

Abstract
Cellular senescence is a feature of most somatic cells. It is characterized by an irreversible cell cycle arrest and by the ability to secrete a plethora of mediators of inflammation and growth factors, which can alter the senescent cell’s microenvironment. Senescent cells accumulate in tissues over time and contribute to both aging and the development of age-associated diseases. Senescent cells have antagonistic pleiotropic roles in cancer. Given the inability of senescent cells to proliferate, cellular senescence is a powerful tumor suppressor mechanism in young individuals. However, accumulation of senescent stromal cells during aging can fuel cancer cell growth in virtue of their capacity to release factors that stimulate cell proliferation. Caveolin-1 is a structural protein component of caveolae, invaginations of the plasma membrane involved in a variety of cellular processes, including signal transduction. Mounting evidence over the last 10–15 years has demonstrated a central role of caveolin-1 in the development of a senescent phenotype and the regulation of both the anti-tumorigenic and pro-tumorigenic properties of cellular senescence. In this review, we discuss the cellular mechanisms and functions of caveolin-1 in the context of cellular senescence and their relevance to the biology of cancer.
Keywords: Caveolin, Stress-induced senescence, Oncogene-induced senescence, Cancer, Aging, p53
1. Introduction
Caveolae were first described as invaginations of the plasma membrane in the 1950s by electron microscopy [1, 2]. However, the functional significance and biological role of caveolae have emerged only after the discovery of caveolin-1 in the early 1990s. Since then, compelling in vitro and in vivo evidence, as well as human studies, has revealed a causal link between caveola/caveolin-mediated functions and a number of fundamental cellular processes and the pathogenesis of multiple human diseases. Cellular senescence has antagonistic pleiotropic properties in cancer biology [3–6]. On the one hand, cellular senescence prevents cells carrying potentially tumor-promoting genetic mutations from proliferating and forming a tumor in young organisms. On the other hand, the age-dependent accumulation of senescent cells has been linked to tumorigenesis given their ability to release growth factors and mediators of inflammation that stimulate the growth of transformed cells. Interestingly, caveolin-1 has emerged as a key regulator of different forms of cellular senescence. Here, we provide a comprehensive description of the signaling pathways and molecular mechanisms through which caveolin-1 controls the development of a senescent phenotype and the role of caveolin-1-mediated cellular senescence in cancer.
2. Caveolin-1 and caveolae
Caveolin-1 is a ~ 20-kDa protein that is required for the formation of caveolae in most cell types [7]. Caveolae are 50–100-nm flask-shaped invaginations of the plasma membrane enriched in cholesterol and glycosphingolipids [8]. Caveolae can exist as individual invaginations or can be found in detached grape-like clusters and long tubular structures derived from the fusion of single caveola. The caveolin-1 protein sequence consists of four domains: (i) an NH2-terminal domain, which includes both phosphorylation (Y14) and ubiquitination sites; (ii) a caveolin scaffolding domain (CSD; residues 82–101) carrying a cholesterol recognition/interaction consensus sequence (CRAC) and an ubiquitination site; (iii) a membrane domain, which interacts with membrane lipids; and (iv) a COOH-terminal domain, which possesses three palmitoylation sites. Oligomeric caveolin-1 complexes, embedded in cholesterol-enriched membranes, travel from the Golgi to the plasma membrane where they interact with cavins to form caveolae [9, 10]. The CSD mediates direct protein-protein interactions between caveolin-1 and a variety of signaling molecules carrying the caveolin-binding domain (CBD: ΦXΦXXXXΦ, ΦXXXXΦXXΦ, or ΦXΦXXXXΦXXΦ where Φ represents an aromatic amino acid and X represents any amino acid) [11–13]. The direct interaction with caveolin-1 generally results in the sequestration of a given signaling molecule within caveolar membranes and modulation of its signaling activity. As such, caveolae are major regulators of signal transduction processes. Signaling transduction proteins that are associated with caveolae include the following: (i) kinases such as Src-family kinases, protein kinase A (PKA), protein kinase C (PKC), p38 and p42/44 mitogen-activated protein kinases; (ii) ion channels and ion exchangers, including Ca2+-ATPase, L-type Ca2+, Na+/K+-ATPase, and voltage-gated K+; (iii) receptors, including G protein-coupled receptors, receptor tyrosine kinases (RTKs), steroid hormone receptors, inositol 1,4,5-triphosphate receptor (IP3R), and transforming growth factor-β receptors; and (iv) nitric oxide synthase (NOS) [14–28]. However, the role of caveolae in eukaryotic cells is not limited to signal transduction mechanisms. In fact, data show that caveolae mediate endocytosis, transcytosis, mechanoprotection, cellular metabolism, and lipid homeostasis [29–35].
Although caveolin-1 and caveolae regulate important and numerous cellular functions, the caveolin-1 null mouse is viable and fertile. However, a lack of caveolin-1 in mice has been linked to a variety of phenotypes such as pulmonary and cardiac defects, vascular abnormalities, atherosclerosis, metabolic disorders, and cancer [33, 36–39]. They are mostly the consequence of a lack of caveola-mediated functions in pulmonary, cardiac, vascular, and adipose tissues. In support of these data, caveolin-1 gene mutations have been linked to a number of pathologies in humans. They include pulmonary arterial hypertension [40], hypertriglyceridemia [41], and cancer [42–44]. Together, these animal and human data provide direct evidence that caveolae play key physiological roles.
3. Cellular senescence
3.1. Definitions and biochemical properties
Most cells cannot divide indefinitely due to a process termed cellular senescence [45–51]. Cellular senescence is a stable form of cell cycle arrest that occurs in somatic cells, with the exception of most tumor cells and certain stem cells. Cellular senescence was originally described in vitro by Hayflick and colleagues when they observed that human diploid fibroblasts permanently stopped dividing after a finite number of cell divisions, becoming irreversibly arrested in the G1 phase of the cell cycle [52]. In contrast to quiescent cells, senescent cells do not re-enter the cell cycle in response to external stimuli, such as growth factors, and they are resistant to apoptotic stimuli [53, 54]. However, senescent cells remain viable and metabolically active despite their non-proliferative state [55].
Cellular senescence can be divided into two categories: replicative senescence (RS) and stress-induced premature senescence (SIPS). Replicative senescence is dependent on the number of divisions the cell has completed. This type of senescence is spontaneously achieved by somatic cells. It is now known that the cell division counting is controlled by telomere shortening, an unavoidable consequence of genome duplication [50, 56–59]. Senescence can be accelerated by a number of stressful stimuli such as oncogene activation, DNA damage, cytotoxic drugs, and oxidative stress [60–64]. This type of senescence is referred to as stress-induced premature senescence. SIPS is independent of telomere status but shares many molecular and functional features with replicative senescence.
Although senescent cells do not express one specific biomarker, a number of features and molecular markers, which represent hallmarks of senescence, can be used to identify senescent cells. Senescent cells can be experimentally identified by their enlarged and flattened morphology, as well as positive staining for β-galactosidase activity at pH 6 [46]. Their inability to proliferate in a pro-mitogenic environment is associated with increased expression of cell cycle inhibitors, including p16INK4a, p21WAF1/CIP1, and hypo-phosphorylated retinoblastoma gene protein (pRb) [45–49]. Senescent cells can also display elevated expression of p19ARF, plasminogen activator inhibitor-1 (PAI-1), and p53 as well as a chronic DNA damage response with DNA damage foci [65] and senescence-associated heterochromatin foci [66] formation. Moreover, loss of lamin B1 [67], accumulation of lipofuscin [68], expression of decoy death receptor 2 (DCR2), and embryonic chondrocyte-expressed 1 (DEC1) [69] have been associated with cellular senescence. Importantly, a key feature of senescent cells is their ability to secrete a variety of factors, including growth factors, mediators of inflammation, and proteases, known as the senescence-associated secretory phenotype (SASP), which can alter the tissue microenvironment [5, 70] (Table 1).
Table 1.
Different forms of cellular senescence
| RS | Replicative senescence | Senescence induced by telomere shortening |
| SIPS | Stress-induced premature senescence | Senescence induced by cellular stress |
| OIS | Oncogene-induced senescence | Senescence induced by oncogene expression |
| LTSIS | Loss of tumor suppressor gene-induced senescence | Senescence induced by loss of tumor suppressor gene expression |
| SASP | Senescent-associated secretory phenotype | Cytokines, chemokines, growth factors, and metalloproteinases secreted by senescent cells |
3.2. The tumor suppressor role of cellular senescence
Cellular senescence is a basic cellular mechanism developed by organisms to prevent the propagation of cells with damaged DNA and potentially carrying oncogenic mutations. Therefore, cellular senescence is considered a powerful tumor suppressor mechanism. The anti-tumorigenic role of cellular senescence is evident from data showing how either (i) oncogene expression or (ii) loss of tumor suppressor genes triggers the development of a senescent phenotype.
Oncogene-induced senescence (OIS) was originally described in human fibroblasts in which over-expression of oncogenic H-Ras (H-RasG12V) induced a permanent cell cycle arrest [63]. K-Ras is a GTPase that plays an important role in normal tissue signaling. Wild-type K-Ras is activated by signals that promote the exchange of bound GDP to GTP. This is a transient activation due to the intrinsic ability of K-Ras to hydrolyze GTP and therefore turn itself off. However, single point mutations generate an oncogenic mutant form of K-Ras with a constitutively activated GTP-bound state. Oncogenic K-Ras can also promote oncogene-induced senescence through the elevation of reactive oxygen species [63, 71, 72]. The in vivo tumor suppressive role of OIS is supported by evidence showing that oncogenic K-Ras induces senescence in pre-malignant lung lesions in mice and that senescent cells are absent in malignant lung adenocarcinomas [63, 69, 73–75]. Tumor cell senescence is not restricted to mouse models, but has been reported in human premalignant lesions as well [69, 76–81]. Thus, oncogenic Ras over-expression promotes transformation and progression to higher grades of malignancy only when oncogenic Ras-expressing cells evade/bypass senescence checkpoints through either the expression of additional oncogenes or the additional loss of tumor suppressor genes. For example, co-expression of oncogenes such as c-Myc, E1A, or dead ringer-like-1 (DRIL1) bypasses K-RasG12V-induced cellular senescence [82], and oncogenic K-Ras-induced senescence is evaded by the inactivation of the tumor suppressors p19ARF, p53, p16INK4a, pRb, and pro-myelocytic leukemia (Pml) protein [63, 83]. Ras is not the only oncogene that induces OIS. For example, constitutively active forms of BRAF can also promote OIS in cells and induce the formation of pre-malignant skin lesions, i.e., melanocytic nevi, in vivo, which carry senescent cells [84]. Over-expression of oncogenes such as epidermal growth factor receptor (EGF-R), phosphoinositide 3-kinase (PI3K), and human epidermal growth factor receptor 2 (HER2) can also induce senescence [85, 86], strengthening the concept that when the cell is hit by a sustained mitogenic stress, due to oncogene over-expression, it undergoes senescence to limit tumor growth.
In addition to oncogene activation, cellular senescence can also be induced by the loss of tumor suppressor genes (i.e., loss of tumor suppressor gene-induced senescence [LTSIS]). A well-established example is loss of the tumor suppressor phosphatase and tensin homolog (PTEN). It leads to cellular senescence associated with increased expression of p16INK4A as well as p53, which occurs in a mammalian target of rapamycin (mTOR)-dependent mechanism and through the inhibition of the oncogene mouse double minute 2 homolog (Mdm2) [77]. Loss of PTEN in a mouse model of prostate cancer promotes the formation of prostate intraepithelial neoplasia, pre-malignant prostate tumor lesions carrying senescent cells. Interestingly, senescence induced by loss of PTEN requires p53 function as these benign and senescent-positive lesions progress to senescence-negative invasive prostate cancer upon p53 inactivation [77, 87]. Finally, data show that inactivating mutations of neurofibromin 1 (NF1) [88], Rb [89], and von Hippel-Lindau [90] are associated with the development of cellular senescence and benign tumor lesions.
3.3. The tumor suppressor and tumor promoter roles of the SASP
The SASP acts as a double-edged sword in cancer. On the one hand, SASP factors, including interleukins such as IL-1α [91], IL-6 [92], and IL-8 [93], as well as vascular endothelial growth factor (VEGF), transforming growth factor beta (TGF-β), chemokine (C-C motif), ligand 2 (CCL2) and ligand 20 (CCL20) [94], can induce senescence in neighboring cells, including cancer cells, in a paracrine fashion, therefore inhibiting tumor growth. Interestingly, the release of IL-6 can also act in an autocrine manner to promote oncogene-induced senescence [92, 95] and the depletion of IL-6 causes cells to bypass OIS [92]. Finally, chemokines and cytokines, which are secreted by senescent cells, inhibit tumor development by activating immune surveillance and promoting clearance of pre-neoplastic and cancer cells [96].
On the other hand, the release of pro-stimulatory SASP factors by senescent cells within the tumor microenvironment can promote tumor growth [97–100]. In fact, data show that senescent cells, which accumulate during the aging process [46, 101–104], can release factors that stimulate the proliferation of pre-malignant and malignant epithelial cells. The release of pro-tumorigenic SASP factors also occurs when chemotherapeutic drugs are used as cancer treatment. Studies in animals and humans have shown that although chemotherapy is initially advantageous in virtue of its ability to either kill cancer cells by apoptosis or induce permanent cell cycle arrest of cancer cells by inducing cancer cell senescence [105, 106], it can subsequently fuel cancer development given the capacity of chemotherapeutic drugs to induce senescence of immune, stromal, and cancer cells. Senescence of immune cells blunts the anti-tumorigenic properties of the immune system while senescent stromal and cancer cells can release SASP factors that stimulate the proliferation of non-senescent cancer cells that survived chemotherapy [107]. In addition, the SASP can attract immune-suppressive myeloid-derived suppressor cells (MDSCs) into the tumor [107].
4. Regulation of cellular senescence by caveolin-1
Mounting evidence over the last 10–15 years has shown that caveolin-1-mediated signaling is causally linked to the development of a senescent phenotype and regulates senescent-dependent physiological and pathological outcomes. In the section below, we describe the molecular mechanisms and signaling pathways through which caveolin-1 regulates cellular senescence with specific emphasis on the functional role of caveolin-1-mediated senescence in cancer.
4.1. Caveolin-1 and replicative senescence
Caveolin-1 expression and the number of caveolae are upregulated in replicative senescent human diploid fibroblasts, where the unresponsiveness of senescent cells to epidermal growth factor (EGF) stimulation is linked to the inhibitory role of caveolin-1 on EGF-R signaling [108, 109]. Interestingly, downregulation of caveolin-1 expression in senescent human diploid fibroblasts by siRNA and antisense oligonucleotides restores extracellular signal-regulated kinase (Erk) activation, DNA synthesis, and cell cycle progression in response to EGF stimulation, with the concomitant decrease of p53 and p21 expression [110]. Moreover, downregulation of caveolin-1 in senescent human diploid fibroblasts results in morphological changes to a non-senescent-like shape possibly through altered focal adhesion and actin stress fiber formation due to focal adhesion kinase (FAK) and Rho family GTPase regulation [111]. It remains to be determined how downregulation of caveolin-1 can overcome/bypass the pro-senescent signaling initiated by telomere shortening. Consistent with these findings, caveolin-1 expression is upregulated in replicative senescent bone marrow mesenchymal ST2 cells [112] and over-expression of caveolin-1 in ST2 cells elevates the expression of the senescence markers p53 and p21 [112]. Similarly, caveolin-1 expression is upregulated in senescent human mesenchymal stem cells (hMSCs) and the over-expression of caveolin-1 in young hMSCs suppresses their adipogenic differentiation potential [113]. Finally, caveolin-1 expression is increased in replicative senescent bone marrow stromal cells [114], old macrophages [115], and senescent retinal pigment epithelium [116] and the upregulation of caveolin-1 positively correlates with shortening of chondrocyte replicative lifespan after treatment with interleukin (IL)-1β [117]. The activity of phosphatidylcholine-specific phospholipase C (PC-PLC) is dramatically increased during replicative senescence of bone marrow stromal cells (BMSCs) [114]. Inhibition of PC-PLC activity by the specific inhibitor D609 reduces the upregulation of caveolin-1 expression and the number of replicative senescent BMSCs, suggesting that PC-PLC mediates senescence of BMSCs possibly with a mechanism involving caveolin-1-dependent signaling [114].
Senescent cells accumulate in mammalian tissues with age. Animal studies show that reduction of senescent cells in mice inhibits the development of age-associated phenotypes. Clearance of p16-expressing cells in the spindle assembly checkpoint protein BubR1 progeroid mouse background delays the development of aging-associated disorders [118]. Clearance of naturally occurring p16-positive cells in mice of two distinct genetic backgrounds attenuates age-related deterioration of several organs and extends lifespan [119]. Similarly, clearance of p16-positive cells in mice mitigates age-associated intervertebral disc degeneration [120]. Consistent with a pro-senescent role of caveolin-1, its expression is upregulated in the lung, spleen, and brain of old rats [108, 121] and in skeletal muscles of old mice [122]. Upregulation of caveolin-1 in skin fibroblasts in both chronological and UV-induced aging is linked to age-related alterations of the skin, including reduction of the dermal thickness, altered levels of hyaluronan, increase dermal white adipose tissue, reduced collagen expression, and increased inflammation [123, 124]. Treatment with methyl-β-cyclodextrin (MβCD) reduces caveolin-1 expression and upregulates collagen activity with increased skin thickness [124]. Caveolin-1 is upregulated in the old rat hippocampus and cerebral cortex, and data show a role of caveolin-1 in beta amyloid precursor protein (APP) processing by beta-secretase [121]. In Caenorhabditis elegans, the insulin/insulin-like growth factor-1 (IGF-1) signaling upregulates caveolin-1 expression and insulin/IGF-1 signaling reduction downregulates caveolin-1 expression and neuronal caveolae [125]. Reduced caveolin-1 expression extends lifespan of Caenorhabditis elegans [125]. In humans, caveolin-1 expression is elevated with age in both the smooth muscle and epithelium of the prostate [126] and in the elderly cerebral cortex [121]. Moreover, increased caveolin-1 protein level in the hippocampus and caveolin-1 mRNA in the frontal cortex were described in patients with Alzheimer’s disease [127]. Cavin-1 (PTRF) is a component of caveolae. Directly supporting a key role of caveolae in the signaling leading to cellular senescence, data show that PTRF expression is upregulated in replicative senescent human fibroblasts, over-expression of PTRF elevates the number of caveolae and promotes premature senescence, and downregulation of PTRF extends replicative lifespan [11].
4.2. Caveolin-1 and stress-induced premature senescence
Data linking caveolin-1 to the development of stress-induced premature senescence began to emerge in the early 2000s. Numerous independent reports, using both cell culture and animal models, provide compelling evidence causally linking caveolin-1-mediated signaling to the acquisition of a senescent phenotype when cells are hit with a stressful stimulus (Fig. 1). Our laboratory was the first to demonstrate that either subcytotoxic levels of hydrogen peroxide or UV-C light up-regulate endogenous caveolin-1 expression and induce premature senescence in fibroblasts [128]. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements [129]. Treatment with the anti-oxidants quercetin and vitamin E prevents upregulation of caveolin-1 following hydrogen peroxide stimulation and the development of a premature senescent phenotype [128]. Importantly, oxidative stress-induced premature senescence is inhibited in NIH 3T3 cells harboring antisense caveolin-1 and in mouse embryonic fibroblasts (MEFs) derived from caveolin-1 null mice, which lack caveolin-1 expression [128, 130]. In support of these findings, studies show that over-expression of caveolin-1 is sufficient to block mouse embryonic fibroblasts (MEFs) in the G0/G1 phase of the cell cycle through modulation of the p53/p21waf1/Cip1 pathway [131]. MEFs transgenically over-expressing caveolin-1 have a shorter proliferative lifespan, as compared with MEFs derived from control littermate embryos, display a large, flat morphology, and express high levels of senescence-associated β-galactosidase activity [128]. Moreover, oxidative stress, following treatment with tert-butyl hydroperoxide, upregulates caveolin-1 mRNA and protein expression in nucleus pulposus cells while caveolin-1 downregulation by shRNA in these cells inhibits SIPS [132]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a major source of oxidative stress. Data show that oxidized low-density lipoprotein (oxLDL) upregulates caveolin-1 expression and promotes translocation/activation of the NADPH oxidase subunit p47phox into caveolae in macrophages, resulting in the development of a ROS-dependent senescent phenotype [133]. Studies show that endothelial cells isolated from chronic smokers with premature atherosclerosis display senescent features, increased oxidative stress, and elevated caveolin-1 expression, as compared with endothelial cells isolated from non-smokers [134]. Consistent with these data, cellular senescence in endothelial cells isolated from patients with severe coronary artery disease is accelerated by oxidative stress associated with risk factors for cardiovascular diseases [135]. In these cells, caveolin-1 expression is elevated [135]. Induction of senescence in endothelial cells by either hydrogen peroxide or ARHGAP18/SENEX is associated with upregulation of caveolin-1 expression and an increased number of caveolae. The ability of ARHGAP18/SENEX to induce senescence in endothelial cells isolated from caveolin-1 null mice is significantly compromised [136].
Fig. 1.
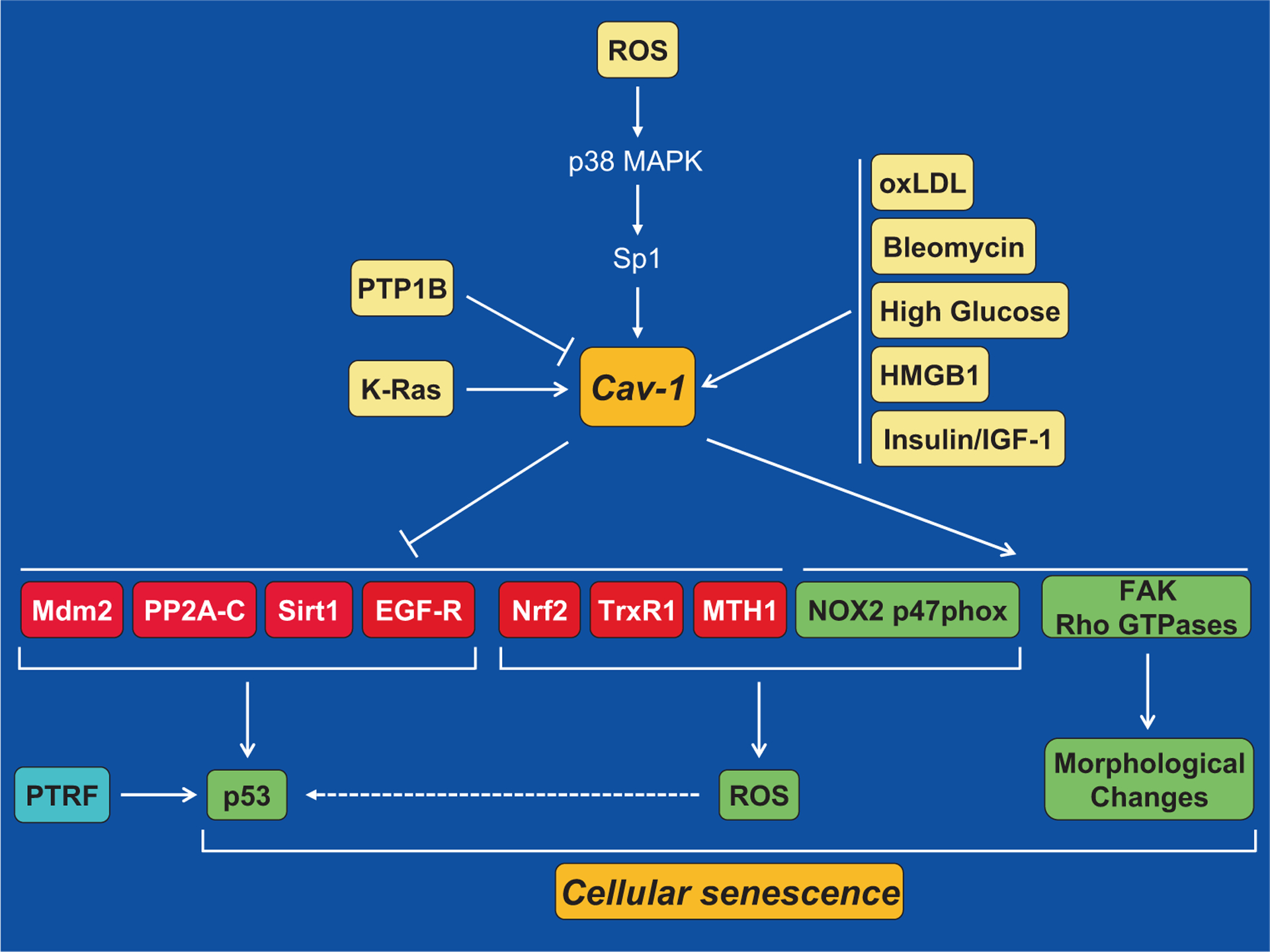
Molecular mechanisms underlying caveolin-1-mediated cellular senescence. Oxidative stress activates the caveolin-1 gene promoter in a p38 MAPK-Sp1-dependent mechanism and upregulates caveolin-1 expression. Oncogenic K-Ras, oxLDL, bleomycin, high levels of glucose, HMGB1, the insulin/IGF-1 pathway, and downregulation of PTP1B induce senescence through caveolin-1-mediated signaling. Upregulation of caveolin-1 promotes cellular senescence by activating the p53/p21Waf1/Cip1 pathway. Activation of p53 occurs through multiple mechanisms: (1) sequestration of Mdm2 into caveolar membranes, away from p53, prevents the Mdm2-dependent degradation of p53, which stabilizes p53 expression; (2) sequestration of PP2A-C into caveolar membranes activates ATM, which in turns phosphorylates and activates p53; (3) the interaction of caveolin-1 with Sirt1 inhibits Sirt1 activity leading to increased acetylation/activation of p53; (4) inhibition of EGF-R by caveolin-1 has been linked to activation of p53; (5) inhibition of Nrf2, TrxR1, and MTH1, through direct interaction with caveolin-1, and activation of NOX2 p47phox activate p53 through ROS production. PTRF can also promote senescence through activation of p53. Finally, activation of FAK and Rho GTPases by caveolin-1 promotes morphological changes that are typical of senescent cells
Pro-inflammatory genes such as cyclooxygenase-2 (COX-2) have been proposed to play an important role in the aging process. Data show that NS-398, a selective COX-2 inhibitor, prevents the upregulation of caveolin-1 expression and the development of cellular senescence in human dermal fibroblasts [137]. Since a selective COX-2 inhibitor has been shown to prevent the Sp1 proteins from binding to SP1 sites [138], inhibition of COX-2 may inhibit the caveolin-1-mediated development of premature senescence following oxidative stress by limiting the Sp1-mediated activation of the caveolin-1 promoter. Endothelial-specific protein tyrosine phosphatase-1B (PTP1B) null mice display increased neointima formation. Genetic knockdown or pharmacological inhibition of PTP1B in endothelial cells induces oxidative stress, upregulation of caveolin-1, and cellular senescence, which can be inhibited by downregulation of caveolin-1 expression [139]. Together, these data support the idea that caveolin-1 may link oxidative stress-induced premature senescence to cardiovascular diseases.
Bleomycin and glucose are additional stimuli that can induce senescence. Bleomycin induces DNA strand breaks and is used to treat certain types of cancer [140]. Bleomycin upregulates caveolin-1 expression and downregulation of caveolin-1 by shRNA inhibits bleomycin-induced senescence in A549 human lung adenocarcinoma epithelial cells [141]. Epithelial cell senescence and pulmonary fibrosis induced by the intratracheal instillation of bleomycin are reduced in caveolin-1 null mice [142]. Treatment with high concentrations of glucose induces senescence in glomerular mesangial cells, which is abrogated by caveolin-1 siRNA [143]. Increased oxidative stress, upregulation of caveolin-1 expression, and induction of cellular senescence occur in fibroblasts derived from patients with type 2 diabetes [18]. Oxidative stress and cellular senescence are observed in diabetic wounds in mice, and downregulation of caveolin-1 inhibits diabetes/oxidative stress-induced senescence and accelerates tissue repair [144].
Finally, a causal link between caveola-mediated signaling and the development of SIPS is further strengthened by data showing that oxidative stress upregulates PTRF protein expression, promotes the interaction of PTRF with caveolin-1, and induces the formation of caveolae [145]. Downregulation of PTRF expression by shRNA inhibits SIPS [145].
4.3. Caveolin-1 promotes stress-induced senescence through activation of the p53-p21 signaling pathway
Activation of p53 can be a major contributor to the development of a senescent phenotype after cellular stress. Data demonstrate that activation of the p53-p21 pathway is a key signaling through which caveolin-1 promotes senescence. After our initial report that over-expression of caveolin-1 is sufficient to induce premature senescence and activate the p53-p21 pathway [128, 131], we and others have shown that downregulation or a lack of caveolin-1 expression inhibits oxidative stress-induced activation of the p53-p21 signaling pathway and SIPS [130, 132, 141, 146, 147]. A number of studies have discovered various molecular mechanisms through which caveolin-1 activates p53 in the context of SIPS. More specifically, data show that caveolin-1 activates the p53/p21 pathway and induces premature senescence in virtue of its ability to directly interact with Mdm2, PP2A-C (the catalytic subunit of protein phosphatase PP2A), sirtuin 1 (Sirt1), nuclear erythroid 2 p45-related factor-2 (Nrf2), thioredoxin reductase 1 (TrxR1), and MutT homolog 1 (MTH1), and to sequester them into caveolar membranes after oxidative stress (Fig. 1).
Mdm2 is a negative regulator of p53 that promotes p53 degradation. Sequestration of Mdm2 into caveolae results in inhibition of Mdm2 activity, leading to stabilization of p53 expression and induction of premature senescence [130]. In support of these findings, upregulation of caveolin-1 and PTRF expression in fibroblasts from patients with type 2 diabetes results in sequestration of Mdm2 away from p53 and activation of the p53-p21 pathway [144]. Moreover, replicative senescent bone marrow mesenchymal ST2 cells display a strong association between caveolin-1 and Mdm2 and over-expression of caveolin-1 in these cells upregulates p53 and p21 [112]. Consistent with these data, PTRF expression is necessary for the oxidant-induced sequestration of Mdm2 into caveolar membranes, away from p53, and activation of the p53-p21-senescence pathway [145]. Expression of a mutant form of PTRF, which fails to localize to caveolar membranes after oxidative stress, inhibits oxidative stress-induced activation of p53 [145].
Ataxia telangiectasia-mutated (ATM) is a positive regulator of p53. PP2A is a negative regulator of ATM autophosphorylation and activity in vivo [148]. Caveolin-1-mediated localization of PP2A-C into caveolae, after oxidative stress, inhibits PP2A and therefore promotes the ATM-dependent phosphorylation/activation of p53 and induction of cellular senescence [146].
Sirt1 is a class III histone deacetylase that regulates a variety of cellular processes, including cellular senescence. Deacetylation of p53 by Sirt1 inactivates p53. Oxidative stress promotes the interaction of Sirt1 with caveolin-1, which leads to inactivation of Sirt1 activity and the consequent acetylation/activation of p53 and induction of premature senescence [149]. Data show that phosphorylation of caveolin-1 on tyrosine 14 is a post-translational modification, induced by oxidative stress in a p38 MAPK-dependent manner, which promotes the oxidant-induced localization of Sirt1 in caveolae resulting in the activation of the p53/senescence pathway in fibroblasts [149]. Consistent with these data, expression of a phosphomimetic mutant form of caveolin-1 (Caveolin-1 Y14D) in MDA-MB-435 cancer cells activates p53, inhibits cellular proliferation, and inhibits tumor growth in xenograft studies in vivo [150]. One can speculate that the phospho-caveolin-1-dependent sequestration of Sitrt1 in caveolae might contribute to the activation of p53 and the development of the phenotypes observed in these cancer cells. Finally, downregulation of caveolin-1 in rats fed high-fat diet supplemented with soy protein isolate results in Sirt1 activation and deacetylation of p53 [151].
Nrf2 is a transcription factor that mediates cytoprotective responses against stress. Under resting conditions, Nrf2 localizes with caveolin-1 in caveolar membranes [152]. After oxidative stress, caveolin-1 limits the translocation of Nrf2 to the nucleus and therefore inhibits the activation of an anti-oxidant response [152]. As a result, the p53-p21 pathway is activated and cells undergo cellular senescence [152]. A mutant form of Nrf2 that fails to interact with caveolin-1 hyperactivates Nrf2 target genes and inhibits both oxidant-induced activation of the p53-p21 pathway and induction of SIPS [152]. In epithelial Beas-2B cells, caveolin-1 inhibits Nrf-2-mediated transcription by promoting the interaction of Nrf2 with its inhibitor Kelch-like ECH-associated protein 1 (Keap1) [153]. Downregulation of caveolin-1 in vascular endothelial cells decreases Keap1 protein levels and increases Nrf2 activity [154]. In breast cancer cells, caveolin-1 loss correlates with activation of Nrf2 and rescue of caveolin-1 expression in these cells inhibits Nrf2-mediated transcription [155].
TrxR1 is an essential anti-oxidant enzyme that controls cellular redox homeostasis [156]. It reduces thioredoxin by using nicotinamide adenine dinucleotide phosphate [157]. TrxR1 is a caveolar membrane-resident protein [158] and caveolin-1 is an endogenous inhibitor of TrxR1 [158]. Data show that oxidative stress fails to activate the p53/p21Waf1/Cip1 pathway and induce premature senescence in cells expressing a mutant form of TrxR1 that cannot bind to caveolin-1 and is constitutively active [158].
MTH1 is the major mammalian detoxifier of the oxidized DNA precursor 8-oxo-dGTP. MTH1 prevents the incorporation of 8-oxoguanine into the DNA, by removing it from the dNTP pool, and therefore inhibits the initiation of a DNA damage/senescence response [159–164]. Interestingly, oncogenic K-Ras promotes the interaction between caveolin-1 and MTH1, which results in MTH1 inhibition, activation of the p53/p21 pathway, and induction of cellular senescence [165].
It is worth mentioning that a few studies have also linked caveolin-1-mediated regulation of epidermal growth factor receptor (EGF-R), NOX2 p47phox, focal adhesion kinase (FAK), and small Rho GTPase pathways to the development of replicative cellular senescence (Fig. 1), as discussed elsewhere in this review.
4.4. Caveolin-1-induced senescence and age-related diseases
Pulmonary emphysema is an age-related lung disease mostly caused by chronic exposure to cigarette smoking. Cigarette smoke is enriched in oxidants and oxidative stress is believed to play a major role in the pathogenesis of emphysema [166]. Cell culture studies show that cigarette smoke induces senescence of lung fibroblasts, which can be inhibited by anti-oxidant treatments [146, 167]. Interestingly, lung fibroblasts obtained from patients with emphysema are positive for the senescence marker senescence-associated β-galactosidase activity and display a reduced proliferation capacity [168, 169]. Data show that caveolin-1 plays a central role linking cigarette smoke-induced oxidative stress to the development of cellular senescence. More precisely, cigarette smoke treatment upregulates endogenous caveolin-1 expression in lung fibroblasts in cell culture studies and in mice [146]. In addition, development of cellular senescence is inhibited in caveolin-1-lacking lung fibroblasts after exposure to cigarette smoke extracts in cell culture studies or when caveolin-1 null mice are chronically exposed to cigarette smoke [146]. A very intriguing finding is that the development of pulmonary emphysema is inhibited in caveolin-1 null mice after 6 months of exposure to cigarette smoking [146], possibly linking oxidative stress-induced and caveolin-1-mediated cellular senescence to the pathogenesis of pulmonary emphysema. Data suggest that cigarette smoke-initiated and caveolin-1-mediated signaling leads to senescence by enforcing an ATM-dependent DNA damage response. In fact, cigarette smoke exposure, a well-known cause of DNA damage, activates ATM and upregulates both p53 and p21 protein expression in wild-type lung fibroblasts but to a much lesser extent in caveolin-1-lacking lung fibroblasts [146]. Mechanistically, caveolin-1 activates ATM by sequestering PP2A-C into caveolar membranes [146]. EGF-R is known to promote the resolution of γH2AX damage foci [170]. Since caveolin-1 can act as an inhibitor of EGF-R [108], an additional mechanism through which caveolin-1 promotes a DNA damage response is by blocking EGF-R signaling, which would support the chronic DNA damage response that is seen in senescent cells. Interestingly, caveolin-1 might also enhance DNA damage itself by elevating the levels of free radicals through inhibition of anti-oxidants such as TrxR1 [158] and Nrf2 [152].
Age-related intervertebral disc (IVD) degeneration is a cause of chronic low back pain [171]. Cellular senescence contributes to IVD degeneration [120, 172–174]. Human cells from the nucleus pulposus (NP) of degenerate discs show evidence of cellular senescence and express high levels of caveolin-1 [173, 175], suggesting that caveolin-1-mediated cellular senescence may be involved in the pathogenesis of IVD degeneration. Treatment of articular chondrocytes with IL-1β and hydrogen peroxide upregulates caveolin-1 mRNA and protein expression and induces cellular senescence [147]. Downregulation of caveolin-1 expression with antisense oligonucleotides prevents the development of chondrocyte senescence induced by these stimuli [147]. Thus, caveolin-1 may play a role in the pathogenesis of osteoarthritis by mediating chondrocyte senescence.
4.5. Downregulation of caveolin-1 can induce premature senescence
Although abundant data causally link increased caveolin-1 expression and caveolin-1-mediated signaling to replicative senescence and SIPS, reports exist showing that downregulation of caveolin-1 in unstimulated cells can also induce a senescent phenotype. For example, either knockout or knockdown of caveolin-1 induces senescence in resting human diploid fibroblasts, HCT116, A549, and MEFs, which correlates with mitochondrial dysfunction [176]. Consistent with these data, caveolin-1 maintains mitochondrial integrity and function by promoting the mitochondrial localization of matrix-oriented AAA (m-AAA) protease and its quality control functions [177]. Moreover, downregulation of caveolin-1 promotes cellular senescence in resting human diploid fibroblasts through formation of the primary cilium following the proteasomal-dependent degradation of aurora kinase A [178]. These results are consistent with data showing accumulation of senescence markers in human breast cancer-associated fibroblasts (CAFs) in which caveolin-1 expression is significantly downregulated [179, 180]. Thus, caveolin-1 may represent a pleiotropic regulator of cellular senescence: elevated caveolin-1-mediated signaling contributes to the development of replicative senescence and premature senescence following cellular stress while caveolin-1 deficiency promotes premature senescence in resting cells by promoting primary cilium formation and mitochondrial dysfunctions. This pleiotropic function is not unique to caveolin-1, as other signaling molecules, such as members of the DNA damage signaling pathway [76, 181] and E2F transcription factors [182, 183], have been shown to either promote or inhibit senescence depending on their expression levels.
5. Caveolin-1-mediated senescence as an anti-tumorigenic mechanism
Senescent cells are characterized by an irreversible exit from the cell cycle. As such, cellular senescence is a powerful tumor suppressor mechanism. Given the key role that caveolin-1 plays in the development of a senescent phenotype, studies have addressed caveolin-1-mediated senescence as a potential anti-tumorigenic mechanism. Over-expression of caveolin-1 in A549 human lung carcinoma epithelial cells, in which caveolin-1 expression is reduced but not lost [184], enhances SIPS [158]. Consistent with these data, over-expression of caveolin-1 in hydrogen peroxide-treated A549 cells inhibits tumor formation in nude mice [158]. Upon cell transformation, TrxR1 supports tumor cell growth, as shown by elevated TrxR protein levels and activity in many cancer cells [185]. Caveolin-1 is an endogenous inhibitor of TrxR1 [158]. Interestingly, over-expression of caveolin-1 inhibits the ability of TrxR1 to increase the transformed phenotype of A549 cells. Moreover, A549 cells maintained their ability to growth in soft agar when caveolin-1 expression was transiently down-regulated by siRNA at the time of oxidative stress, in contrast to control siRNA-transfected A549 cells, which formed only a few small foci in soft agar assays [158]. Over-expression of caveolin-1 induces premature senescence in A549 and H460 lung cancer cells and inhibits their growth in soft agar even in the absence of oxidative stress [165]. Thus, inhibition of TrxR1 by caveolin-1 induces premature senescence and acts in a tumor-suppressor manner.
Nrf2 signaling has been linked to tumor growth in HCT116 colon cancer cells, which are known to express caveolin-1. Downregulation of caveolin-1 expression by siRNA in HCT116 cells activates anti-oxidant-responsive elements (AREs) while the over-expression of caveolin-1 inhibits the oxidant-induced upregulation of heme oxygenase 1 (HO1) [152], an Nrf2 target [186]. Thus, caveolin-1 acts as an inhibitor of Nrf2 signaling in colon cancer cells. Importantly, the stable over-expression of caveolin-1 in HCT116 cells potentiates oxidative stress-induced premature senescence [152]. Moreover, over-expression of caveolin-1 inhibits the transformed phenotype of HCT116 cells, as assessed by growth in soft agar, after oxidative stress [152]. These data support findings showing that Nrf2 signaling may be beneficial to cancer cells and suggest that caveolin-1-mediated suppression of Nrf2 inhibits cell transformation by promoting premature senescence. Similarly, over-expression of caveolin-1 induces premature senescence in MCF-7 breast cancer cells following oxidative stress [129]. Thus, caveolin-1-mediated signaling promotes the tumor suppressor functions of premature senescence in lung, breast, and colon cancer cell lines.
Induction of oncogene-induced senescence is a well-established function of certain oncogenes, including K-Ras [63, 71, 72]. Cellular senescence has been reported in human premalignant lesions but not in the tumor itself [69, 76, 77, 81, 187], suggesting that oncogene-transformed cells need to either evade or bypass the OIS barrier in order to proliferate and progress to higher grades of malignancy [63, 69, 73, 74]. Data show a direct and causal role of caveolin-1 in the development of OIS. Over-expression of K-RasG12V induces senescence in wild-type MEFs [165]. In contrast, K-RasG12V-induced senescence is inhibited in MEFs lacking caveolin-1 expression [165]. In support of these data, oncogenic K-Ras induces senescence in normal human bronchial epithelial cells; downregulation of caveolin-1 in these cells inhibits K-RasG12V-induced senescence [165]. Thus, K-RasG12V induces cellular senescence in a caveolin-1-dependent manner. Oncogenic K-Ras promotes OIS through oxidative DNA damage [63, 71, 72]. Data show that caveolin-1 promotes K-Ras-induced premature senescence by inhibiting the detoxification function of MTH1. The scaffold kinase suppressor of Ras 1 (KSR1) regulates the activation of the Raf/MEK/ERK pathway. KSR1 interacts with caveolin-1 and promotes the localization of MEK and ERK in caveolin-1-rich domains, which is required for optimal ERK activation [188]. When the interaction between caveolin-1 and KSR1 is prevented, H-RasG12V-mediated senescence is inhibited, suggesting that caveolin-1 can also promote OIS by facilitating the assembly at the plasma membrane of key signaling molecules that transduce Ras signaling, at least when the pathway is activated by H-Ras [188].
K-RasLA2-G12D mice are heterozygous for the K-Ras G12D mutation and develop lung tumors with a histopathology very similar to human disease [189]. The role of caveolin-1 in OIS is supported by in vivo data obtained using the K-RasLA2-G12D/Cav-1−/− mouse, which lacks caveolin-1 expression [165]. The number of cells positive for SA-β-gal activity and the expression of p21 and p16 are reduced in the lung of K-RasLA2-G12D/Cav-1−/− mice, as compared with control K-RasLA2-G12D/Cav-1+/+ mice [165]. Consistent with the tumor suppressor role of OIS, the number of microscopic malignant lesions in the lung of K-RasLA2-G12D/Cav-1−/− mice is increased by 3-fold, as compared with that of K-RasLA2-G12D/Cav-1+/+ mice. Moreover, the total number of surface lung tumors is 2.5-fold higher in K-RasLA2-G12D/Cav-1−/− than in K-RasLA2-G12D/Cav-1+/+ mice. Overall survival and mean age of death of K-RasLA2-G12D/Cav-1−/− mice are lower than those of K-RasLA2-G12D/Cav-1+/+ mice [165]. Thus, caveolin-1 promotes the tumor suppressive function of cellular senescence during lung cancer development in mice.
As mentioned above, K-RasG12D mice (wild type for caveolin-1) show abundant lung cell senescence and only marginal lung tumor formation. However, they do develop lung tumors as they age, albeit to a lesser extent than their caveolin-1-lacking counterparts [165]. Interestingly, endogenous caveolin-1 expression is downregulated during aging in K-RasG12D mice. Similarly, caveolin-1 expression is down-regulated in K-RasG12V-infected MEFs that escape OIS [165]. Thus, a selective pressure appears to exist in cells expressing oncogenic K-Ras, which allows them to escape senescence, possibly through the downregulation of caveolin-1, and promote tumor development. These cell culture and animal data are supported by human studies using lung adenocarcinoma tissue microarrays (TMAs) showing that 86% of human adenocarcinomas have caveolin-1-negative cancer epithelial cells and that 91% of adenocarcinoma cases carrying mutant K-Ras are caveolin-1-negative [165]. Moreover, a significant correlation exists between increased methylation of the caveolin-1 gene and reduced caveolin-1 expression in human lung adenocarcinomas [165]. Finally, gene expression analysis using RNA derived from lung adenocarcinoma patients shows that the rate of death was 60% lower for patients with high caveolin-1 expression [165]. These human data strengthen the notion that caveolin-1 has tumor suppressive properties in cancer. Consistent with a tumor suppressor role of caveolin-1, its expression is down-regulated in other human cancers, such as breast [43, 190], colon [191, 192], ovarian [193], glioblastoma [194], and sarcoma [195] cancer. Moreover, a lack of caveolin-1 increases tumorigenesis in certain mouse models of cancer, such as breast [196] and skin cancer [36].
6. Caveolin-1-mediated senescence as a pro-tumorigenic mechanism
Stromal cells, including fibroblasts, are known to have a pro-found effect on tumor development. As described in Section 3.3, senescent stromal cells, through the secretion of SASP factors, can stimulate the growth of pre-neoplastic and neoplastic cells. Since caveolin-1 promotes both replicative and stress-induced senescence in fibroblasts, studies have been performed to determine whether caveolin-1-mediated senescence can also represent a pro-tumorigenic mechanism. Wild-type MEFs, which are induced to senesce by oxidative stress, but not caveolin-1 null MEFs, enhance the growth of both H-RasG12V-transformed NIH 3T3 and MDA-MB-231 breast cancer cells in co-culture experiments [130]. The caveolin-1-mediated growth-promoting action of senescent fibroblasts is due to their ability to release pro-stimulatory factors since conditioned medium derived from wild-type senescent MEFs, but not caveolin-1 null MEFs, is sufficient to stimulate the growth of RasG12V-transformed NIH 3T3, MDA-MB-231 [130], and PC3 prostate cancer cells [149]. This effect occurs in vivo as well, as shown by the enhanced tumor growth when either H-RasG12V-transformed NIH 3T3 or MDA-MB-231 cells are co-injected with senescent wild-type MEFs, but not caveolin-1 null MEFs, in immunodeficient mice, as compared with injection of cancer cells alone [130].
Data show that IL-6 is a caveolin-1-specific pro-tumorigenic factor. In fact, both IL-6 mRNA expression level in senescent wild-type MEFs and IL-6 protein level in conditioned medium derived from senescent wild-type MEFs are upregulated following oxidative stress, but not in MEFs obtained from caveolin-1 null mice [149]. Downregulation of caveolin-1 by siRNA in fibroblasts inhibits oxidant-induced activation of the IL-6 promoter [149]. Re-expression of caveolin-1 in caveolin-1 null MEFs rescues oxidant-induced upregulation of IL-6 mRNA. The caveolin-1-mediated upregulation of IL-6 is dependent on the ability of caveolin-1 to inhibit Sirt1 since the free radical-induced upregulation of IL-6 mRNA and activation of the IL-6 promoter can be restored in caveolin-1-lacking fibroblasts following downregulation of Sirt1 by siRNA [149]. Importantly, the enhancement of RasG12V-transformed NIH 3T3, MDA-MB-231, and PC3 prostate cancer cell proliferation by the conditioned medium derived from senescent wild-type fibroblasts is inhibited by an IL-6 neutralizing antibody [149]. Thus, the pro-stimulatory properties of IL-6, which is secreted by senescent fibroblasts, are caveolin-1-dependent.
In support of a pro-tumorigenic role of caveolin-1 in stromal fibroblasts, downregulation of caveolin-1 in cancer-associated fibroblasts (CAFs), which were derived from pancreatic cancer patients, reduces the ability of their conditioned medium to stimulate invasiveness of MIAPaCa-2 pancreatic cancer cells [197]. Moreover, caveolin-1 expression in CAFs is associated with pancreatic cancer patients’ poor prognosis [197]. In support of these findings, although caveolin-1 expression is reduced in lung adenocarcinoma epithelial cells, caveolin-1 remains highly expressed in the lung tumor stroma, which is caveolin-1-positive in 82% of cases [165]. These data are in apparent contrast with data showing accumulation of senescence markers in human breast cancer-associated fibroblasts in which caveolin-1 expression is significantly down-regulated [179, 180]. However, as discussed above in Section 4.5, downregulation of caveolin-1 expression can also induce, under certain conditions, cellular senescence. Thus, one can speculate that depending on the cancer type, either excessively high or low caveolin-1 levels in CAFs can have pro-tumorigenic functions through induction of senescence and the release of SASP factors.
7. Conclusive remarks
Senescent cells accumulate with aging and contribute to the development of age-related deterioration of organ functions. An additional detrimental effect of cellular senescence relates to the accumulation of senescent stromal cells within the tumor microenvironment, where they secrete pro-tumorigenic SASP factors that stimulate the growth of pre-neoplastic and transformed epithelial cells. However, cellular senescence represents a natural tumor suppressor mechanism given the permanent exit from the cell cycle of senescent cells. Cancer cells need to either escape or bypass the barrier represented by cellular senescence in order to produce a clinically relevant tumor mass. Thus, the identification of the molecular mechanisms underlying the development and evasion of cellular senescence is key to a better understanding of the pathogenesis of age-related diseases, including cancer.
Multiple and independent findings from a number of laboratories have identified caveolin-1 as a major contributor to the acquisition of a senescent phenotype. Upregulation of caveolin-1 expression has been causally linked to the development of both replicative senescence and stress-induced premature senescence. Cellular stressors that promote caveolin-1-mediated SIPS include oxidative stress, UV-C light, bleomycin, and oncogenic K-Ras. Mechanistically, activation of the p53/p21 pathway is pivotal to the pro-senescent action of caveolin-1. Modulation of EGF-, focal adhesion-, and small Rho GTPase-dependent signaling represents additional regulatory mechanisms that have been linked to caveolin-1-mediated senescence. Downregulation of caveolin-1 in resting cells has also been causally linked to the development of premature senescence through mitochondrial dysfunction and the forced maintenance of the primary cilium, supporting the notion that both “too much” and “too little” of caveolin-1-mediated signaling promote a senescent response.
Oncogenic K-Ras (K-RasG12D)-induced senescence is inhibited in caveolin-1 null mice, which display enhanced lung tumorigenesis after expression of K-RasG12D. Also, both downregulation of endogenous caveolin-1 expression and lung tumor development are age-dependent in K-RasG12D-expressing mice (wild type for caveolin-1). Moreover, caveolin-1 expression is downregulated in MEFs expressing oncogenic K-Ras that escape OIS. As such, downregulation of caveolin-1 expression might represent an early molecular event in lung tumorigenesis through which oncogene-expressing epithelial cells evade/bypass senescence to progress to cancer (Fig. 2). In contrast, caveolin-1-mediated senescence in stromal cells might, through the release of pro-tumorigenic SASP factors, fuel the growth of oncogene-expressing epithelial cells that have already evaded senescence (Fig. 2). This would be consistent with data suggesting that caveolin-1 functions predominantly as a tumor suppressor in early stages of disease while it is linked more to tumor progression and metastasis at later stages [198, 199] and with data showing that caveolin-1 expression in lung cancer patients is lost in cancer epithelial cells but maintained in cancer stromal cells. Thus, caveolin-1-mediated functions contribute to explain the pleiotropic role of senescence in cancer (Fig. 2). Further studies are necessary to determine whether targeted therapeutic interventions aimed at preventing downregulation of caveolin-1 expression in epithelial cells can limit the pro-tumorigenic evasion from oncogene-induced senescence during the early phases of tumorigenesis while those aimed at promoting the downregulation of caveolin-1 in stromal cells can restrain the accumulation of pro-tumorigenic senescent stromal cells in the tumor microenvironment during the later stages of tumorigenesis.
Fig. 2.
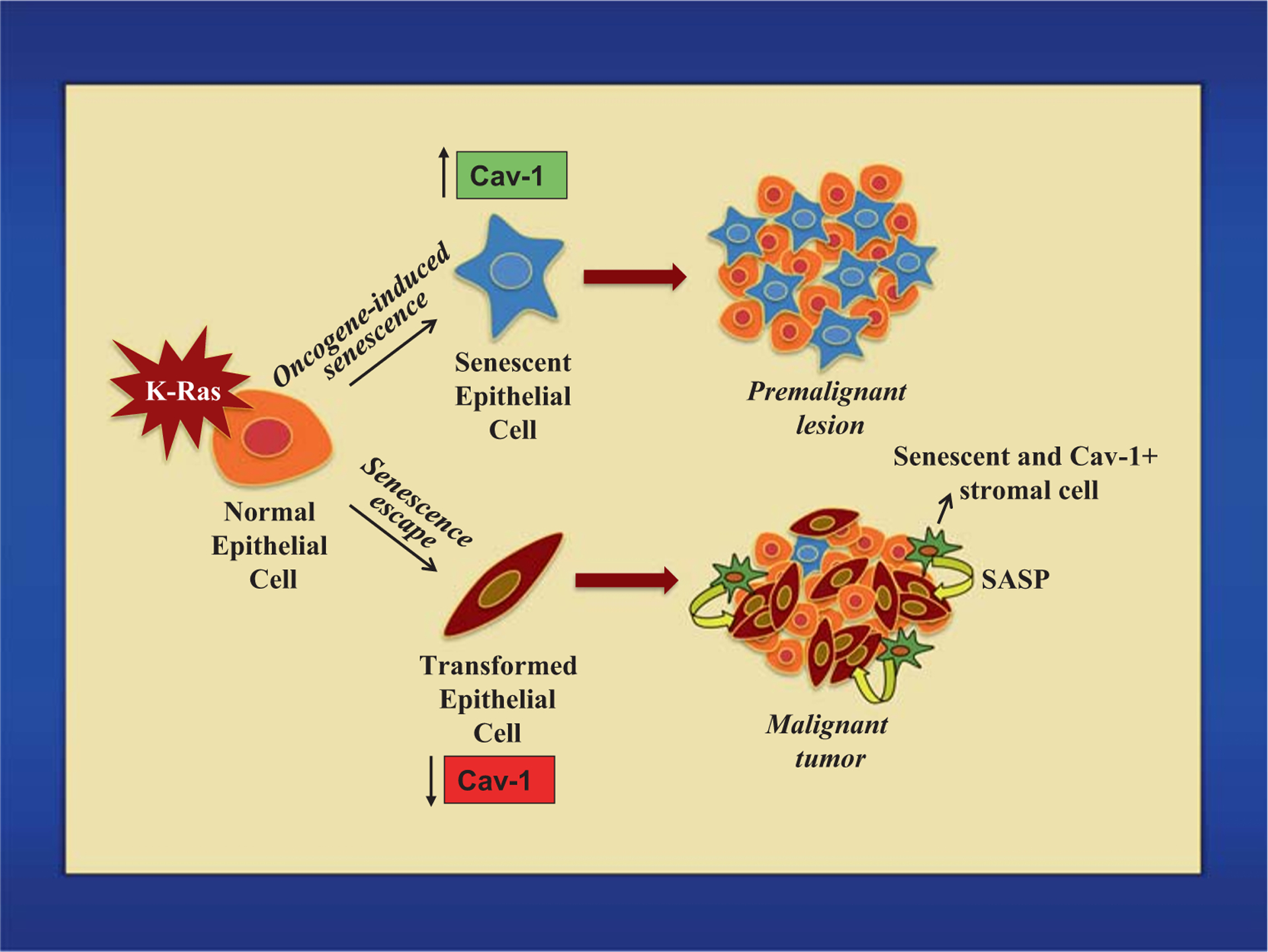
Proposed pleiotropic role of caveolin-1-mediated senescence in cancer. Expression of oncogenic K-Ras induces oncogene-induced senescence in epithelial cells in a caveolin-1-dependent manner. This is an anti-tumorigenic event that prevents premalignant lesions from progressing to cancer. Downregulation of caveolin-1 in oncogenic K-Ras-expressing cells is a pro-tumorigenic event that allows the escape/bypass of OIS and the development of a malignant tumor. Moreover, accumulation of caveolin-1-positive senescent stromal cells in the tumor microenvironment has pro-tumorigenic properties in virtue of their capacity to release growth-promoting factors that fuel the progression and possibly the metastatic features of cancer cells
Funding information
F.G. was supported by grants from the National Cancer Institute (R01-CA205165) and the Aging Institute and Hillman Cancer Center Seed Grant Program; D.V. was supported by a grant from the National Institute on Aging (R21-AG061614).
References
- 1.Palade GE (1953). The fine structure of blood capillaries. Journal of Applied Physics, 24, 1424. [Google Scholar]
- 2.Yamada E (1955). The fine structure of the gall bladder epithelium of the mouse. The Journal of Biophysical and Biochemical Cytology, 1, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campisi J (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell, 120(4), 513–522. [DOI] [PubMed] [Google Scholar]
- 4.Campisi J (2013). Aging, cellular senescence, and cancer. Annual Review of Physiology, 75, 685–705. 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coppe JP, Desprez PY, Krtolica A, & Campisi J (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology, 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodier F, & Campisi J (2011). Four faces of cellular senescence. The Journal of Cell Biology, 192(4), 547–556. 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherer PE, Lewis RY, Volonte D, Engelman JA, Galbiati F, Couet J, Kohtz DS, van Donselaar E, Peters P, & Lisanti MP (1997). Cell-type and tissue-specific expression of caveolin-2. Caveolins 1 and 2 co-localize and form a stable hetero-oligomeric complex in vivo. The Journal of Biological Chemistry, 272, 29337–29346. [DOI] [PubMed] [Google Scholar]
- 8.Pike LJ (2006). Rafts defined: a report on the keystone symposium on lipid rafts and cell function. Journal of Lipid Research, 47(7), 1597–1598. 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Gambin Y, Ariotti N, McMahon KA, Bastiani M, Sierecki E, Kovtun O, et al. (2013). Single-molecule analysis reveals self assembly and nanoscale segregation of two distinct cavin subcomplexes on caveolae. Elife, 3, e01434 10.7554/eLife.01434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayer A, Stoeber M, Bissig C, & Helenius A (2010). Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic, 11(3), 361–382. 10.1111/j.1600-0854.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- 11.Couet J, Li S, Okamoto T, Ikezu T, & Lisanti MP (1997). Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. The Journal of Biological Chemistry, 272, 6525–6533. [DOI] [PubMed] [Google Scholar]
- 12.Jagannadham MV, Sharadadevi A, & Nagaraj R (2002). Effects of deleting a tripeptide sequence observed in muscular dystrophy patients on the conformation of synthetic peptides corresponding to the scaffolding domain of caveolin-3. Biochemical and Biophysical Research Communications, 298(2), 203–206. [DOI] [PubMed] [Google Scholar]
- 13.Song KS, Tang Z-L, Li S, & Lisanti MP (1997). Mutational analysis of the properties of caveolin-1. A novel role for the C-terminal domain in mediating homotypic caveolin-caveolin interactions. The Journal of Biological Chemistry, 272, 4398–4403. [DOI] [PubMed] [Google Scholar]
- 14.Sonnino S, & Prinetti A (2009). Sphingolipids and membrane environments for caveolin. FEBS Letters, 583(4), 597–606. [DOI] [PubMed] [Google Scholar]
- 15.Lisanti MP, Scherer P, Tang Z-L, & Sargiacomo M (1994). Caveolae, caveolin and caveolin-rich membrane domains: a signalling hypothesis. Trends in Cell Biology, 4, 231–235. [DOI] [PubMed] [Google Scholar]
- 16.Song KS, Li S, Okamoto T, Quilliam L, Sargiacomo M, & Lisanti MP (1996). Copurification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent free purification of caveolae membranes. The Journal of Biological Chemistry, 271, 9690–9697. [DOI] [PubMed] [Google Scholar]
- 17.Mineo C, James GL, Smart EJ, & Anderson RGW (1996). Localization of EGF-stimulated Ras/Raf-1 interaction to caveolae membrane. The Journal of Biological Chemistry, 271, 11930–11935. [DOI] [PubMed] [Google Scholar]
- 18.Liu P, Ying Y, & Anderson RG (1997). Platelet-derived growth factor activates mitogen-activated protein kinase in isolated caveolae. Proceedings of the National Academy of Sciences of the United States of America, 94(25), 13666–13670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Okamoto T, Chun M, Sargiacomo M, Casanova JE, Hansen SH, Nishimoto I, & Lisanti MP (1995). Evidence for a regulated interaction of hetero-trimeric G proteins with caveolin. The Journal of Biological Chemistry, 270, 15693–15701. [DOI] [PubMed] [Google Scholar]
- 20.Li S, Couet J, & Lisanti MP (1996). Src tyrosine kinases, G alpha subunits and H-Ras share a common membrane-anchored scaffolding protein, Caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. The Journal of Biological Chemistry, 271, 29182–29190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shenoy-Scaria AM, Dietzen DJ, Kwong J, Link DC, & Lublin DM (1994). Cysteine-3 of Src family tyrosine kinases determines palmitoylation and localization in caveolae. Journal of Cell Biology, 126, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smart EJ, Foster D, Ying Y-S, Kamen BA, & Anderson RGW (1993). Protein kinase C activators inhibit receptor-mediated potocytosis by preventing internalization of caveolae. The Journal of Cell Biology, 124, 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schnitzer JE, Liu J, & Oh P (1995). Endothelial caveolae have the molecular transport machinery for vesicle budding, docking, and fusion including VAMP, NSF, SNAP, annexins, and GTPases. The Journal of Biological Chemistry, 270, 14399–14404. [DOI] [PubMed] [Google Scholar]
- 24.Couet J, Sargiacomo M, & Lisanti MP (1997). Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. The Journal of Biological Chemistry, 272, 30429–30438. [DOI] [PubMed] [Google Scholar]
- 25.Ju H, Zou R, Venema VJ, & Venema RC (1997). Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. The Journal of Biological Chemistry, 272(30), 18522–18525. [DOI] [PubMed] [Google Scholar]
- 26.Feron O, Belhassen L, Kobzik L, Smith TW, Kelly RA, & Michel T (1996). Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. The Journal of Biological Chemistry, 271(37), 22810–22814. [DOI] [PubMed] [Google Scholar]
- 27.Segal SS, Brett SE, & Sessa WC (1999). Codistribution of NOS and caveolin throughout peripheral vasculature and skeletal muscle of hamsters. The American Journal of Physiology, 277(3 Pt 2), H1167–H1177. [DOI] [PubMed] [Google Scholar]
- 28.Saltiel AR, & Pessin JE (2003). Insulin signaling in microdomains of the plasma membrane. Traffic, 4(11), 711–716. 10.1034/j.1600-0854.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- 29.Galbiati F, Razani B, & Lisanti MP (2001). Emerging themes in lipid rafts and caveolae. Cell, 106(4), 403–411. [DOI] [PubMed] [Google Scholar]
- 30.Galbiati F, Razani B, & Lisanti MP (2001). Caveolae and caveolin-3 in muscular dystrophy. Trends in Molecular Medicine, 7(10), 435–441. [DOI] [PubMed] [Google Scholar]
- 31.Parton RG, & Richards AA (2003). Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic, 4(11), 724–738. [DOI] [PubMed] [Google Scholar]
- 32.Razani B, Schlegel A, & Lisanti MP (2000). Caveolin proteins in signaling, oncogenic transformation and muscular dystrophy. Journal of Cell Science, 113(Pt 12), 2103–2109. [DOI] [PubMed] [Google Scholar]
- 33.Williams TM, & Lisanti MP (2004). The Caveolin genes: from cell biology to medicine. Annals of Medicine, 36(8), 584–595. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe M, Yang G, Cao G, Tahir SA, Naruishi K, Tabata K, Fattah EA, Rajagopalan K, Timme TL, Park S, Kurosaka S, Edamura K, Tanimoto R, Demayo FJ, Goltsov AA, & Thompson TC (2009). Functional analysis of secreted caveolin-1 in mouse models of prostate cancer progression. Molecular Cancer Research, 7(9), 1446–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams TM, Hassan GS, Li J, Cohen AW, Medina F, Frank PG, Pestell RG, di Vizio D, Loda M, & Lisanti MP (2005). Caveolin-1 promotes tumor progression in an autochthonous mouse model of prostate cancer: genetic ablation of Cav-1 delays advanced prostate tumor development in tramp mice. The Journal of Biological Chemistry, 280(26), 25134–25145. [DOI] [PubMed] [Google Scholar]
- 36.Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, & Lisanti MP (2003). Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. The American Journal of Pathology, 162(6), 2029–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, Li M, Tang B, Jelicks LA, Scherer PE, & Lisanti MP (2002). Caveolin-1-deficient mice are lean, resistant to diet-induced obesity, and show hypertriglyceridemia with adipocyte abnormalities. The Journal of Biological Chemistry, 277(10), 8635–8647. [DOI] [PubMed] [Google Scholar]
- 38.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. (2001). Caveolin-1 null mice are viable, but show evidence of hyper-proliferative and vascular abnormalities. The Journal of Biological Chemistry, 276(41), 38121–38138. [DOI] [PubMed] [Google Scholar]
- 39.Williams TM, Lee H, Cheung MW, Cohen AW, Razani B, Iyengar P, Scherer PE, Pestell RG, & Lisanti MP (2004). Combined loss of INK4a and caveolin-1 synergistically enhances cell proliferation and oncogene-induced tumorigenesis: role of INK4a/CAV-1 in mammary epithelial cell hyperplasia. The Journal of Biological Chemistry, 279(23), 24745–24756. 10.1074/jbc.M402064200. [DOI] [PubMed] [Google Scholar]
- 40.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA 3rd, et al. (2012). Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circulation. Cardiovascular Genetics, 5(3), 336–343. 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao H, Alston L, Ruschman J, & Hegele RA (2008). Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids in Health and Disease, 7, 3 10.1186/1476-511X-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han SE, Park KH, Lee G, Huh YJ, & Min BM (2004). Mutation and aberrant expression of Caveolin-1 in human oral squamous cell carcinomas and oral cancer cell lines. International Journal of Oncology, 24(2), 435–440. [PubMed] [Google Scholar]
- 43.Li T, Sotgia F, Vuolo MA, Li M, Yang WC, Pestell RG, Sparano JA, & Lisanti MP (2006). Caveolin-1 mutations in human breast cancer: functional association with estrogen receptor alpha-positive status. The American Journal of Pathology, 168(6), 1998–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patani N, Lambros MB, Natrajan R, Dedes KJ, Geyer FC, Ward E, Martin LA, Dowsett M, & Reis-Filho JS (2012). Non-existence of caveolin-1 gene mutations in human breast cancer. Breast Cancer Research and Treatment, 131(1), 307–310. 10.1007/s10549-011-1761-2. [DOI] [PubMed] [Google Scholar]
- 45.Lundberg AS, Hahn WC, Gupta P, & Weinberg RA (2000). Genes involved in senescence and immortalization. Current Opinion in Cell Biology, 12(6), 705–709. [DOI] [PubMed] [Google Scholar]
- 46.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, & Pereira-Smith O (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Sciences of the United States of America, 92(20), 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Black EJ, Clark W, & Gillespie DA (2000). Transient deactivation of ERK signalling is sufficient for stable entry into G0 in primary avian fibroblasts. Current Biology, 10(18), 1119–1122. [DOI] [PubMed] [Google Scholar]
- 48.Sherr CJ, & DePinho RA (2000). Cellular senescence: mitotic clock or culture shock? Cell, 102(4), 407–410. [DOI] [PubMed] [Google Scholar]
- 49.Wynford-Thomas D (1999). Cellular senescence and cancer. The Journal of Pathology, 187(1), 100–111. [DOI] [PubMed] [Google Scholar]
- 50.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, & Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science, 266(5193), 2011–2015. [DOI] [PubMed] [Google Scholar]
- 51.Lee SW, Reimer CL, Oh P, Campbel IDB, & Schnitzer JE (1998). Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene, 16, 1391–1397. [DOI] [PubMed] [Google Scholar]
- 52.Hayflick L, & Moorhead PS (1961). The serial cultivation of human diploid cell strains. Experimental Cell Research, 25, 585–621. [DOI] [PubMed] [Google Scholar]
- 53.Cristofalo VJ, Phillips PD, Sorger T, & Gerhard G (1989). Alterations in the responsiveness of senescent cells to growth factors. Journal of Gerontology, 44(6), 55–62. [DOI] [PubMed] [Google Scholar]
- 54.Wang E (1995). Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Research, 55(11), 2284–2292. [PubMed] [Google Scholar]
- 55.Matsumura T, Zerrudo Z, & Hayflick L (1979). Senescent human diploid cells in culture: survival, DNA synthesis and morphology. Journal of Gerontology, 34(3), 328–334. [DOI] [PubMed] [Google Scholar]
- 56.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, & Wright WE (1998). Extension of life-span by introduction of telomerase into normal human cells. Science, 279(5349), 349–352. [DOI] [PubMed] [Google Scholar]
- 57.Harley CB, Futcher AB, & Greider CW (1990). Telomeres shorten during ageing of human fibroblasts. Nature, 345(6274), 458–460. [DOI] [PubMed] [Google Scholar]
- 58.Lansdorp PM (2000). Repair of telomeric DNA prior to replicative senescence. Mechanisms of Ageing and Development, 118(1–2), 23–34. [DOI] [PubMed] [Google Scholar]
- 59.Martens UM, Chavez EA, Poon SS, Schmoor C, & Lansdorp PM (2000). Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Experimental Cell Research, 256(1), 291–299. [DOI] [PubMed] [Google Scholar]
- 60.Chen Q, Fischer A, Reagan JD, Yan LJ, & Ames BN (1995). Oxidative DNA damage and senescence of human diploid fibroblast cells. Proceedings of the National Academy of Sciences of the United States of America, 92(10), 4337–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Frippiat C, Chen QM, Zdanov S, Magalhaes JP, Remacle J, & Toussaint O (2001). Subcytotoxic H2O2 stress triggers a release of transforming growth factor-beta 1, which induces biomarkers of cellular senescence of human diploid fibroblasts. The Journal of Biological Chemistry, 276(4), 2531–2537. [DOI] [PubMed] [Google Scholar]
- 62.Robles SJ, & Adami GR (1998). Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene, 16(9), 1113–1123. [DOI] [PubMed] [Google Scholar]
- 63.Serrano M, Lin AW, McCurrach ME, Beach D, & Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 88(5), 593–602. [DOI] [PubMed] [Google Scholar]
- 64.Zhu J, Woods D, McMahon M, & Bishop JM (1998). Senescence of human fibroblasts induced by oncogenic Raf. Genes & Development, 12(19), 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, & Passos JF (2012). Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nature Communications, 3, 708 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell, 113(6), 703–716. 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 67.Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, & Goldman RD (2011). The role of nuclear lamin B1 in cell proliferation and senescence. Genes & Development, 25(24), 2579–2593. 10.1101/gad.179515.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, et al. (2013). Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY), 5(1), 37–50. 10.18632/aging.100527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM, Barbacid M, Beach D, & Serrano M (2005). Tumour biology: senescence in premalignant tumours. Nature, 436(7051), 642 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 70.van Deursen JM (2014). The role of senescent cells in ageing. Nature, 509(7501), 439–446. 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, & Chodosh LA (2007). Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nature Cell Biology, 9(5), 493–505. 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 72.Young TW, Mei FC, Yang G, Thompson-Lanza JA, Liu J, & Cheng X (2004). Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Research, 64(13), 4577–4584. 10.1158/0008-5472.CAN-04-0222. [DOI] [PubMed] [Google Scholar]
- 73.Baek KH, Bhang D, Zaslavsky A, Wang LC, Vachani A, Kim CF, Albelda SM, Evan GI, & Ryeom S (2013). Thrombospondin-1 mediates oncogenic Ras-induced senescence in premalignant lung tumors. The Journal of Clinical Investigation, 123(10), 4375–4389. 10.1172/JCI67465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collado M, & Serrano M (2010). Senescence in tumours: evidence from mice and humans. Nature Reviews. Cancer, 10(1), 51–57. 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tran PT, Shroff EH, Burns TF, Thiyagarajan S, Das ST, Zabuawala T, Chen J, Cho YJ, Luong R, Tamayo P, Salih T, Aziz K, Adam SJ, Vicent S, Nielsen CH, Withofs N, Sweet-Cordero A, Gambhir SS, Rudin CM, & Felsher DW (2012). Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS Genetics, 8(5), e1002650 10.1371/journal.pgen.1002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Ørntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, & Gorgoulis VG (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature, 444(7119), 633–637. 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 77.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, & Pandolfi PP (2005). Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature, 436(7051), 725–730. 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, et al. (2006). A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell, 10(6), 459–472. 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR, Ji H, Vojtesek B, Bourdon JC, Lane DP, & Harris CC (2009). p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nature Cell Biology, 11(9), 1135–1142. 10.1038/ncb1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, Marais R, Wynford-Thomas D, & Bennett DC (2006). Cellular senescence in naevi and immortalisation in melanoma: a role for p16? British Journal of Cancer, 95(4), 496–505. 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature, 436(7051), 720–724. 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 82.Peeper DS, Shvarts A, Brummelkamp T, Douma S, Koh EY, Daley GQ, & Bernards R (2002). A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nature Cell Biology, 4(2), 148–153. 10.1038/ncb742. [DOI] [PubMed] [Google Scholar]
- 83.Palmero I, Pantoja C, & Serrano M (1998). p19ARF links the tumour suppressor p53 to Ras. Nature, 395(6698), 125–126. 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- 84.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, & Green MR (2008). Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell, 132(3), 363–374. 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Angelini PD, Zacarias Fluck MF, Pedersen K, Parra-Palau JL, Guiu M, Bernado Morales C, et al. (2013). Constitutive HER2 signaling promotes breast cancer metastasis through cellular senescence. Cancer Research, 73(1), 450–458. 10.1158/0008-5472.CAN-12-2301. [DOI] [PubMed] [Google Scholar]
- 86.Garbers C, Kuck F, Aparicio-Siegmund S, Konzak K, Kessenbrock M, Sommerfeld A, Häussinger D, Lang PA, Brenner D, Mak TW, Rose-John S, Essmann F, Schulze-Osthoff K, Piekorz RP, & Scheller J (2013). Cellular senescence or EGFR signaling induces interleukin 6 (IL-6) receptor expression controlled by mammalian target of rapamycin (mTOR). Cell Cycle, 12(21), 3421–3432. 10.4161/cc.26431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berger AH, Knudson AG, & Pandolfi PP (2011). A continuum model for tumour suppression. Nature, 476(7359), 163–169. 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larribere L, Wu H, Novak D, Galach M, Bernhardt M, Orouji E, Weina K, Knappe N, Sachpekidis C, Umansky L, Beckhove P, Umansky V, de Schepper S, Kaufmann D, Ballotti R, Bertolotto C, & Utikal J (2015). NF1 loss induces senescence during human melanocyte differentiation in an iPSC-based model. Pigment Cell & Melanoma Research, 28(4), 407–416. 10.1111/pcmr.12369. [DOI] [PubMed] [Google Scholar]
- 89.Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T, & Takahashi C (2009). Rb regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell, 15(4), 255–269. 10.1016/j.ccr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 90.Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, Signoretti S, & Kaelin WG Jr. (2008). VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nature Cell Biology, 10(3), 361–369. 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- 91.Di Mitri D, & Alimonti A (2016). Non-cell-autonomous regulation of cellular senescence in cancer. Trends in Cell Biology, 26(3), 215–226. 10.1016/j.tcb.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 92.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, & Peeper DS (2008). Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell, 133(6), 1019–1031. 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 93.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, & Gil J (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell, 133(6), 1006–1018. 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 94.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, & Gil J (2013). A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nature Cell Biology, 15(8), 978–990. 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lopes-Paciencia S, Saint-Germain E, Rowell MC, Ruiz AF, Kalegari P, & Ferbeyre G (2019). The senescence-associated secretory phenotype and its regulation. Cytokine, 117, 15–22. 10.1016/j.cyto.2019.01.013. [DOI] [PubMed] [Google Scholar]
- 96.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, & Zender L (2011). Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature, 479(7374), 547–551. 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 97.Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, & Nelson PS (2006). The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Research, 66(2), 794–802. [DOI] [PubMed] [Google Scholar]
- 98.Krtolica A, Parrinello S, Lockett S, Desprez PY, & Campisi J (2001). Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proceedings of the National Academy of Sciences of the United States of America, 98(21), 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu D, & Hornsby PJ (2007). Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Research, 67(7), 3117–3126. [DOI] [PubMed] [Google Scholar]
- 100.Parrinello S, Coppe JP, Krtolica A, & Campisi J (2005). Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. Journal of Cell Science, 118(Pt 3), 485–496. 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herbig U, Ferreira M, Condel L, Carey D, & Sedivy JM (2006). Cellular senescence in aging primates. Science, 311(5765), 1257. [DOI] [PubMed] [Google Scholar]
- 102.Jeyapalan JC, Ferreira M, Sedivy JM, & Herbig U (2007). Accumulation of senescent cells in mitotic tissue of aging primates. Mechanisms of Ageing and Development, 128(1), 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kishi S (2004). Functional aging and gradual senescence in zebrafish. Annals of the New York Academy of Sciences, 1019, 521–526. [DOI] [PubMed] [Google Scholar]
- 104.Melk A, Kittikowit W, Sandhu I, Halloran KM, Grimm P, Schmidt BM, et al. (2003). Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney International, 63(6), 2134–2143. [DOI] [PubMed] [Google Scholar]
- 105.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. (2017). Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discovery, 7(2), 165–176. 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ewald JA, Desotelle JA, Wilding G, & Jarrard DF (2010). Therapy-induced senescence in cancer. Journal of the National Cancer Institute, 102(20), 1536–1546. 10.1093/jnci/djq364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Toso A, Di Mitri D, & Alimonti A (2015). Enhancing chemotherapy efficacy by reprogramming the senescence-associated secretory phenotype of prostate tumors: a way to reactivate the antitumor immunity. Oncoimmunology, 4(3), e994380 10.4161/2162402X.2014.994380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park WY, Park JS, Cho KA, Kim DI, Ko YG, Seo JS, et al. (2000). Up-regulation of caveolin attenuates epidermal growth factor signaling in senescent cells. The Journal of Biological Chemistry, 275(27), 20847–20852. [DOI] [PubMed] [Google Scholar]
- 109.Wheaton K, Sampsel K, Boisvert FM, Davy A, Robbins S, & Riabowol K (2001). Loss of functional caveolae during senescence of human fibroblasts. Journal of Cellular Physiology, 187(2), 226–235. [DOI] [PubMed] [Google Scholar]
- 110.Cho KA, Ryu SJ, Park JS, Jang IS, Ahn JS, Kim KT, et al. (2003). Senescent phenotype can be reversed by reduction of caveolin status. The Journal of Biological Chemistry, 278(30), 27789–27795. [DOI] [PubMed] [Google Scholar]
- 111.Cho KA, Ryu SJ, Oh YS, Park JH, Lee JW, Kim HP, et al. (2004). Morphological adjustment of senescent cells by modulating caveolin-1 status. The Journal of Biological Chemistry, 279(40), 42270–42278. [DOI] [PubMed] [Google Scholar]
- 112.Zhang J, Lazarenko OP, Blackburn ML, Badger TM, Ronis MJ, & Chen JR (2014). Soy protein isolate downregulates caveolin-1 expression to suppress osteoblastic cell senescence pathways. The FASEB Journal, 28(7), 3134–3145. 10.1096/fj.13-243659. [DOI] [PubMed] [Google Scholar]
- 113.Park JS, Kim HY, Kim HW, Chae GN, Oh HT, Park JY, et al. (2005). Increased caveolin-1, a cause for the declined adipogenic potential of senescent human mesenchymal stem cells. Mechanisms of Ageing and Development, 126(5), 551–559. [DOI] [PubMed] [Google Scholar]
- 114.Sun C, Wang N, Huang J, Xin J, Peng F, Ren Y, et al. (2009). Inhibition of phosphatidylcholine-specific phospholipase C prevents bone marrow stromal cell senescence in vitro. Journal of Cellular Biochemistry, 108(2), 519–528. [DOI] [PubMed] [Google Scholar]
- 115.Lim JS, Nguyen KC, Nguyen CT, Jang IS, Han JM, Fabian C, et al. (2015). Flagellin-dependent TLR5/caveolin-1 as a promising immune activator in immunosenescence. Aging Cell, 14(5), 907–915. 10.1111/acel.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sun S, Cai B, Li Y, Su W, Zhao X, Gong B, et al. (2019). HMGB1 and Caveolin-1 related to RPE cell senescence in age-related macular degeneration. Aging (Albany NY), 11(13), 4323–4337. 10.18632/aging.102039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yudoh K, Shi Y, & Karasawa R (2009). Angiogenic growth factors inhibit chondrocyte ageing in osteoarthritis: potential involvement of catabolic stress-induced overexpression of caveolin-1 in cellular ageing. International Journal of Rheumatic Diseases, 12(2), 90–99. [DOI] [PubMed] [Google Scholar]
- 118.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature, 479(7372), 232–236. 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature, 530(7589), 184–189. 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Patil P, Dong Q, Wang D, Chang J, Wiley C, Demaria M, et al. (2019). Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell, 18(3), e12927 10.1111/acel.12927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kang MJ, Chung YH, Hwang CI, Murata M, Fujimoto T, Mook-Jung IH, et al. (2006). Caveolin-1 upregulation in senescent neurons alters amyloid precursor protein processing. Experimental & Molecular Medicine, 38(2), 126–133. [DOI] [PubMed] [Google Scholar]
- 122.Oh YS, Khil LY, Cho KA, Ryu SJ, Ha MK, Cheon GJ, et al. (2008). A potential role for skeletal muscle caveolin-1 as an insulin sensitivity modulator in ageing-dependent non-obese type 2 diabetes: studies in a new mouse model. Diabetologia, 51(6), 1025–1034. [DOI] [PubMed] [Google Scholar]
- 123.Kruglikov IL, Zhang Z, & Scherer PE (2019). Caveolin-1 in skin aging - from innocent bystander to major contributor. Ageing Research Reviews, 55, 100959 10.1016/j.arr.2019.100959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee JA, Choi DI, Choi JY, Kim SO, Cho KA, Lee JB, et al. (2015). Methyl-beta-cyclodextrin up-regulates collagen I expression in chronologically-aged skin via its anti-caveolin-1 activity. Oncotarget, 6(4), 1942–1953. 10.18632/oncotarget.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roitenberg N, Bejerano-Sagie M, Boocholez H, Moll L, Marques FC, Golodetzki L, et al. (2018). Modulation of caveolae by insulin/IGF-1 signaling regulates aging of Caenorhabditis eglegans. EMBO Reports, 19(8). 10.15252/embr.201745673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Herbert Z, Botticher G, Aschoff A, Sendemir E, Zermann DH, Arnold R, et al. (2007). Changing caveolin-1 and oxytocin receptor distribution in the ageing human prostate. Anatomia, Histologia, Embryologia, 36(5), 361–365. [DOI] [PubMed] [Google Scholar]
- 127.Gaudreault SB, Dea D, & Poirier J (2004). Increased caveolin-1 expression in Alzheimer’s disease brain. Neurobiology of Aging, 25(6), 753–759. [DOI] [PubMed] [Google Scholar]
- 128.Volonte D, Zhang K, Lisanti MP, & Galbiati F (2002). Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Molecular Biology of the Cell, 13(7), 2502–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dasari A, Bartholomew JN, Volonte D, & Galbiati F (2006). Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Research, 66(22), 10805–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bartholomew JN, Volonte D, & Galbiati F (2009). Caveolin-1 regulates the antagonistic pleiotropic properties of cellular senescence through a novel Mdm2/p53-mediated pathway. Cancer Research, 69(7), 2878–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galbiati F, Volonte D, Liu J, Capozza F, Frank PG, Zhu L, et al. (2001). Caveolin-1 expression negatively regulates cell cycle progression by inducing G(0)/G(1) arrest via a p53/p21(WAF1/Cip1)-dependent mechanism. Molecular Biology of the Cell, 12(8), 2229–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ding L, Zeng Q, Wu J, Li D, Wang H, Lu W, et al. (2017). Caveolin1 regulates oxidative stressinduced senescence in nucleus pulposus cells primarily via the p53/p21 signaling pathway in vitro. Molecular Medicine Reports, 16(6), 9521–9527. 10.3892/mmr.2017.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang J, Bai Y, Zhao X, Ru J, Kang N, Tian T, et al. (2018). oxLDL-mediated cellular senescence is associated with increased NADPH oxidase p47phox recruitment to caveolae. Bioscience Reports, 38(3). 10.1042/BSR20180283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Farhat N, Thorin-Trescases N, Voghel G, Villeneuve L, Mamarbachi M, Perrault LP, et al. (2008). Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Canadian Journal of Physiology and Pharmacology, 86(11), 761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, et al. (2007). Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mechanisms of Ageing and Development, 128(11–12), 662–671. [DOI] [PubMed] [Google Scholar]
- 136.Powter EE, Coleman PR, Tran MH, Lay AJ, Bertolino P, Parton RG, et al. (2015). Caveolae control the anti-inflammatory phenotype of senescent endothelial cells. Aging Cell, 14(1), 102–111. 10.1111/acel.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kim SR, Park JH, Lee ME, Park JS, Park SC, & Han JA (2008). Selective COX-2 inhibitors modulate cellular senescence in human dermal fibroblasts in a catalytic activity-independent manner. Mechanisms of Ageing and Development, 129(12), 706–713. [DOI] [PubMed] [Google Scholar]
- 138.Wu CC, Lin JC, Yang SC, Lin CW, Chen JJ, Shih JY, et al. (2008). Modulation of the expression of the invasion-suppressor CRMP-1 by cyclooxygenase-2 inhibition via reciprocal regulation of Sp1 and C/EBPalpha. Molecular Cancer Therapeutics, 7(6), 1365–1375. [DOI] [PubMed] [Google Scholar]
- 139.Jager M, Hubert A, Gogiraju R, Bochenek ML, Munzel T, & Schafer K (2019). Inducible knockdown of endothelial protein tyrosine phosphatase-1B promotes neointima formation in obese mice by enhancing endothelial senescence. Antioxidants & Redox Signaling, 30(7), 927–944. 10.1089/ars.2017.7169. [DOI] [PubMed] [Google Scholar]
- 140.Blasiak J (2017). DNA-damaging anticancer drugs - a perspective for DNA repair-oriented therapy. Current Medicinal Chemistry, 24(15), 1488–1503. 10.2174/0929867324666170124145557. [DOI] [PubMed] [Google Scholar]
- 141.Linge A, Weinhold K, Blasche R, Kasper M, & Barth K (2007). Downregulation of caveolin-1 affects bleomycin-induced growth arrest and cellular senescence in A549 cells. The International Journal of Biochemistry & Cell Biology, 39(10), 1964–1974. [DOI] [PubMed] [Google Scholar]
- 142.Shivshankar P, Brampton C, Miyasato S, Kasper M, Thannickal VJ, & Le Saux CJ (2012). Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. American Journal of Respiratory Cell and Molecular Biology, 47(1), 28–36. 10.1165/rcmb.2011-0349OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Feng X, Gao W, & Li Y (2017). Caveolin-1 is involved in high glucose accelerated human glomerular mesangial cell senescence. The Korean Journal of Internal Medicine, 32(5), 883–889. 10.3904/kjim.2015.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bitar MS, Abdel-Halim SM, & Al-Mulla F (2013). Caveolin-1/PTRF upregulation constitutes a mechanism for mediating p53-induced cellular senescence: implications for evidence-based therapy of delayed wound healing in diabetes. American Journal of Physiology. Endocrinology and Metabolism, 305(8), E951–E963. 10.1152/ajpendo.00189.2013. [DOI] [PubMed] [Google Scholar]
- 145.Volonte D, & Galbiati F (2011). Polymerase I and transcript release factor (PTRF)/cavin-1 is a novel regulator of stress-induced premature senescence. The Journal of Biological Chemistry, 286(33), 28657–28661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Volonte D, Kahkonen B, Shapiro S, Di Y, & Galbiati F (2009). Caveolin-1 expression is required for the development of pulmonary emphysema through activation of the ATM-p53-p21 pathway. The Journal of Biological Chemistry, 284(9), 5462–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dai SM, Shan ZZ, Nakamura H, Masuko-Hongo K, Kato T, Nishioka K, et al. (2006). Catabolic stress induces features of chondrocyte senescence through overexpression of caveolin 1: possible involvement of caveolin 1-induced down-regulation of articular chondrocytes in the pathogenesis of osteoarthritis. Arthritis and Rheumatism, 54(3), 818–831. [DOI] [PubMed] [Google Scholar]
- 148.Goodarzi AA, Jonnalagadda JC, Douglas P, Young D, Ye R, Moorhead GB, et al. (2004). Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. The EMBO Journal, 23(22), 4451–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Volonte D, Zou H, Bartholomew JN, Liu Z, Morel PA, & Galbiati F (2015). Oxidative stress-induced inhibition of Sirt1 by caveolin-1 promotes p53-dependent premature senescence and stimulates the secretion of interleukin 6 (IL-6). The Journal of Biological Chemistry, 290(7), 4202–4214. 10.1074/jbc.M114.598268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Joshi B, Pawling J, Shankar J, Pacholczyk K, Kim Y, Tran W, et al. (2019). Caveolin-1 Y14 phosphorylation suppresses tumor growth while promoting invasion. Oncotarget, 10(62), 6668–6677. 10.18632/oncotarget.27313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen JR, Lazarenko OP, Blackburn ML, Badger TM, & Ronis MJ (2015). Soy protein isolate inhibits high-fat diet-induced senescence pathways in osteoblasts to maintain bone acquisition in male rats. Endocrinology, 156(2), 475–487. 10.1210/en.2014-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Volonte D, Liu Z, Musille PM, Stoppani E, Wakabayashi N, Di YP, et al. (2013). Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Molecular Biology of the Cell, 24(12), 1852–1862. 10.1091/mbc.E12-09-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li W, Liu H, Zhou JS, Cao JF, Zhou XB, Choi AM, et al. (2012). Caveolin-1 inhibits expression of antioxidant enzymes through direct interaction with nuclear erythroid 2 p45-related factor-2 (Nrf2). The Journal of Biological Chemistry, 287(25), 20922–20930. 10.1074/jbc.M112.352336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Petriello MC, Han SG, Newsome BJ, & Hennig B (2014). PCB 126 toxicity is modulated by cross-talk between caveolae and Nrf2 signaling. Toxicology and Applied Pharmacology, 277(2), 192–199. 10.1016/j.taap.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hart PC, Ratti BA, Mao M, Ansenberger-Fricano K, Shajahan-Haq AN, Tyner AL, et al. (2016). Caveolin-1 regulates cancer cell metabolism via scavenging Nrf2 and suppressing MnSOD-driven glycolysis. Oncotarget, 7(1), 308–322. 10.18632/oncotarget.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Behne D, & Kyriakopoulos A (2001). Mammalian selenium-containing proteins. Annual Review of Nutrition, 21, 453–473. [DOI] [PubMed] [Google Scholar]
- 157.Mustacich D, & Powis G (2000). Thioredoxin reductase. The Biochemical Journal, 346(Pt 1), 1–8. [PMC free article] [PubMed] [Google Scholar]
- 158.Volonte D, & Galbiati F (2009). Inhibition of thioredoxin reductase 1 by caveolin 1 promotes stress-induced premature senescence. EMBO Reports, 10(12), 1334–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, et al. (2014). MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature, 508(7495), 215–221. 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- 160.Huber KV, Salah E, Radic B, Gridling M, Elkins JM, Stukalov A, et al. (2014). Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature, 508(7495), 222–227. 10.1038/nature13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Patel A, Burton DG, Halvorsen K, Balkan W, Reiner T, Perez-Stable C, et al. (2015). MutT Homolog 1 (MTH1) maintains multiple KRAS-driven pro-malignant pathways. Oncogene, 34(20), 2586–2596. 10.1038/onc.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rai P, Onder TT, Young JJ, McFaline JL, Pang B, Dedon PC, et al. (2009). Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proceedings of the National Academy of Sciences of the United States of America, 106(1), 169–174. 10.1073/pnas.0809834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rai P, Young JJ, Burton DG, Giribaldi MG, Onder TT, & Weinberg RA (2011). Enhanced elimination of oxidized guanine nucleotides inhibits oncogenic RAS-induced DNA damage and premature senescence. Oncogene, 30(12), 1489–1496. 10.1038/onc.2010.520. [DOI] [PubMed] [Google Scholar]
- 164.Rai P (2012). Human Mut T Homolog 1 (MTH1): a roadblock for the tumor-suppressive effects of oncogenic RAS-induced ROS. Small GTPases, 3(2), 120–125. 10.4161/sgtp.19556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Volonte D, Vyas AR, Chen C, Dacic S, Stabile LP, Kurland BF, et al. (2018). Caveolin-1 promotes the tumor suppressor properties of oncogene-induced cellular senescence. The Journal of Biological Chemistry, 293(5), 1794–1809. 10.1074/jbc.M117.815902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.MacNee W (2005). Pulmonary and systemic oxidant/antioxidant imbalance in chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society, 2(1), 50–60. [DOI] [PubMed] [Google Scholar]
- 167.Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, & Hunninghake GW (2006). Cigarette smoke induces cellular senescence. American Journal of Respiratory Cell and Molecular Biology, 35(6), 681–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Holz O, Zuhlke I, Jaksztat E, Muller KC, Welker L, Nakashima M, et al. (2004). Lung fibroblasts from patients with emphysema show a reduced proliferation rate in culture. The European Respiratory Journal, 24(4), 575–579. [DOI] [PubMed] [Google Scholar]
- 169.Muller KC, Welker L, Paasch K, Feindt B, Erpenbeck VJ, Hohlfeld JM, et al. (2006). Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respiratory Research, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Meyn RE, Munshi A, Haymach JV, Milas L, & Ang KK (2009). Receptor signaling as a regulatory mechanism of DNA repair. Radiotherapy and Oncology, 92(3), 316–322. 10.1016/j.radonc.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Luoma K, Riihimaki H, Luukkonen R, Raininko R, Viikari-Juntura E, & Lamminen A (2000). Low back pain in relation to lumbar disc degeneration. Spine (Phila Pa 1976), 25(4), 487–492. [DOI] [PubMed] [Google Scholar]
- 172.Gruber HE, Ingram JA, Norton HJ, & Hanley EN Jr. (2007). Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine (Phila Pa 1976), 32(3), 321–327. [DOI] [PubMed] [Google Scholar]
- 173.Le Maitre CL, Freemont AJ, & Hoyland JA (2007). Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Research & Therapy, 9(3), R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Roberts S, Evans EH, Kletsas D, Jaffray DC, & Eisenstein SM (2006). Senescence in human intervertebral discs. European Spine Journal, 15(Suppl 3), S312–S316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Heathfield SK, Le Maitre CL, & Hoyland JA (2008). Caveolin-1 expression and stress-induced premature senescence in human intervertebral disc degeneration. Arthritis Research & Therapy, 10(4), R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yu DM, Jung SH, An HT, Lee S, Hong J, Park JS, et al. (2017). Caveolin-1 deficiency induces premature senescence with mitochondrial dysfunction. Aging Cell, 16(4), 773–784. 10.1111/acel.12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Volonte D, Liu Z, Shiva S, & Galbiati F (2016). Caveolin-1 controls mitochondrial function through regulation of m-AAA mitochondrial protease. Aging (Albany NY), 8(10), 2355–2369. 10.18632/aging.101051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jeffries EP, Di Filippo M, & Galbiati F (2018). Failure to reabsorb the primary cilium induces cellular senescence. The FASEB Journal, fj201801382R. 10.1096/fj.201801382R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. (2012). Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle, 11(12), 2285–2302. 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Minkeu A, et al. (2008). Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: implications for the response to hormonal therapy. Cancer Biology & Therapy, 7(8), 1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mallette FA, Gaumont-Leclerc MF, & Ferbeyre G (2007). The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes & Development, 21(1), 43–48. 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Maehara K, Yamakoshi K, Ohtani N, Kubo Y, Takahashi A, Arase S, et al. (2005). Reduction of total E2F/DP activity induces senescence-like cell cycle arrest in cancer cells lacking functional pRB and p53. The Journal of Cell Biology, 168(4), 553–560. 10.1083/jcb.200411093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Park C, Lee I, & Kang WK (2006). E2F-1 is a critical modulator of cellular senescence in human cancer. International Journal of Molecular Medicine, 17(5), 715–720. [PubMed] [Google Scholar]
- 184.Yang C-P, Galbiati F, Volonte’ D, Horwitz SB, & Lisanti MP (1998). Upregulation of caveolin-1 and caveolae organelles in Taxol-resistant A549 cells. FEBS Letters, 439, 368–372. [DOI] [PubMed] [Google Scholar]
- 185.Berggren M, Gallegos A, Gasdaska JR, Gasdaska PY, Warneke J, & Powis G (1996). Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Research, 16(6B), 3459–3466. [PubMed] [Google Scholar]
- 186.Kim TH, Hur EG, Kang SJ, Kim JA, Thapa D, Lee YM, et al. (2011). NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1alpha. Cancer Research, 71(6), 2260–2275. [DOI] [PubMed] [Google Scholar]
- 187.Krtolica A, & Campisi J (2002). Cancer and aging: a model for the cancer promoting effects of the aging stroma. The International Journal of Biochemistry & Cell Biology, 34(11), 1401–1414. [DOI] [PubMed] [Google Scholar]
- 188.Kortum RL, Fernandez MR, Costanzo-Garvey DL, Johnson HJ, Fisher KW, Volle DJ, et al. (2014). Caveolin-1 is required for kinase suppressor of Ras 1 (KSR1)-mediated extracellular signal-regulated kinase 1/2 activation, H-RasV12-induced senescence, and transformation. Molecular and Cellular Biology, 34(18), 3461–3472. 10.1128/MCB.01633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. (2001). Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature, 410(6832), 1111–1116. 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 190.Hayashi K, Matsuda S, Machida K, Yamamoto T, Fukuda Y, Nimura Y, et al. (2001). Invasion activating caveolin-1 mutation in human scirrhous breast cancers. Cancer Research, 61(6), 2361–2364. [PubMed] [Google Scholar]
- 191.Bender FC, Reymond MA, Bron C, & Quest AF (2000). Caveolin-1 levels are down-regulated in human colon tumors, and ectopic expression of caveolin-1 in colon carcinoma cell lines reduces cell tumorigenicity. Cancer Research, 60(20), 5870–5878. [PubMed] [Google Scholar]
- 192.Torrejon B, Cristobal I, Rojo F, & Garcia-Foncillas J (2017). Caveolin-1 is markedly downregulated in patients with early-stage colorectal cancer. World Journal of Surgery, 41(10), 2625–2630. 10.1007/s00268-017-4065-9. [DOI] [PubMed] [Google Scholar]
- 193.Wiechen K, Diatchenko L, Agoulnik A, Scharff KM, Schober H, Arlt K, et al. (2001). Caveolin-1 is down-regulated in human ovarian carcinoma and acts as a candidate tumor suppressor gene. The American Journal of Pathology, 159(5), 1635–1643. 10.1016/S0002-9440(10)63010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Quann K, Gonzales DM, Mercier I, Wang C, Sotgia F, Pestell RG, et al. (2013). Caveolin-1 is a negative regulator of tumor growth in glioblastoma and modulates chemosensitivity to temozolomide. Cell Cycle, 12(10), 1510–1520. 10.4161/cc.24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wiechen K, Sers C, Agoulnik A, Arlt K, Dietel M, Schlag PM, et al. (2001). Down-regulation of caveolin-1, a candidate tumor suppressor gene, in sarcomas. The American Journal of Pathology, 158(3), 833–839. 10.1016/S0002-9440(10)64031-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Williams TM, Cheung MW, Park DS, Razani B, Cohen AW, Muller WJ, et al. (2003). Loss of caveolin-1 gene expression accelerates the development of dysplastic mammary lesions in tumor-prone transgenic mice. Molecular Biology of the Cell, 14(3), 1027–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yamao T, Yamashita YI, Yamamura K, Nakao Y, Tsukamoto M, Nakagawa S, et al. (2019). Cellular senescence, represented by expression of caveolin-1, in cancer-associated fibroblasts promotes tumor invasion in pancreatic cancer. Annals of Surgical Oncology, 26(5), 1552–1559. 10.1245/s10434-019-07266-2. [DOI] [PubMed] [Google Scholar]
- 198.Nunez-Wehinger S, Ortiz RJ, Diaz N, Diaz J, Lobos-Gonzalez L, & Quest AF (2014). Caveolin-1 in cell migration and metastasis. Current Molecular Medicine, 14(2), 255–274. 10.2174/1566524014666140128112827. [DOI] [PubMed] [Google Scholar]
- 199.Quest AF, Gutierrez-Pajares JL, & Torres VA (2008). Caveolin-1: an ambiguous partner in cell signalling and cancer. Journal of Cellular and Molecular Medicine, 12(4), 1130–1150. 10.1111/j.1582-4934.2008.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]