

Abstract
Enterotoxigenic E.coli is causing diarrheal illness in children as well as adults with the majority of the cases occurring in developing countries. To reduce the number of cases occurring worldwide, the development of an effectual vaccine against these bacteria can be the only prevention. This conjectural work was performed using modern bioinformatics tools for investigation of proteome of ETEC strain E24377A. Different computational vaccinology approaches were deployed to assess several parameters including antigenicity, allergenicity, stability, localization, molecular weight and toxicity of the predicted epitopes required for good vaccine candidate to elicit immune response against diarrhea. We estimated two known control antigens, epitope 141STLPETTVV149 of Hepatitis B virus and epitope 265ILRGSVAHK273 of H1N1 Nucleoprotein in an attempt to corroborate our research work. Furthermore molecular docking was performed to evaluate the interaction between HLA allele and peptide, the peptide QYGGGNSAL and peptide LPYFELRWL were considered to be the most promiscuous T cell epitopes with the highest binding energy value of −2.09 kcal/mol and −1.84 kcal/mol, respectively. In addition, dynamic simulation revealed good stability of the vaccine construct as well as population coverage analysis exhibits the highest population coverage in the regions of East Asia, India, Northeast Asia, South Asia and North America. Therefore, these two epitopes can be further synthesized for wet lab analysis and could be considered as a promising vaccine against diarrhea.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13721-021-00287-6.
Keywords: Epitope, Immunoinformatics, Docking, Vaccine
Introduction
Enterotoxigenic Escherichia coli (ETEC) bacteria are the major cause of diarrheal disease among children and adults globally. It was estimated that ETEC cause 280-400 million diarrheal cases in children under 5 years of age and 100 million cases in children above 5 years annually (WHO 2018). Diarrheal infection is primarily caused by the ingestion of contaminated food or water that allows to enter the strain into body (Seo et al. 2019). ETEC bacterium colonizes intestinal epithelial cells and releases two enterotoxins, heat-labile toxin (LT) and heat-stable toxin (ST) that results in the disruption of fluid and electrolyte homeostasis leading to fluid hypersecretion and watery diarrhea (Huang et al. 2018). The first cause of enterotoxigenic E. coli diarrhea was observed in 1956 in Kolkata and from then it continues to be a global killer in countries like Dhaka, Bangladesh, U.S. and India (Isidean et al. 2011). ETEC diarrhea is also referred to as Traveler’s diarrhea as it is more vulnerable to travelers who are travelling to developing countries and it remains to be endemic whole year but mostly in warm season (Qadri et al. 2005).
There is no vaccine for ETEC diarrhea till date; therefore, developing an ETEC vaccine could induce broad-spectrum immune response against ETEC strains. From the previous literature, it depicts that ETEC vaccine has been proven to be challenging in the past few years and primarily all the research studies have engrossed on known virulence factors, mainly CF/CS antigens and heat-labile toxin (Holmgren et al. 2017). However, comprehensive studies indicate the mode of ETEC infection is beyond these antigens and a new approach targeting multiple antigens apart from these classical known antigens could provide significant protection (Zhang and Sack 2015). Antibiotic treatment is not suggested for diarrhea since it can lead to antibiotic resistance in ETEC as well as costly; therefore, it has become one of the major public health problems all over the world (Shaheen et al. 2003). ETEC has always been associated with post-diarrheal long-term effect causing malnutrition, growth stunting and impaired cognitive development and therefore remains the leading cause among the children (Chakraborty et. al 2019). All the research work has focused to target known proteins, that is, involve in causing bacteria and few previous studies also depicted common proteins between shared between two or three strains using Omics data technology (Mehla and Ramana 2016). But the use of upgraded tools for docking and simulation with better accuracy has led towards the identification of unknown proteins of strain ETEC E24377A with better result.
In the last few years, reports suggest the occurrence of many diseases due to the several outbreaks of different viruses and bacterial infection. So, the requirement of vaccine is crucial which can provide a better prevention or treatment against these diseases. But to minimize the cost of developing vaccine, different technologies have been identified that include computational immunology approach. Therefore, the present study employed the use of an immunoinformatic approach for scrutinization of the immunogenic epitopes from the most various pathogenic and widespread ETEC strain causing diarrhea globally. The decline in the traditional experimental vaccine development approach is due to high cost, time-consuming and practical limitations of feasibility had led to the use of computational approach which involves the epitope-based prediction methods in search of novel vaccine candidates from the whole proteomes (Khan et al. 2019). This approach can be termed as “reverse vaccinology” which involves the characterization of vaccine candidates by screening complete proteome sequence analysis of the targeted pathogen. Here, in our study, only MHC-I T cell epitopes are calculated as accuracy of different tools for predicting MHC II restricted epitopes are much lower in comparison to MHC-I tools. And therefore selecting particular tool for accurate result for MHC II T cell epitopes has become concern (Zawawi et al. 2020).
To stimulate the different arms of the immune system, prediction of potential epitopes with the different computational tools was employed (Kumar et al. 2013). The proteome of the most pathogenic strain E24377A can be helpful in identifying T cell epitopes in designing the vaccine candidates (Khan et al. 2018). Furthermore, allergic and toxicity prediction, modeling of epitopes with HLA alleles can be studied and the docking of the MHC molecules and identified peptides will be performed.
Methodology
Sequence retrieval and identification of antigenic proteins
Here, in our analysis, complete proteome of the most widespread and pathogenic strain Escherichia coli E24377A has been selected. This whole proteome comprises 6 plasmids and a total of 4915 protein-coding sequences (Rasko et al. 2008). The FASTA file of these protein sequences of the proteome id UP000001122 was isolated from Uniprot database (Morgat et al. 2019). Uniprot database is a very informational database that stores protein sequence and functional information. Vaxign tool was applied to screen antigens that show adhesin value ≥ 0.51 (Xiang and He 2009). This tool not only determines the adhesion values of protein but also their localization, orthologs and transmembrane properties. By characterizing the adhesion probability of the antigenic protein, we will be identifying the binding ability of the pathogen to the host and thus targeting on the mode of action of bacteria. The flowchart to depict the complete methodology of the steps involved in the analysis of vaccine development is shown in Fig. 1.
Fig. 1.
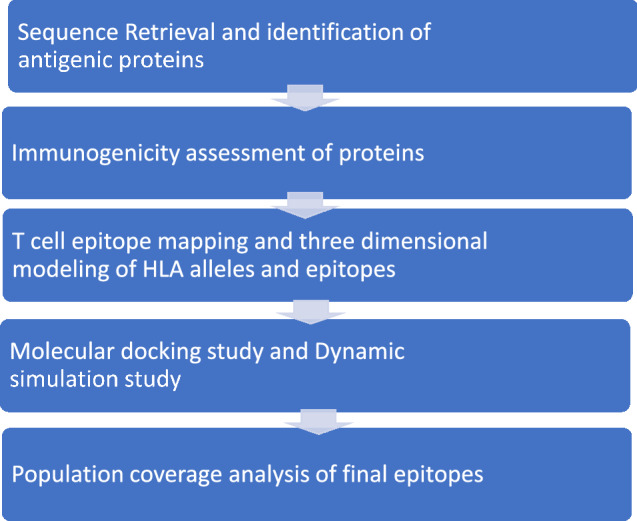
Flowchart to represent the methodology used in the analysis for T cell epitope designing
Immunogenicity assessment of proteins
Antigenicity of the filtered proteins was analyzed by VaxiJen server. VaxiJen is the most reliable server used for calculation of antigens irrespective of sequence length and alignment in reference to three models, i.e. bacteria, virus and tumour (Doytchinova and Flower 2007). These three models have shown remarkable stability and therefore the threshold value for the analysis was adjusted to 0.51 and the proteins having above this value were marked as antigenic proteins. Proteins that show VaxiJen scores above the cut-off value will only be selected for further study and low-score peptides will be eliminated. Allergen FP v.1.0 tool was used to differentiate between allergens and non-allergens proteins (Dimitrov et al. 2014).
T cell epitope mapping
Cytotoxic T lymphocyte (CTL) epitopes play an essential role in rational vaccine design; therefore, prediction of CTL epitopes can minimize the experimental effort needed to identify epitopes. Identification of the T cell epitopes binding with higher affinity with HLA class I alleles was predicted using Net CTL 1.2 tool (Larsen et al. 2007). NetCTL is a web-based tool designed to predict human CTL epitopes in any given protein sequence. This tool is used for the predictions of proteasomal cleavage, TAP transport efficiency, and MHC class I affinity. This upgraded version of Net CTL 1.2 showed an improved performance and specificity as compared to the older version of Net CTL 1.0.
To screen the best immunogenic epitopes with higher accuracy, we present the shortlisted peptides to VaxiJen v2.0 server. The peptides with high scores were selected for further analysis and those epitopes showing score ≥ 1.4 were selected for the toxicity prediction. Further, the toxicity of the peptides was calculated by Toxin Pred. This tool removes all the toxin peptides on the basis of toxin score and categorize only non-toxin peptides for analysis (Gupta et al. 2013).
Three-dimensional modeling of HLA alleles and epitopes
The three-dimensional mapping of the selected epitopes will be done with the help of PEPstrMOD tool (Kaur et al. 2007), a tool that well defines the 3D structures of the small peptide. The 3-D structures of the alleles HLA-A*11:01, HLA-B*15:02 and HLA-B*15:03 were modeled by Modeler 9.18 with their sequence downloaded from IPDIMGT/HLA Database (Robinson et al. 2015). Modeler relies on several parameters for loop modelling and predicts the quality of model using the DOPE score. This score is referred to as Z-score which differentiates between poor and native models, positive scores are considered to be poor-quality models but the scores less than − 1 are good-quality models. Modeler 9.18 tool generates five models, out of which the best model with was selected on the basis of DOPE score. Validation of the model was done using Ramachandran Plot analysis.
Molecular docking study
To initiate an appropriate immune response, the interaction between antigenic epitope with the receptor is mandatory. So, the next step in the process is docking of the designed peptides with HLA alleles was performed and for this molecular docking, AutoDock 4.2 (Goodsell et al. 1998) was undertaken. AutoDock is an excellent user-friendly, non-commercial program that is widely being used by researchers and experts for vaccine construct (Morris et al. 2009). AutoDock works on the principle of Lamarckian genetic algorithm that combines with free-energy force field which enables the stability of the ligand and macromolecule, thus initiates fast prediction of the bound conformations. It uses a grid-based method and autodock tools are embedded in an object-oriented programming language Python. This tool generates 10 trial conformations and the best output was finalized on the basis of higher binding energy score. The Chimera 1.2 tool was used for visualization of the docked complex structure orientations (Pettersen et al. 2004). This tool allows the user to visualize multiple sequence alignments, displays volumetric data and visualizes molecular dynamic trajectories of the docked models.
Dynamic simulation study
Molecular dynamics (MD) study was performed to study the molecular interactions and RMSD values using MDWeb server. It enables a user-friendly setup to run simulations and works on different operations within guided interface which involves Amber, NAMD, and Gromacs full MD setup and analysis can be carried out using any standard trajectory format. It performs various functions like Trajectory manipulation, analysis per residue and flexibility analysis. The structure model, chain and residues locations have been checked in the initial step only as minor error can also lead to the unstable trajectories.
Evaluation of population coverage
To estimate the HLA allele distribution among the world population, an IEDB tool was used (Bui et al. 2006). In this study, population coverage analysis of the potential epitopes with their corresponding MHC-I alleles was analyzed through IEDB tool. This tool generates the results in the form of histogram that shows the HLA gene frequencies on the specific populations. This is the best population coverage tool which is publicly accessible to all the users providing the most accurate result by covering 115 countries and help in the development of a T cell epitope-based vaccines.
Results and discussion
Highest protein selection and evaluation
The genome of selected strain E24377A comprises 4.9 Mb and has 5305 coding sequences and 67 RNAs and the complete genome of this strain was sequenced (Ashok et al. 2015). For the construction of vaccine candidates, a total number of 222 protein sequences were shortlisted from the complete proteome of E24377A strain through Vaxign on the basis of default adhesion cut-off value. In the next step, with the help of VaxiJen server, 95 proteins were selected and further presented to AllergenFP tool to distinguish between allergic and non-allergic proteins. Only proteins that were non-allergic in nature were shortlisted for further study and a total of 9 selected proteins are A7ZKX4, A7ZU80, A7ZJC8, A7ZK46, A7ZHN2, A7ZKR1, A7ZVJ1, A7ZRC0 and A7ZKE5(Table 1).
Table 1.
Allergen prediction, adhesion probability and vaxiJen prediction of selected proteins to find out the best epitopes with good scores
| S.no | Uniprot Id | Protein | AllergenFP prediction | Adhesin(probability) | VaxiJen prediction |
|---|---|---|---|---|---|
| 1 | A7ZKX4 | Putative outer membrane autotransporter | Non -Allergen | 0.682 | 0.8216 |
| 2 | A7ZU80 | Oligogalacturonatespecific porin protein | Non -Allergen | 0.581 | 0.8345 |
| 3 | A7ZJC8 | Uncharacterized protein | Non -Allergen | 0.545 | 1.1646 |
| 4 | A7ZK46 | Fimbrial protein | Non -Allergen | 0.599 | 0.9987 |
| 5 | A7ZHN2 | Fimbrial protein | Non -Allergen | 0.689 | 0.8400 |
| 6 | A7ZKR1 | Putative phage tail domain protein | Non -Allergen | 0.702 | 0.8577 |
| 7 | A7ZVJ1 | Antigen 43 | Non -Allergen | 0.718 | 0.8807 |
| 8 | A7ZRC0 | Antigen 43 | Non -Allergen | 0.713 | 0.8658 |
| 9 | A7ZKE5 |
Major curlin protein CsgA |
Non -Allergen | 0.713 | 1.3095 |
Characterization of MHC restricted T cell epitopes
To assess the amino acid sequences of the selected protein, these were subjected to NetCTL 1.2 for the prediction of cytotoxic T-cell epitope prediction. This method predicts MHC restricted epitopes on the basis of proteasomal C terminal cleavage and TAP transport efficiency and is restricted to 12 MHC class I supertype. The threshold value for epitope identification was 0.75 while all the other parameters were set to its default value. The results were sorted on the basis of combined score and epitopes that show good binding score were considered to be a good vaccine candidate (Table 2). Previous literature work depicts the importance of MHC restricted T cell epitopes in the design of vaccine and how it can reduce the time and effort through computational algorithm (Singh et al. 2015).
Table 2.
Cytotoxic T cell epitopes binding with different MHC class I HLA alleles by NetCTL 1.2 tool
| (a) Binding of epitopes with A supertypes | |||||
|---|---|---|---|---|---|
| Uniprot Id | A1 supertype | A2 supertype | A3 supertype | A24 supertype | A26 supertype |
| A7ZKX4 | TTDGSTG VY (202: 3.6227) | AMNNSVWNV (480: 1.4176) | RLSDVMPLY (734: 1.6370) |
NYLNVGYLL (668: 1.8324) |
TTADAGGNY (661:2.2210) |
| A7ZU80 | WLDRNVEPY(207:2.6424) | FVPWFNLTV (114:1.2937) | RINEHWLPY (194: 1.6676) | VYLDVNYKF (106: 2.0443) | EIEGWYPLF (73: 1.4596) |
| A7ZJC8 | MTKLATLFL (3: 0.8453) | KLATLFLTA (5: 1.1647) | SMNNDGMTK (78: 1.4199) | LFLTATLSL (9: 1.0996) | – |
| A7ZK46 | NTLKYQLRY (149:2.8381) | ILFFSILNI (11:1.2648) | TLKYQLRYK (150:1.2712) | IFQCVILFF(6:1.7285) | NTLKYQLRY (149:1.5472) |
| A7ZHN2 | DATTKSAVY (155:0.9496) | AMVAGTASA (15:1.1206) | VLNDTVGAK (68: 1.1170) | VYDFKASYV (162:0.8954) | AVYDFKASY (161:1.7411) |
| A7ZKR1 | WTDRGRYAY(595:3.4422) |
YVDGAAFPV (633: 1.2950) |
SSASTATTK (332: 1.3553) | SYRSYYQRI (1061: 1.7247) | DTYILVNFY (847: 2.0818) |
| A7ZVJ1 | MTISTGLEY (83: 3.0722) | KTWLAFTNV (546: 1.0965) | VLEGHSAWK (351: 1.4060) | SYRLVWNHI (8: 1.7014) | ATPESSGSY (674: 2.0319) |
| A7ZRC0 | MTISTGLEY (83: 3.0722) | SLGGYLNLV (733: 1.2681) | VLEGHSAWK (351: 1.4060) | SYRLVWNHI (8: 1.7014) | ATPESSGSY (674: 2.0319) |
| A7ZKE5 | NSELNIYQY (42: 2.7269) | KVAAIAAIV (5: 0.9260) | ALAGVVPQY (18: 1.2059) | VAAIAAIVF (6: 0.7849) | ALAGVVPQY (18: 1.4166) |
| (b) Binding of epitopes with B supertypes | |||||||
|---|---|---|---|---|---|---|---|
| Uniprot Id | B7 superty pe | B8 superty pe | B27 supertype | B39 superty pe | B44 supertype | B58 supertype | B62 Supertype |
| A7ZKX4 | AARFTATDL(222:1.3500) | FGQVSANAL (271:1.5620) | GRASMILGY (869:1.8085) | IHLNHYESL (859: 1.7093) | LEAGQR FNL (818: 1.7855) | KASRQKNSF (787: 1.6034) | AQYNKQHTF (928:1.4697) |
| A7ZU80 | LPYFELRWL (200:1.2920) | MKKINAIIL (1:0.8472) | IRIGTKYFF (222:1.5998) | HHWEITNTF(183:1.9338) | NEIEGWYPL(72:1.9421) | SSNGKDHHW(177:1.8025) | RINEHWLPY(194:1.466) |
| A7ZJC8 | APDARENVA (46:0.8286) | MKMTKLATL (1:0.7435) | GRCPDI NKK (93:0.7380) | – | RENVAP NNV (50:1.3014) | KMTKLATLF (2:1.3815) | KMTKLATLF (2:1.1804) |
| A7ZK46 | IPLNQVQPL(133:1.2402) | CVILFFSIL (9:0.7544) | YQLRYKSTK (153:1.3636) | TGGNATAVL (165:1.0651) | AQSQQEIPL (127:0.9957) | KAGDNTLKY (145:1.2547) | LQNIHIGDF (50:1.3263) |
| A7ZHN2 | KASYVRAVA (166:0.7911) | MSKK LGFAL (1:1.7999) | - | PSDGVNIAL (122:1.1292) | – | KMTFGSVFF (98:1.0853) | AVYDFKASY (161:1.3631) |
|
A7ZKR 1 |
KPASGRAVL (957:1.8071) | KPASGRAVL (957:1.2028) | GRYSMDVEY (47:1.6740) | TRSGDTYIL (843:2.1758) | GELVIGTKL (767:1.9252) | IAADVILDF (677:1.7426) | QVSPETSSY (1054:1.3888) |
| A7ZVJ1 | SPSRNGTSL (896:1.7761) | RGKRTGVAV (30:1.6057) | KRTGVAVAL (32:1.8212) | FHKLTTSNL (506:1.9676) | GETVSGGTL (60:1.7351) | MTISTGLEY (83:1.5972) | LMLEPQLQY (792:1.4476) |
| A7ZRC0 | SPSRNGTSL (896:1.7761) | YASMLTQAM (622:1.5902) | KRAGVAIAL (32:1.8353) | FHKLTTSNL (506:1.9676) | LENGGSFTV (418:1.6935) | MTISTGLEY (83:1.5972) | GMSLTTGVY (698:1.4694) |
| A7ZKE5 | AIVFSGSAL (11:1.3029) | MKLL KVAAI (1:0.9024) | – | QYGG GNSAL (49:1.2572) | – | VAAIAAIVF (6:1.7817) | ALAGVVPQY (18:1.3000) |
In addition to selecting the most potential immunogenic epitope, the epitopes selected from NetCTL 1.2 were further presented to VaxiJen tool to predict the peptides showing high scores ≥ 1.75 were selected. Peptides binding to MHC class I alleles (IC50) ≤ 500 nM were shortlisted for the further analysis and therefore we found four peptides with highest scores.
LPYFELRWL, CVILFFSIL, QYGGGNSAL and SSASTATTK with scores showing 2.4136, 1.9547, 1.7562 and 1.8430, respectively (Supplementary Table 1). To validate the result, we included two control epitopes 141STLPETTVV149 of Hepatitis B virus (Accession number-CAA59535) and epitope 265ILRGSVAHK273 of H1N1 Nucleoprotein (Accession Number- P03466).
Evaluation of antigenic properties of vaccine candidates
To evaluate the potential predicted epitopes as a good immunogen for vaccine candidates, rigorous methods have applied to view their antigenic nature. These peptides were characterized to be non-toxic in nature as predicted by ToxinPred tool (Table 3). All the parameters are kept in mind while selecting the proteins for potential candidates and therefore proteins that show more than two transmembrane regions are not considered as a good antigen for vaccine due to the difficulty in cloning, expressing and purifying. Hence, we applied TMHMM method which classified all the selected antigens having less than 1 transmembrane region as it is shown in Table 3. The mechanism of ETEC infection is primarily dependent on its binding ability to the membrane and therefore, all the 9 antigens were declared as adhesions by SPAAN program (Sachdeva et al. 2005). To check the adhesion capability of pathogens to host through experimental method is a rigorous and time-consuming process, so computational algorithm SPAAN has shown specificity of 100% to determine adhesions.
Table 3.
Prediction of toxicity, number of transmembrane regions and molecular weight of the potential peptide to calculate the tendency of peptides as vaccine candidates
| Peptide | Toxicity Score | Toxicity prediction | Number of predicted transmembrane regions | Molecular weight |
|---|---|---|---|---|
| LPYFELRWL | − 1.17 | Non-Toxin | 0 | 1236.61 |
| CVILFFSIL | − 0.75 | Non-Toxin | 0 | 1054.49 |
| QYGGGNSAL | − 0.52 | Non-Toxin | 0 | 866.03 |
| SSASTATTK | − 0.89 | Non-Toxin | 0 | 853 |
| STLPETTVV( +) | − 1.26 | Non-Toxin | 0 | 946.19 |
| ILRGSVAHK( +) | − 1.12 | Non-Toxin | 0 | 980.31 |
Three-dimensional modeling and validation of alleles and epitopes
Modeling of the three-dimensional structure of the MHC-I HLA alleles-HLA-A*1101 and HLA-B*1502 corresponding to the highest scoring epitopes was generated using Modeler 9.18 (Table 4). The three-dimensional structural knowledge of proteins plays an essential role in depicting the complete information of their molecular functions as well as identification of their binding sites (Sali et al. 1995). According to the DOPE scores of the selected epitopes and alleles’ model, the highest negative value model was selected. To further validate the overall quality of modeled structure of these HLA alleles, it was subjected to RAMPAGE server which calculates the four-modeled structure’s residues are > 90% in favored region and approved the quality of the models. In 2014, this paper stated the fact that epitopes are enough to trigger the strong immune response in comparison to the whole protein sequences (Huber et al. 2014). Therefore, distribution of HLA alleles on a specific set of population is very important to understand the potential of vaccine worldwide.
Table 4.
Homology modeling of HLA I alleles binding to the selected epitopes using Modeller tool
| S. no | HLA Allele | Chain | Template (PDB database ID) | DOPE Score | Crystal Structure / model |
|---|---|---|---|---|---|
| 1 | HLA-A*1101 | B | 1Q94 | − 29,084.093 | Model |
| 2 | HLA-B*1502 | A | 1XR8 | − 29,724.101 | Model |
Molecular docking of HLA allele and epitope
The docking of the potential three epitopes with their corresponding HLA allele was performed using Autodock 4.2. Different conformations were generated and the best conformation was selected on the basis of binding energy score, lower the binding affinity, the stronger is the interaction between HLA allele and epitope. The interactions between peptide QYGGGNSAL corresponding to HLA-A*11:01 and HLA-B*15:02, peptide LPYFELRWL corresponding to HLA-A*11:01and HLA-B*15:02 and peptide CVILFFSIL corresponding to HLA-A*11:01 and HLA-B*15:02 were considered to be the most effective T cell epitopes with highest binding energy of − 2.09 kcal/mol, − 1.84 kcal/mol and − 1.30 kcal/mol, respectively (Table 5). Out of the three epitopes, two epitopes with best binding energies were selected and therefore QYGGGNSAL and LPYFELRWL were analyzed by Python Molecular viewer as shown in the Figs. 2 and 3. The ligand-enzyme complex is stabilized mainly by hydrogen bonds and hydrophobic interactions. The binding affinity of the ligand and protein molecule can also be affected if the bulk water around it has more strong bonds. Therefore, strong hydrogen bond between docked complexes can lead to a better binding energies with better stability in simulation also.
Table 5.
Binding energy calculation of the best identified epitopes interacting with HLA alleles using Autodock 4.2
| Peptide | HLA allele | Binding Energy (kcal/mol) | IntermolecularEnergy (kcal/mol) | Internal Energy (kcal/mol) | Torsiona l Energy (kcal/mo) | Vander wal energy (kcal/ml) | Electro static energy (kcal/mol) |
|---|---|---|---|---|---|---|---|
| QYGGGNSAL | A*1101 | − 2.09 | − 8.82 | − 6.15 | 8.05 | − 7.48 | − 1.34 |
| QYGGGN SAL | B*1502 | − 1.74 | − 9.79 | + 0.00 | 8.05 | − 8.13 | − 1.66 |
| LPYFELRWL | A*1101 | − 0.15 | − 10.59 | + 0.00 | 10.44 | − 10.53 | − 0.06 |
| LPYFELRWL | B*1502 | − 1.84 | − 12.28 | − 8.21 | 10.44 | − 11.53 | − 0.75 |
| CVILFFSIL | B*1502 | − 1.30 | − 10.85 | − 4.68 | 9.55 | − 10.83 | − 0.02 |
| CVILFFSIL | A*1101 | − 1.07 | − 10.62 | + 0.00 | 9.55 | − 10.36 | − 0.26 |
Here in this table the binding ability of peptide with the corresponding alleles will indicate the most complex docked structure for stability check in vaccine development
Fig. 2.
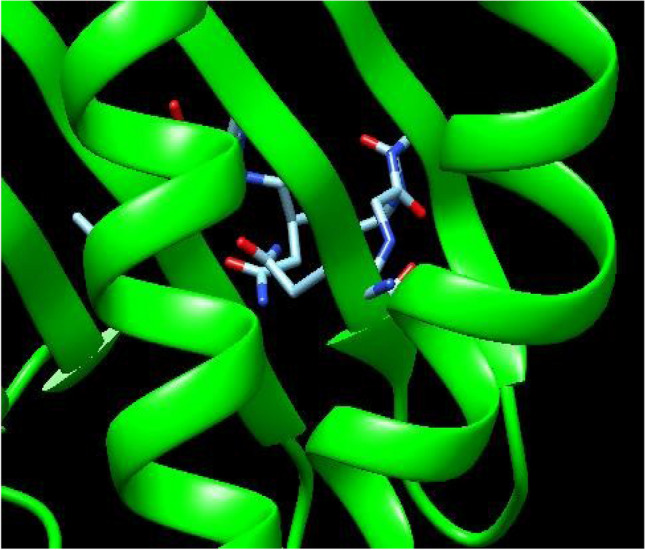
Docked complex of QYGGGNSAL with HLA class I allele A*11:01 visualized through Chimera 1.12
Fig. 3.
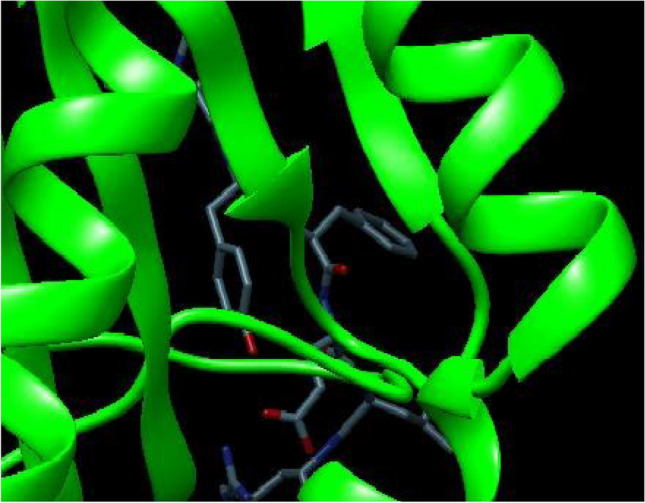
Docked complex of LPYFELRWL with HLA class I allele B*15:02 visualized through Chimera 1.12
Dynamic simulation of the designed complex
The RMSD values were calculated to check the stability of the selected epitopes interacting with their corresponding alleles. For preparing the complex for simulation, there are several steps that is required to get better result. In the first step, it removes crystallographic water molecules and adds hydrogen atoms and missing side chains. In the further steps, it restrains the heavy atoms to their position with a pressure of 500 KJ/mol*nm2 and then a truncated box of TIP3P water molecules (Amber Force) or other Forces at a distance of 15 Å around the molecules. To validate the stability of the vaccine construct over a period of time, simulation analysis was carried out. However, two epitopes, i.e. QYGGGNSAL corresponding to HLA-A*11:01 and LPYFELRWL corresponding to HLA-B*15:02, were selected and presented to the server. The result from Root Mean Square deviation (RMSD) per residue analysis indicates the regions that have less mobility or loops, that means the flexibility and stability of the structure remain almost same during the whole simulation as it is shown in Figs. 4 and 5.
Fig. 4.

RMSd analysis for LPYFELRWL corresponding to HLA-B*15:02
Fig. 5.

RMSd analysis for QYGGGNSAL corresponding to HLA-A*11:01
Worldwide population coverage analysis
Population coverage analyses were predicted through IEDB tool, these two epitopes QYGGGNSAL and LPYFELRWL were found to be most immunogenic with the highest population coverage > 50% in the regions of East Asia, India, Northeast Asia and South Asia. Figures 6 and 7 indicate the maximum coverage was observed to be 71.44% in Northeast Asia and 50.99% in India for epitope QYGGGNSAL and 66.56% in Northeast Asia and 50.71% in India for epitope LPYFELRWL. The results from population coverage analysis indicate that epitopes cover different populations effectively in all the countries but are mainly high population coverage in the places highlighted in both the figures. Thus, the potential vaccine candidates are covering maximum number of population worldwide and can benefit large masses of ethnic groups.
Fig. 6.

Population coverage analysis of T cell epitope QYGGGNSAL
Fig. 7.

Population coverage data of T cell epitope LPYFELRWL
Conclusion
We have predicted two epitopes QYGGGNSAL and LPYFELRWL for designing vaccine against diarrhea. These epitopes were selected on the basis of different parameters like antigenicity, docking score, Vaxijen scores, population coverage data and stability over a period of time. Apart from several advantages of Immunoinformatics methods are valuable in terms of reducing time and cost in vaccine design, there are few limitations as well which cannot be neglected. First, variation in results may occur in different softwares, and second, multi-epitopes vaccine can offer better immunity than single-based approach. Therefore, these epitopes identified in our analysis could be further tested in experimental laboratory for its successful outcome as a vaccine that could provide protection worldwide.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors gratefully acknowledge the necessary computational facilities and sound supervision provided through the research work by the Department of Biotechnology, Faculty of Engineering & Technology, Rama University, Kanpur, U.P., India for their generous support during the research work.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006;7:153. doi: 10.1186/1471-2105-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Randall A, Vickers TJ, et al. Interrogation of a live-attenuated enterotoxigenicEscherichia coli vaccine highlights features unique to wild-type infection. Vaccines. 2019;4:37. doi: 10.1038/s41541-019-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846–851. doi: 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8:4. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell DS, Morris GM, Halliday RS, Huey R, Belew RK, Olson AJ. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J ComputChem. 1998;19:1639–1662. [Google Scholar]
- Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GPS, Open Source Drug Discovery Consortium In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8(9):e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren J, Parashar UD, Plotkin S, Louis J, Ng SP, Desauziers E, Picot V, Saadatian EM. Correlates of protection for enteric vaccines. Vaccine. 2017;35:3355–3363. doi: 10.1016/j.vaccine.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Duan Q, Zhang W. Significance of enterotoxigenic Escherichia coli (ETEC) heat-labile toxin (LT) enzymatic subunit epitopes in LT enterotoxicity and immunogenicity. Appl Environ Microbiology. 2018;84(15):e00849–e18. doi: 10.1128/AEM.00849-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S, van Beek J, de Jonge J, Luytjes W, van Baarle D. T cell responses to viral infections—opportunities for Peptide vaccination. Front Immunol. 2014;5:171. doi: 10.3389/fimmu.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidean SD, Riddle MS, Savarino SJ, Porter CKA. systematic review of ETEC epidemiology focusing on coloniz r and toxin expression. Vaccine. 2011;29(37):6167–6178. doi: 10.1016/j.vaccine.2011.06.084. [DOI] [PubMed] [Google Scholar]
- Kaur H, Garg A, Raghava GP. PEPstr: a de novo method for tertiary structure prediction of small bioactive peptides. Protein Pept Lett. 2007;14(7):626–631. doi: 10.2174/092986607781483859. [DOI] [PubMed] [Google Scholar]
- Khan F, Srivastava V, Kumar A. Epitope based peptide prediction from proteome of enterotoxigenicE. coli. Int J Pept Res Ther. 2018;24:323–336. doi: 10.1007/s10989-017-9617-1. [DOI] [Google Scholar]
- Khan F, Srivastava V, Kumar A. Computational identifcation and characterization of potential T-cell epitope for the utility of vaccine design against enterotoxigenic Escherichia coli. Int J Pept Res Ther. 2019;25(1):289–302. doi: 10.1007/s10989-018-9671-3. [DOI] [Google Scholar]
- Kumar A, Jain A, Shraddha Verma SK. Screening and structure-based modeling of T-cell epitopes of Marburg virus NP, GP and VP40: an immunoinformatic approach for designing peptide-based vaccine. Trends Bioinform. 2013;6:10–16. doi: 10.3923/tb.2013.10.16. [DOI] [Google Scholar]
- Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M(2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 8: 424–10.1186/1471-2105-8-424. [DOI] [PMC free article] [PubMed]
- Mehla K, Ramana J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: a comparative genomics and immunoinformatics approach. MolBiosystem. 2016;12(3):890–901. doi: 10.1039/c5mb00745c. [DOI] [PubMed] [Google Scholar]
- Morgat A, Lombardot T, Coudert E, Axelsen K, Neto TB, Gehant S, Bansal P, Bolleman J, Gasteiger E, de Castro E, Baratin D, Pozzato M, Xenarios I, Poux S, Redaschi N, Bridge A UniProt Consortium (2019) Enzyme annotation in UniProtKB using Rhea. Bioinformatics 36(6):1896-1901 [DOI] [PMC free article] [PubMed]
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J ComputatChem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 251605–1612 [DOI] [PubMed]
- Qadri F, Svennerholm AM, Faruque AS, Sack RB. Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin Microbiol Rev. 2005;18:465–483. doi: 10.1128/CMR.18.3.465-483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, et al. Thepangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG.(2015) The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43(Database issue):423–31. [DOI] [PMC free article] [PubMed]
- Sachdeva G, Kumar K, Jain P, Ramachandran S. SPAAN: a software program for prediction of adhesins and adhesin-like proteins using neural networks. Bioinformatics. 2005;21:483–491. doi: 10.1093/bioinformatics/bti028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- Seo H, Nandre RM, Nietfeld J, Chen Z, Duan Q, Zhang W. Antibodies induced by enterotoxigenicEscherichia coli (ETEC) adhesin major structural subunit and minor tip adhesin subunit equivalently inhibit bacteria adherence in vitro. PLoS ONE. 2019;14(5):e0216076. doi: 10.1371/journal.pone.0216076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen HI, Kamal KA, Wasfy MO, El-Ghorab NM, Lowe B, Steffen R, Kodkani N, Amsler L, Waiyaki P, David JC, Khalil SB, Peruski LF. Phenotypic diversity of enterotoxigenicEscherichia coli (ETEC) isolated from cases of travelers’ diarrhoea in Kenya. Int J Infect Dis. 2003;7:35–38. doi: 10.1016/S1201-9712(03)90040-3. [DOI] [PubMed] [Google Scholar]
- Singh A, Mitra M, Sampath G, Venugopal P, Rao JV, Krishnamurthy B, Gupta MK, Sri Krishna S, Sudhakar B, Rao NB, Kaushik Y, Gopinathan K, Hegde NR, Gore MM, Krishna Mohan V, Ella KM. A Japanese encephalitis vaccine from India induces durable and cross-protective immunity against temporally and spatially wide-ranging global field strains. J Infect Dis. 2015;212(5):715–725. doi: 10.1093/infdis/jiv023. [DOI] [PubMed] [Google Scholar]
- WHO (2018) https://www.who.int/en/news-room/fact-sheets/detail/diarrhoeal-disease
- Xiang Z, He Y. Vaxign: a web-based vaccine target design program for reverse vaccinology. Procedia Vaccinol. 2009;1(1):23–29. doi: 10.1016/j.provac.2009.07.005. [DOI] [Google Scholar]
- Zawawi A, Forman R, Smith MI, Jibril M, Albaqshi MH, Brass A, Derrick JP, Else KJ. In silico design of a T-cell epitope vaccine candidate for parasitic helminth infection. PLoSPathog. 2020;16(3):e1008243. doi: 10.1371/journal.ppat.1008243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Sack DA. Current progress in developing subunit vaccines against enterotoxigenicEscherichia coli-associated diarrhea. Clin Vaccine Immunol. 2015;22:983–991. doi: 10.1128/CVI.00224-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.