

Abstract
Background
Duchenne muscular dystrophy (DMD) causes severe disability of children and death of young men, with an incidence of approximately 1/5000 male births. Symptoms appear in early childhood, with a diagnosis made mostly around 4 years old, a time where the amount of muscle damage is already significant, preventing early therapeutic interventions that could be more efficient at halting disease progression. In the meantime, the precise moment at which disease phenotypes arise—even asymptomatically—is still unknown. Thus, there is a critical need to better define DMD onset as well as its first manifestations, which could help identify early disease biomarkers and novel therapeutic targets.
Methods
We have used both human tissue‐derived myoblasts and human induced pluripotent stem cells (hiPSCs) from DMD patients to model skeletal myogenesis and compared their differentiation dynamics with that of healthy control cells by a comprehensive multi‐omic analysis at seven time points. Results were strengthened with the analysis of isogenic CRISPR‐edited human embryonic stem cells and through comparisons against published transcriptomic and proteomic datasets from human DMD muscles. The study was completed with DMD knockdown/rescue experiments in hiPSC‐derived skeletal muscle progenitor cells and adenosine triphosphate measurement in hiPSC‐derived myotubes.
Results
Transcriptome and miRnome comparisons combined with protein analyses demonstrated that hiPSC differentiation (i) leads to embryonic/foetal myotubes that mimic described DMD phenotypes at the differentiation endpoint and (ii) homogeneously and robustly recapitulates key developmental steps—mesoderm, somite, and skeletal muscle. Starting at the somite stage, DMD dysregulations concerned almost 10% of the transcriptome. These include mitochondrial genes whose dysregulations escalate during differentiation. We also describe fibrosis as an intrinsic feature of DMD skeletal muscle cells that begins early during myogenesis. All the omics data are available online for exploration through a graphical interface at https://muscle‐dmd.omics.ovh/.
Conclusions
Our data argue for an early developmental manifestation of DMD whose onset is triggered before the entry into the skeletal muscle compartment, data leading to a necessary reconsideration of dystrophin roles during muscle development. This hiPSC model of skeletal muscle differentiation offers the possibility to explore these functions as well as find earlier DMD biomarkers and therapeutic targets.
Keywords: Duchenne muscular dystrophy, Myogenesis, Human pluripotent stem cells, Omics
Introduction
Duchenne muscular dystrophy (DMD) is a rare genetic disease, but it is the most common form of myopathy affecting approximately one in 5000 male births and very rarely female. In this recessive X‐linked monogenic disorder, mutations in the DMD gene lead to the loss of functional dystrophin protein, resulting in a progressive—yet severe—muscle wasting phenotype. 1 In patients, symptoms usually appear in early childhood (2–5 years old) and worsen with age, imposing the use of wheelchair before 15 and leading to premature death by cardiac and/or respiratory failure(s) mostly around 30 years of age. 2
At the age of diagnosis (mostly around 4 years old), muscles of DMD patients have already suffered from the pathology. 3 , 4 Several reviews pointed out the limitations of current disease biomarkers, which fail to detect the development of DMD specifically and at an early age. 5 , 6 Meanwhile, no treatment is available to stop this degenerative disease yet. Developing therapies aim at restoring the expression of dystrophin in muscle cells, but so far, the level stays too low to be beneficial to patients. 7 The absence of both reliable biomarkers and effective therapies stresses the need of better defining the first steps of DMD in humans to be able to increase diagnosis sensitivity and therefore improve patient management by accelerating their access to better healthcare as well as develop alternative therapeutic approaches by finding targets that compensate the lack of dystrophin and complement current attempts at restoring its expression. 8
In 2007, a seminal publication reported that the gene expression profile of muscles from asymptomatic DMD children younger than 2 years old is already distinguishable from healthy muscles, suggesting that DMD molecular dysregulations appear before disease symptomatic manifestations. 4 Evidence obtained in multiple animal models, such as neonatal GRMD dogs, 9 DMD zebrafish, 10 and mdx mouse embryos, 11 as well as in human foetuses, 12 , 13 , 14 even suggest that DMD starts before birth, during prenatal development. Our team recently identified the embryonic dystrophin isoform Dp412e expressed in early mesoderm‐committed cells, 15 another indication that DMD can start in utero. Further exploring DMD onset in human foetuses is extremely challenging for obvious ethical and practical reasons. A way to overcome these issues is to develop a human DMD model in vitro, recapitulating embryonic development from human pluripotent stem cells to skeletal muscle lineage.
To our knowledge, none of the existing human DMD in vitro models, either based on tissue‐derived myoblasts 16 or on the differentiation of induced pluripotent stem cells, 17 , 18 , 19 , 20 , 21 have been used for studying DMD during the ontogeny of the skeletal muscle lineage. Moreover, original protocols for in vitro myogenesis from human pluripotent stem cells (reviewed in Kodaka et al. 22 ) use transgene overexpression or/and cell sorting procedures and thereby miss the steps preceding skeletal muscle commitment, for example, paraxial mesoderm and myotome. Novel protocols have recently used transgene‐free directed differentiation to recapitulate human embryonic development in a dish, giving theoretical access to the developmental steps. 19 , 23 , 24 , 25
Using one of these protocols, 23 we compared the myogenic differentiation dynamics of healthy and DMD human induced pluripotent stem cells (hiPSCs) using a multi‐omic approach to identify early disease manifestations in vitro. DMD cells showed marked transcriptome dysregulations from Day 10, before the detection of skeletal muscle regulatory factors at Day 17. Specifically, we identified the dysregulation of mitochondrial genes as one of the earliest detectable phenotypes. These alterations escalated over the course of muscle specification. In addition, we showed an early induction of Sonic hedgehog (SSH) signalling pathway, followed by collagens as well as fibrosis‐related genes suggesting the existence of an intrinsic fibrotic process solely driven by DMD muscle cells. Overall, our data highlight that human pluripotent stem cells are a suitable cell model to study the ontogeny of skeletal muscle lineage in both healthy and disease conditions. In the context of DMD, they strongly argue for the existence of early disease manifestations during somite development.
Materials and methods
Ethics, consent, and permissions
The collection of primary myoblasts was established from patient muscle biopsies at the Cochin Hospital/Cochin Institute as part of the medical diagnostic procedure of neuromuscular disorders. A signed and informed consent was obtained by the Cochin Hospital cell bank/Assistance Publique‐Hôpitaux de Paris for each patient included in this study to collect, establish, and study primary cultures of fibroblasts and myoblasts. This collection of myoblasts was declared to (i) legal and ethical authorities at the Ministry of Research (declaration number 701, modified declaration number 701–1) via the medical hosting institution (Assistance Publique‐Hôpitaux de Paris) and to (ii) the ‘Commission Nationale de l'Informatique et des Libertés’ (declaration number 1154515).
Cells
Human primary adult myoblasts from healthy individuals and DMD patients were provided by Celogos laboratory and Cochin Hospital/Cochin Institute (Supporting Information, Table S1). DMD cells carry the following mutations: in‐frame duplication exons 3–26 (DMD Myoblast 1), out‐of‐frame deletion exons 8–43 (DMD Myoblast 2), and stop exon 7 c (DMD Myoblast 3). In Celogos laboratory, cell preparation was done according to patent US2010/018873 A1.
Cell culture
Human tissue‐derived myoblasts
Primary myoblasts were maintained in a myoblast medium: DMEM/F‐12, HEPES (31330–038, Thermo Fisher Scientific) supplemented with 10% foetal bovine serum (Hyclone, Logan, UT), 10 ng/mL fibroblast growth factor 2 (FGF2, 100‐18B, Peprotech), and 50 nM dexamethasone (D4902, Sigma‐Aldrich) on 0.1% gelatin‐coated (G1393, Sigma‐Aldrich) cultureware.
Human tissue‐derived myotubes
Primary myoblasts were differentiated into myotubes. Cells were seeded at 600 cells/cm2 on 0.1% gelatin‐coated cultureware in myoblast medium containing 1 mM acid ascorbic 2P (A8960, Sigma‐Aldrich).
Human induced pluripotent and embryonic stem cells
Primary myoblasts were reprogrammed into hiPSCs following the protocol described in Massouridès et al., 15 using the Yamanaka's factors POU5F1, SOX2, cMYC and KLF4 transduction by ecotropic or amphotropic vectors (Table S1). HiPSCs and human embryonic stem cells (hESCs) were adapted and maintained with mTeSR™1 culture medium (05850, Stemcell Technologies) on Corning® Matrigel® Basement Membrane Matrix coated cultureware (354234, Corning Incorporated). Cells were then seeded at 20 000 cells/cm2, passaged, and thawed each time with 10 μM StemMACS™ Y27632.
Human embryonic stem cell‐derived and human induced pluripotent stem cell‐derived cells
Six hiPSCs (three healthy and three DMD) were differentiated three times towards skeletal muscle lineage using commercial media designed from the work of Caron et al. 23 (Skeletal Muscle Induction medium SKM01, Myoblast Cell Culture Medium SKM02, Myotube Cell Culture Medium SKM03, AMSbio). This protocol is a 2D directed differentiation that uses three consecutive defined media (SKM01 from Day 0 to 10, SKM02 from Day 10 to 17, and SKM03 from Day 17 to 25) and only one cell passage at Day 10. Cells were seeded at 3500 cells/cm2 at Day 0 and Day 10 on BioCoat™ Collagen I cultureware (356485, Corning Incorporated). Part of the cell culture was frozen at Day 17 for further experiments such as DNA extraction. These cells were then thawed at 30 000 cells/cm2 and cultured in SKM02 for 3 days and SKM03 for 3 additional days to get myotubes. Two isogenic hESCs (one healthy and one DMD) were differentiated two times using the same protocol.
Gene editing
Exon 52 of the DMD gene was removed by gene editing. Benchling and Crispor software were used to design the single guide (sg) RNAs upstream and downstream of DMD exon 52 (sgRNA sequences are followed by scaffold sequences, which are underlined; upstream: GTTTGGTGATTCTTACGGACGTTTCAGAGCTATGCTGGAAACAGCATAGCAAGTTGAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT; downstream: GCCCACCCTACTACGGCATAGTTTCAGAGCTATGCTGGAAACAGCATAGCAAGTTGAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT). SgRNAs were diluted at 150 pmol/μL in TE 1X buffer and then at 30 pmol/μL in nuclease‐free water. The ribonucleoprotein complex was formed in 25 μL, with 20 pmol of Cas9 2NLS nuclease (S. pyrogenes, Synthego) mixed to 90 pmol of each diluted sgRNAs in P3 solution (V4XP‐3024, Lonza), incubated at room temperature for 10 min before being stored at 4°C. After an amplification in mTeSR™1 culture medium (05850, Stemcell Technologies) on Corning® Matrigel® Basement Membrane Matrix coated cultureware, SA001 hESCs (Cellartis) were harvested and passed through a sieve (40 μm, Falcon). A total of 150 000 cells were centrifuged for 5 min at 90 g, resuspended in 5 μL of P3 solution, and mixed to the complex. The resulting 30 μL were then transferred to a 16‐well Nucleocuvette™ Strip processed with the 4D‐Nucleofector System™ (Lonza) to introduce the complex into cells by electroporation (CD118 program). Cells were then seeded at a non‐clonal density in pre‐warmed mTeSR™1 culture medium with 10 μM Rock Inhibitor on Corning® Matrigel® Basement Membrane Matrix coated 24‐well cultureware. One well was used 48 h later to do DNA extraction (QuickExtract™ DNA, Lucigen) and validate the deletion by PCR [using Q5® High‐Fidelity DNA Polymerase (New England Biolabs) and DMD Delta exon 52 primers (Table S2); with the program: 95°C for 5 min; for 35 cycles: 95°C for 30 s, 64°C for 30 s, 72°C for 39 s; and 72°C for 2 min; amplicon of 893 pb]. Seventy‐six hours after the CRISPR, one well was used for cell banking and another was used to be seeded into 96‐well plates at a clonal density with cloneR (#05888, Stemcell Technologies) addition and a medium change after 2 days. The first half of each colony was used after 15 days of culture for amplification, while the other half was processed for PCR analysis. Eurofins Genomics performed the sequencing of the selected clone. The resulting sequence was analysed with Benchling, NCBI (BLASTn), and Biomanda: the DMD Delta52 cell line had a deletion of 1161 base pair (bp) containing the targeted exon 52 (118 pb) and an insertion of a random sequence of 6 bp.
DNA and RNA experiments
RNA extraction and quality
RNA extraction was done in the six cell lines at seven different time points: tissue‐derived myoblast and tissue‐derived myotube, as well as during hiPSC differentiation at Days 0, 3, 10, 17, and 25 (hiPSC‐derived myotube) using the miRNeasy Mini kit (217004, QIAgen) on the QIAcube instrument. RNAs coming from part A of the extraction protocol was used for mRNA‐seq and RT‐qPCR. RNAs coming from part B of the extraction protocol was used for miRseq. PartA RNA was quantified by Nanodrop spectrophotometer (ND‐1000, Thermo Fisher Scientific) and purity/quality (RIN ≥ 7) was assessed with the 2200 TapeStation using the Agilent RNA ScreenTape (5067‐5576/5067‐5577/5067‐5578, Agilent). PartB RNA was quantified and purity/quality was assessed with the 2100 Agilent Bioanalyzer using the Agilent small RNA kit (5067‐1548, Agilent).
Reverse transcription
Total RNA (500 ng) were reverse transcribed with random primers (48190–011, Thermo Fisher Scientific), oligo (dT) (SO131, Thermo Fisher Scientific), and deoxynucleotide (dNTP, 10297–018, Thermo Fisher Scientific) using Superscript® III reverse transcriptase (18080–044, Thermo Fisher Scientific). Thermocycling conditions were 10 min, 25°C; 60 min, 55°C; and 15 min, 75°C.
Quantitative PCR
Total DNA and cDNA were amplified using primers (Thermo Fisher Scientific) designed with Primer blast tool (http://www.ncbi.nlm.nih.gov/tools/primer‐blast) and listed in Table S2. The amplification efficiency of each primer set was preliminarily determined by running a standard curve. Detection was performed using a QuantStudio™ 12K Flex Real‐Time PCR System (Thermo Fisher Scientific). Reactions were carried out in a 384‐well plate, with 10 μL containing 2.5 μL of 1/10 cDNA or 6.25 ng/uL total DNA, 0.2 μL of mixed forward and reverse primers at 10 μM each, and 5 μL of 2X Luminaris Color HiGreen qPCR Master Mix Low Rox (K0973, Thermo Fisher Scientific). Thermocycling conditions were 50°C during 2 min and 95°C during 10 min, followed by 45 cycles including 15 s at 95°C and 1 min at 60°C plus a dissociation stage. All samples were measured in triplicate. Experiments were normalized using UBC as reference gene and relative quantification was done with the ΔΔCt method.
Bulk mRNA sequencing
Libraries were prepared with the TruSeq Stranded mRNA kit protocol according to supplier recommendations. Briefly, the first step was the purification of PolyA‐containing mRNA molecules from 1 μg of total RNA using poly‐T oligomers attached to magnetic beads, followed by a fragmentation using divalent cations under elevated temperature to obtain approximately 300 bp fragments. Then, double‐stranded cDNA was synthesized, Illumina adapters were ligated, and the cDNA library was amplified by PCR for sequencing. Finally, paired‐end 100 bp/75 bp sequencing was carried out with an Illumina HiSeq 4000 sequencer.
The RNA‐seq analysis workflow was designed using snakemake 3.5.4 26 for read quality estimation, mapping, and differential expression analysis. Quality estimation was obtained with FastQC 0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Mapping to the human genome assembly Ensembl GRCh37.87 (43 695 transcripts) was performed with STAR 2.5.0a. 27 According to STAR manual and for more sensitive novel junction discovery, the junctions detected in a first round of mapping were used in a second mapping round. Read strandness was confirmed using RSeQC. 28 Analysis results were summarized using MultiQC 1.0. 29 Normalized counts (median ratio normalization) and differential expression analysis were performed with DESeq2 1.16.1, 30 considering pairwise comparisons with all developmental stages and comparing DMD vs. healthy cells within developmental stages. BiomaRt 2.30.0 31 was used to fetch gene annotations from Ensembl. Transcripts with |log2FoldChange| ≥ 0.4 (equivalent of DMD/healthy ratio ≤0.76 or ≥1.32) and adjusted P value ≤ 0.05 were considered differentially expressed. RNA‐seq data have been deposited in the ArrayExpress database 32 at EMBL‐EBI under accession number E‐MTAB‐8321 (https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8321).
Single‐cell mRNA sequencing
One cryotube (1.7 million cells) of the M398 healthy cell line was thawed in a T75 BioCoat™ Collagen I cultureware (356485, Corning Incorporated) with SKM02 medium (AMSbio). Cells were harvested with trypsin the following day. A total of 1.5 million cells were centrifuged at 300 g for 5 min. Cells were resuspended at 1500 cells/μL in DMEM F‐12 with 10% serum and processed according to the 10X Genomics single‐cell RNA sequencing protocol. Briefly, the library was prepared with Chromium™ Single Cell 3' Library & Gel Bead Kit v2. Six thousand cells with a viability of 94% were processed to finally sequence 3625 of them. Sequencing was done on Illumina HiSeq 4000 sequencer. Bioinformatics analysis was performed using 10X Genomics recommendations. Briefly, fastq files were created with cellranger mkfastq, and cellranger count was used to perform alignment, filtering, barcode counting, and unique molecular identifier counting. RNA‐seq data have been deposited in the ArrayExpress database 32 at EMBL‐EBI under accession number E‐MTAB‐9510 (https://www.ebi.ac.uk/arrayexpress/experiments/MTAB‐9510).
miRNA sequencing
miRNAs (10 ng) were reverse transcribed using the Ion Total RNA‐seq kit v2 (4475936, Thermo Fisher Scientific) following the manufacturer's protocol for small RNA libraries. The cDNA libraries were amplified and barcoded using Ion Total RNA‐seq kit v2 and Ion Xpress RNA‐seq Barcode Adapters 1‐16 Kit (Thermo Fisher Scientific). Amplicons were quantified using Agilent High Sensitivity DNA kit before samples pooling in sets of 15. Emulsion PCR and enrichment were performed on the Ion OT2 system instrument using the Ion PI Hi‐Q OT2 200 kit (A26434, Thermo Fisher Scientific). Samples were loaded on an Ion PI v3 Chip and sequenced on the Ion Proton System using Ion PI Hi‐Q sequencing 200 kit chemistry (200 bp read length; A26433, Thermo Fisher Scientific). Sequencing reads were trimmed with Prinseq 33 (v0.20.4) (‐‐trim‐right 20) and filtered by average quality score (‐‐trim‐qual 20). Reads with a size less than 15 bp were removed and reads with a size greater than 100 bp were trimmed with Cutadapt (v1.16). 34 Mapping to the human genome assembly Ensembl GRCh37.87 (3111 transcripts) was performed with STAR 2.5.3a. 27 Normalized counts (median ratio normalization) and differential expression analysis were performed with DESeq2 1.16.1, 30 considering pairwise comparisons with all developmental stages and comparing DMD vs. healthy cells within developmental stages. Transcripts with |log2FoldChange| ≥ 0.4 (equivalent of DMD/healthy ratio ≤0.76 or ≥1.32) and P value ≤ 0.05 were considered differentially expressed. The use of P value instead of adjusted P value was justified by biological meaning 35 (i.e. well‐known regulated/dysregulated miRNAs had a P value ≤ 0.05 but not an adjusted P value ≤ 0.05). miRNA‐seq data have been deposited in the ArrayExpress database 32 at EMBL‐EBI under accession number E‐MTAB‐8293 (https://www.ebi.ac.uk/arrayexpress/experiments/E‐MTAB‐8293).
High‐throughput data analyses
Graphs were realized using RStudio. Viridis 0.5.1 library 36 was used for the colour palette to ease reading with colour blindness and print well in grey scale. Normalized counts were standardized for unsupervised analyses with the scale function (centre = TRUE, scale = TRUE) and plotted with the corrplot function from corrplot 0.84 library. 37 Spearman correlation was done with the cor function (method = ‘spearman’, use = ‘pairwise.complete.obs’) on standardized data. Hierarchical clustering and heatmap were performed with gplots 3.0.3 library 38 heatmap.2 function on standardized data. Gene enrichment data were retrieved from DAVID database using RDAVIDWebService 1.24.0 library 39 on supervised list of mRNAs [mRNA‐seq data: adjusted P value ≤ 0.01, normalized counts ≥ 5 in at least one sample, ratio ≤ 0.5 or ≥2 for myogenesis (Figure S1B) and ratio ≤ 0.76 or ≥1.32 for DMD phenotype (Figure S1C); enrichment data: Benjamini value ≤ 0.05, enrichment ≥ 1.5]. Only Gene Ontology terms were processed. Spearman correlations for the transcriptomics vs. proteomics comparison at Day 17 and for comparisons with published omics datasets were performed using two‐tailed non‐parametric Spearman correlation by GraphPad Prism software.
Exon skipping
One million cells were transfected after 17 days of culture by electroporation with a phosphorodiamidate morpholino oligomer (PMO) targeting exon 7 (custom oligo PMO7 5′‐ATGTTGAATGCATGTTCCAGTCGTTGTGTG‐3′, Gene Tools) or 51 (custom oligo PMO51 5′‐CTCCAACATCAAGGAAGATGGCATTTCTAG‐3′, Gene Tools) of the DMD gene in 100 μL solution from the P3 Primary Cell 4D‐Nucleofector® X Kit L (V4XP‐3024, Lonza) using the CB150 program on the 4D‐Nucleofector™ System (Lonza). PMO7 was transfected into healthy M180 cells at concentrations of 0.1, 0.25, 0.5, 0.75, 1, 5, 10, or 100 μM. PMO51 was transfected into DMD52 cells at concentrations of 0.1, 0.5, 20, 50, 60, 75, or 100 μM. A PMO control (standard control 5′‐CCTCTTACCTCAGTTACAATTTATA‐3′, Gene Tools) was transfected at a concentration of 100 μM. Cells were seeded at a density of 100 000 cells/cm2. RNA extraction was carried on transfected cells 24, 48, and 72 h later followed by a reverse transcription as described above. PCR was done on 1 μL of cDNA using 10 μM of forward and reverse primers (Table S2, Life technologies) and 1 unit of Taq DNA polymerase (10342, Life technologies) as described by the manufacturer's instructions, for a final reaction volume of 25 μL. PCR reaction started by a step at 94°C for 3 min, followed by 27 cycles at 94°C for 45 s, 55°C for 45 s, and 72°C for 45 s and a final step at 72°C for 5 min. Exon skipping was analysed using the DNA 1000 kit (5067, Agilent) with the Agilent 2100 Bioanalyzer. Full‐length PCR product was 372 bp and exon skipped length PCR product was 253 bp for M180 cells and 422 bp and 189 bp for DMD52 cells. Results were computed by the Agilent 2100 Bioanalyzer software v3.81. Spearman correlations were performed using two‐tailed non‐parametric Spearman correlation by GraphPad Prism software.
Protein experiments
Immunolabelling
Cells (healthy hiPSC 1/DMD hiPSC 2, Table S1) at Day 17 of culture were thawed and seeded at 10 000 cells/cm2 in SKM02 medium in Falcon® 96‐well microplate (353219, Corning) coated with 0.1% gelatin (G1393, Sigma‐Aldrich) and 2.4 μg/mL laminin (23017015, Thermo Fisher Scientific) in phosphate‐buffered saline (PBS) 1X (D8537, Sigma‐Aldrich). Cells were switched to DMEM/F‐12, HEPES (31330038, Thermo Fisher Scientific) with 2% horse serum (H1270, Sigma‐Aldrich) after 4 days of culture. Cells were fixed 15 min at 4°C with paraformaldehyde 4% (15710, Euromedex) after 7 days of culture. A first quick PBS 1X tablets (P4417, Sigma‐Aldrich) wash was done, followed by another lasting 10 min. Then, a solution with PBS 1X, Triton™ X‐100 0.25% (T8787, Sigma‐Aldrich) and bovine serum albumin 2.5% (A9418, Sigma‐Aldrich) was added and incubated 30 min at room temperature. Primary antibody diluted in the same buffer (α‐actinin 1/500, A7811, Sigma‐Aldrich) was finally added overnight at 4°C. Two quick PBS 1X washes followed by a third incubated 10 min at room temperature were done the next day. An incubation was done 45 min at room temperature with a mix of 4′,6‐diamidine‐2′‐phenylindole dihydrochloride (DAPI, 1 μg/mL, 10236276001, Sigma‐Aldrich) and the secondary antibody Donkey anti‐Mouse Alexa Fluor 555 in PBS 1X, (1/1000, A‐31570, Thermo Fisher Scientific). Finally, two quick PBS 1X washes were followed by a third incubated 10 min at room temperature. Stained cells were kept in PBS 1X at 4°C before imaging with a Zeiss LSM880 Airyscan confocal and Zen software (Black edition).
Western blotting
Culture of tissue‐derived myotubes were washed three times with cold PBS 1X (w/o Ca2+ and Mg2+, D8537, Sigma‐Aldrich) and proteins were isolated by scraping (010154, Dutscher) cultured cells with an extraction protein buffer [NaCl 150 mM, Tris 50 mM, EDTA 10 mM (AM9260G, Thermo Fisher Scientific), Triton 1X, 1/100 Protease Inhibitor Cocktail (P8340, Sigma‐Aldrich), PhosSTOP tablet (04906845001, Roche Diagnostics)]. Cell pellets of hiPSC‐derived myotubes were rinsed once with cold PBS 1X, spun 5 min at 300 g, and resuspended in the same extraction protein buffer. Protein extracts were centrifuged at 4°C 10 min at 16 000 g and supernatants were kept at −80°C. Quantitation of total protein was done with Pierce BCA protein assay kit (23225, Thermo Fisher Scientific). Protein extracts were mixed before gel loading with 9 μL of loading buffer [urea 4 M, sodium dodecyl sulfate (SDS) 3.8%, glycerol 20%, Tris 75 mM pH 6.8, 5% β‐mercaptoethanol, 0.1 mg/mL bromophenol blue] and completed with 28 μL of extraction protein buffer (for one well) and then heated once 5 min at 95°C. Western blots for RYR1 (1/1000, MA3‐925, Thermo Fisher Scientific), MF20 (1/500, DSHB, concentrate), Manex50 (1/30, DSHB), α‐sarcoglycane (1/150, A‐SARC‐L‐CE, Leica biosystems), and γ‐sarcoglycane (1/150, G‐SARC‐CE, Leica biosystems) were performed with Criterion™ XT Tris‐Acetate Precast Gels 3–8% (3450130, Bio‐Rad, Hercules, CA), XT Tricine running buffer (161–0790, Bio‐Rad) and ran at room temperature for 1 h and 15 min at 150 V. Western blots for CaV1.1 (1/1000, MA3‐920, Thermo Fisher Scientific), ATP5A (1/1,000, ab14748, ABCAM), Semaphorin 6A (1/55, AF1146, R&D systems), and GLI3 (1/200, AF3690, R&D systems) were performed with 4–15% Criterion™ TGX™ Precast Midi Protein Gel (5671084, Bio‐Rad), 10x Tris/Glycine/SDS Running Buffer (1610772), and ran at room temperature for 1 h at 200 V. Gels were rinsed once in water and blotted either with ‘high molecular weight’ or ‘mixed molecular weight’ program of Trans‐Blot® Turbo™ transfer system (Bio‐Rad) using Trans‐Blot® Turbo™ Midi Nitrocellulose Transfer Packs (170–4159, Bio‐Rad). Blots were then processed with the SNAP i.d.® 2.0 Protein Detection System following the manufacturer's protocol, using Odyssey® Blocking Buffer (927‐40003, LI‐COR) for blocking and 0,2% Tween® for antibody dilutions (28829.296, VWR). Washes were done with PBS tween buffer (PBS 1X tablets, P4417, Sigma‐Aldrich; 0.1% Tween® 20). Each primary antibody was pooled with either α‐actinin (1/12,500, sc‐17829, Santa Cruz or 1/7000, A7811, Sigma‐Aldrich) or α‐tubulin (1/6666, Ab7291, Abcam). Either IRDye 800CW donkey anti‐mouse and/or IRDye® 680RD donkey anti‐goat (1/5000‐1/10000, 926‐32212, 926‐68074, LI‐COR) were used as secondary antibodies. After completion of SNAP i.d.® general protocol, two PBS 1X washes were finally done with the membrane still in the blot holder before band visualization with Odyssey® CLx Imaging System and quantification with Image Studio Lite software (Version 5.2). Statistical analysis was performed using unpaired t‐test by GraphPad Prism software.
Tandem mass tag isobaric quantitative proteomics
Samples preparation
Cells were collected after 17 days of culture and resuspended in 90% foetal bovine serum (Hyclone), 10% DMSO (A3672.0050, VWR), cooled down until −90°C with the CryoMed™ device (Thermo Fisher Scientific), before storage in liquid nitrogen. Cells were then thawed and washed five times with cold PBS and air was replaced by Argon to thoroughly dry the pellet that was flash frozen in liquid nitrogen. Five to ten times the approximate cell pellet volume of 0.5 M triethyl ammonium bicarbonate with 0.05% SDS was added to the cell pellet for protein extraction. Cell pellet was resuspended and triturated by passing through a 23‐gauge needle and 1 mL syringe for 30 times. Samples were then sonicated on ice at amplitude of 20% for 30 × 2 s bursts and centrifuged at 16 000 g for 10 min at 4°C. Supernatant was transferred to a fresh Eppendorf tube. Protein was quantified by Nanodrop spectrophotometer. A total of 100–150 μg of protein was aliquoted for each individual sample and 2 μL TCEP (50 mM tris‐2‐carboxymethyl phosphine) was added for every 20 μL of protein used for reducing the samples. After 1 h of incubation at 60°C, 1 μL MMTS (200 mM methylmethane thiosulphonate) was added for every 20 μL of protein used for alkylating/‘blocking’ the samples. Finally, after a 10 min incubation at room temperature, samples were trypsinized by addition of 6–7.5 μL of 500 ng/μL trypsin. The ratio between enzyme:substrate was 1:40. Samples were incubated overnight at 37°C in the dark.
Tandem mass tag labelling
When tandem mass tag (TMT) reagents reached room temperature, 50 μL of isopropanol/[acetonitrile] was added to each TMT 11‐plex reagent and was incubated at room temperature for 2 h, in the dark; 8 μL of 5% hydroxylamine was added to neutralize the reaction. Each sample was separately lyophilized at 45°C. Samples have been stored at −20°C or used immediately.
Offline C4 high‐performance liquid chromatography
All eight samples were pooled together in 60 μL of 97% mobile phase A (99.92% % H2O, 0.08% NH4OH) and 3% mobile phase B (99.92% % acetonitrile, 0.02% NH4OH) by serially reconstituting each sample. Extra 40 μL of mobile phase was added to Sample 1; after sample has been well vortexed, its content was transferred to the tube with Sample 2 (and serially repeated until all samples were pooled). Final volume of samples needed to be 100 μL. After sample was centrifuged at 13 000 g for 10 min, supernatant was collected with a high‐performance liquid chromatography (HPLC) injection syringe; 100 μL was injected onto the sample loop. Fractions were collected in a peak‐dependent manner. Finally, fractions were lyophilized at 45°C and stored at −20°C until required. The used column was a Kromasil C4 column 100 Å pore size, 3.5 μm particle size, 2.1 mm inner diameter, and 150 mm length. The gradient for C4 separation was (RT in min—%B): 0‐3, 10‐3, 11‐5, 16‐5, 65‐20, 100‐30, 15‐80, 120‐80, and 125‐3.
Solid phase extraction cleaning of peptides fractions
A GracePureTMT SPE C18‐Aq cartridge was used for pre‐cleaning of samples (support: silica, % carbon: 12.5%, with endcapping, surface area: 518 m2/g, particle size: 50 μm, pore size: 60 Å, water‐wettable). Samples were reconstituted using in total 400 μL of 1% Formic Acid (ACN) and 0.01% formic acid (FA). Cartridge was washed with 600 μL of ACN. ACN was then completely flushed out of the column at dropwise speed. This activated the ligands. Then, 1% ACN and 0.01% FA (600 μL) was flushed through the cartridge to equilibrate the sorbent; 400 μL of the sample was loaded in the cartridge. It was then very slowly flushed through the cartridge and recovered into a fresh tube. This process was repeated three times. Two volumes of 250 μL of 1% ACN and 0.01% FA were used to clean and de‐salt the sample. It was flushed through very slowly. Two volumes (250 μL each) were used per step (2% ACN, 10% ACN, 30% ACN, 50% ACN, 70% ACN). This cycle was repeated twice. Each particular concentration was pooled in one tube. Samples were dried to dryness in a Speedvac at room temperature overnight and stored at −20°C. Like previously, samples were pooled with 100 μL of 97% mobile phase A (99.92% % H2O, 0.08% NH4OH) and 3% mobile phase B (99.92% acetonitrile, 0.02% NH4OH) and injected onto the sample loop. Fractions were collected in a peak‐dependent manner. The gradient for SPE‐cleaned peptides C4 separation (RT in min—%B): 0‐2, 10‐2, 20‐5, 25‐5, 35‐20, 55‐35, 60‐35, 70‐80, 75‐80, and 80‐3.
Online C18 high‐precision liquid chromatography
Thirty microliters of loading phase (2% acetonitrile, 1.0% formic acid) was added to each fraction‐containing Eppendorf tube. Samples were vortexed and centrifuged. Blanks (30 μL mobile phase) were added into wells A1 to A12. Thirty microliters of Sample 1 was pipetted into well B1, Sample 2 in well B2, and so on. An orthogonal 2D‐LC–MS/MS analysis was performed with the Dionex Ultimate 3000 UHPLC system coupled with the ultra‐high‐resolution nano ESI LTQ‐Velos Pro Orbitrap Elite mass spectrometer (Thermo Fisher Scientific).
Data analysis
Higher‐energy collisional dissociation (HCD) and collision‐induced dissociation (CID) tandem mass spectra were collected and submitted to Sequest search engine implemented on the Proteome Discoverer software version 1.4 for peptide and protein identifications. All spectra were searched against the UniProtKB SwissProt. The level of confidence for peptide identifications was estimated using the Percolator node with decoy database searching. False discovery rate was set to 0.05, and validation was based on the q value. Protein ratios were normalized to protein median and peptides with missing TMT values were rejected from protein quantification. Phosphorylation localization probability was estimated with the phosphoRS node. Protein ratios were transformed to log2 ratios and significant changes were determined by two‐tailed one‐sample t‐test with the Benjamini method for multiple testing corrections. To reduce the impact of possible false positive identifications, more parameters were set: (i) only proteins with more than two quantified unique peptides and (ii) DMD/healthy ratio ≥1.32 or ≤0.76 and 3 only false discovery rate corrected P value ≤ 0.05 were retained for bioinformatics analysis. The list of proteins quantified in the six samples is in Table S3. Proteomic data have been deposited in the PRIDE Archive database 40 at EMBL‐EBI under accession number PXD015355 (https://www.ebi.ac.uk/pride/archive/projects/PXD015355).
Adenosine triphosphate experiments
Two healthy (M180 and M398) and two DMD (M202 and M418) cell lines after 17 days of culture were seeded in 384‐well plates at a density of 30 000 cells/cm2. Living cells were stained with HOECHST at a concentration of 1/300 6 days later for cell quantification (nuclei per well were counted using the CX7 imaging system, Thermo Fisher Scientific). Adenosine triphosphate (ATP) measurement was done using the CellTiter‐Glo™ Luminescent Cell Viability Assay Kit (Promega) following the manufacturer's protocol and normalized by the cell quantification. Statistical analysis was performed using two‐tailed one‐sample t‐test by GraphPad Prism software (each healthy cell line was compared with each DMD cell line).
Results
To establish the early/developmental impact of DMD gene mutations, hiPSCs from three DMD patients and three healthy individuals were generated as described previously. 15 These cells were subjected to a standardized differentiation protocol without utilization of feeder cells, cell sorting, or gene overexpression resulting in elongated and plurinucleated myotubes within 25 days, 23 with an amplification fold of 2918 ± 480 (mean ± SEM). Skeletal muscle progenitor cells after 10 and 17 days of differentiation could be cryopreserved (Figure S2A). Whole transcriptome and miRnome profiles were compared at seven differentiation time points (tissue‐derived myoblasts and myotubes, as well as hiPSC‐derived cells at Days 0, 3, 10, 17, and 25) and complemented by single‐cell transctriptomics, TMT proteomics and western blot analyses (Table S4).
We analysed gene expression variations to estimate the impact of using genetically unmatched cells. Variability between cell lines was greater than within cell lines for each genotype (1.7 ± 0.4 times on average for healthy cells and 3.9 ± 0.7 times for DMD cells, Figure S3A). Variability within each cell line was equivalent in healthy and DMD cells (Figure S3B), while variability between healthy cell lines was lower than between DMD cell lines (Figure S3C). Figure S4 gives detailed gene expression of SOX2, SOX5, PAX3, SGCA, and MYH3 to illustrate gene expression variations between cell differentiation and between cell lines.
Duchenne muscular dystrophy is initiated prior to the expression of skeletal muscle markers
First, the expression profile of the DMD variants was studied by RT‐qPCR in healthy and DMD hiPSCs during the differentiation process described in Figure S2A. The Dp427m variant, which is normally observed in muscle cells, 41 appeared from Day 3 and was increased at Day 17, in contrast with Dp412e—the embryonic variant of dystrophin present in mesoderm cells 15 —which was expressed from Day 0, increased at differentiation Day 3, and disappeared from Day 10. Therefore, the expression of the DMD locus is initiated in the very first steps of the differentiation protocol, well before the entry into the skeletal muscle lineage. The ubiquitous variant Dp71‐40 was detected at every time point, in contrast with Dp116 (Schwann cell variant 42 ), Dp140 (kidney and foetal brain variant 43 ) Dp427p1p2 (Purkinje cell variant 44 ), and Dp427c, which were either undetected or expressed at very low levels (Figure S2B). Interestingly, Dp260 (retinal variant 45 ) followed a similar expression pattern than Dp427m.
A strong correlation in the transcriptomic data was observed by mRNA‐seq and miRNA‐seq between samples collected at an individual time point, as opposed to samples from two distinct time points. In addition, the correlation coefficient between samples taken at two successive time points increased as differentiation progressed (Figure 1A). Differential expression analysis in healthy controls between two successive collection days (Days 3/0, Days 10/3, Days 17/10, Days 25/17) showed that the proportion of regulated genes decreased from 26% to 18% of the whole transcriptome through the course of differentiation (8080 to 5320 mRNAs, adjusted P value ≤ 0.01, Figure S1A). These observations demonstrate the robustness of the differentiation protocol and are in agreement with an early specialization and a later refinement of the transcriptome as cells quickly exit pluripotency and become progressively restricted to the skeletal muscle lineage.
Figure 1.
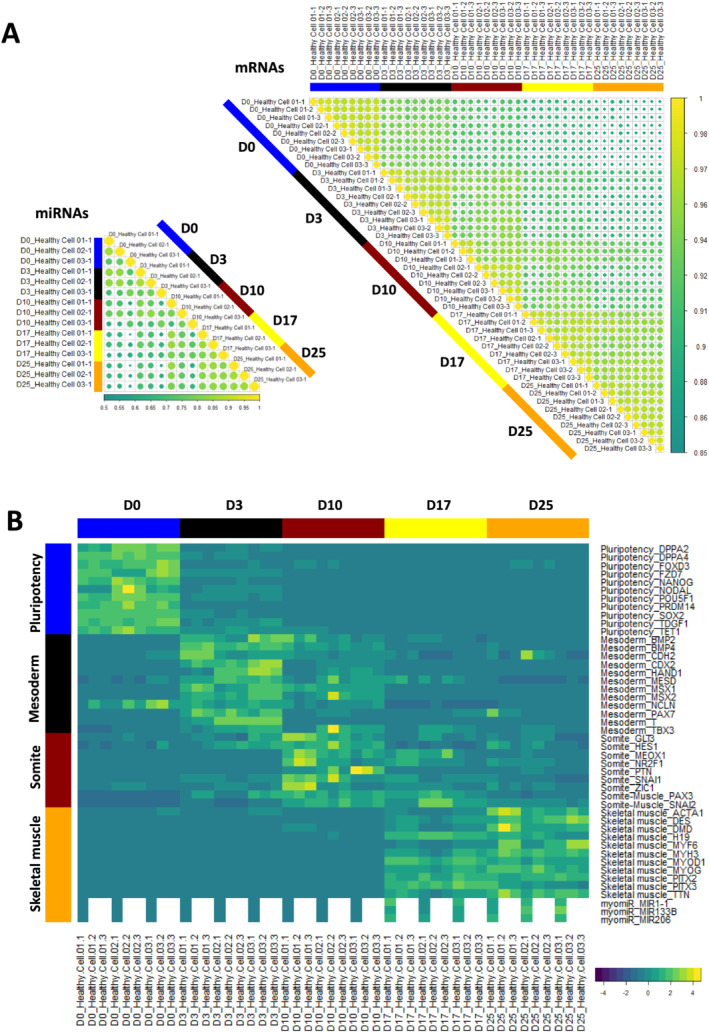
Differentiation dynamics of hiPSCs (D0) into MyoT (D25) in healthy cells at the transcriptomic level. (A) Spearman correlation matrix of transcriptomes (mRNAs, right) and miRnomes (miRNAs, left). Yellow dots indicate a stronger correlation. (B) Gene expression heatmap of selected differentiation markers (D: day; hiPSC: human induced pluripotent stem cell; MyoT: myotube).
To characterize the developmental stages achieved by the cells, the expression of lineage‐specific markers (both mRNAs and miRNAs) was determined at each time point, together with gene ontology enrichment analyses (Figures 1B, 2A, S1B, and S1C; Table S5).
Figure 2.
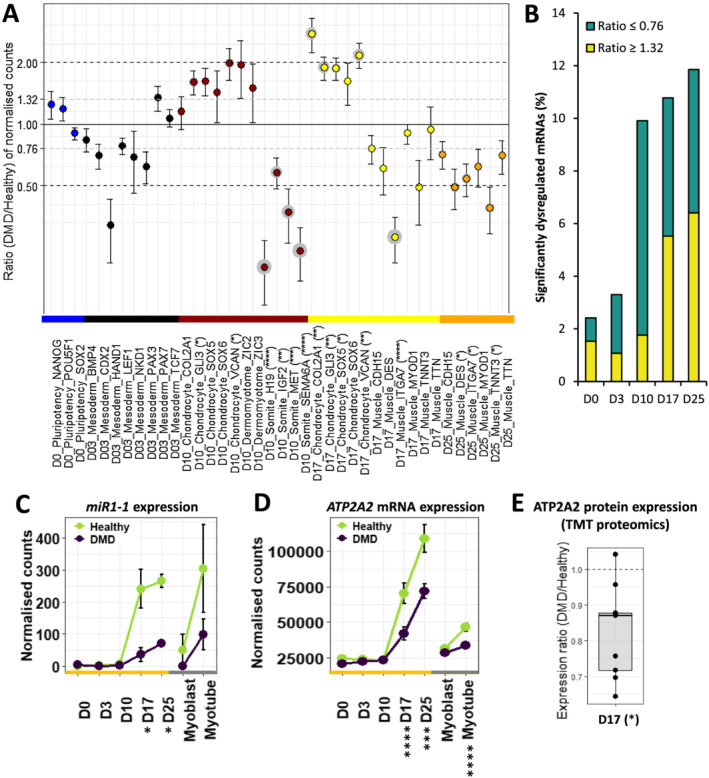
Differentiation dynamics of hiPSCs (D0) into MyoT (D25) in Duchenne muscular dystrophy (DMD) cells. (A) Dotplot of DMD/healthy expression ratios of selected differentiation markers. Statistical differences are indicated in brackets after gene names and grey circles around the corresponding dots. (B) Proportions of significantly dysregulated mRNAs (adjusted P value ≤ 0.05) in DMD cells at each time point. Expression of (C) MIR1–1 and (D) ATP2A2 mRNA during differentiation. (E) ATP2A2 protein level at D17 (*adjusted P value ≤ 0.05, **adjusted P value ≤ 0.01, ***adjusted P value ≤ 0.001, ****adjusted P value ≤ 0.0001; D: day; hiPSC: human induced pluripotent stem cell; MyoT: myotube).
Pluripotency was similarly maintained in healthy and DMD cells at Day 0 (Figure 2A, Table S5), as already shown by our group. 15 At Day 3, cells lost pluripotency and became paraxial mesoderm cells expressing marker genes such as PAX3 and PAX7 (Figures 2A and S5A, Table S5). Importantly, markers of lateral plate (e.g. GATA4) and intermediate mesoderm (e.g. PAX8) were not up‐regulated at this stage (Table S5). Similarly, earlier markers of primitive streak (e.g. TBX6) and mesendoderm (e.g. MIXL1), as well as markers of the other germ layers, endoderm (e.g. SOX17) and ectoderm (e.g. SOX2), were either not expressed, greatly down‐regulated, or expressed at very low levels (Table S5), suggesting cell homogeneity in the differentiation process. Pluripotency markers as well as transgenes were properly down‐regulated/silenced all along cell differentiation (Figure S5)—except for MYC at the later stages in one healthy cell line. However, the transgene partial reactivation did not have a quantifiable impact neither on total MYC expression (Figure S5B) nor on global transcriptomics data, as shown by Spearman correlation coefficients between the three healthy lines at D17 and D25 (Figure S5D).
At that early time point, DMD‐associated gene dysregulation represented less than 3% of the entire transcriptome (adjusted P value ≤ 0.05, Figure 2B) but already contained genes important for development (e.g. MEIS2) and muscle formation (e.g. ACTA1). However, mesoderm markers were not significantly dysregulated, attesting that mesoderm commitment was mostly unimpaired (Figure 2A, Table S5). No increase in the expression of primitive streak, mesendoderm, endoderm, or ectoderm markers was detected, suggesting no differences in the differentiation process of DMD cells at that stage (Table S5).
In contrast, a sharp increase in the proportion of dysregulated genes appeared at Day 10, mostly including gene down‐regulations (DMD/healthy expression ratio ≤ 0.76, adjusted P value ≤ 0.05). This concerned almost 10% of the transcriptome at Day 10 (against 3% at Day 3) and remained stable from 10% to 12% (1226 mRNAs) until Day 25 (Figure 2B). At Day 10, healthy cells expressed genes typically observed during somitogenesis, such as PAX3, NR2F2, PTN, MET, H19, and IGF2 (Table S5). More precisely, their transcriptome exhibits a mixed profile between dermomyotome (expression of GLI3 and GAS1 but not ZIC3) and myotome (expression of MET and EPHA4 but not LBX1) (Table S5). Neither markers of presomitic mesoderm cells (e.g. FGF8) and neural plate cells (e.g. FOXD3) nor markers of sclerotome (e.g. PAX1) and dermatome (e.g. EGFL6) were up‐regulated (Table S5) in both healthy and DMD cells. In the meantime, several somite markers were down‐regulated, including H19, IGF2, MET, and SEMA6A (validated at the protein level for SEMA6A, Figures 2A and S6A, Table S5), while a slight up‐regulation of chondrocyte markers was highlighted and confirmed at the protein level for GLI3 (Figure S6B), together with a significant enrichment of the gene ontology term ‘nervous system development’, suggesting potential lineage bifurcations at Day 10 (Figures 2A and S1C, Table S5).
The study of differentiation dynamics presented above highlights that mesoderm commitment is not impaired by the absence of dystrophin and shows that DMD onset takes place at the somite cell stage, before the expression of the skeletal muscle program and especially before the up‐regulation of Dp427m expression.
Duchenne muscular dystrophy skeletal muscle progenitor cells exhibit specific muscle gene dysregulations
Healthy and DMD cells were in the skeletal muscle compartment at Day 17, as evidenced by the expression of multiple lineage‐specific genes, such as transcription factors (e.g. MYOD1), cell surface markers (e.g. CDH15), sarcomere genes (e.g. TNNC2), dystrophin‐associated protein complex (DAPC) genes (e.g. SGCA), calcium homeostasis genes (e.g. RYR1), and muscle‐specific miRNAs (myomiR, e.g. MIR1‐1, MIR206, and MIR133). This was also observed at the protein level for CDH15, TNNC2, and RYR1 (Figure 1B, Table S5). They both showed an embryonic/foetal phenotype characterized by ERBB3 expression, in contrast with tissue‐derived myoblasts that expressed NGFR. PAX7 and CD34, two markers of skeletal muscle stem cells, were 12 and 23 times less expressed in hiPSC‐derived cells than in their tissue‐derived myoblasts (Figure S7, Table S5). Here again, alternative cell lineages were absent or greatly down‐regulated, such as tenocytes (e.g. MKX), chondrocytes (e.g. SOX5), osteoblasts (e.g. SPP1), or nephron progenitors (e.g. SALL1) (Table S5). Purity of the cell cultures was confirmed by single‐cell RNA‐seq in one healthy cell line (Figure S8). For instance, expression of SOX2 (a reprogramming factor) and OLIG2 (a moto‐neuron marker) was barely detected, while a large majority of cells expressed the skeletal muscle markers MYOD1 and ACTC1.
Interestingly, DMD cells did not show a significant dysregulation of skeletal muscle transcription factors (Table S5). However, several myomiRs were found down‐regulated (e.g. MIR1‐1, Figure 2C), together with genes related to calcium homeostasis (e.g. ATP2A2, at both mRNA and protein level, Figure 2D and 2E) as well as members of the DAPC (e.g. SNTA1) (Table S5). Concerning cell lineages, there was no visible difference when compared with healthy controls, except an up‐regulation of markers associated with chondrocytes, which was confirmed at the protein level for GLI3 (Figure S6C), and a significant enrichment of the gene ontology term ‘nervous system development’ previously seen at Day 10, together with ‘kidney development’ and ‘ossification’ (Figures 2A and S1C, Table S5). To further consolidate the findings from hiPSC‐derived cells, a pair of isogenic hESCs was differentiated up to the myotube stage. Like in the DMD hiPSC‐derived cells, DMD was down‐regulated in the DMD Delta52 hESC‐derived cells (Figure S9A). The absence of dystrophin was confirmed by western blot (Figure S9B). Dysregulations of a selection of 10 genes were similar to those observed with the hiPSC model (Spearman correlation of r = 0.89, P value = 0.0012, Figure S9C).
Duchenne muscular dystrophy‐specific dysregulations were further queried at the protein level using TMT proteomics. A total of 3826 proteins were detected in the six processed samples (three healthy and three DMD, Table S3). Among this list, 185 proteins (139 + 46) were found significantly dysregulated in DMD and 375 (329 + 46) of the corresponding mRNAs were previously detected dysregulated in the RNA‐seq analysis, the overlap between protein and mRNA identified dysregulations being 46 (|log2FoldChange| ≥ 0.4 and adjusted P value ≤ 0.05, Figure S6D and S6E, Table S6). Moreover, among the total of 514 genes represented in Figure S6F, 98 were dysregulated alike in both datasets (56 up‐regulated + 42 down‐regulated) against 13 (12 + 1) in the opposite direction (|log2FoldChange| ≥ 0.4, Figure S6F, Table S6) resulting in a Spearman correlation of r = 0.49 and P value < 0.0001. In this mRNA/protein comparison, the mRNA experiment was more sensitive than protein experiment and could also be considered as a good proxy for proteins.
To better characterize the most direct consequences of the loss of DMD in muscle cells, DMD expression was either knocked down or rescued at Day 17 by transient exon skipping using a specific PMO targeting DMD exon 7 or 51. Treatment with PMO7 in a healthy hiPSC line resulted in significant exon skipping, which was correlated with reduced DMD expression up to 94% (Spearman r = −0.88, analysed pairs = 59, P value < 0.0001, Figure S10A, S10D, and S10E) and reduced dystrophin protein levels (up to 81%, Figure S10C). In parallel, treatment with PMO51 in DMD Delta52 hESC‐derived cells also resulted in significant exon skipping (100% and 92.4%, respectively, with the 100 μM PMO concentration, Figure S10B). This was correlated with an increased expression of DMD (Spearman r = 0.65, analysed pairs = 27, P value = 0.0002 for the DMD hiPSC line; r = 0.89, analysed pairs = 20, P value < 0.0001 for the DMD Delta52 hESC‐derived cells, Figure S10D and S10E) and the expression of the dystrophin protein (Figure S10C). The expression of specific transcripts was measured by RT‐qPCR the 3 following days (Figure S10D): expression of MYH3, MYOG, and SGCA was significantly affected by PMO treatment, while transcripts coding for DES and ITGA7 were not.
Therefore, DMD cells efficiently enter the skeletal muscle compartment at Day 17 but exhibit dysregulations in several features typically associated with dystrophic muscles, which could be a consequence of the early manifestations of DMD detected at Day 10. Some of these identified dysregulations were mimicked by transient DMD knockdown and partly abolished by transient DMD rescue.
Human induced pluripotent stem cell differentiation leads to embryonic/foetal myotubes that reproduce Duchenne muscular dystrophy phenotypes
As previously described, 23 both healthy and DMD hiPSC‐derived myotubes (Day 25) were able to twitch spontaneously in culture, and fluorescent staining of nuclei and α‐actinin confirmed cell fusion and the formation of striation patterns typical of muscle fibres in vivo (Figure 3A). Western blot analyses on protein extracts from DMD cells confirmed that dystrophin was either undetectable or slightly expressed (Figure 3B), as in the corresponding patient muscle biopsies (data not shown).
Figure 3.
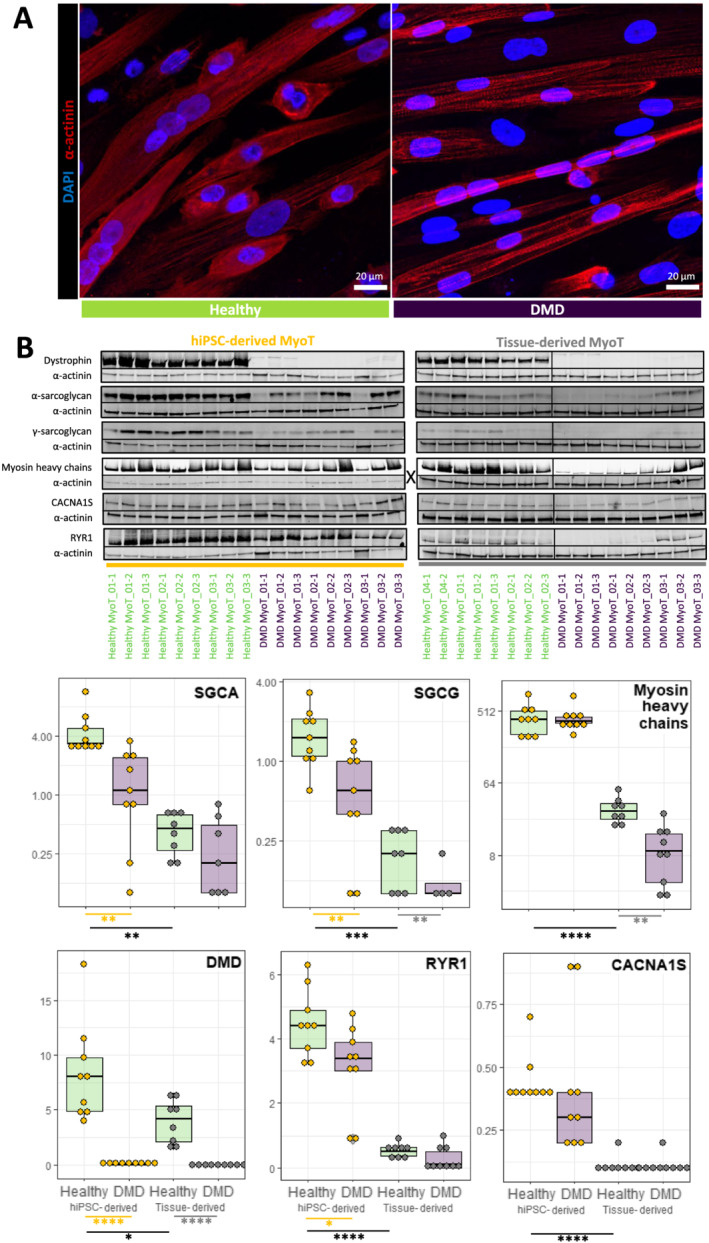
Comparison of healthy and Duchenne muscular dystrophy (DMD) MyoT from hiPSCs and tissues at the protein level. (A) hiPSC‐derived MyoT immunolabelling of α‐actinin (red) and nuclei (DAPI, blue) in healthy (left) and DMD cells (right). (B) Representative western blots and related quantifications of DMD, SGCA, SGCG, myosin heavy chains, CACNA1S, and RYR1 from protein extracts in healthy and DMD hiPSC‐derived and tissue‐derived MyoT (X: 0.25 μg of total protein was used in hiPSC‐derived MyoT instead of 7 μg in tissue‐derived MyoT—*P value ≤ 0.05, **P value ≤ 0.01, ***P value ≤ 0.001, ****P value ≤ 0.0001) (hiPSC: human induced pluripotent stem cell; MyoT: myotube).
We selected representative mRNAs and miRNAs and showed that both hiPSC‐derived and tissue‐derived myotubes have exited the cell cycle and up‐regulated genes expressed in skeletal muscles (Figures S4A and S12A, Table S5). This included skeletal muscle myomiRs (MIR1‐1, MIR133, and MIR206), transcription factors involved in skeletal myogenesis including those of the muscle regulatory factor family (e.g. MYOD, MYOG), and specific muscle cell surface markers (e.g. CDH15, ITGA7), as well as genes involved in the formation of the DAPC (e.g. SGCA, DTNA), sarcomeres (e.g. TNNC2, TNNT3), myofibril organization (e.g. UNC45B, NACA), and the triggering of excitation–contraction coupling at the neuromuscular junction (NMJ, e.g. MUSK, DOK7) (Figure 4A, Table S5). Neither visual nor transcriptional cell death was noticed in DMD myotubes (Figure S11).
Figure 4.
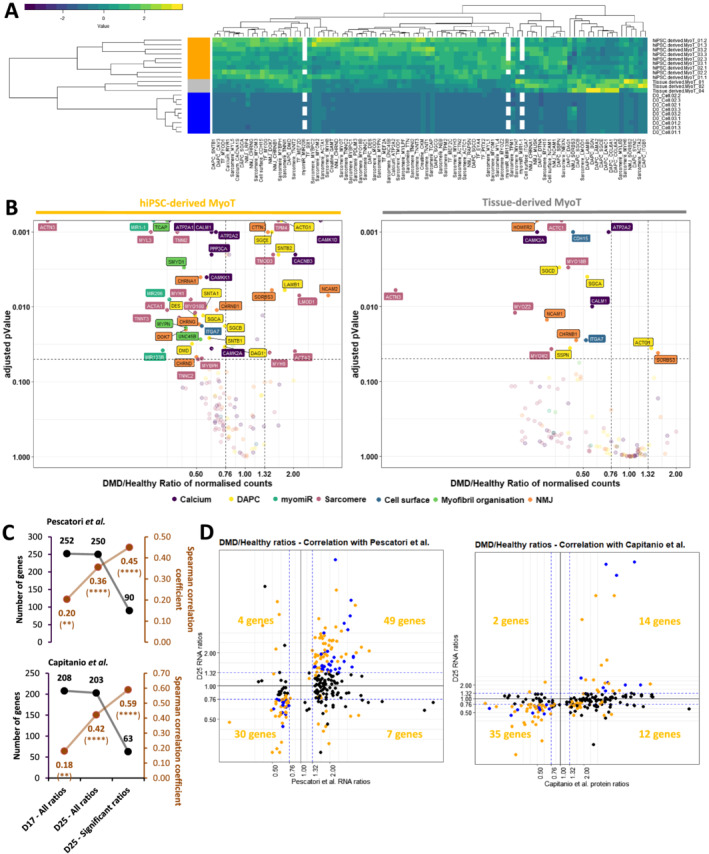
Manifestation of the Duchenne muscular dystrophy (DMD) phenotype in the transcriptome and miRnome of myotubes derived from hiPSCs and tissues. (A) Hierarchical clustering and heatmap in healthy hiPSCs (D0), hiPSC‐derived MyoT, and tissue‐derived MyoT with selected skeletal muscle transcripts and miRNAs. (B) Volcano plots of dysregulated mRNAs/miRNAs in hiPSC‐derived MyoT (left) and tissue‐derived MyoT (right)—vertical grey dashed lines represent DMD/healthy ratio thresholds at 0.76 or 1.32, and the horizontal grey dashed line represents the adjusted P value threshold at 0.05. Comparisons of DMD/healthy expression ratios at D17 and D25 with published omics data from muscle biopsies 4 , 46 : (C) number of genes in black and Spearman correlation coefficients in brown found in common with Pescatori et al.'s mRNA data (top) and Capitanio et al.'s protein data (bottom) as well as (D) correlation graphs of the D25 data compared with Pescatori et al.'s mRNA data (left) and Capitanio et al.'s protein data (right). Genes with |log2FoldChange| ≥ 0.4 are in blue if adjusted P value ≥ 0.05 and yellow if adjusted P value ≤ 0.05 (DAPC: dystrophin‐associated protein complex; hiPSC: human induced pluripotent stem cell; MyoT: myotube; NMJ: neuromuscular junction; TF: transcription factor MyoT—*P value ≤ 0.05, **P value ≤ 0.01, ***P value ≤ 0.001, ****P value ≤ 0.0001).
Even though global analysis showed that hiPSC‐derived myotubes were similar to their tissue‐derived counterparts in term of lineage commitment, they displayed an embryonic/foetal phenotype—as suggested in progenitors at Day 17. This can be illustrated by the expression of the embryonic/foetal myosin heavy/light chains MYH3, MYH8, MYL4, and MYL5 but not the postnatal transcripts MYH1 and MYH2, which were detected in tissue‐derived myotubes. Myotubes derived from hiPSCs had also higher levels of IGF2, which is down‐regulated at birth, 47 and expressed DLK1, which is known to be extinct in adult muscles 48 (Figure S12B).
Despite the embryonic/foetal phenotype, hiPSC‐derived myotubes showed evidence of terminal differentiation and cellular maturation. First, their total level of myosin heavy chain proteins was significantly higher than in tissue‐derived myotubes, as confirmed by western blotting (Figure 3B). RNAs and proteins involved in DAPC formation (e.g. DMD, SGCA, and SGCG), as well as in excitation–contraction coupling (e.g. RYR1 and CACNA1S/CAV1.1), were also present at higher levels (Figures 3B and 4A). Finally, higher expression of skeletal muscle transcription factors (e.g. MEF2C) and of multiple genes involved in muscle contraction (e.g. TNNT3), NMJ formation (e.g. RAPSN), and creatine metabolism (e.g. CKM) indicates that hiPSC‐derived cells expressed features of fully differentiated muscle cells (Figure 4A). Similar to previous time points, Day 25 cells were negative for markers of alternative muscle lineages, that is, cardiac (MIR208a, MYL7, and RYR2) and smooth muscle cells (MYH11, CNN1, and CHRNA3/B2/B4) (Table S5).
In DMD cells, unbiased mRNA‐seq analysis highlighted striking transcriptome dysregulations with 3578 differentially expressed genes in hiPSC‐derived myotubes including well‐known muscle genes. There was a global trend towards down‐regulation of muscle transcription factors, which was only significant for MEF2A and MEF2D in hiPSC‐derived myotubes and EYA4 and MYOD1 in tissue‐derived myotubes (Figure S12C). In addition, myomiRs previously associated with muscle dystrophy (dystromiRs, e.g. MIR1‐1, MIR206, and MIR133, Figures 2C and 4B) were found down‐regulated (Table S5). Similarly, a global down‐regulation phenotype was observed in both tissue‐derived and hiPSC‐derived DMD myotubes and concerned multiple mRNAs and/or proteins associated with known disease phenotypes, such as cell surface markers (e.g. ITGA7), DAPC organization (e.g. both SGCA mRNA and protein as well as SGCG protein), myofibril organization (e.g. UNC45B), sarcomere formation (e.g. MYO18B), NMJ function (e.g. CHRNB1), and calcium homeostasis (e.g. ATP2A2 mRNA and RYR1 protein) (Figure 3B for protein data, Figure 4B for transcript data, and Figure S1C for enrichment data). Like at Day 17, dysregulations of a selection of 10 genes were similar in the DMD Delta52 hESC‐derived cells and in the hiPSC model (Spearman correlation of r = 0.92, P value = 0.0004, Figure S9C).
Then, we compared the DMD/healthy expression ratios at Day 25 to two sets of published omics data from healthy and DMD muscle biopsies: one obtained at the mRNA level in pre‐symptomatic DMD patients younger than 2 years old 4 and another at the protein level in patients aged from 9 months to 8 years old. 46 Both datasets were closer to Day 25 cells (hiPSC‐derived myotubes) than Day 17 cells as expected. Our hiPSC‐derived myotubes expressed 250 of the 261 dysregulated genes and 203 of the 226 dysregulated proteins found in these respective studies (Spearman correlations of r = 0.36 and r = 0.42, P value < 0.0001, Figure 4C, Table S6). Among these, respectively 90 and 63 genes were also significantly dysregulated in our dataset (|log2FoldChange| ≥ 0.4, adjusted P value ≤ 0.05): 88% (79/90 genes) of the identified genes from the mRNA dataset and 78% (49/63 genes) of the identified genes from the protein dataset were dysregulated in the same direction, resulting in Spearman correlation of r = 0.45 and r = 0.59, respectively (P value ≤ 0.0001, Figure 4C and 4D, Table S6).
Altogether, these data indicate that hiPSC‐derived myotubes recapitulate a full skeletal muscle differentiation program and exhibit an embryonic/foetal phenotype. Despite that, it shows that DMD phenotypes are already detectable at the transcriptional level and correlated with those found in human patients. This validates the relevance of this cell system to model the DMD pathology.
Markers of fibrosis are intrinsic to Duchenne muscular dystrophy human induced pluripotent stem cell‐derived myotubes
As presented above, the up‐regulation of chondrocyte markers in DMD cells, although already present at Day 10, became significant from Day 17 (Figure 2A, Table S5). It was accompanied by the up‐regulations of the SHH signalling pathway and of multiple collagens (Figure 5A, Table S5). Genes encoding the P4H collagen synthases were not dysregulated, while RRBP1 (that stimulates collagen synthesis) together with PLOD1 and PLOD2 (that stabilize collagens) were significantly up‐regulated. Moreover, SETD7, a gene known for activating collagenases, was significantly down‐regulated.
Figure 5.
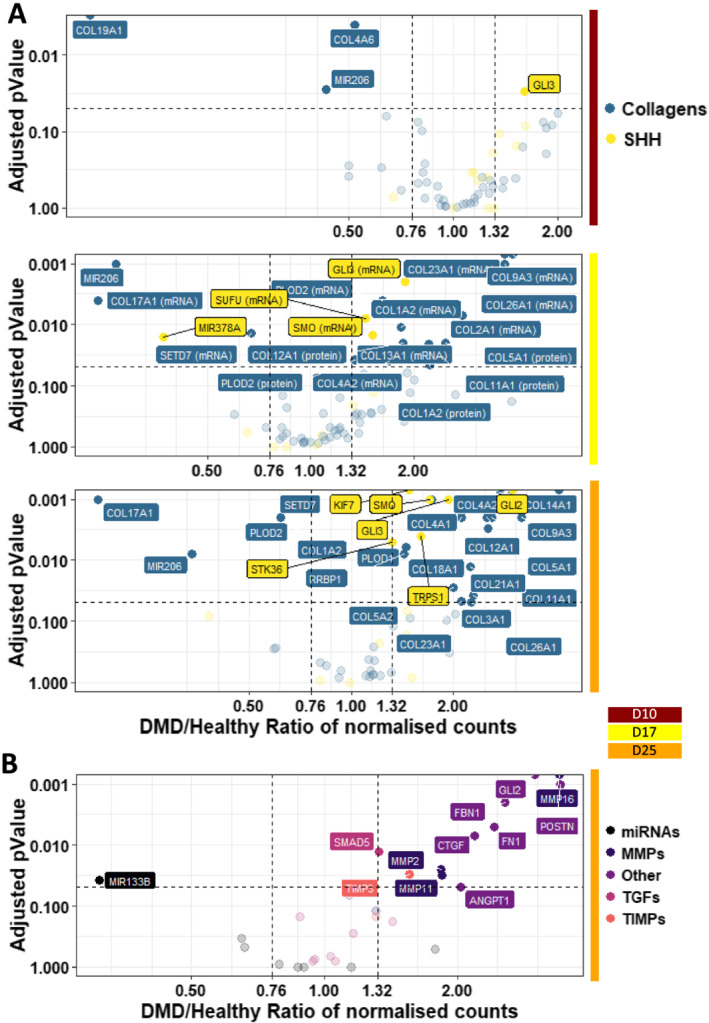
Illustration of the fibrosis phenotypes in Duchenne muscular dystrophy (DMD) cells. Volcano plots of dysregulated mRNAs/miRNAs related to (A) the SHH pathway and collagen metabolism at D10/17/25 and (B) fibrosis at D25—vertical grey dashed lines represent DMD/healthy ratio thresholds at 0.76 or 1.32, and the horizontal grey dashed line represents the adjusted P value threshold at 0.05 (D: day; MMP: matrix metallopeptidase; SHH: sonic hedgehog pathway; TIMP: tissue inhibitor of metallopeptidase; TGF: transforming growth factor).
At the myotube stage, a fibrosis‐related gene set was clearly up‐regulated in DMD cells, as illustrated by the overexpression of ANGPT1, CTGF, collagens (e.g. COL1A2), matrix metallopeptidases (MMPs), and tissue inhibitors of metallopeptidase (TIMPs) (Figure 5B). Conversely, the myomiR MIR133 that controls CTGF expression 49 was repressed (Table S5). Interestingly, gene members of the transforming growth factor (TGF)‐β pathway, a well‐known inducer of fibrosis, 50 were not found dysregulated (Figure 5B, Table S5).
Altogether, these data argue for fibrosis as an intrinsic feature of DMD skeletal muscle cells, rather than a process solely driven by interstitial cell populations in the niche. Furthermore, this muscle‐driven fibrosis seems independent of the TGF‐β pathway, and could rather depend on the SHH pathway, together with an intrinsic up‐regulation of chondrocyte markers and collagens.
Genes involved in mitochondrial metabolism are drastically dysregulated in Duchenne muscular dystrophy human induced pluripotent stem cell‐derived myotubes
As previously described 51 and illustrated on Figure S13A, genes involved in the energy metabolism of DMD hiPSC‐derived myotubes were dysregulated at the creatine and carbohydrate levels, up to the respiration (Figures 6A, 6B, and S1C; Table S5). The creatine transporter was not impacted, while mRNAs coding for enzymes of both creatine and creatine phosphate biosynthesis were underrepresented. Neither glucose nor glutamate transporter expression were impaired. However, genes involved in glutamine biosynthesis (followed by gluconeogenesis that feeds glycolysis from glutamine) as well as glycogenesis (followed by glycogenolysis that feeds glycolysis from glycogen) were all down‐regulated, together with genes coding for glycolysis itself. In contrast, genes coding for the pentose phosphate pathway (which is in parallel to glycolysis) were up‐regulated, especially the oxidative part. Gene expression for pyruvate decarboxylation and generation of acetyl‐CoA to feed the tricarboxylic acid cycle was also impaired. Finally, the genes involved in the tricarboxylic acid cycle itself (Figures 6A and S1C) and the mitochondrial electron transport chain were down‐regulated (Figures 6B and S1C). This is particularly reinforced by lower levels of a member of the ATP synthase complex ATP5A1 at both mRNA and protein levels (Figure 6C and 6D). These mRNA and protein data were completed by the measurement of ATP levels, which were significantly decreased in DMD hiPSC‐derived myotubes (Figure 6E). Moreover, transcripts encoded by the mitochondrial DNA and mitochondrial DNA itself were decreased in DMD hiPSC‐derived myotubes at Day 25 (Figure S13B–E).
Figure 6.
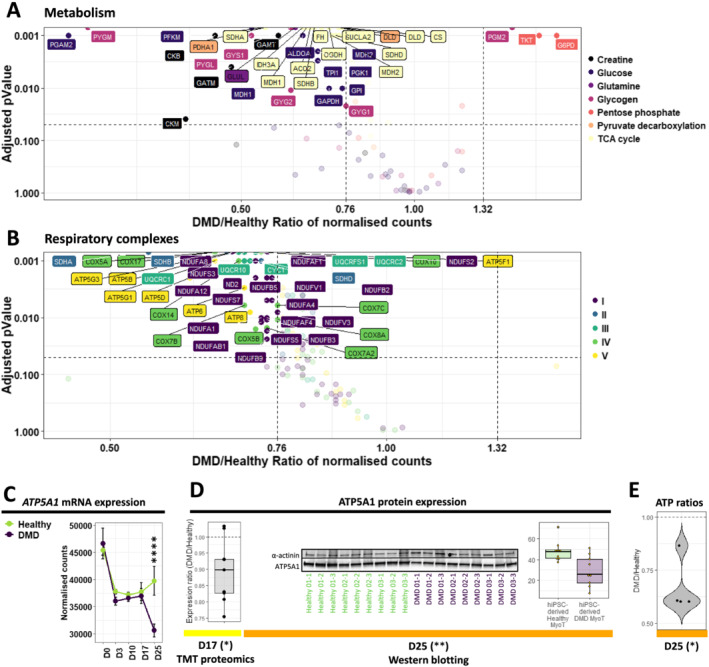
Illustration of the metabolic and mitochondrial phenotypes in Duchenne muscular dystrophy (DMD) cells. Volcano plots of dysregulated mRNAs/miRNAs related to (A) principal metabolic pathways and (B) the constitution of the five mitochondrial respiratory complexes in DMD hiPSC‐derived MyoT—vertical grey dashed lines represent DMD/healthy ratio thresholds at 0.76 or 1.32, and the horizontal grey dashed line represents the adjusted P value threshold at 0.05. Quantification of ATP5A1 expression (C) at the mRNA level during differentiation and (D) at the protein level at D17 (TMT proteomic data, left) and D25 (western blot data, right). (E) Measure of ATP levels in DMD cell lines, relative to healthy controls (*adjusted P value ≤ 0.05, **adjusted P value ≤ 0.01, ***adjusted P value ≤ 0.001, ****adjusted P value ≤ 0.0001) (D: day; hiPSC: human induced pluripotent stem cell, MyoT: myotube)
In the presented cell model, a significant down‐regulation of an mRNA set coding for mitochondrial proteins was primarily observed at Day 10 with the down‐regulation of 11% (12 mRNAs, DMD/healthy expression ratio ≤ 0.76, adjusted P value ≤ 0.05) of the mitochondrial outer membrane genes and amplified during the differentiation of DMD cells (Figure 7A). Therefore, defects depicted at Day 25 rooted before the expression of the skeletal muscle program at Day 17. Among them, mRNA down‐regulation of TSPO, a channel‐like molecule involved in the modulation of mitochondrial transition pore, 52 occurred from Day 10 to Day 25. This down‐regulation was also observed at the protein level at Day 17 (Figure 7B). Moreover, the protein import system was affected from Day 17 at both mRNA and protein levels (Figure S13C–F). Simultaneously, mRNAs involved in mitochondrial genome transcription started to be down‐regulated, followed by genes involved in mitochondrial DNA replication at Day 25 (Figure S13D–G). This progressive increase of dysregulations was also observed at the level of the entire mRNA set related to mitochondria (around 1000 mRNAs) as illustrated by the volcano plots as well as the gene ontology enrichments (Figures 7C and S1C).
Figure 7.
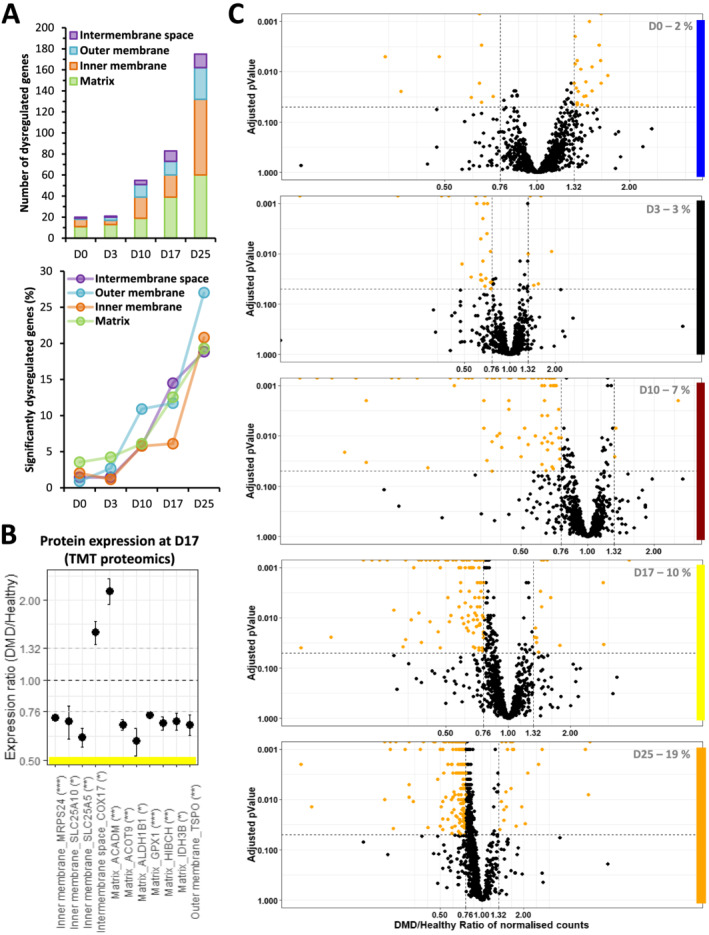
Mitochondrial dysregulations in Duchenne muscular dystrophy (DMD) cells during differentiation. (A) Absolute (top) and relative numbers (%, bottom) of dysregulated genes from the different mitochondrial compartments over the course of DMD hiPSC differentiation. (B) Expression ratios of selected mitochondrial proteins. Statistical differences are indicated in brackets (*adjusted P value ≤ 0.05, **adjusted P value ≤ 0.01, ***adjusted P value ≤ 0.001, ****adjusted P value ≤ 0.0001). (C) Volcano plots of mitochondria‐related genes over the course of DMD hiPSC differentiation. Statistical differences are symbolized with orange dots—vertical grey dashed lines represent DMD/healthy ratio thresholds at 0.76 or 1.32, and the horizontal grey dashed line represents the adjusted P value threshold at 0.05. The percentage of significantly dysregulated genes is indicated at the bottom right in grey (D: day).
Our data highlight early impairments in genes coding for mitochondria that start at the somite stage and increase with the differentiation in an orderly manner. These elements complete the mitochondrial DMD phenotype described above at the myotube stage.
Altogether, our study demonstrates that DMD starts prior to the expression of well‐described markers of muscle differentiation. It shows that hiPSC‐based experimental models of DMD can help identify early disease manifestations and stratify multiple pathological features over the course of muscle development.
Additional references describing the expression of marker genes at specific differentiation steps can be found in the Data S1.
Discussion
Since the discovery of the DMD gene in 1987, 1 DMD cellular phenotypes were considered under the unique scope of a ‘mechanical hypothesis’ in which dystrophin deficiency led to membrane leakage and ultimately muscle cell rupture. However, over the last 15–20 years, studies have brought unequivocal evidence that multiple additional factors are in play, such as calcium intracellular overloads, 53 , 54 excessive oxidative stress, 55 , 56 metabolic switches, 57 , 58 as well as an overall tissue context where aberrant interactions between resident cells lead to inflammation and fibro‐adipogenesis. 59 , 60 , 61 This has progressively led to a complex picture involving interdependent homeostatic perturbations, and to date, the identification of prevalent pathological features driving the initiation of DMD is hardly feasible.
The skeletal myogenesis modelled here by the differentiation of hiPSCs, without gene overexpression or cell sorting, homogeneously and robustly recapitulates key developmental steps—pluripotency, mesoderm, somite, and skeletal muscle—without any trace of other lineages. As shown by the analysis of gene expression variations between triplicates, variability between cell lines was greater than within cell lines for each genotype. The different genetic backgrounds can explain this. In the meantime, healthy cells exhibited less variability between each other than DMD cells that bear distinct DMD mutations. We can therefore argue that our phenotypical analysis will have more false negatives than false positives. It can also be considered closer to clinical heterogeneity. Therefore, it is a suitable dynamic model for studying human skeletal muscle development in both healthy and DMD cells, offering the possibility to clarify the consequences of the absence of dystrophin at each step of the differentiation process, as well as to explore dystrophin functions and find earlier and more specific disease biomarkers.
As previously observed with pluripotent stem cells, 62 hiPSC‐derived myotubes at Day 25 displayed an embryonic/foetal gene expression profile. However, a clear distinction must be made between the nature of the expressed isoforms—embryonic/foetal/postnatal—and the degree of differentiation. For instance, hiPSC‐derived myotubes expressed multiple markers of terminally differentiated muscles at levels higher than those measured in tissue‐derived myotubes. With the idea of exploring human DMD phenotypes during muscle development, we argued that generating embryonic/foetal myotubes from hiPSCs would not be a limitation.
In qualitative terms, DMD hiPSC‐derived myotubes showed an overall morphology similar to healthy controls, with cell fusion and clear striation patterns, suggesting that the potential impact of dystrophin during in vitro differentiation is subtle and does not prevent myotube formation. However, our unbiased mRNA‐seq analysis highlighted striking transcriptome dysregulations at Day 25. This includes numerous genes that can be linked to previously described DMD phenotypes such as (i) DAPC dissociation, 63 (ii) rupture of calcium homeostasis, 53 (iii) myomiR down‐regulation, 64 , 65 (iv) sarcomere destabilization, 66 , 67 (v) mitochondrial and metabolism dysregulations, 57 , 58 (vi) NMJ fragmentation, 68 , 69 and (vii) fibrosis. 61 , 70 It is interesting to note that these phenotypes are already detected at the transcriptional level in embryonic/foetal myotubes, while they usually appear postnatally in human patients and other animal models. In addition, most of them are often considered as consequences of degeneration–regeneration cycles typical of DMD muscles in vivo, 71 , 72 , 73 which are absent in our in vitro model, indicating that a part of these defects is primarily due to the absence of dystrophin itself. In particular, our data suggest that fibrosis is an intrinsic feature of DMD skeletal muscle cells, and therefore, it does not absolutely require a specific tissue context or additional cell populations to be detected in vitro. Fibrosis is a major hallmark of DMD pathophysiology, and the regulation of this process has been largely investigated in the past. 50 , 74 A long‐debated question is the implication of the TGFβ signalling pathway. 75 , 76 In this study, TGFβ signalling was inhibited up to Day 17 by specific molecules contained in the cell culture media, and TGFβ‐related genes were not up‐regulated at Day 25, suggesting that the observed up‐regulation of fibrosis‐related markers is TGFβ‐independent.
Because several studies in human patients and animal models had described dystrophic phenotypes in DMD foetuses/infants, 9 – 14 we investigated the precise timing of disease onset in our hiPSC‐derived cells. First, the absence of dystrophin does not modify the capacity of cells derived from adult tissue biopsies to be reprogramed using the approach developed by Takahashi et al. 77 Both healthy and DMD cells retained pluripotency and the capacity to enter the mesoderm compartment at Day 3. At that time, the embryonic dystrophin Dp412e is expressed and only marginal dysregulations are observed in DMD cells, a priori unrelated to cell fate choice as cells only express paraxial mesoderm markers at levels similar to healthy controls.
Duchenne muscular dystrophy dysregulations are greatly increased at Day 10, when cells express somite markers. At that time, we noticed few significant dysregulations of cell lineage markers, which became more prevalent at Days 17 and 25. This might be an indication that to some extent, cell fate is misguided in DMD cells, where skeletal muscle markers are underexpressed and replaced by markers of alternative lineages, such as chondrocytes.
First visible at Day 10, we identified the dysregulation of mitochondrial genes as one of the key processes happening in an orderly manner. Interestingly, early observations prior to the discovery of the DMD gene had hypothesized that DMD was a mitochondrial/metabolic disease based on protein quantifications and enzyme activities. 57 , 78 Later, mitochondria was identified as a key organelle in DMD, responsible for metabolic perturbations but also calcium accumulation and generation of reactive oxygen species. 53 – 56 In this study, numerous genes coding for proteins located in the outer mitochondrial membrane start to be down‐regulated from Day 10 in DMD cells, such as the benzodiazepine receptor TSPO, a member of the controversial mitochondrial permeability transition pore (mPTP). 52 The mPTP is a multiprotein complex whose members are not all precisely identified, and several studies suggest that it might be involved in DMD pathophysiology. 79 , 80 A chicken‐and‐egg question currently debated relates to the initiation of these homeostatic breakdowns, as positive feedbacks exist between mitochondria, oxidative stress, and calcium homeostasis dysregulations. 54 , 55 At the transcriptome level, dysregulations of genes controlling calcium homeostasis were detected after Day 10, suggesting that mitochondrial impairment starts early and has predominant consequences in DMD, as hypothesized by Timpani et al. 51 Further experiments are needed to better evaluate the impact of mitochondrial dysregulations at the functional level.
Day 17 marks the entry into the skeletal muscle compartment with the expression of specific transcription factors, cell surface markers, myomiRs, as well as the increase of skeletal muscle variant of dystrophin (Dp427m). It also marks the initiation of the skeletal muscle gene dysregulations observed at the myotube stage (i.e. down‐regulation of genes involved in DAPC and calcium homeostasis). For instance, the up‐regulation of fibrosis‐related genes observed in DMD myotubes at Day 25 is already visible at Day 17, with the up‐regulation of the SHH pathway as well as collagen‐related genes. In this study, it is seen as an early indicator of DMD physiopathology, confirming previous observations in DMD infants, both transcriptionally 4 and histologically. 81 , 82
Moreover, several myomiRs were found down‐regulated at Days 17 and 25 and seem to play a central part in multiple DMD phenotypes. Besides their role in myogenesis, 83 , 84 myomiRs can be involved in calcium homeostasis, 85 metabolism and mitochondrial functions, 86 , 87 and fibrosis. 49 , 88 In particular, MIR1‐1 and MIR206 are known to target key genes such as CACNA1C, 85 CTGF, 49 RRBP1, 88 several regulators of the pentose phosphate pathway, 86 and even transcripts encoded by the mitochondrial genome. 87 Even though the functional consequences of the multiple gene and myomiR dysregulations highlighted in this study are virtually impossible to anticipate, we believe that myomiRs can be key players in DMD physiopathology.
Previous studies in mice suggest early functional roles of muscular dystrophin in the development of striated muscles 89 , 90 and show that dystrophin participates in regulating satellite cell polarity, asymmetric division, and possibly commitment. 91 , 92 , 93 Few other studies in DMD hiPSC‐derived myoblasts 18 and in DMD human primary myoblasts 94 argued that DMD starts before the expression of the muscular dystrophin protein. Our data suggest that Dp427m is actually expressed before muscle commitment but at a lower level. This fact might explain why disease phenotypes seem to be initiated at the somite stage. This early initiation could also be explained by the deficit in other dystrophin isoforms expressed before Day 10, such as Dp412e at Day 3, 15 as well as by the decrease or loss of other RNA products expressed from the DMD locus, such as the ubiquitous isoform Dp71‐40 or long non‐coding RNAs. 95 The lack of knowledge around these additional products from the DMD locus contrasts with the extensive amount of data on the structure and function of the main muscular isoform Dp427m whose most studied role is to stabilize muscle cell membrane during contraction. 96 DMD knockdown results at Day 17 in a healthy cell line with partial mimicking of DMD phenotype could suggest a dynamic process in DMD: some dysregulations might not be reproduced by removing DMD after muscle commitment, highlighting the fact that absence of DMD locus expression during development could have impacts before cells becoming muscles and therefore before Dp427m having its well‐known role in muscles, as it is shown by our multi‐omic study. The role of Dp427m in non‐muscle cells could also be questioned. Other tissue‐specific isoforms have been described, for example, in the retina (Dp260 45 ) and in the brain (Dp427c, 97 Dp427p, 44 and Dp140 43 ), some of which are also slightly expressed in skeletal muscles under certain circumstances, 98 but their role remains mostly unknown. Interestingly, in our data, the expression of Dp260 follows the same pattern of expression as Dp427m. It has been shown that the expression of Dp260 in mdx/utrnK/K mice can rescue the mdx phenotype, 99 indicating overlapping functions between Dp427m and Dp260. On the other hand, it is now well established that a third of DMD patients display cognitive deficiencies—which might be correlated with mutations affecting Dp140 100 —attesting that dystrophin can be involved in other cell functions.
To date, DMD diagnosis is made mostly around 4 years old when the first physical symptoms appear, meaning the muscles are already greatly damaged. Our study gives an argument for newborn screening. Moreover, the standard of care for DMD patients helps mitigate and delay some of the most severe symptoms but remains insufficient to have a curative effect. Despite decades of work with the mdx mouse model, only a few pharmacological candidate molecules have moved forward to clinical trials, with variable efficiency. As several gene therapy trials have been recently initiated with promising preliminary data, we believe that our human in vitro model system might be useful for the development of combination therapies. Recent studies have proved that the association of two different therapeutic approaches could have a synergistic effect on the overall treatment outcome and can be used for instance to boost the effect of dystrophin re‐expression by antisense oligonucleotides or gene therapy. 8 , 101 , 102 Here, our extensive RNA‐seq data could help identify relevant therapeutic targets for pharmacological intervention, such as CTGF—involved in fibrosis and found up‐regulated in DMD myotubes—which can be inhibited by monoclonal antibodies, 103 or TSPO receptor—a receptor potentially member of the mPTP down‐regulated in DMD cells—targetable with benzodiazepines. 104 In addition, our model might also be used as a platform to screen pharmacological compounds in an unbiased high‐throughput manner. Indeed, skeletal muscle progenitor cells at Day 17 can be robustly amplified, cryopreserved, and plated in a 384‐well plate format (data not shown). Thus, they could be an interesting tool to highlight pharmacological compounds to be used alone or in combination with gene therapy.
To summarize, the directed differentiation of hiPSCs without gene overexpression or cell sorting homogeneously and robustly recapitulates key developmental steps of skeletal myogenesis and generates embryonic/foetal myotubes without any trace of other lineages. The absence of dystrophin does not compromise cell reprogramming, pluripotency, or the entry into the mesoderm compartment. While a very low amount of the long muscular dystrophin isoform is expressed, a significant transcriptome dysregulation can be observed at the somite stage that implicates mitochondria prior to dysregulations of genes controlling calcium homeostasis. Despite their ability to enter the skeletal muscle lineage compartment and become myotubes, DMD cells exhibit an imbalance in cell fate choice as they express lower amounts of key muscle proteins and retain basal expression of marker genes from other lineages, leading to the well‐characterized DMD phenotypes including muscle features and metabolism dysregulations as well as fibrosis. Altogether, these data argue for (i) a deficit and not a delay in DMD differentiation, (ii) seeing DMD as a progressive developmental disease as well as a metabolic pathology whose onset is triggered before the entry into the skeletal muscle compartment, and (iii) fibrosis as an intrinsic feature of DMD muscle cells. Future studies could explore the additional roles of DMD locus products and the impact of their loss during skeletal muscle development, as well as find earlier and more specific disease biomarkers and develop combination therapeutic strategies using high‐throughput drug screening.
Data availability statement
All the omics data are available online for exploration through a graphical interface (https://muscle‐dmd.omics.ovh/). For additional information on this interface, please send an email to shiny@virginie‐mournetas.fr.
Funding
We thank the Fondation Maladies Rares (GenOmics grant), Labex Revive (Investissement d'Avenir; ANR‐10‐LABX‐73), and the AFM Téléthon for funding this project.
Conflicts of interest
Spiros D. Garbis is Founder, President and CEO of Proteas Bioanalytics, Inc.
Supporting information
Data S1. Supporting Information
Table S1. Supporting Information
Table S2. Supporting Information
Table S3. Supporting Information
Table S4. Supporting Information
Table S5. Supporting Information
Table S6. Supporting Information
Figure S1. Supporting Information
Figure S2. Supporting Information
Figure S3. Supporting Information
Figure S4. Supporting Information
Figure S5. Supporting Information
Figure S6. Supporting Information
Figure S7. Supporting Information
Figure S8. Supporting Information
Figure S9. Supporting Information
Figure S10. Supporting Information
Figure S11. Supporting Information
Figure S12. Supporting Information
Figure S13. Supporting Information
Acknowledgements
The bulk and single‐cell RNA sequencing libraries were processed and sequenced by Integragen (Evry, France). We especially thank them for their help on the single‐cell RNA sequencing experiment. We gratefully acknowledge support from the PSMN (Pôle Scientifique de Modélisation Numérique) of the ENS de Lyon for the computing resources. We thank Dr Nacira Tabti, Dr Elisabeth Le Rumeur, Dr Nathalie Deburgrave, and Dr Malgorzata Rak for providing us with specific reagents and antibodies. We thank Dr Linda Popplewell for designing and validating the PMO7 sequence as well as for providing us the PMO51 sequence. We thank Dr Matteo Bovolenta for his helpful guidance on gene editing. We thank Dr David Israeli for his feedback on the manuscript and overall discussion on our project.
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 105
Mournetas V., Massouridès E., Dupont J.‐B., Kornobis E., Polvèche H., Jarrige M., Dorval A. R. L., Gosselin M. R. F., Manousopoulou A., Garbis S. D., Górecki D. C., and Pinset C. (2021) Myogenesis modelled by human pluripotent stem cells: a multi‐omic study of Duchenne myopathy early onset, Journal of Cachexia, Sarcopenia and Muscle, 12, 209–232, doi: 10.1002/jcsm.12665
References
- 1. Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the duchenne muscular dystrophy locus. Cell 1987;51:919–928. [DOI] [PubMed] [Google Scholar]
- 2. Koeks Z, Bladen CL, Salgado D, Van Zwet E, Pogoryelova O, McMacken G, et al. Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT‐NMD DMD global database. J Neuromuscul Dis 2017;4:293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu M, Chino N, Ishihara T. Muscle damage progression in Duchenne muscular dystrophy evaluated by a new quantitative computed tomography method. Arch Phys Med Rehabil 1993;74:507–514. [DOI] [PubMed] [Google Scholar]
- 4. Pescatori M, Broccolini A, Minetti C, Bertini E, Bruno C, D'amico A, et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J 2007;21:1210–1226. [DOI] [PubMed] [Google Scholar]
- 5. Al‐Khalili Szigyarto C, Spitali P. Biomarkers of Duchenne muscular dystrophy: current findings. Degener Neurol Neuromuscul Dis, vol 2018;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Moat SJ, Bradley DM, Salmon R, Clarke A, Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur J Hum Genet 2013;21:1049–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crone M, Mah JK. Current and emerging therapies for Duchenne muscular dystrophy. Curr Treat Options Neurol 2018;20:31. [DOI] [PubMed] [Google Scholar]
- 8. Lu‐Nguyen N, Malerba A, Popplewell L, Schnell F, Hanson G, Dickson G. Systemic antisense therapeutics for dystrophin and myostatin exon splice modulation improve muscle pathology of adult mdx mice. Mol Ther ‐ Nucleic Acids 2017;6:15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nguyen F, Cherel Y, Guigand L, Goubault‐Leroux I, Wyers M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J Comp Pathol 2002;126:100–108. [DOI] [PubMed] [Google Scholar]
- 10. Bassett DI, Bryson‐Richardson RJ, Daggett DF, Gautier P, Keenan DG, Currie PD. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003;130:5851–5860. [DOI] [PubMed] [Google Scholar]
- 11. Merrick D, Stadler LKJ, Larner D, Smith J. Muscular dystrophy begins early in embryonic development deriving from stem cell loss and disrupted skeletal muscle formation. DMM Dis Model Mech 2009;2:374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emery AEH. Muscle histology and creatine kinase levels in the foetus in Duchenne muscular dystrophy [31]. Nature 1977;266:472–473. [DOI] [PubMed] [Google Scholar]
- 13. Toop J, Emery AEH. Muscle histology in fetuses at risk for Duchenne muscular dystrophy. Clin Genet 1974;5:230–233. [DOI] [PubMed] [Google Scholar]
- 14. Vassilopoulos D, Emery AEH. Muscle nuclear changes in fetuses at risk for Duchenne muscular dystrophy. J Med Genet 1977;14:13–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Massouridès E, Polentes J, Mangeot PE, Mournetas V, Nectoux J, Deburgrave N, et al. Dp412e: a novel human embryonic dystrophin isoform induced by BMP4 in early differentiated cells. Skelet Muscle Dec. 2015;5:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nesmith AP, Wagner MA, Pasqualini FS, O’Connor BB, Pincus MJ, August PR, et al. A human in vitro model of Duchenne muscular dystrophy muscle formation and contractility. J Cell Biol 2016;215:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shoji E, Sakurai H, Nishino T, Nakahata T, Heike T, Awaya T, et al. Early pathogenesis of Duchenne muscular dystrophy modelled in patient‐derived human induced pluripotent stem cells. Sci Rep 2015;5:12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi IY, Lim HT, Estrellas K, Mula J, Cohen TV, Zhang Y, et al. Concordant but varied phenotypes among Duchenne muscular dystrophy patient‐specific myoblasts derived using a human iPSC‐based model. Cell Rep 2016;15:2301–2312. [DOI] [PubMed] [Google Scholar]
- 19. Chal J, Oginuma M, Al Tanoury Z, Gobert B, Sumara O, Hick A, et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol 2015;33:962–969. [DOI] [PubMed] [Google Scholar]
- 20. Young CS, Hicks MR, Ermolova NV, Nakano H, Jan M, Younesi S, et al. A single CRISPR‐Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC‐derived muscle cells. Cell Stem Cell 2016;18:533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hicks MR, Hiserodt J, Paras K, Fujiwara W, Eskin A, Jan M, et al. ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat Cell Biol 2018;20:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kodaka Y, Rabu G, Asakura A. Skeletal muscle cell induction from pluripotent stem cells. Stem Cells Int 2017;2017:1376151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caron L, Kher D, Lee KL, McKernan R, Dumevska B, Hidalgo A, et al. A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy‐affected skeletal muscles. Stem Cells Transl Med 2016;5:1145–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shelton M, Metz J, Liu J, Carpenedo RL, Demers SP, Stanford WL, et al. Derivation and expansion of PAX7‐positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Rep 2014;3:516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xi H, Fujiwara W, Gonzalez K, Jan M, Liebscher S, Van Handel B, et al. In vivo human somitogenesis guides somite development from hPSCs. Cell Rep 2017;18:1573–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Köster J, Rahmann S. Snakemake‐a scalable bioinformatics workflow engine. Bioinformatics 2012;28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 27. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics Jan. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang L, Wang S, Li W. RSeQC: quality control of RNA‐seq experiments. Bioinformatics Aug. 2012;28:2184–2185. [DOI] [PubMed] [Google Scholar]
- 29. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics Oct. 2016;32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 2009;4:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kolesnikov N, Hastings E, Keays M, Melnichuk O, Tang YA, Williams E, et al. ArrayExpress update‐simplifying data submissions. Nucleic Acids Res 2015;43, no. D1:D1113–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011;27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnetjournal 2011;17:10. [Google Scholar]
- 35. Amrhein V, Greenland S, McShane B. Scientists rise up against statistical significance. Nature 2019;567:305–307. [DOI] [PubMed] [Google Scholar]
- 36. Garnier S. viridis: default color maps from ‘matplotlib,’. Cranio 2018;1–6.29364088 [Google Scholar]
- 37. Wei T, Simko V, Levy M, Xie Y, Jin Y, Zemla J. R package ‘corrplot’: visualization of a correlation matrix. Stat 2017;56:316–324. [Google Scholar]
- 38. Gregory R, Warnes BBL. gplots: various R programming tools for plotting data. R package version 260 2008. [Google Scholar]
- 39. Fresno C, Fernández EA. RDAVIDWebService: a versatile R interface to DAVID. Bioinformatics 2013;29:2810–2811. [DOI] [PubMed] [Google Scholar]
- 40. Vizcaíno JA, Csordas A, Del‐Toro N, Dianes JA, Griss J, Lavidas I, et al. update of the PRIDE database and its related tools. Nucleic Acids Res 2016;44, no. D1:D447–D456, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Monaco AP, Neve RL, Colletti‐Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986;323:646–650. [DOI] [PubMed] [Google Scholar]
- 42. Byers TJ, Lidov HGW, Kunkel LM. An alternative dystrophin transcript specific to peripheral nerve. Nat Genet 1993;4:77–81. [DOI] [PubMed] [Google Scholar]
- 43. Lidov HGW, Selig S, Kunkel LM. Dp140: A novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet 1995;4:329–335. [DOI] [PubMed] [Google Scholar]
- 44. Górecki DC, Monaco AP, Derry JMJ, Walker AP, Barnard EA, Barnard PJ. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet 1992;1:505–510. [DOI] [PubMed] [Google Scholar]
- 45. D'Souza V. N., Man N. T., Morris G. E., Karges W., Pillers D. A. M., and Ray P. N., “A novel dystrophin isoform is required for normal retinal electrophysiology,” Hum Mol Genet, vol. 4, no. 5, pp. 837–842, 1995. [DOI] [PubMed] [Google Scholar]
- 46. Capitanio D, Moriggi M, Torretta E, Barbacini P, De Palma S, Viganò A, et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: changes contributing to preserve muscle function in Becker muscular dystrophy patients. J Cachexia Sarcopenia Muscle 2020;11:547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rotwein P, Pollock KM, Watson M, Milbrandt JD. Insulin‐like growth factor gene expression during rat embryonic development. Endocrinology 1987;121:2141–2144. [DOI] [PubMed] [Google Scholar]
- 48. Andersen DC, Laborda J, Baladron V, Kassem M, Sheikh SP, Jensen CH. Dual role of delta‐like 1 homolog (DLK1) in skeletal muscle development and adult muscle regeneration. Development 2013;140:3743–3753. [DOI] [PubMed] [Google Scholar]
- 49. Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, Van Der Made I, et al. MiR‐133 and miR‐30 regulate connective tissue growth factor: implications for a role of micrornas in myocardial matrix remodeling. Circ Res 2009;104:170–178. [DOI] [PubMed] [Google Scholar]
- 50. Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, et al. Fibrinogen drives dystrophic muscle fibrosis via a TGFβ/alternative macrophage activation pathway. Genes Dev 2008;22:1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Timpani CA, Hayes A, Rybalka E. Revisiting the dystrophin‐ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med Hypotheses 2015;85:1021–1033. [DOI] [PubMed] [Google Scholar]
- 52. Šileikyte J, Blachly‐Dyson E, Sewell R, Carpi A, Menabò R, Di Lisa F, et al. Regulation of the mitochondrial permeability transition pore by the outer membrane does not involve the peripheral benzodiazepine receptor (translocator protein of 18 kDa (TSPO)). J Biol Chem 2014;289:13769–13781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Emery AEH, Burt D. Intracellular calcium and pathogenesis and antenatal diagnosis of Duchenne muscular dystrophy. Br Med J 1980;280:355–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch Eur J Physiol 2009;458:915–928. [DOI] [PubMed] [Google Scholar]
- 55. Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch‐induced damage in the mdx mouse. PLoS ONE 2010;5:e15354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rodriguez MC, Tarnopolsky MA. Patients with dystrophinopathy show evidence of increased oxidative stress. Free Radic Biol Med 2003;34:1217–1220. [DOI] [PubMed] [Google Scholar]
- 57. Scholte HR, Busch HFM. Early changes of muscle mitochondria in duchenne dystrophy. Partition and activity of mitochondrial enzymes in fractionated muscle of unaffected boys and adults and patients. J Neurol Sci 1980;45:217–234. [DOI] [PubMed] [Google Scholar]
- 58. Sharma U, Atri S, Sharma MC, Sarkar C, Jagannathan NR. Skeletal muscle metabolism in Duchenne muscular dystrophy (DMD): an in‐vitro proton NMR spectroscopy study. Magn Reson Imaging 2003;21:145–153. [DOI] [PubMed] [Google Scholar]
- 59. Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, Fiore D, et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF‐mediated apoptosis of fibro/adipogenic progenitors. Nat Med 2015;21:786–794. [DOI] [PubMed] [Google Scholar]
- 60. Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidbal JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 2009;18:482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Desguerre I, Mayer M, Leturcq F, Barbet JP, Gherardi RK, Christov C. Endomysial fibrosis in duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol 2009;68:762–773. [DOI] [PubMed] [Google Scholar]
- 62. Sakai‐Takemura F, Narita A, Masuda S, Wakamatsu T, Watanabe N, Nishiyama T, et al. Premyogenic progenitors derived from human pluripotent stem cells expand in floating culture and differentiate into transplantable myogenic progenitors. Sci Rep 2018;8:6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsumura K, Tome FMS, Ionasescu V, Ervasti JM, Anderson RD, Romero NB, et al. Deficiency of dystrophin‐associated proteins in Duchenne muscular dystrophy patients lacking COOH‐terminal domains of dystrophin. J Clin Invest 1993;92:866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuasa K, Hagiwara Y, Ando M, Nakamura A, Takeda S, Hijikata T. MicroRNA‐206 is highly expressed in newly formed muscle fibers: Implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct Funct 2008;33:163–169. [DOI] [PubMed] [Google Scholar]
- 65. Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, et al. Common micro‐RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J 2009;23:3335–3346. [DOI] [PubMed] [Google Scholar]
- 66. Cullen MJ, Fulthorpe JJ. Stages in fibre breakdown in duchenne muscular dystrophy. an electron‐microscopic study. J Neurol Sci 1975;24:179–200. [DOI] [PubMed] [Google Scholar]
- 67. Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J Appl Physiol 2004;96:633–638. [DOI] [PubMed] [Google Scholar]
- 68. Kong J, Anderson JE. Dystrophin is required for organizing large acetylcholine receptor aggregates. Brain Res 1999;839:298–304. [DOI] [PubMed] [Google Scholar]
- 69. Kong J, Yang L, Li Q, Cao J, Yang J, Chen F, et al. The absence of dystrophin rather than muscle degeneration causes acetylcholine receptor cluster defects in dystrophic muscle. Neuroreport 2012;23:82–87. [DOI] [PubMed] [Google Scholar]
- 70. Bell CD, Conen PE. Histopathological changes in Duchenne muscular dystrophy. J Neurol Sci 1968;7:529–544. [DOI] [PubMed] [Google Scholar]
- 71. Brouilly N, Lecroisey C, Martin E, Pierson L, Mariol MC, Mounier N, et al. Ultra‐structural time‐course study in the C. elegans model for Duchenne muscular dystrophy highlights a crucial role for sarcomere‐anchoring structures and sarcolemma integrity in the earliest steps of the muscle degeneration process. Hum Mol Genet 2015;24:6428–6445. [DOI] [PubMed] [Google Scholar]
- 72. Haddix SG, Lee YI, Kornegay JN, Thompson WJ. Cycles of myofiber degeneration and regeneration lead to remodeling of the neuromuscular junction in two mammalian models of Duchenne muscular dystrophy. PLoS ONE 2018;13, no. 10:e0205926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Luz MAM, Marques MJ, Santo Neto H. Impaired regeneration of dystrophin‐deficient muscle fibers is caused by exhaustion of myogenic cells. Braz J Med Biol Res 2002;35:691–695. [DOI] [PubMed] [Google Scholar]
- 74. Zhou L, Porter JD, Cheng G, Gong B, Hatala DA, Merriam AP, et al. Temporal and spatial mRNA expression patterns of TGF‐β1, 2, 3 and TβRI, II, III in skeletal muscles of mdx mice. Neuromuscul Disord 2006;16:32–38. [DOI] [PubMed] [Google Scholar]
- 75. Bernasconi P, Torchiana E, Confalonieri P, Brugnoni R, Barresi R, Mora M, et al. Expression of transforming growth factor‐β1 in dystrophic patient muscles correlates with fibrosis. Pathogenetic role of a fibrogenic cytokine. J Clin Invest 1995;96:1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Caputo L, Granados A, Lenzi J, Rosa A, Ait‐Si‐Ali S, Puri PL, et al. Acute conversion of patient‐derived Duchenne muscular dystrophy iPSC into myotubes reveals constitutive and inducible over‐activation of TGFβ‐dependent pro‐fibrotic signaling. Skelet Muscle 2020;10:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 78. Dreyfus JC, Schapira G, Schapira F. Biochemical study of muscle in progressive muscular dystrophy. J Clin Invest 1954;33:794–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ascah A, Khairallah M, Daussin F, Bourcier‐Lucas C, Godin R, Allen BG, et al. Stress‐induced opening of the permeability transition pore in the dystrophin‐deficient heart is attenuated by acute treatment with sildenafil. Am J Physiol Heart Circ Physiol 2011;300:H144–H153. [DOI] [PubMed] [Google Scholar]
- 80. Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 2012;181:583–592. [DOI] [PubMed] [Google Scholar]
- 81. Pearson CM. Histopathological features of muscle in the preclinical stages of muscular dystrophy. Brain 1962;85:109–120. [DOI] [PubMed] [Google Scholar]
- 82. Bradley WG, Hudgson P, Larson PF, Papapetropoulos TA, Jenkison M. Structural changes in the early stages of Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry 1972;35:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, et al. The role of microRNA‐1 and microRNA‐133 in skeletal muscle proliferation and differentiation. Nat Genet 2006;38:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hak KK, Yong SL, Sivaprasad U, Malhotra A, Dutta A. Muscle‐specific microRNA miR‐206 promotes muscle differentiation. J Cell Biol 2006;174:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, et al. Misregulation of miR‐1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol 2011;18:840–845. [DOI] [PubMed] [Google Scholar]
- 86. Singh A, Happel C, Manna SK, Acquaah‐Mensah G, Carrerero J, Kumar S, et al. Transcription factor NRF2 regulates miR‐1 and miR‐206 to drive tumorigenesis. J Clin Invest 2013;123:2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 2014;158:607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fry CS, Kirby TJ, Kosmac K, McCarthy JJ, Peterson CA. Myogenic progenitor cells control extracellular matrix production by fibroblasts during skeletal muscle hypertrophy. Cell Stem Cell 2017;20:56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Seno MMG, Graham IR, Athanasopoulos T, Trollet C, Pohlschmidt M, Crompton MR, et al. RNAi‐mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum Mol Genet 2008;17:2622–2632. [DOI] [PubMed] [Google Scholar]
- 90. Ghahramani Seno MM, Trollet C, Athanasopoulos T, Graham IR, Hu P, Dickson G. Transcriptomic analysis of dystrophin RNAi knockdown reveals a central role for dystrophin in muscle differentiation and contractile apparatus organization. BMC Genomics 2010;11:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dumont NA, Wang YX, Von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, et al. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med 2015;21:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang YX, Feige P, Brun CE, Hekmatnejad B, Dumont NA, Renaud JM, et al. EGFR‐Aurka signaling rescues polarity and regeneration defects in dystrophin‐deficient muscle stem cells by increasing asymmetric divisions. Cell Stem Cell 2019;24, no. 3:419–432.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang NC, Sincennes MC, Chevalier FP, Brun CE, Lacaria M, Segalés J, et al. The dystrophin glycoprotein complex regulates the epigenetic activation of muscle stem cell commitment. Cell Stem Cell 2018;22, no. 5:755–768.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 1983;80, no. 15 I:4856–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bovolenta M, Erriquez D, Valli E, Brioschi S, Scotton C, Neri M, et al. The DMD locus harbours multiple long non‐coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS One, vol 2012;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ervasti JM, Campbell KP. A role for the dystrophin‐glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol 1993;122:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nudel U, Zuk D, Einat P, Zeelon E, Levy Z, Neuman S, et al. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature 1989;337:76–78. [DOI] [PubMed] [Google Scholar]
- 98. Muntoni F, Melis MA, Ganau A, Dubowitz V. Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X‐linked dilated cardiomyopathy. Am J Hum Genet 1995;56:151–157. [PMC free article] [PubMed] [Google Scholar]
- 99. Warner LE, DelloRusso CT, Crawford RW, Rybakova IN, Patel JR, Ervasti JM, et al. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin–glycoprotein complex and partially prevents dystrophy. Hum Mol Genet 2002;11:1095–1105. [DOI] [PubMed] [Google Scholar]
- 100. Doorenweerd N, Mahfouz A, Van Putten M, Kaliyaperumal R, T'Hoen PAC, Hendriksen JGM, et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep, vol 2017;1:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lu‐Nguyen N, Ferry A, Schnell FJ, Hanson GJ, Popplewell L, Dickson G, et al. Functional muscle recovery following dystrophin and myostatin exon splice modulation in aged mdx mice. Hum Mol Genet 2019;28:3091–3100. [DOI] [PubMed] [Google Scholar]
- 102. Peccate C, Mollard A, Le Hir M, Julien L, McClorey G, Jarmin S, et al. Antisense pre‐treatment increases gene therapy efficacy in dystrophic muscles. Hum Mol Genet 2016;25:3555–3563. [DOI] [PubMed] [Google Scholar]
- 103. Morales MG, Gutierrez J, Cabello‐Verrugio C, Cabrera D, Lipson KE, Goldschmeding R, et al. Reducing CTGF/CCN2 slows down mdx muscle dystrophy and improves cell therapy. Hum Mol Genet 2013;22:4938–4951. [DOI] [PubMed] [Google Scholar]
- 104. Gatliff J, Campanella M. TSPO: Kaleidoscopic 18‐kDa amid biochemical pharmacology, control and targeting of mitochondria. Biochem J 2016;473:107–121. [DOI] [PubMed] [Google Scholar]
- 105. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information
Table S1. Supporting Information
Table S2. Supporting Information
Table S3. Supporting Information
Table S4. Supporting Information
Table S5. Supporting Information
Table S6. Supporting Information
Figure S1. Supporting Information
Figure S2. Supporting Information
Figure S3. Supporting Information
Figure S4. Supporting Information
Figure S5. Supporting Information
Figure S6. Supporting Information
Figure S7. Supporting Information
Figure S8. Supporting Information
Figure S9. Supporting Information
Figure S10. Supporting Information
Figure S11. Supporting Information
Figure S12. Supporting Information
Figure S13. Supporting Information
Data Availability Statement
All the omics data are available online for exploration through a graphical interface (https://muscle‐dmd.omics.ovh/). For additional information on this interface, please send an email to shiny@virginie‐mournetas.fr.