

Abstract
In the present investigation, a totally of 673 newly designed 2,6-diphenyl piperidin-4-one derivatives are docked with 2B7N protein of Helicobacter pylori which causes peptic ulcer disease. The docking studies revealed that 2,6-bis(3,4-dihydroxyphenyl)-3-phenethylpiperidin-4-one (BDPO) is identified as the most promising new compound with active nature against 2B7N with a binding affinity value of -8.0 Kcal/mol. The molecular structure of BDPO has been analyzed by DFT based theoretical calculations at the B3LYP/6-311++G(d,p) level of theory using the Gaussian 16W program package. The molecular electrostatic potential, Frontier molecular energy gap, and Mulliken population analysis have been used to understand the reactive site of the molecule. The stability and hyper-conjugative interactions were studied by natural bond orbital analysis (NBO) based on a second-order perturbation approach. The thermodynamic properties like thermal energy, specific heat capacity, and entropy at different temperatures were also calculated. The calculated first-order hyper polarizability results show that the title compound is 25.3 times greater than that of standard reference material, urea. So the title molecule is a good non-linear material. Also, this molecule has Van der Waals attraction and steric effect. It undergoes local excitation for the first five excitations.
Keywords: Docking; 2B7N; DFT; NBO; NLO; 2,6-bis(3,4-dihydroxyphenyl)-3-phenethylpiperidin-4-one
Docking; 2B7N; DFT; NBO; NLO; 2,6-bis(3,4-dihydroxyphenyl)-3-phenethylpiperidin-4-one
1. Introduction
In the past and recent years, heterocyclic compounds like piperidin-4-one nucleus having a large number of biological properties. Piperidine and its derivatives are having many applications in the field of medicinal chemistry, material chemistry, agricultural chemistry, and others. Piperidin-4-one is an important drug derivative as well as an intermediate in the synthesis of certain chemicals and pharmaceutical drugs [1]. Piperidin-4-one has a wide range of biological applications such as antimicrobial activity [2, 3], analgesic, antipyretic [4, 5], antiviral, antitumor [6], antihypertensors [7], anticancer [8], local anesthetic [7, 9], anti-tubercular activity [10], anti-Alzheimer activity [11], anti-inflammatory, central nervous system [12, 13], and antifungal activity [14, 15] etc., Piperine, present in black pepper and white pepper is used to cure gastric ulcers [16]. Piperine has piperidine moiety. This high degree of biological activities of piperidin-4-one compounds attracts us to do the present study. Peptic ulcer disease is the most deadly bacterial disease, that affects a large population of the world caused by species of bacteria known as Helicobacter pylori [17, 18]. So the need for a novel compounds with potential anti-Helicobacter pylori activity is very essential. The theoretical and computational approach is a very effective method for designing new drugs to achieve a successful result and it reduces the cost. The objective of the present work is to promise newly designed piperidin-4-one derivatives as anti-ulcer agents since it specifically binds with the target protein 2B7N perfectly.
2. Materials and methods
2.1. Preparation of protein
The crystal structure of quinolinic acid phosphoribosyltransferase from Helicobacter pylori (PDB ID: 2B7N) used in the study was obtained from the protein data bank (http://www.rcsb.org.pdb) [19]. The 2B7N was obtained by X-ray diffraction method. Discovery Studio 4.0 [20] visualizer was used to remove all solvent molecules and bounded substances (ligands and cofactors) with the receptor and then it was saved in PDB format.
2.2. Design
The various aromatic aldehydes collected from ASPIN CO (http://www.apin.co.uk/?op=groups&group=Aromatic+Aldehydes+%26+Derivatives+(substituted) and Open Molecule website (http://www.openmolecules.org/name2structure.html). Based on the structures, the 2D structures of 673 piperidone derivatives are drawn by using Chemsketch [21] software.
The parent piperidine-4-one structure was modified in the aromatic ring part (substituting Cl, Br, I, NH2, OH, OMe, NO2, etc., in various positions as mono, di, or tri substitutions) and R1, R2 positions. The modification was also carried out at –NH position (N–CH3, N–NH2, N–OH, etc.) in the piperidone ring. Finally, the best docking model was selected for further studies.
2.3. Preparation of the ligands
The general preparation procedure of piperidone [22] is shown in Scheme 1, All the piperidone derivatives are minimized by Avogadro [23] tool. The minimized structures were then saved in PDB format.
Scheme 1.

Ligand designing root.
2.4. Docking process
A series of designed 673 piperidin-4-one ligands and prepared protein 2B7N were docked using PyRx [24] software with AutoDock tool [25]. The docking results were visualized and analyzed by PyMOL [26] software.
2.5. Computational details
The quantum computational calculations have been carried out for BDPO by DFT with Becke's the three-parameter hybrid function (B3) combined with Lee- Yang- Parr (LYP) [27] correlation level using Gaussian 16W program package [28] with the standard 6-311++G(d,p) basis set. Gaussview 06 [29] is a molecular visualization software that is used to observe the properties of the molecule. The structural parameters like bond length, bond angle, and dihedral angles have been calculated theoretically. The molecular electrostatic potential, Mulliken population analysis, and natural bond orbital analysis of the target molecule have been calculated by DFT with the B3LYP/6-311++G(d,p) method. The chemical hardness, nucleophilicity index, electrophilicity index, net electrophilicity, optical softness, chemical potential, and various quantum chemical parameters have been calculated using the HOMO and LUMO values. DFT is an excellent alternative method for predicting NLO property of the new material [30]. Dipole moment, polarizability, anisotropy of polarizability, first-order hyper polarizability, and vector component of the target molecule also calculated by the density functional theory computations. Multiwfn 3.7 [31] program is used to determine NCI, Surface projection map, Electron excitation studies, etc.
3. Results and discussion
3.1. Molecular docking studies
In the present docking analysis, a newly designed series of 673 ligands were docked with 2B7N protein of Helicobacter pylori using PyRx software. The ligand, BDPO is found to have a good binding affinity value of -8.0 Kcal/mol. It is higher than usual ulcer drugs (given in supplementary material Table S1). It has a good drug-likeness value and it was found to be 1.17 by Molsoft prediction (http://www.molsoft.com/) and given in Table 1.
Table 1.
Binding energy and drug likeness value of BDPO.
| Name of the lead molecule | Structure | Binding affinity (Kcal/mol) | Drug likeness score |
|---|---|---|---|
| 2,6-bis(3,4-dihydroxyphenyl)-3-phenethylpiperidin-4-one | 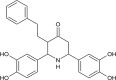 |
-8.0 | 1.17 |
3.2. ADMET property prediction
The physicochemical properties and bioavailability of the lead BDPO was predicted by SwissADME [32] (Absorption, Distribution, Metabolism, and Excretion) program. The ligand BDPO obeys Lipinski's rule of five [33], Veber (GSK) [34], Egan (Pharmacia) [35], Ghose [36] and Muegge (Bayer) [37] are shown in Table 2. The BDPO molecule fulfils a drug-like character and can be considered as an excellent candidate for drug development.
Table 2.
Physicochemical property and drug like characters.
| Method | Rules | Druglikeness |
|---|---|---|
| Lipinski | MW ≤ 500 | Yes |
| MLOGP ≤4.15 | ||
| N or O ≤ 10 | ||
| NH or OH ≤ 5 | ||
| Ghose | 160 ≤ MW ≤ 480 | Yes |
| -0.4 ≤ WLOGP ≤5.6 | ||
| 40 ≤ MR ≤ 130 | ||
| 20 ≤ atoms ≤70 | ||
| Veber | Rotatable bonds ≤10 | Yes |
| TPSA ≤140 | ||
| Egan | WLOGP ≤5.88 | Yes |
| TPSA ≤131.6 | ||
| Muegge | 200 ≤ MW ≤ 600 | Yes |
| -2 ≤ XLOGP ≤5 | ||
| TPSA ≤150 | ||
| Num. rings ≤7 | ||
| Num. carbon >4 | ||
| Num. heteroatoms >1 | ||
| Num. rotatable bonds ≤15 | ||
| H-bond acc. ≤ 10 | ||
| H-bond don. ≤ 5 |
3.3. Docking result
The protein-ligand interaction was predicted by PyMOL software. The binding results reveal that four hydrogen bonds are formed between residues ASP 153, ARG 148, HIS 147, ASN 146 with hydroxyl and carbonyl groups of BDPO ligand and shown in Figure 1. Docking results reveal that the hydrogen bond interactions fix the ligand tightly and firmly in the active site. It is very important for the perfect fitting of the ligand within the protein enzyme [38].The docking score is found to be -8.0 Kcal/mol conclude that newly designed BDPO have excellent binding capability with 2.2 Å, 2.7 Å, 2.6 Å, and 2.2 Å respectively with the above-said amino acids.
Figure 1.

Docking pattern and hydrogen bond interactions of the title compound with chain A of 2B7N protein.
The lead like molecule under investigation is docked with some of the other proteins of H.Pylori and tabulated in Table 3. From the docking scores, we can conclude that the docking of the molecule under investigation with 2B7N protein gives a better result. So it is concluded that the lead-like molecule would be able to inhibit the specified target or act as a ligand on the target protein.
Table 3.
Binding affinity of BDPO with various proteins of H.Pylori.
| S.No | Various Proteins of H.pylori | Binding affinity (Kcal/mol) | S.No | Various Proteins of H.pylori | Binding affinity (Kcal/mol) |
|---|---|---|---|---|---|
| 1 | 2B7N | -8.0 | 8 | 1Z8M | -6.9 |
| 2 | 5XRW | -7.8 | 9 | 3H1E | -6.6 |
| 3 | 4FQ0 | -7.5 | 10 | 1ZHC | -6.5 |
| 4 | 3RPF | -7.3 | 11 | 2MX0 | -6.3 |
| 5 | 6GW5 | -7.3 | 12 | 6GIF | -6.3 |
| 6 | 2OTR | -7.2 | 13 | 5J4G | -5.7 |
| 7 | 1X93 | -6.9 | 14 | 1YG0 | -5.5 |
3.4. Molecular geometry
In this present study, all the quantum computational calculations were carried out by DFT and B3LYP with triple split valence basis set 6-311++G(d,p) level using Gaussian 16W program utilizing gradient geometrical optimization of the target molecule. The 3D optimized structure of the target molecule is shown in Figure 2 which was obtained from the Gaussian 16W program. The molecular formula of the target molecule is C25H25NO5. It contains 56 atoms and 222 electrons. It is a neutral molecule with a singlet system.
Figure 2.

Optimized structure of BDPO at the B3LYP/6-311++G(d,p) level theory.
3.4.1. Optimization and convergence
The molecule was drawn and subjected to optimization process with a B3LYP/6-311++G(d,p) basis set. Imaginary frequency calculations were also performed to confirm the absence of negative frequencies. The molecule was fully optimized and converged. The Gaussum software tool [39] was used to perform this. Figure 3 explains the complete optimization and convergence of the target molecule.
Figure 3.

(a) & (b): Optimization and convergence of the BDPO molecule by B3LYP/6-311++G(d,p).
The list of atoms of the target molecule was generated by Gaussview 06 software and reported in Table 4. The molecule contains one nitrogen atom and five oxygen atoms as hetero atoms. The carbon atoms 1, 2, 3, 5, 6 are present in the piperidone ring and the other carbon atoms except for C19 and C20 are present in the three phenyl rings.
Table 4.
Atom list of the optimized BDPO molecule.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| C | C | C | N | C | C | C | C |
| 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| C | C | C | C | C | C | C | C |
| 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 |
| C | C | C | C | C | C | C | C |
| 25 | 26 | 27 | 28 | 29 | 30 | 31 | 32 |
| C | C | O | O | O | O | O | H |
| 33 | 34 | 35 | 36 | 37 | 38 | 39 | 40 |
| H | H | H | H | H | H | H | H |
| 41 | 42 | 43 | 44 | 45 | 46 | 47 | 48 |
| H | H | H | H | H | H | H | H |
| 49 | 50 | 51 | 52 | 53 | 54 | 54 | 56 |
| H | H | H | H | H | H | H | H |
3.4.2. Optimized parameter of the target molecule
The optimization of the target molecule is obtained from the Gaussian 16W program. The optimized parameters like bond length, bond angle, dihedral angle of the target molecule are obtained by higher basis set B3LYP/6-311++G(d,p) method and given in Table S2. The distance between the nuclei of two bonded atoms in a molecule is called bond distance. The distance between C5 – C6 is having the highest value with a bond distance of 1.5613 Å and O28 – H53 is having the shortest bond distance of 0.9626 Å. The distance between C - H in the piperidone ring shows ≈1.09 Å. At the same time, the C - H bond distance in the phenyl ring lays ≈1.08 Å. The two phenyl rings present in the 2nd and 6th positions of piperidone contains four hydroxyl groups. The bond distance between the O29 – H54, O28 – H53, O30 – H55, O31 – H56 are found to be 0.9659 Å, 0.9626 Å, 0.9659 Å, and 0.9629 Å respectively. The bond distance of the title compound has good agreement with previously reported similar compound 3-methyl 2,6- diphenyl-piperidin-4-one as discussed by G. Velrajet.al [40].
The shortest bond angle is seen at C6 – C5 – H36 with an angle of 103.4075° and the largest bond angle is observed at C6 – C13 – C14 atoms with an angle of 125.0891°. In the piperidone ring, the bond angle around N4 i.e., C2 – N4 – C6; C2 – N4 – H35; and C4 – N4 – H35 are found to be 119.9882°, 110.4614° and 109.7222° respectively. The C - N – C bond angle is larger than the N - C – C bond angle. The C1 – C3 – O27 and C5 – C3 – O27 bond angles are found to be 122.4185° and 123.4959° respectively. In three phenyl rings, the C - C – C bond angle is found to be approximately 120°. The bond angle and dihedral angle of the target molecule are in a good manner with earlier reports [40].
3.4.3. Molecular electrostatic potentials
The molecular electrostatic potential is a very useful method to predict the reactive sites for the investigated target molecule. It is also used to explain structure–activity relationship, hydrogen bonding, the reactivity of molecules including bio molecules and drugs [41]. The different surface colours represent the different values of electrostatic potential. The most negative electrostatic region is represented by a red colour. The positive electrostatic region is represented by blue colour. Green represents the zero electrostatic potential regions. The increasing order of electrostatic potential is red < orange < yellow < green < blue [42]. The MEP surface has been plotted for the target molecule with the help of 6–311++G(d,p) basis set using Gaussview 06 software and given in Figure 4. The electrostatic potential of the target molecule ranges from -6.296 e−2 to 6.296 e−2. The most electronegative potential region is seen around the O30 and O31 and it is denoted by red colour. This is the possible site of electrophilic reactivity. The very light red colour area is seen around the carbonyl carbon represent the axial attack is possible for this site [43]. The blue colour has appeared around H53, H55, and H56 which represent the most electropositive potential regions. It is the possible sites for the nucleophilic reaction. The yellow colour is seen around the nucleus of the benzene ring attached as a side chain to the central six-member alicyclic ring which bears nitrogen.
Figure 4.

Molecular electrostatic potential map of BDPO.
3.4.4. Contour diagram of the target molecule
The contour diagram of the target molecule is shown in Figure 5. The contour line divided exactly the aromatic benzene ring which bears hydroxyl group and passes through H33, C3, C18, C19, and C21. C=O is present at the lower part and NH appears above the contour line. O38 is present above the plane whereas the remaining hydroxyl group appears lower part of the contour line.
Figure 5.

Contour map of BDPO.
3.4.5. Frontier molecular orbital
The Frontier molecular orbital of the target molecule have been calculated by DFT method B3LYP/6-311++G(d,p) level of theory. The chemical reactivity and kinetic stability of a molecule are highly dependent on the energy gap between the highest molecular orbital and lowest unoccupied molecular orbital. Higher the energy gap indicates the harder and more stable the molecule. Lower the energy gap indicates the soft and more polarizable nature of the molecule and having low kinetic stability and high chemical reactivity [44]. The Frontier molecular orbital energy gap of the target molecule is found to be 4.6880 eV and it is shown in Figure 6.
Figure 6.

Frontier molecular orbital and their energy gap obtained from DFT/B3LYP/6-311++G(d,p) level of the target molecule.
3.4.6. Quantum chemical parameter
Using HOMO and LUMO values the quantum chemical parameters such as ionization potential (I), electron affinity (A), electronegativity (χ), chemical hardness (η), Global softness (S), chemical potential (μ), electrophilicity index (ω), net electrophilicity (), electron accepting (ω+) and donating capability (ω−), Chemical softness (σ), nucleophilicity index (N), additional electronic charges (ΔNmax) and optical softness (σ0) [45-47] have been calculated with the help of Eqs. (1), (2), (3), (4), (5), (6), (7), (8), (9), (10), (11), (12), (13), (14), and (15) and given in Table 5.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
Table 5.
Calculated quantum chemical parameters at B3LYP/6-311++G(d,p) level.
| S. No | Physical Parameter | Charge (eV) |
|---|---|---|
| 1 | HOMO | -5.9171 |
| 2 | LUMO | -1.2291 |
| 3 | Energy gap (ΔE) | 4.6880 |
| 4 | Ionization potential (I) | 5.9171 |
| 5 | Electron affinity (A) | 1.2291 |
| 6 | Electro negativity (χ) | 3.5731 |
| 7 | Chemical hardness (η) | 2.3440 |
| 8 | Global softness (S) | 0.2133 |
| 9 | Chemical potential (μ) | -3.5731 |
| 10 | Electrophilicity index (ω) | 2.7233 |
| 11 | Electron donating capability (ω−) | 4.8029 |
| 12 | Electron accepting capability (ω+) | 1.2298 |
| 13 | Net electrophilicity ( | 6.0327 |
| 14 | Chemical softness (σ) | 0.4266 |
| 15 | Nucleophilicity index (N) | 0.3672 |
| 16 | Additional electronic charges (ΔNmax) | 1.5244 |
| 17 | Optical softness (σ0) | 0.2133 |
The energy of HOMO and LUMO directly related to ionization potential and electron affinity of the molecule. Larger the ω+ value corresponds to the better capability of accepting charge and smaller the ω− value makes it to the better capability of electron-donating system [46]. Nucleophilicity index and electrophilicity index are also related to the biological activity of a molecule. The reactivity of the molecule increasing with increased nucleophilicity index [45].
3.4.7. Mulliken population analysis
In quantum mechanical calculation, the Mulliken population analysis plays an important application in summarizing the distribution of charge over the skeleton of the whole molecule [48]. The charge distribution of the target molecule is shown in Figure 7 which was calculated by DFT/B3LYP/6-311++G(d,p) level in the gas phase and listed in Table 6.
Figure 7.

The Mulliken atomic charge distribution of target molecule.
Table 6.
The Mulliken atomic charge distribution of target molecule.
| Atom no | Mulliken charge (a.u) | Atom no | Mulliken charge (a.u) |
|---|---|---|---|
| 1 C | -0.716521 | 29 O | -0.208253 |
| 2 C | -0.004510 | 30 O | -0.213286 |
| 3 C | -0.222875 | 31 O | -0.204969 |
| 4 N | -0.021105 | 32 H | 0.191675 |
| 5 C | 0.103674 | 33 H | 0.207700 |
| 6 C | -0.430502 | 34 H | 0.241163 |
| 7 C | 1.111097 | 35 H | 0.248121 |
| 8 C | -0.004848 | 36 H | 0.246232 |
| 9 C | -0.393625 | 37 H | 0.257275 |
| 10 C | -0.697400 | 38 H | 0.173708 |
| 11 C | -0.234086 | 39 H | 0.124963 |
| 12 C | -0.277082 | 40 H | 0.198370 |
| 13 C | 0.722859 | 41 H | 0.235563 |
| 14 C | 0.019851 | 42 H | 0.130124 |
| 15 C | -0.137788 | 43 H | 0.155154 |
| 16 C | -0.066764 | 44 H | 0.228273 |
| 17 C | -0.554994 | 45 H | 0.160979 |
| 18 C | -0.417326 | 46 H | 0.154786 |
| 19 C | -0.670200 | 47 H | 0.154948 |
| 20 C | -0.733180 | 48 H | 0.142787 |
| 21 C | 1.354816 | 49 H | 0.147424 |
| 22 C | -0.159006 | 50 H | 0.180230 |
| 23 C | -0.352616 | 51 H | 0.170956 |
| 24 C | -0.433527 | 52 H | 0.150753 |
| 25 C | -0.354890 | 53 H | 0.283699 |
| 26 C | -0.348239 | 54 H | 0.269684 |
| 27 O | -0.163669 | 55 H | 0.261228 |
| 28 O | -0.268999 | 56 H | 0.262163 |
Carbon 21 is having a higher positive value with 1.354816 a.u. and C2 is having a lower value with -0.02110 a.u. All the hetero atoms like O27, O28, O29, O30, O31, and N4 are having negative charges. The O28 is having more negative charge (-0.268999 a.u.) and O27 shows less negative charge (-0.163669 a.u.). Most of the carbons are having a negative charge. The negative region in the target molecule behaves as electron acceptors and it is related to electrophilic reactivity. The carbon atoms C21, C5, C13, C14, and C7 are having a positive charge. All the hydrogen atoms in the target molecule are having positive charges. H53 is having a more positive charge due to the presence of more electronegative nearby oxygen and H35 having a less positive charge. In this positive region of the target molecule behaves as an electron donor and it is related to nucleophilic reactivity. The charge distribution of the target molecule is having both donor and acceptors. It suggests that the target compound more reactive towards substitution reactions.
3.4.8. Nonlinear optical effect
In recent years, organic non-linear molecules have more potential applications in various photonic technologies. In the presence of applied electric fields, the NLO properties of such compounds attributed to two factors. In the ground state, charge distribution is altered by donor moiety and acceptor moiety and it enhances the π electronic charge distribution through the π conjugation. The intra-molecular charge transfer is associated with the first-order hyper polarizability as the result, the electron cloud moves from an electron donor to electron acceptor group through the π conjugated framework [49]. The electron cloud interacts with an external electric field and it alters the first hyper polarizability and dipole moment.
The first hyper polarizability of this molecular system and its related properties like dipole moment, mean polarizability, anisotropy of polarizability, and vector component of the target molecule were calculated by using the B3LYP/6-311++G(d,p) method based on finite field approach. In the presence of an applied electric field, the energy of a system is a function of the electric field. The first order hyper polarizability is the third rank tensor that is described by 3 × 3 × 3 matrices. Totally 27 component of the 3-dimensional matrix can be reduced into 10 component due to the Kleinman symmetry [50].
The dipole moment, mean polarizability, anisotropy of polarizability, first hyper polarizability and βvec [51] using xyz components are defined as follows(Eqs. (17), (18), (19), (20), and (21)).
| (16) |
| (17) |
| (18) |
| (19) |
| (20) |
The dipole moment value for the target molecule is found to be 2.1122 D. The highest value of dipole moment is present in the compound μx (0.3647 D). The higher polarizability and first hyper polarizability values for the target molecule are found to be 326.469 a.u. and 625.498 a.u. for αxx and βzzz respectively.
The μ, α, β values are obtained by the Gaussian 16W output file and it is reported in atomic units. The calculated values are converted into e.s.u. (for α: 1a.u. = 0.1482 × 10−24 e.s.u; for β 1a.u = 8.6393 × 10−33 e.s.u.) [52]. The calculated μTot, Δα, αTot,βTot, βvec values are given in Table 7. The μTot, αTot, βTot values are found to be 2.1122 D, 45.4305 x 10−24 e.s.u. and 9.4378 x 10−30 e.s.u. respectively. The total dipole moment of the target molecule is 1.5 times greater than that of urea and β is 25.3 times greater than urea. (Urea μ Tot = 1.3732D, β Tot = 0.3728 × 10−30e.s.u.).
Table 7.
The mean polarizability, Anisotropy of polarizability, Dipole moment and First hyper polarizability and Vector component for target molecule.
| Parameters | B3LYP/6-311++G(d,p) | Parameters | B3LYP/6-311++G(d,p) |
|---|---|---|---|
| μx | 0.3647 | βxxx | 332.553 |
| μy | -1.7256 | βxyy | 28.1225 |
| μz | -1.1622 | βxzz | 169.130 |
| αxx | 326.469 | βyyy | 76.6903 |
| αyy | 275.883 | βxxy | -30.1071 |
| αzz | 317.294 | βyyz | 208.576 |
| αxy | 7.65703 | βzzz | 625.498 |
| αxz | 0.21.8633 | βxxz | 119.386 |
| αyz | 9.12301 | βyzz | 13.7068 |
| μTot | 2.1122 Debye | βxyz | -16.9650 |
| Δα | 84.0863 × 10−24 e.s.u. | βvec | 5.6627 × 10−30 e.s.u. |
| αTot | 45.4305 × 10−24 e.s.u. | βTot | 9.4378 × 10−30 e.s.u. |
3.4.9. NBO analysis
NBO analysis is used to study the inter and intra-molecular interaction among bonds with the help of interaction in both filled and unfilled orbital's. It is also used to investigate conjugative interaction or charge transfer in a molecular system [53]. The second-order perturbation theory analysis of the Fock matrix in NBO analysis of the title molecule is carried out by the DFT level B3LYP/6-311++G(d,p) method. This method is carried out for possible interactions between all the filled NBOs (Lewis or donor) to unfilled (non-Lewis or acceptor) NBOs as a result of the loss of occupancy from donor to acceptor. For each donor (i) and acceptor (j) NBO, and the stabilization energy between donor and acceptor E(2) associated with delocalization of i→j is estimated as [53].
| (21) |
Where qi → donor orbital occupancy; Ei - Ej → diagonal element, and F(i,j) → off-diagonal NBO Fock matrix elements.
The second-order Fock matrix analysis is reported in Table 8, it explains the electron donor and electron acceptor orbital and the interacting stabilization energy of the target molecule. The more intensive interaction takes place between electron donor to electron acceptors having large E(2) value. This more tendency of electron donor to acceptor leads to the greater extent of conjugation in the whole system [54, 55].
Table 8.
Second order perturbation theory analysis of Fock matrix in NBO basis.
| NBO NO (Donor) | Donor (i) | ED/e | NBO NO (Acceptor) | Acceptor (j) | ED/e | E(2) kcal/mol | E(j)-E(i) a.u. | F(i,j) a.u. |
|---|---|---|---|---|---|---|---|---|
| 4 | σ C 1 - H 33 | 1.98028 | 794 | σ∗ N 4 - C 6 | 0.03414 | 171.77 | 0.30 | 0.203 |
| 7 | σ C 2 - H 34 | 1.96215 | 794 | σ∗ N 4 - C 6 | 0.03414 | 17.86 | 0.21 | 0.055 |
| 16 | σ C 6 - C 13 | 1.97312 | 794 | σ∗ N 4 - C 6 | 0.03414 | 28.64 | 0.35 | 0.089 |
| 17 | σ C 6 - H 37 | 1.96643 | 794 | σ∗ N 4 - C 6 | 0.03414 | 15.33 | 0.21 | 0.051 |
| 20 | π C 7 - C 9 | 1.67193 | 805 | π∗ C 8 - C 10 | 0.02406 | 20.11 | 0.27 | 0.067 |
| 20 | π C 7 - C 9 | 1.67193 | 812 | π∗ C 11 - C 12 | 0.38884 | 20.31 | 0.27 | 0.068 |
| 22 | π C 8 - C 10 | 1.69351 | 803 | π∗ C 7 - C 9 | 0.38037 | 19.31 | 0.31 | 0.070 |
| 22 | π C 8 - C 10 | 1.69351 | 812 | π∗ C 11 - C 12 | 0.38884 | 18.32 | 0.29 | 0.067 |
| 26 | σ C 10 - C 12 | 1.97135 | 794 | σ∗ N 4 - C 6 | 0.03414 | 110.89 | 0.50 | 0.211 |
| 26 | σ C 10 - C 12 | 1.97135 | 852 | σ∗ O 31 - H 56 | 0.00729 | 26.17 | 4.16 | 0.296 |
| 29 | π C 11 - C 12 | 1.65053 | 803 | π∗ C 7 - C 9 | 0.38037 | 20.08 | 0.30 | 0.069 |
| 29 | π C 11 - C 12 | 1.65053 | 805 | π∗ C 8 - C 10 | 0.02406 | 20.96 | 0.28 | 0.069 |
| 33 | π C 13 - C 14 | 1.67992 | 821 | π∗ C 15 - C 17 | 0.39062 | 19.55 | 0.27 | 0.066 |
| 33 | π C 13 - C 14 | 1.67992 | 824 | π∗ C 16 - C 18 | 0.38728 | 19.82 | 0.27 | 0.067 |
| 34 | σ C 13 - C 15 | 1.96850 | 794 | σ∗ N 4 - C 6 | 0.03414 | 28.66 | 0.43 | 0.099 |
| 35 | σ C 14 - C 16 | 1.97310 | 794 | σ∗ N 4 - C 6 | 0.03414 | 223.74 | 0.56 | 0.317 |
| 35 | σ C 14 - C 16 | 1.97310 | 852 | σ∗ O 31 - H 56 | 0.00729 | 25.89 | 4.22 | 0.297 |
| 38 | π C 15 - C 17 | 1.69693 | 816 | π∗ C 13 - C 14 | 0.36835 | 19.23 | 0.31 | 0.070 |
| 38 | π C 15 - C 17 | 1.69693 | 824 | π∗ C 16 - C 18 | 0.38728 | 17.60 | 0.29 | 0.065 |
| 39 | σ C 15 - H 42 | 1.97694 | 794 | σ∗ N 4 - C 6 | 0.03414 | 10.30 | 0.25 | 0.045 |
| 40 | σ C 16 - C 18 | 1.97702 | 794 | σ∗ N 4 - C 6 | 0.03414 | 185.91 | 0.43 | 0.253 |
| 41 | π C 16 - C 18 | 1.68267 | 816 | π∗ C 13 - C 14 | 0.36835 | 19.24 | 0.30 | 0.069 |
| 41 | π C 16 - C 18 | 1.68267 | 821 | π∗ C 15 - C 17 | 0.39062 | 18.72 | 0.28 | 0.066 |
| 42 | σ C 16 - H 43 | 1.97789 | 794 | σ∗ N 4 - C 6 | 0.03414 | 161.65 | 0.25 | 0.179 |
| 43 | σ C 17 - C 18 | 1.96710 | 794 | σ∗ N 4 - C 6 | 0.03414 | 67.89 | 0.43 | 0.152 |
| 44 | σ C 17 - O 30 | 1.99341 | 794 | σ∗ N 4 - C 6 | 0.03414 | 11.37 | 0.63 | 0.076 |
| 45 | σ C 18 - O 31 | 1.99368 | 794 | σ∗ N 4 - C 6 | 0.03414 | 450.50 | 0.54 | 0.443 |
| 45 | σ C 18 - O 31 | 1.99368 | 835 | σ∗ C 21 - C 22 | 0.02466 | 20.56 | 1.34 | 0.149 |
| 45 | σ C 18 - O 31 | 1.99368 | 852 | σ∗ O 31 - H 56 | 0.00729 | 131.33 | 4.20 | 0.663 |
| 52 | σ C 21 - C 22 | 1.97289 | 794 | σ∗ N 4 - C 6 | 0.03414 | 61.77 | 0.44 | 0.148 |
| 54 | π C 21 - C 23 | 1.65053 | 839 | π∗ C 22 - C 24 | 0.33011 | 19.61 | 0.28 | 0.066 |
| 54 | π C 21 - C 23 | 1.65053 | 846 | π∗ C 25 - C 26 | 0.33762 | 35.84 | 0.17 | 0.070 |
| 56 | π C 22 - C 24 | 1.66908 | 837 | π∗ C 21 - C 23 | 0.34412 | 19.98 | 0.32 | 0.072 |
| 60 | σ C 24 - C 26 | 1.97938 | 794 | σ∗ N 4 - C 6 | 0.03414 | 27.13 | 0.42 | 0.096 |
| 63 | π C 25 - C 26 | 1.66755 | 837 | π∗ C 21 - C 23 | 0.34412 | 19.34 | 0.29 | 0.067 |
| 63 | π C 25 - C 26 | 1.66755 | 839 | π∗ C 22 - C 24 | 0.33011 | 20.68 | 0.28 | 0.068 |
| 69 | σ O 31 - H 56 | 1.98720 | 794 | σ∗ N 4 - C 6 | 0.03414 | 815.41 | 0.34 | 0.472 |
| 69 | σ O 31 - H 56 | 1.98720 | 835 | σ∗ C 21 - C 22 | 0.02466 | 27.80 | 1.14 | 0.159 |
| 69 | σ O 31 - H 56 | 1.98720 | 850 | σ∗ O 29 - H 54 | 0.00904 | 10.50 | 2.66 | 0.149 |
| 69 | σ O 31 - H 56 | 1.98720 | 852 | σ∗ O 31 - H 56 | 0.00729 | 191.15 | 4.00 | 0.782 |
| 103 | LP (2) O 27 | 1.89124 | 785 | σ∗ C 1 - C 3 | 0.06047 | 19.81 | 0.66 | 0.103 |
| 103 | LP (2) O 27 | 1.89124 | 791 | σ∗ C 3 - C 5 | 0.06997 | 20.67 | 0.66 | 0.105 |
| 105 | LP (2) O 28 | 1.89798 | 794 | σ∗ N 4 - C 6 | 0.03414 | 18.65 | 0.06 | 0.031 |
| 105 | LP (2) O 28 | 1.89798 | 805 | π∗ C 8 - C 10 | 0.02406 | 23.35 | 0.36 | 0.089 |
| 107 | LP (2) O 29 | 1.87605 | 812 | π∗ C 11 - C 12 | 0.38884 | 26.62 | 0.35 | 0.092 |
| 109 | LP (2) O 30 | 1.88000 | 821 | π∗ C 15 - C 17 | 0.39062 | 27.16 | 0.35 | 0.093 |
| 110 | LP (1) O 31 | 1.97909 | 794 | σ∗ N 4 - C 6 | 0.03414 | 509.62 | 0.31 | 0.355 |
| 110 | LP (1) O 31 | 1.97909 | 835 | σ∗ C 21 - C 22 | 0.02466 | 15.92 | 1.11 | 0.119 |
| 110 | LP (1) O 31 | 1.97909 | 846 | π∗ C 25 - C 26 | 0.33762 | 47.19 | 0.52 | 0.152 |
| 110 | LP (1) O 31 | 1.97909 | 851 | σ∗ O 30 - H 55 | 0.00716 | 13.15 | 2.76 | 0.171 |
| 110 | LP (1) O 31 | 1.97909 | 852 | σ∗ O 31 - H 56 | 0.00729 | 121.34 | 3.97 | 0.622 |
| 111 | LP (2) O 31 | 1.87823 | 824 | π∗ C 16 - C 18 | 0.38728 | 27.12 | 0.35 | 0.093 |
| 111 | LP (2) O 31 | 1.87823 | 850 | σ∗ O 29 - H 54 | 0.00904 | 14.38 | 2.36 | 0.169 |
E (2) → Energy of hyper conjugative interaction (Stabilization energy).
E (j)-E (i) → The energy difference between the donor (i) and acceptor (j) NBO orbital.
F (i,j) → Fock Matrix element between i and j NBO orbital.
ED/e → Electron density.
The intra-molecular interaction takes place by the orbital overlap between nonbonding LP (2) O27 donor to σ∗ C1 – C3 acceptor and LP (2) O 27 donor to σ∗ C3 – C5 acceptor are found to be 19.81 and 20.67 Kcal/mol respectively. These interaction values around the piperidone ring indicate the large bioactivity of the target molecule [40].
The largest stabilization energy is developed between σ O31 – H56 to σ∗ N4 – C6 donor-acceptor system (815.41 Kcal/mol). It helps the molecule to stabilize. The stabilization energy of 509.62 Kcal/mol, due to the interaction between the lone pair of O31- σ∗ N4 – C6 also helps for stabilization. The contribution of C18 – O31 to σ∗ N4 – C6 system also helps to stabilize the molecule with the energy of 450.50 Kcal/mol. The presence of π bonds present in the ring also helps to stabilize the molecule further. The contribution of σ C14 – C16, to σ C16 – C18 and σ C1 – H33 to σ∗ N4 – C6 with the energy of 223.74, 185.91, and 171.77 Kcal/mol are non-negligible.
3.4.10. Thermodynamic properties
The thermodynamic properties like thermal energy (E), specific heat capacity (cv), and entropy (S) for the title compound was obtained by DFT with the B3LYP/6-311++G(d,p) method and presented in Table 9. The statistical thermodynamic analysis of the target molecule was carried out at room temperature of 298.15 K and 1 atm pressure. The total energy of a molecule is the sum of translational, rotational, vibrational, and electronic energies [49].
| (22) |
Table 9.
Thermodynamic properties of the title compound B3LYP/6-311++G(d,p).
| Temperature (Kelvin) | E (thermal) Kcal/mol |
cv cal/mol-Kelvin |
Entropy cal/mol-Kelvin |
|---|---|---|---|
| 100 | 287.638 | 41.875 | 111.625 |
| 200 | 293.489 | 75.468 | 152.227 |
| 298.15 | 302.585 | 109.941 | 189.561 |
| 300 | 302.789 | 110.587 | 190.260 |
| 400 | 315.538 | 143.701 | 227.280 |
| 500 | 331.354 | 171.673 | 262.899 |
| 600 | 349.691 | 194.231 | 296.631 |
| 700 | 370.053 | 212.374 | 328.290 |
| 800 | 392.054 | 227.170 | 357.912 |
| 900 | 415.403 | 239.438 | 385.633 |
| 1000 | 439.877 | 249.749 | 411.618 |
The thermodynamic properties such as thermal energy (E), specific heat capacity (cv), and entropy (S) for various temperature ranges from 100 K to 1000 K are graphically represented in Figure 8. All the thermodynamic properties are increasing with an increase in temperature ranges from 100 K to 1000 K. This is due to the fact that the molecular vibrational intensities increase with temperature [40]. The corresponding fitting factors (R2) for the thermodynamic properties are found to be 0.973, 0.962, and 0.995 respectively.
Figure 8.

Variation of thermodynamic properties with temperature of BDPO.
All the thermodynamic properties of the target molecule are done on the gaseous phase and these data provide very helpful information for further investigation of the title compound. All the data provides thermodynamic energies to evaluate the directions of chemical reactions according to the second law of thermodynamics in the thermo chemical field.
3.5. Non covalent interaction (NCI) analysis
The NCI interaction, which is also known as reduced density gradient (RDG) is a famous method to investigate weak interaction like Van der Waals forces, hydrogen bond, halogen bond, and steric effect present in the molecule. Multiwfn 3.7 is used to calculate the interaction and the result is viewed by VMD 1.9.3 tool [56]. The RDG is calculated by the following relation
| (23) |
The RDG for the BDPO molecule is studied and shown in Figure 9. Here sign (λ2)ρ(a.u) is plotted against RDG. The spikes near -0.010 to +0.010 a.u. confirm the presence of Van der Waals forces within the molecule. The spikes near 0.020 a.u. confirm the presence of a steric effect within the molecule.
Figure 9.

(a) & (b): NCI interaction and isosurface of BDPO.
The cube files are generated by the Multiwfn 3.7 tool and viewed by VMD 1.9.3 tool to confirm the above statement. Many Van der Waals force places are identified (green dots) around the middle six-membered ring. The red spheres present inside all the rings suggest the presence of a steric effect. Even though this molecule has two sets of two nearby –OH groups, they do not have hydrogen bonds but electrostatic force can be seen between these moieties.
3.6. Surface projection map
Localized orbital locator (LOL) is an important function to locate high localization region like ELF, which is defined by Schmider and Becke [57].
| (24) |
where,
D0(r) for spin-polarized and close shell systems which are defined in the same manner as in ELF. LOL also has similar relation compared to ELF.
The surface projected map calculation for the BDPO is investigated and shown in Figure 10. From Figure 10 it is clear that LOL is seen in the molecule around the aromatic rings, which bears OH groups (red colour circles). Also, an electron depleted area is seen inside the molecule (blue coloured circles).
Figure 10.

Shaded surface map with projection of LOL of BDPO molecule.
3.7. Electron excitation analysis
The presence of electron donor groups (OH) and π-linker (ϕ) and electron withdrawing groups (C=O) are present in the molecule, it is decided to carry out electron excitation analysis for first five excitations with the help of the Multiwfn 3.7 tool. The results obtained are shown in Table 10.
Table 10.
Electron excitation analysis of BDPO.
| Excitation | D (Å) | Sm (a.u.) | Sr(a.u.) | H (Å) | τ (Å) | Excitation energy (eV) | Coulomb attractive energy (eV) |
|---|---|---|---|---|---|---|---|
| S0 → S1 | 0.437 | 0.28113 | 0.54275 | 1.914 | -0.768 | 4.293 | 7.262503 |
| S0 → S2 | 0.616 | 0.51800 | 0.79799 | 2.612 | -1.118 | 4.835 | 5.350274 |
| S0 → S3 | 0.427 | 0.58251 | 0.84582 | 2.500 | -0.998 | 4.958 | 5.598035 |
| S0 → s4 | 1.591 | 0.17985 | 0.42236 | 3.647 | -0.559 | 5.152 | 3.661583 |
| S0 → S5 | 1.472 | 0.19635 | 0.42703 | 3.608 | -0.571 | 5.202 | 3.694333 |
The molecule was optimized by the B3LYP/6-311++G(d,p) level of theory. TDDFT calculation was carried out with CAM-B3LYP IOp (9/40 = 4) keyword.
The D index value for S0→ S1 and S0→ S3 is very small since it is even less than half the length of a typical C - C bond. The D index value for S0→ S4 and S0→ S5 is more than 1, it may undergo charge transfer (CT). But the negative τ values confirm that these excitations are of LE (Local excitation) only.
The Sr index reaches above 0.5 (the theoretical upper limit 1.0) for first three excitations and found to be large value implies that about half part of a hole and electron has perfectly matched. Particularly Sr index for S0→ S2 and S0→ S3 reach 0.8 reveals the presence of highly localized π-π∗ type of excitation on benzene ring.
H-index refers to breadth of the average distribution of hole and electron. From the Table 10 it is clear that, except S0 → S1, all the other excitations evidently wider and hence the H indices are larger.
If the τ index value is positive, indicating that the separation of hole and electron is obvious, and it can be considered as a charge transfer excitation. Since all the τ values are negative, confirms the absence of CT type of excitation.
The excitation energy gradually increases from S0 → S1 to S0 → S5. The hole-electron Coulomb attractive energy shown in Table 10 is closely related to the electron excitation property. It is always related to the D index. It is very easy to understand, that the high value of the D index is the farther the distance between the main distribution region of hole and electron. So the Coulomb attractive energy is weaker for that particular excitation. In this case, S0 → S4 has a high D index value (1.591 Ǻ) and low Coulomb attractive energy (3.661583eV).
The Coulomb attractive energy is very large for S0 → S1 and the H index for the same excitation are found to be very low. Since the spatial extent of their hole and electron is very narrow, they have strong Coulomb attraction energy. From the above all points, we can conclude the following facts.
S0 → S1 LE excitation of n-π∗ type.
S0 → S2 LE excitation of π-π∗ type.
S0 → S3 LE excitation of π-π∗ type.
S0 → S4 LE excitation of n-π∗ type.
S0 → S5 LE excitation of n-π∗ type.
4. Conclusion
In this present work, the docking studies of 2,6-bis(3,4-dihydroxyphenyl)-3-phenethylpiperidin-4-one (BDPO) molecule confirms that it could be an attractive object for future medicinal and pharmacological studies to cure ulcers. Computational theoretical studies using density functional theory have been done to get the optimized structure of the 2,6-bis(3,4-dihydroxyphenyl)-3-phenethylpiperidin-4-one molecule. The HOMO-LUMO energy gap of the piperidone derivative (4.6880 eV) provides the stability of the molecule. Molecular electrostatic potential and Mulliken population analysis explains the nucleophilic and electrophilic centers present in the target molecule. Natural bond orbital analysis confirms the molecule is stable. The higher first-order hyper polarizability value suggests that the molecule is a good NLO material. The thermodynamic properties like total energy, specific heat capacity, entropy increase with increasing temperature. There is no intermolecular hydrogen bonding is seen and the first five excitations are of LE type only.
Declarations
Author contribution statement
J. Winfred Jebaraj: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Contributed reagents, materials, analysis tools or data; Wrote the paper.
T. Sasitha: Conceived and designed the experiments; Performed the experiments; Analyzed and interpreted the data; Wrote the paper.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability statement
Data included in article/supplementary material/referenced in article.
Declaration of interests statement
The authors declare no conflict of interest.
Additional information
No additional information is available for this paper.
Acknowledgements
The authors are thankful to the management of St. John's College, Palayamkottai for providing the computer and software facilities.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
Supplementary materials
References
- 1.SankaraPandian N., Siva Prakash B., ThillaiNatarajan M., Ramalingan C., VelayuthamPillai M. Computational aspects of (E)-O-carbomethoxy methyl oxime ether of 1,3-dimethyl 2,6-diphenylpiperidin-4-one. Int. J. Innov. Tech. Expl. Eng. 2019;9(2S2):701–706. [Google Scholar]
- 2.Ramalingan C., Park Y.T., Kabilan S. Synthesis, stereochemistry, and antimicrobial evaluation of substituted piperidin-4-one oxime ethers. Eur. J. Med. Chem. 2006;41:683–696. doi: 10.1016/j.ejmech.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Aridoss G., Balasubramanian S., Parthiban P., Kabilan S. Synthesis, stereochemistry and antimicrobial evaluation of some N-morpholinoacetyl-2,6-diarylpiperidin-4-ones. Eur. J. Med. Chem. 2007;42:851–860. doi: 10.1016/j.ejmech.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Aridoss G., Parthiban P., Ramachandran R., Prakash M., Kabilan S., Tae Jeong Y. Synthesis and spectral characterization of a new class of N-(N-methylpiperazinoacetyl)-2,6-diarylpiperidin-4-ones:Antimicrobial, analgesic and antipyretic studies. Eur. J. Med. Chem. 2009;44:577–592. doi: 10.1016/j.ejmech.2008.03.031. [DOI] [PubMed] [Google Scholar]
- 5.Purnima T., Avinash C.T., Viney C., Shailendra K.S. Syntheses, characterization and evaluation of novel 2,6-diarylpiperidin-4-ones as potential analgesic-antipyretic agents. Eur. J. Med. Chem. 2014;82:439–448. doi: 10.1016/j.ejmech.2014.05.080. [DOI] [PubMed] [Google Scholar]
- 6.El-Subbagh H.I., Abu-Zaid S.M., Mahran M.A., Badria F.A., Al-Obaid A.M. Synthesis and biological evaluation of certain α,β-unsaturated ketones and their corresponding fused pyridines as antiviral and cytotoxic agents. J. Med. Chem. 2000;43(15):2915–2921. doi: 10.1021/jm000038m. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L., Yang M., Song Y., Sun Z., Peng Y., Qu K., Zhu H. Antihypertensive effect of 3,3,5,5-tetramethyl-4-piperidone, a new compound extracted from Marasmiusandrosaceus. J. Ethnopharm. 2009;123:34–39. doi: 10.1016/j.jep.2009.02.033. [DOI] [PubMed] [Google Scholar]
- 8.Baracu V. Dobre, Duvaz I.N. Potential anticancer agents. XXVI. Spin labelled nitrosoureas. J. Prakt. Chem. 1985;327(4):667–674. [Google Scholar]
- 9.Rameshkumar N., Veena A., Ilavarasan R., Adiraj M., Shanmugapandiyan P., Sridhar S.K. Synthesis and biological activities of 2,6-Diaryl-3-methyl-4 piperidone Derivatives. Biol. Pharm. Bull. 2003;26(2):188–193. doi: 10.1248/bpb.26.188. [DOI] [PubMed] [Google Scholar]
- 10.Aridoss G., Amirthaganesan S., Kumar N.A., Kim J.T., Lim K.T., Kabilan S., Jeong Y.T. A facile synthesis, antibacterial, and antitubercular studies of some piperidin-4-one and tetrahydropyridine derivatives. Bioor. Med. Chem. Lett. 2008;18:6542–6548. doi: 10.1016/j.bmcl.2008.10.045. [DOI] [PubMed] [Google Scholar]
- 11.Pavadai P., Ramalingam S., Panneerselvam T., Kunjiappan S., Perumal P., Mani V., Saravanan G., Alagarsamy V., Ammunje D.N., Chimakurthy J. Synthesis of piperidine-4-one derivative containing dipeptides: an acetylcholinesterase and β-secretase inhibitor. Anti-Infec. Agents. 2019;17(1):1–9. [Google Scholar]
- 12.Ganellin C.R., Spickett R.G.W. Compounds affecting the central nervous system. I. 4-Piperidones and related compounds. J. Med. Chem. 1965;8(5):619–625. doi: 10.1021/jm00329a015. [DOI] [PubMed] [Google Scholar]
- 13.Aridoss G., Balasubramanian S., Parthiban P., Kabilan S. Synthesis and in vitro microbiological evaluation of imidazo(4,5-b)pyridinylethoxypiperidones. Eur. J. Med. Chem. 2006;41:268–275. doi: 10.1016/j.ejmech.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Ouf S.A., Gomha S.M., Eweis M., Ouf A.S., Sharawy I.A. Efficiency of newly prepared thiazole derivatives against some cutaneous fungi. Bioorg. Med. Chem. 2018;26:3287–3295. doi: 10.1016/j.bmc.2018.04.056. [DOI] [PubMed] [Google Scholar]
- 15.Ouf S.A., Gomha S.M., Ewies M.M., Sharawy I.A.A. Synthesis, characterization, and antifungal activity evaluation of some novel arylazothiazoles. J. Heterocycl. Chem. 2018;55(1):258–264. [Google Scholar]
- 16.Boddupalli B.M., Ramani R., Subramaniam B., Anisetti R.N. In vitro and invivo evaluation of hepato protection and anti ulcer activities of piperine gastro retentive micropspheres. Asian Pac. J. Trop. Biomed. 2012;2(3):S1237–S1240. [Google Scholar]
- 17.Sutton P., Mitchell H.M. CAB International; 2010. Helicobacter pylori in the 21st Century; pp. 1–302. [Google Scholar]
- 18.Muh’d M.B., Uzairu A., Shallangwa G.A., Uba S. Molecular docking and quantitative structure-activity relationship study of anti-ulcer activity of quinazolinone derivatives. J. King Saud Univ. Sci. 2020;32:657–666. [Google Scholar]
- 19.Sussman J.L., Lin D., Jiang J., Manning N.O., Prilusky J., Ritter O., Abola E.E. Protein data bank (PDB): database of three-dimensional structural information of biological macromolecules. ActaCryst. 1998;D54:1078–1084. doi: 10.1107/s0907444998009378. [DOI] [PubMed] [Google Scholar]
- 20.Discovery Studio Visualizer Software, Version 4.0. 2012. http://www.accelrys.com [Google Scholar]
- 21.ACD/ChemSketch, Version 2020.1.2. Advanced Chemistry Development, Inc.; Toronto, ON, Canada: 2020. www.acdlabs.com [Google Scholar]
- 22.Noller C.R., Baliah V. The preparation of some piperidine derivatives by the mannich reaction. J. Am. Chem. Soc. 1948;70(11):3853–3855. doi: 10.1021/ja01191a092. [DOI] [PubMed] [Google Scholar]
- 23.Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012;4(17):1–33. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dallakyan S., Olson A. Small-molecule library screening by docking with PyRx. J. Methods Mol Biol. 2015;1263:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- 25.Trott O., Olson A.J. Software news and update AutoDockVina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeLano W.L. 2002. The PyMOL User's ManuaDeLano Scientific, San Carlos, CA, USA. [Google Scholar]
- 27.Parr R.G., Yang W. Oxford University Press, New York, Clarendon Press; Oxford: 1989. Density Functional Theory of Atoms and Molecules. [Google Scholar]
- 28.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Peterson G.A., Nakatsuji H., Li X., Caricato M., Marenich A.V., Bloino J., Janesko B.G., Comperts R., Mennucci B., Hratchian H.P., Oritz J.V., Izmaylov A.F., Sonnenberg J.L., Williams-Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vereven T., throssell K., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M.J., Heyd J.J., Borhters E.N., Kudin K.N., Staroverov V.N., Keith T.A., Kobayashi R., Normand J., Raghavachari K., Rendell A.P., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Millam J.M., Klene M., Adamo C., Cammi R., Ochterski J.W., Martin R.L., Morokuma K., Farkas O., Forseman J.B., Fox D.J. Gaussian, Inc.; Wallingford CT: 2016. Gaussian 16, Revision B.01. [Google Scholar]
- 29.Dennington R., Keith T.A., Millam J.M. Semichem Inc.; Shawnee Mission, KS: 2016. GaussView, Version 6.1. [Google Scholar]
- 30.Arockia doss M., Savithiri S., Rajarajan G., Thanikachalama V., Anbuselvana C. Synthesis, electronic structure investigation of 3-pentyl-2,6-di(furan- 2-yl)piperidin-4-one by FT-IR, FT-Raman and UV–visible spectral studies and ab initio/DFT calculations. Spectrochim. Acta A: Mol. Biomol. Spect. 2015:1–48. doi: 10.1016/j.saa.2015.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Lu T., Chen F. Multiwfn: a multifunctional wavefunctionanalyzer. J. Comput. Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 32.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, druglikeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;42717:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1-3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 34.Veber D.F., Johnson S.R., Cheng H., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of Drug Candidates. J. Med. Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 35.Egan W.J., Merz K.M., Baldwin J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 36.Ghose A.K., Viswanadhan V.N., Wendoloski J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery, A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 37.Muegge I., Heald S.L., Brittelli D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001;44(12):1841–1846. doi: 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- 38.Kataria R., Khatkar A. Molecular docking, synthesis, kinetics study, structure–activity relationship and ADMET analysis of morin analogous as Helicobacterpylori urease inhibitors, Kataria and Khatkar. BMC Chem. 2019;45:1–17. doi: 10.1186/s13065-019-0562-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaussum N.M., O'Boyle, Tenderholt A.L., Langner K.M. Software news and updates cclib: a library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008;29:839–845. doi: 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- 40.Velraj G., Soundharam S., Sridevi C. Structure, vibrational, electronic, NBO and NMR analyses of 3-methyl-2,6-diphenylpiperidin-4-one (MDPO) by experimental and theoretical approach. J. Mol. Str. 2014;1060:156–165. doi: 10.1016/j.saa.2014.08.075. [DOI] [PubMed] [Google Scholar]
- 41.Srivastava A., Tandon P., Ayala A.P., Jain S. Solid state characterization of an antioxidant alkaloid boldine using vibrational spectroscopy and quantum chemical calculations. Vib. Spect. 2011;56:82–88. [Google Scholar]
- 42.Abbaz T., Bendjeddou A., Villemin D. Theoretical investigations on molecular structure, homo, lumo, nbo analysis and hyperpolarizability calculations of mono and extended-tetrathiafulvalenes. Indo. Am. J. Pharm. Sci. 2018;5(4):2488–2498. [Google Scholar]
- 43.Hehre W.J. Wavefunction, Inc.; Irvine, California: 2003. A Guide to Molecular Mechanics and Quantum Chemical Calculations, Printed in the United States of America; pp. 40–47. [Google Scholar]
- 44.Bendjeddou A., Abbaz T., Gouasmia A., Villemin D. Molecular structure, first order hyperpolarizability, homo and lumo analysis, mep and nbo analysis of MonohydroxymethylTTFSBY density functional theory. Glob. J. Eng. Sci. Res. 2017;4(4):21–35. [Google Scholar]
- 45.Azad I., Akhter Y., Khan T., Irfan Azad M., Chandra S., Singh P., Kumar D., Nasibullah M. Synthesis, quantum chemical study, AIM simulation, in silicoADMET profileanalysis, molecular docking and antioxidant activity assessment of aminofuran derivatives. J. Mol. Str. 2020;1203:127285. [Google Scholar]
- 46.Chattaraj P.K., Chakraborty A., Giri S. Net Electrophilicity. J. Phys. Chem. A. 2009;113:10068–10074. doi: 10.1021/jp904674x. [DOI] [PubMed] [Google Scholar]
- 47.Fatma S., Bishnoi A., Kumar Vermaa A. Synthesis, spectral analysis (FT-IR, 1HNMR, 13CNMR and UV-visible) and quantum chemical studies on molecular geometry, NBO, NLO, chemical reactivity and thermodynamic properties of novel 2-amino-4-(4-(dimethylamino)phenyl)-5-oxo-6-phenyl-5,6-dihydro-4H-pyrano[3,2-c]quinoline-3-carbonitrile. J. Mol. Str. 2015;1095:112–124. [Google Scholar]
- 48.Demircioglu Z., AlbayrakKastas C., Buyukgungor O. Theoretical analysis (NBO, NPA, Mulliken Population Method) and molecular orbital studies (hardness, chemical potential, electrophilicity and Fukui function analysis) of (E)-2-((4-hydroxy- 2-methylphenylimino)methyl)-3-methoxyphenol. J. Mol. Str. 2015;1091:183–195. [Google Scholar]
- 49.Suresh M., Syed Ali Padusha M., Bharanidharan S., Saleem H., Dhandapani A., Manivarman S. Synthesis, spectral characterization and density functional theory exploration of 1-(quinolin-3-yl)piperidin-2-ol. Spectrochim. Acta A: Mol. Biomol. Spect. 2015;144:243–257. doi: 10.1016/j.saa.2015.02.018. [DOI] [PubMed] [Google Scholar]
- 50.Kleinmen D.A. Nonlinear dielectric polarization in op-tical media. Phys. Rev. 1962;126:1977–1979. [Google Scholar]
- 51.Sambathkumar K., Ravichandran G. FTIR, FT-RAMAN, AB INITIO and DFT studies on 2-Methoxy-6-Methyl Pyridine. Elixir. Comp. Chem. 2016;91:38077–38086. [Google Scholar]
- 52.Rajasekhar B., Muhammad Hijaz P.K., Swu T. Computational study on non-linear optical property of wittig based schiff-base ligands (both Z & E isomers) & copper(II) complex. J. Mol. Str. 2018;1168:212–222. [Google Scholar]
- 53.Xavier S., Periandy S., Ramalingam S. NBO, conformational, NLO, HOMO–LUMO, NMR and electronic spectral study on 1-phenyl-1-propanol by quantum computational methods. Spectrochim. Acta A: Mol. Biomol. Spect. 2015;137:306–320. doi: 10.1016/j.saa.2014.08.039. [DOI] [PubMed] [Google Scholar]
- 54.Suvitha A., Periandy S., Boomadevi S., Govindarajan M. Vibrational frequency analysis, FT-IR, FT-Raman, ab initio, HF and DFT studies, NBO, HOMO–LUMO and electronic structure calculations on pycolinaldehydeoxime. Spectrochim. Acta A: Mol. Biomol. Spect. 2014;117:216–224. doi: 10.1016/j.saa.2013.07.080. [DOI] [PubMed] [Google Scholar]
- 55.Erdogdu Y., Unsalan O., Amalanathan M., Hubert Joe I. Infrared and Raman spectra, vibrational assignment, NBO analysis and DFT calculations of 6-aminoflavone. J. Mol. Str. 2010;980:24–30. [Google Scholar]
- 56.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 57.Schmider H.L., Becke A.D. Chemical content of the kinetic energy density. J. Mol. Str. (Theochem). 2000;527(1–3):51–61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials
Data Availability Statement
Data included in article/supplementary material/referenced in article.