3‐regulated mechanisms as a main affected pathway in Daun and Etop‐treated muscle cells. Nonetheless, we speculate that beyond transcriptional process, Daun and Etop might influence other mechanisms towards influencing muscle cell function. Consistent with this notion, previously, it was shown that in the breast cancer stem cells, chemotherapy‐induced calcium releases through modulating ryanodine receptor type 1 (RyR1) channel. 52 RyR1 releases calcium from sarcoplasmic reticulum and is a critical player in skeletal muscle contraction. Thus, further investigation will be key to establish broader understanding of molecular mechanisms—apart from transcriptional regulation—relevant for chemotherapeutic agents affecting skeletal muscle function.
Conclusions
In summary, our study revealed an unanticipated link of chemotherapeutic drugs targeting mechanisms governed by the component of SUMO pathway in muscle cells. In addition to skeletal muscle, we hypothesize that the similar pathway could also be targeted in cardiac cells because the sarcomere machinery and fundamentals of cell contraction mechanisms are similar in both types of striated muscles. This rationale is consistent with the reports showing doxorubicin (a variant of Daun) inducing cardiac muscle atrophy. 53 , 54 , 55 Our findings established a conceptual framework for further careful investigation and assessment of current chemotherapeutic drugs in order to improve efficacy and avoid profoundly toxic side effects on muscle. Therapeutic interventions to treat cachexia remains a critical yet unresolved challenge. The drugs mainly alter synergistic action of histone‐modifying enzymes in muscle cells. The initial trigger was destabilization of SENP3. As we found SENP3 as a prime target of Daun and Etop, we reason in favour of pharmacological intervention of SUMO isopeptidases that might serve as an important tool to alleviate conditions associated with chemotherapy‐induced cachexia in striated muscle.
Funding
This research was partly supported by a grant from Deutsche Forschungsgemeinschaft (DFG) to A.N. (NA 1565/2‐1), and M.A. was supported by a DFG grant (AM/507/1‐1) and a grant from Fritz Thyseen Stiftung (10.19.1.009MN).
Conflict of interest
The authors declare no conflict of interests.
Author contributions
A.N. conceptualized the project. A.N. and M.A. designed and performed most of the experiments and the data analysis. G.P. and C.L. isolated satellite stem cells from mice and prepared primary myotubes and performed immunostainings. T.H. performed immunostainings associated with C2C12 myotubes. A.N. and A.L.D. performed myotube contraction experiments. A.N. and M.A. wrote and edited the manuscript.
Ethics statement
Wild‐type C57BL/6J mice were used, male and female, 30 days old. All the experimental procedures were performed under the ethical approval of the Italian Ministry of Health and the Institutional Animal Care and Use Committee (authorization no. 83/2019‐PR). The animals were maintained in an authorized facility at San Raffaele Hospital, Milan (authorization no. N. 127/2012‐A). The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. 56
Supporting information
Movie S1: The control myotubes contract following electrical stimulation. The excitation‐contraction coupling was monitored following 1HZ electric stimulation of the myotubes cultured on the coverslips. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Movie S2. The AraC treated myotubes contract following electrical stimulation. The excitation‐contraction coupling was monitored following 1HZ electric stimulation of the myotubes. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Movie S3. Majority of the Daun treated myotubes did not contract following electrical stimulation at 1HZ frequency. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Movie S4. A large fraction of the Etop treated myotubes did not contract following electrical stimulation at 1HZ frequency. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Figure S1. (A) The excitation‐contraction coupling of the myotubes was monitored at 1 Hz excitation frequency. The kymographs (time vs. distance plot) shows the movement of intracellular structures of the myotubes in response to the electrical stimulation. The kymographs were plotted by drawing a line across the myotube/s to monitor the changes in the intensity along the plotted line over a stack of 100 images or frames. The change in the intensity indicative of change in the position of respective organelle or structure represents the contraction. As seen for control and AraC treated cells, along the vertical axis, the change in the intensities with horizontal shifts (peaks) at regular intervals indicate contraction. For Daun and Etop however, no intensity shifts along the vertical axis was observed, thus indicating lack of contractions.(B) Same as in Figure 1A except, lower magnification confocal images shows effects of Ara‐C, Daun and Etop treatment on sarcomere organization. Scale bar −50 μm.
Figure S2.(A) Same as in Figure 2A except, the changes in transcript levels are shown as fold difference for the regulated genes upon Daun treatment. Green bars –upregulation, red bars –downregulation, and blue bars –for changes in the expression. The normalized data represent average (+/‐SEM) of three biological experiments with technical duplicates in each experiment. Unaltered expression of other sarcomeric genes such as troponins were also shown. (B) Total RNA with indicated drug treatment (used in Figure S2A), representative from two independent experiments were loaded in agarose gel to monitor quality of RNA preparation. Two distinct bands of rRNA (28S rRNA and 18sRNA) are present, indicating high quality of total RNA with no visible degradation. (C) Same as in Figure 2F except, the examples of another independent experiment is shown.
Figure S3. Same as in Figure 3B except, ChIP assays were performed with chromatin from Etop treated myotubes. Anti‐SETD7 antibodies were used for ChIP.6
Figure S4. Similar to Figure 4B and C except, ChIP assays were performed with chromatin from Etop treated myotubes. Anti‐H3 Ac and anti‐H4 Ac antibodies were used for ChIP.
Figure S5. Western blots shows level of global SUMO2/3 conjugated proteins after Daun and Etop treatments, lysates corresponding to the Figure 6F. Different concentrations of drugs were tested.
Figure S6. Similar to Figure 7A; additional examples of primary myotubes are shown.
Figure S7. (A) Two days differentiated myotubeswere transfected either with control or 120 pmolof SETD7 siRNA. Three days after siRNA transfection (5‐days after differentiation), total RNA was isolated and RTqPCR were performed to monitor the indicated mRNA. The data represents average (+/‐SEM) from two biological experiments with technical replicates in each experiment. P‐values originated from unpaired t‐test, are indicated in the figure. (B) siRNA efficiency of Figure S7A was checked by monitoring the level of indicated proteins. (C) Similar to Figure 3C except, ChIPswere performed with the control siRNA and SETD7 depleted myotubes. P‐values originated from unpaired t‐test, are indicated in the figure. (D)Depletion of SETD7 protein levels following SiRNA treatment (of Figure S7C) was monitored by western blot. (E, F)Similar to figure 7C except, indicated amount of Sinefungin (SETD7 inhibitor) was added to satellite stem cell derived primary myotubesfor 20 hours before total RNA was prepared. Sinefungintreatment influenced the Myh1and Myh2 expression levels.
Acknowledgements
We thank Prof. Theresia Kraft for providing her expertise in muscle physiology and critical comments on the manuscript. We thank Prof. Stefan Müller and Dr Joachim Meissner for critical reading and comments on the manuscript.
Amrute‐Nayak M., Pegoli G., Holler T., Lopez‐Davila AJ., Lanzuolo C., and Nayak A. (2021) Chemotherapy triggers cachexia by deregulating synergetic function of histone‐modifying enzymes, Journal of Cachexia, Sarcopenia and Muscle, 12, 159–176, doi: 10.1002/jcsm.12645
References
- 1. Janssen I, Heymsfield SB, Wang ZM, Ross R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J Appl Physiol (1985) 2000;89:81–88. [DOI] [PubMed] [Google Scholar]
- 2. Luther PK. The vertebrate muscle Z‐disc: sarcomere anchor for structure and signalling. J Muscle Res Cell Motil 2009;30:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ehler E, Gautel M. The sarcomere and sarcomerogenesis. Adv Exp Med Biol 2008;642:1–14. [DOI] [PubMed] [Google Scholar]
- 4. Bossola M, Marzetti E, Rosa F, Pacelli F. Skeletal muscle regeneration in cancer cachexia. Clin Exp Pharmacol Physiol 2016;43:522–527. [DOI] [PubMed] [Google Scholar]
- 5. Banduseela V, Ochala J, Lamberg K, Kalimo H, Larsson L. Muscle paralysis and myosin loss in a patient with cancer cachexia. Acta Myol 2007;26:136–144. [PMC free article] [PubMed] [Google Scholar]
- 6. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 7. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 8. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge DC, et al. Cachexia: a new definition. Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 9. Peterson SJ, Mozer M. Differentiating sarcopenia and cachexia among patients with cancer. Nutr Clin Pract 2017;32:30–39. [DOI] [PubMed] [Google Scholar]
- 10. Pin F, Barreto R, Couch ME, Bonetto A, O'Connell TM. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J Cachexia Sarcopenia Muscle 2019;10:140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arcamone FM. Fifty years of chemical research at Farmitalia. Chemistry 2009;15:7774–7791. [DOI] [PubMed] [Google Scholar]
- 12. Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999;57:727–741. [DOI] [PubMed] [Google Scholar]
- 13. Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009;9:338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hain BA, Xu H, Wilcox JR, Mutua D, Waning DL. Chemotherapy‐induced loss of bone and muscle mass in a mouse model of breast cancer bone metastases and cachexia. JCSM Rapid Commun 2019;2:1–12. [PMC free article] [PubMed] [Google Scholar]
- 15. Coletti D. Chemotherapy‐induced muscle wasting: an update. Eur J Transl Myol 2018;28:7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care 2010;13:225–229. [DOI] [PubMed] [Google Scholar]
- 17. Gilliam LA, St Clair DK. Chemotherapy‐induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid Redox Signal 2011;15:2543–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guigni BA, Callahan DM, Tourville TW, Miller MS, Fiske B, Voigt T, et al. Skeletal muscle atrophy and dysfunction in breast cancer patients: role for chemotherapy‐derived oxidant stress. Am J Physiol Cell Physiol 2018;315:C744–C756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hiensch AE, Bolam KA, Mijwel S, Jeneson JAL, Huitema ADR, Kranenburg O, et al. Doxorubicin‐induced skeletal muscle atrophy: elucidating the underlying molecular pathways. Acta Physiol 2019;229:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schimmel J, Eifler K, Sigurðsson JO, Cuijpers SA, Hendriks IA, Verlaan‐De Vries M, et al. Uncovering SUMOylation dynamics during cell‐cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol Cell 2014;53:1053–1066. [DOI] [PubMed] [Google Scholar]
- 21. Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 2010;11:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hay RT. SUMO: a history of modification. Mol Cell 2005;18:1–12. [DOI] [PubMed] [Google Scholar]
- 23. Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 2013;82:357–385. [DOI] [PubMed] [Google Scholar]
- 24. Nacerddine K, Lehembre F, Bhaumik M, Artus J, Cohen‐Tannoudji M, Babinet C, et al. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev Cell 2005;9:769–779. [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Dasso M. SUMOylation and deSUMOylation at a glance. J Cell Sci 2009;122:4249–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu R, Yu S, Zhu D, Huang X, Xu Y, Lao Y, et al. hCINAP regulates the DNA‐damage response and mediates the resistance of acute myelocytic leukemia cells to therapy. Nat Commun 2019;10:3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu K, Guo C, Lao Y, Yang J, Chen F, Zhao Y, et al. A fine‐tuning mechanism underlying self‐control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy 2019;2:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seifert A, Schofield P, Barton GJ, Hay RT. Proteotoxic stress reprograms the chromatin landscape of SUMO modification. Sci Signal 2015;8:rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niskanen EA, Malinen M, Sutinen P, Toropainen S, Paakinaho V, Vihervaara A, et al. Global SUMOylation on active chromatin is an acute heat stress response restricting transcription. Genome Biol 2015;16:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liebelt F, Sebastian RM, Moore CL, Mulder MPC, Ovaa H, Shoulders MD, et al. SUMOylation and the HSF1‐regulated chaperone network converge to promote proteostasis in response to heat shock. Cell Rep 2019;26:236–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kunz K, Piller T, Muller S. SUMO‐specific proteases and isopeptidases of the SENP family at a glance. J Cell Sci 2018;131:jcs211904. [DOI] [PubMed] [Google Scholar]
- 32. Yeh ET. SUMOylation and De‐SUMOylation: wrestling with life's processes. J Biol Chem 2009;284:8223–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nayak A, Muller S. SUMO‐specific proteases/isopeptidases: SENPs and beyond. Genome Biol 2014;15:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Finkbeiner E, Haindl M, Muller S. The SUMO system controls nucleolar partitioning of a novel mammalian ribosome biogenesis complex. EMBO J 2011;30:1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yun C, Wang Y, Mukhopadhyay D, Backlund P, Kolli N, Yergey A, et al. Nucleolar protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J Cell Biol 2008;183:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raman N, Nayak A, Muller S. mTOR signaling regulates nucleolar targeting of the SUMO‐specific isopeptidase SENP3. Mol Cell Biol 2014;34:4474–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nayak A, Viale‐Bouroncle S, Morsczeck C, Muller S. The SUMO‐specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiation. Mol Cell 2014;55:47–58. [DOI] [PubMed] [Google Scholar]
- 38. Nayak A, Lopez‐Davila AJ, Kefalakes E, Holler T, Kraft T, Amrute‐Nayak M. Regulation of SETD7 methyltransferase by SENP3 is crucial for sarcomere organization and cachexia. Cell Rep 2019;27:2725–2736. [DOI] [PubMed] [Google Scholar]
- 39. Shishmarev D. Excitation–contraction coupling in skeletal muscle: recent progress and unanswered questions. Biophys Rev 2020;12:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pang B, de Jong J, Qiao X, Wessels LF, Neefjes J. Chemical profiling of the genome with anti‐cancer drugs defines target specificities. Nat Chem Biol 2015;11:472–480. [DOI] [PubMed] [Google Scholar]
- 41. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018;563:131–136. [DOI] [PubMed] [Google Scholar]
- 42. Bossis G, Sarry JE, Kifagi C, Ristic M, Saland E, Vergez F, Bossis G, et al. The ROS/SUMO axis contributes to the response of acute myeloid leukemia cells to chemotherapeutic drugs. Cell Rep 2014;7:1815–1823. [DOI] [PubMed] [Google Scholar]
- 43. Frank D, Kuhn C, Katus HA, Frey N. The sarcomeric Z‐disc: a nodal point in signalling and disease. J Mol Med (Berl) 2006;84:446–468. [DOI] [PubMed] [Google Scholar]
- 44. Tao Y, Neppl RL, Huang ZP, Chen J, Tang RH, Cao R, et al. The histone methyltransferase Set7/9 promotes myoblast differentiation and myofibril assembly. J Cell Biol 2011;194:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee J, Shao NY, Paik DT, Wu H, Guo H, Termglinchan V, et al. SETD7 drives cardiac lineage commitment through stage‐specific transcriptional activation. Cell Stem Cell 2018;22:428–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang H, Cao R, Xia L, Erdjument‐Bromage H, Borchers C, Tempst P, et al. Purification and functional characterization of a histone H3‐lysine 4‐specific methyltransferase. Mol Cell 2001;8:1207–1217. [DOI] [PubMed] [Google Scholar]
- 47. Damrauer JS, Stadler ME, Acharyya S, Baldwin AS, Couch ME, Guttridge DC. Chemotherapy‐induced muscle wasting: association with NF‐κB and cancer cachexia. Eur J Transl Myol 2018;28:7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, et al. Mediation of IGF‐1‐induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 2001;3:1009–1013. [DOI] [PubMed] [Google Scholar]
- 49. Gallot YS, Durieux AC, Castells J, Desgeorges MM, Vernus B, Plantureux L, et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res 2014;74:7344–7356. [DOI] [PubMed] [Google Scholar]
- 50. Liu D, Qiao X, Ge Z, Shang Y, Li Y, Wang W, et al. IMB0901 inhibits muscle atrophy induced by cancer cachexia through MSTN signaling pathway. Skelet Muscle 2019;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, et al. Chemotherapy‐induced Ca 2+ release stimulates breast cancer stem cell enrichment. Cell Rep 2017;18:1946–1957. [DOI] [PubMed] [Google Scholar]
- 52. Zhang S, Liu X, Bawa‐Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin‐induced cardiotoxicity. Nat Med 2012;18:1639–1642. [DOI] [PubMed] [Google Scholar]
- 53. Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin‐induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 2012;52:1213–1225. [DOI] [PubMed] [Google Scholar]
- 54. Willis MS, Parry TL, Brown DI, Mota RI, Huang W, Beak JY. Doxorubicin exposure causes subacute cardiac atrophy dependent on the striated muscle‐specific ubiquitin ligase MuRF1. Circ Heart Fail 2019;12:e005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 2017;18:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1: The control myotubes contract following electrical stimulation. The excitation‐contraction coupling was monitored following 1HZ electric stimulation of the myotubes cultured on the coverslips. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Movie S2. The AraC treated myotubes contract following electrical stimulation. The excitation‐contraction coupling was monitored following 1HZ electric stimulation of the myotubes. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Movie S3. Majority of the Daun treated myotubes did not contract following electrical stimulation at 1HZ frequency. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Movie S4. A large fraction of the Etop treated myotubes did not contract following electrical stimulation at 1HZ frequency. The movies were acquired at 5.8 frames/s and are played at 30 images/s. The scale bar −10 μm.
Figure S1. (A) The excitation‐contraction coupling of the myotubes was monitored at 1 Hz excitation frequency. The kymographs (time vs. distance plot) shows the movement of intracellular structures of the myotubes in response to the electrical stimulation. The kymographs were plotted by drawing a line across the myotube/s to monitor the changes in the intensity along the plotted line over a stack of 100 images or frames. The change in the intensity indicative of change in the position of respective organelle or structure represents the contraction. As seen for control and AraC treated cells, along the vertical axis, the change in the intensities with horizontal shifts (peaks) at regular intervals indicate contraction. For Daun and Etop however, no intensity shifts along the vertical axis was observed, thus indicating lack of contractions.(B) Same as in Figure 1A except, lower magnification confocal images shows effects of Ara‐C, Daun and Etop treatment on sarcomere organization. Scale bar −50 μm.
Figure S2.(A) Same as in Figure 2A except, the changes in transcript levels are shown as fold difference for the regulated genes upon Daun treatment. Green bars –upregulation, red bars –downregulation, and blue bars –for changes in the expression. The normalized data represent average (+/‐SEM) of three biological experiments with technical duplicates in each experiment. Unaltered expression of other sarcomeric genes such as troponins were also shown. (B) Total RNA with indicated drug treatment (used in Figure S2A), representative from two independent experiments were loaded in agarose gel to monitor quality of RNA preparation. Two distinct bands of rRNA (28S rRNA and 18sRNA) are present, indicating high quality of total RNA with no visible degradation. (C) Same as in Figure 2F except, the examples of another independent experiment is shown.
Figure S3. Same as in Figure 3B except, ChIP assays were performed with chromatin from Etop treated myotubes. Anti‐SETD7 antibodies were used for ChIP.6
Figure S4. Similar to Figure 4B and C except, ChIP assays were performed with chromatin from Etop treated myotubes. Anti‐H3 Ac and anti‐H4 Ac antibodies were used for ChIP.
Figure S5. Western blots shows level of global SUMO2/3 conjugated proteins after Daun and Etop treatments, lysates corresponding to the Figure 6F. Different concentrations of drugs were tested.
Figure S6. Similar to Figure 7A; additional examples of primary myotubes are shown.
Figure S7. (A) Two days differentiated myotubeswere transfected either with control or 120 pmolof SETD7 siRNA. Three days after siRNA transfection (5‐days after differentiation), total RNA was isolated and RTqPCR were performed to monitor the indicated mRNA. The data represents average (+/‐SEM) from two biological experiments with technical replicates in each experiment. P‐values originated from unpaired t‐test, are indicated in the figure. (B) siRNA efficiency of Figure S7A was checked by monitoring the level of indicated proteins. (C) Similar to Figure 3C except, ChIPswere performed with the control siRNA and SETD7 depleted myotubes. P‐values originated from unpaired t‐test, are indicated in the figure. (D)Depletion of SETD7 protein levels following SiRNA treatment (of Figure S7C) was monitored by western blot. (E, F)Similar to figure 7C except, indicated amount of Sinefungin (SETD7 inhibitor) was added to satellite stem cell derived primary myotubesfor 20 hours before total RNA was prepared. Sinefungintreatment influenced the Myh1and Myh2 expression levels.