

Abstract
Mast cells have been shown to release extracellular vesicles (EVs) in vitro. However, EV‐mediated mast cell communication in vivo remains unexplored. Primary mast cells from GFP‐transgenic and wild type mice, were grown in the presence or absence of lipopolysaccharide (LPS), and the secreted EVs were separated from the conditioned media. Mast cell‐derived EVs were next cultured with LPS‐naïve mast cells, and the induction of TNF‐α expression was monitored. In addition, primary mast cells were seeded in diffusion chambers that were implanted into the peritoneal cavities of mice. Diffusion chambers enabled the release of GFP+ mast cell‐derived EVs in vivo into the peritoneal cavity. Peritoneal lavage cells were assessed for the uptake of GFP+ EVs and for TNF‐α production. In vitro, LPS‐stimulated mast cell‐derived EVs were efficiently taken up by non‐stimulated mast cells, and induced TNF‐α expression in a TLR4, JNK and P38 MAPK dependent manner. In vivo, using implanted diffusion chambers, we confirmed the release and transmission of mast cell‐derived EVs to other mast cells with subsequent induction of TNF‐α expression. These data show an EV‐mediated spreading of pro‐inflammatory response between mast cells, and provide the first in vivo evidence for the biological role of mast cell‐derived EVs.
Keywords: mast cell, extracellular vesicles, LPS, TNF‐α, in vitro, in vivo
1. INTRODUCTION
Mast cells (MCs) are important players of the immune system. Although they are best known for their role in hypersensitivity reactions, they are also important participants of the immune response against pathogens. Similarly to dendritic cells (DCs) and macrophages, MCs express toll like receptors (TLRs) including TLR2, TLR3, TLR4, TLR6, TLR7 and TLR9 that recognize invading pathogens (Marshall, King, & McCurdy, 2003). TLR4 dependent stimulation of MCs with lipopolysaccharide (LPS) induces TNF‐α, IL‐1b, IL‐6 and IL‐13 but not IL‐4 or IL‐5 secretion (Supajatura et al., 2002). We and others have shown that MCs play a crucial role in the clearance of bacterial infections such as Bordetella pertussis or Escherichia coli (Mielcarek et al., 2001; Wierzbicki & Brzezinska‐Blaszczyk, 2009) by secretion of TNF‐α (Vukman et al., 2012; Vukman, Ravida, Aldridge, & O'Neill, 2013).
Nowadays, it is obvious that beside cytokines, MCs also secrete extracellular vesicles (EVs), conveyors of messages in cell‐to‐cell communication (Ekstrom et al., 2012; Stassen, Hartmann, Delgado, Dehmel, & Braun, 2019; Vukman, Forsonits, Oszvald, Toth, & Buzas, 2017). MC‐derived EVs have been shown to induce both Th1 and Th2 immune responses (Skokos et al., 2001) and influence the function of other immune cells such as DCs (Skokos et al., 2003) or B‐cells (Mion et al., 2014; Skokos et al., 2001). During infection, MCs produce EVs that contain increased levels of ICAM1, TGF‐β and TNF‐precursors, thus, might influence the function other immune cells during inflammation (Al‐Nedawi, Szemraj, & Cierniewski, 2005; Hugle, Hogan, White, & van Laar, 2011; Nakae et al., 2006). There is an increasing number of evidence that EVs are also involved in the MC‐MC communication. MC‐derived EVs contain functional mRNA, shuttle it to other MCs cells and alter their function (Eldh et al., 2010; Valadi et al., 2007).
Although MCs have been suggested to serve as conductor cells in the immune response, they are present in tissues in a surprisingly low number. Here we addressed the question whether EVs could spread MC‐derived messages upon sensing the bacterial ligand LPS thus compensating for the low frequency of MCs. The data presented in this report provide evidence for both in vitro and in vivo spread of a proinflammatory response by MC‐derived EVs.
2. MATERIALS AND METHODS
2.1. Mice
C57BL/6 and GFP‐expressing C57BL/6‐Tg(UBC‐GFP)30Scha/J mice (designated in this manuscript as “GFP mice”) were both purchased from the Jackson Laboratory, and were bred in the specific pathogen‐free animal facility at the Department of Genetics, Cell‐ and Immunobiology, Semmelweis University. Ethics approval for mouse experiments was obtained from local ethical committee (PE/EA/561‐7/2019, PE/EA/562‐7/2019).
2.2. Differentiation and expansion of bone marrow‐derived MCs (BMMCs) and peritoneal cell‐derived cultured MCs (PCMCs)
BMMCs were generated from femoral and tibial bone marrow cells of C57BL/6 and GFP mice. Cells were cultured in complete IMDM (Gibco) in the presence of 10% heat‐inactivated foetal bovine serum (FBS, Gibco, Life Technologies), 100 u/ml penicillin/streptomycin (Sigma‐Aldrich). As a source of murine IL‐3, cells were grown in 30% WEHI‐3 conditioned IMDM medium (TIB‐68; American Type Culture Collection, Manassas, VA, USA) for 4 weeks (Vukman, Adams, Metz, Maurer, & O'Neill, 2013). PCMCs were obtained as described previously (Vukman et al., 2013). Briefly, C57BL/6 mice were injected intraperitoneally with 10 ml sterile PBS. The peritoneal lavage cells were cultured in RPMI‐1640 medium, supplemented with 10% FCS and 100 u/ml penicillin/streptomycin, l‐glutamine (2 mM; Sigma‐Aldrich), 10 ng/ml mouse rIL‐3 (Calbiochem, Merck), and 30 ng/ml recombinant mouse stem cell factor (Sigma‐Aldrich) at 37°C. Forty‐eight hours later, non‐adherent cells were discarded and fresh culture medium was added for a further 7 days. In both MC preparations, > 95% of the total cells were identified as MCs on the basis of cell‐surface expressions of c‐Kit (Clone 2B8; eBioscience) and FcɛRI (Clone MAR1; eBioscience) and Kimura staining (0.05% toluidine blue solution + saturated saponin solution + phosphate buffer pH6.4) (Kimura, Moritani, & Tanizaki, 1973). Cell number and viability were monitored using trypan blue staining (Sigma‐Aldrich) and by flow cytometry using propidium iodide, annexin V‐APC, 7AAD or TO‐Pro3 as described by the manufacturer (ThermoFischer).
β‐Hexosaminidase release from MCs was measured in a 96 well plate colorimetric assay as described previously (Vukman et al., 2012). Briefly, BMMCs or PCMCs were washed twice with PBS, resuspended in Tyrode's buffer and seeded in a 96‐well round‐bottom plate at a density of 4 × 105 cells/200 μl followed by stimulation with stimulants, LPS (100 ng/ml ), A23187 (0.5 μM) or PBS for 1 h at 37°C. Cell pellets were solubilized in Tyrode's buffer supplemented with 1% Triton X‐100, and the cell lysates and supernatants were incubated separately with p‐nitrophenyl‐N‐acetil‐b‐d‐glucosamide (p‐NAG) substrate for 1 h at 37°C. The production of b‐hexosaminidase‐mediated p‐nitrophenol was terminated by adding 150 μl 0.2 M glycine (pH10.7) and quantified by absorbance at 405 nm with an ELISA plate reader. The percentage of β‐hexosaminidase release indicating the extent of degranulation was calculated as the ratio of p‐nitrophenol absorbance in the supernatants and the sum of absorbance in the supernatant and cell lysates.
2.3. EV separation from conditioned cell culture medium
BMMCs (20 × 106 cells) or PCMCs (2 × 106 cells) were incubated for 24 h in EV‐depleted complete, IL‐3 containing MC culture medium as described above in the presence or absence of 100 ng/ml LPS (Sigma). For co‐culture and in vivo studies, after 1 h incubation with LPS, cells were washed twice and were placed in fresh medium to avoid the carry‐over of LPS in the released EV populations. As positive control of induction of EV release, 0.5 μM A23187 (Sigma) was used, as upon degranulation, MCs secrete huge amount of EVs as described previously (Amanda Carroll‐Portillo, Cambi, Lidke, & Wilson, 2012). EVs were separated with minor modifications of the method described previously by us (Osteikoetxea et al., 2015). Briefly, cells were removed by centrifugation at 300 × g for 10 min at room temperature (RT), and then the supernatant was filtered by gravity through a 5 μm filter (Millipore, Billerica, MA). The filtrate was next submitted to a 2,000 × g centrifugation for 20 min at 4°C to pellet lEVs (lEVs) (Avanti J‐XP26 centrifuge, JA 25.15 rotor, Beckman Coulter Inc.). Afterwards, the supernatant was filtered by gravity through a 0.8 μm filter (Millipore) to remove any remaining lEVs, and centrifuged at 12,500 × g for 40 min at 4°C to pellet medium sized EVs (mEVs) (Avanti J‐XP26 centrifuge, JA 25.15 rotor, Beckman Coulter Inc.). Finally, the supernatant was filtered by gravity through a 0.22 μm filter (Millipore) and ultracentrifuged in an Optima MAX‐XP bench top ultracentrifuge with MLA‐55 rotor (Beckman Coulter Inc.) at 100,000 × g for 70 min at 4°C to pellet small EVs (sEVs). Each EV pellet was re‐suspended once in phosphate‐buffer‐saline (PBS), and re‐centrifuged under the same conditions as what has been used originally for pelleting. Three different, size‐based subpopulations were isolated including lEVs, mEVs and sEVs by the above combination of gravity driven filtration and differential centrifugation.
2.4. Mast cell – mast cell co‐culture
BMMCs from wild type C57BL/6 mice and GFP‐BMMCs or PKH67 (SIGMA)‐stained BMMCs were either cultured alone or were co‐cultured at 1:1 ratio in EV‐free complete medium (see above) in the presence or absence of LPS (100 ng/ml). A23187 (0.5 μM) was used as positive control. After 24 h, fluorescence intensity (GFP) was measured by flow cytometry. Mean fluorescence intensity (MFI) of unstained wild type cells were calculated using FlowJo after exclusion of GFP‐BMMCs by gating. Results were normalized to MFI values of unstained wild type cells cultured alone. Results were confirmed by confocal microscopy (Leica SP8).
2.5. Exposure of MCs to EVs secreted by unstimulated or LPS‐stimulated MCs
GFP‐BMMCs and PKH67‐stained BMMCs were cultured in complete medium supplemented with EV‐depleted serum in the presence or absence of LPS (100 ng/ml) in the first hour. After washing, cells were grown for 24 h in EV‐depleted complete medium. A23187 (0.5 μM) and PBS were used as positive and negative controls, respectively. EVs were separated from the conditioned medium as described above. Unstained non‐stimulated BMMCs derived from C57BL/6 mice and were cultured in (i) EV‐free medium (control), (ii) conditioned medium (SN), (iii) EV‐depleted conditioned medium (EV‐dep SN), (iv) EV‐reconstituted medium (EV‐rec SN) or (v) non‐conditioned medium supplemented by separated EVs (EV). We kept the EV donor MC numbers the same as the acceptor ones. Fluorescence intensity was measured by flow cytometry. EV uptake was monitored by fluorescent and confocal microscopy (Nikon Eclipse 80i). TNF‐α was measured by commercially available ELISA kit (R&D Systems).
In inhibition experiments, cells were treated with TAK242 (0.2 μM), dynasore (80 μM) or cytochalasin D (10 μg/ml) prior to adding separated EVs to cells. To exclude a potential LPS carry‐over, EVs from unstimulated BMMCs were incubated in 100 ng/ml LPS for 2 h, and we used GW4869 (10 μM), a neutral sphingomyelinase inhibitor to block small EV generation, 30 min before LPS stimulation of donor cells. Then EVs were submitted to washing by PBS before addition to naïve (non‐stimulated) BMMCs. The inhibitory effect of GW4869 was monitored by NTA (as manufacturer protocol) on separated sEVs.
2.6. Cell‐based ELISA measurements
To investigate if exposure to EVs induced the activation of MAPK‐signalling, BMMCs were cultured for 24 h in EV‐free complete medium in the presence of isolated EVs derived from LPS‐stimulated and unstimulated BMMCs (donor cell numbers were the same as naïve MC numbers). Cells were washed twice, lysed and investigated for phosphorylation of ERK1/2, JNK and P38 by RayBio Cell‐based phosphorylation ELISA kits following the protocol of the manufacturers (RayBiotech PEL‐Erk‐T202‐T‐01, PEL‐JNK‐T183‐T‐1 and PEL‐P38‐T180‐T‐1).
2.7. Determination of size distribution and concentration of EVs
Size distribution and concentration of MC‐derived EVs in the conditioned medium or in the case of separated EVs were determined by tuneable resistive pulse sensing (TRPS) using a qNano instrument (IZON Science) as we described previously (Osteikoetxea et al., 2015). Briefly, EV samples from 10 ml supernatant of 20 × 106 cells were diluted in 0.2 μm‐filtered 0.01% Triton X‐100 (Sigma) containing PBS (in 1:50 and 1:100). At least 500 particles were counted using 3–5 mbar pressure using NP200, NP400, NP800 and NP2000 nanopore membranes. Calibration was performed with known concentration of beads CPC100, CPC200, CPC300, CPC800, CPC1000 and CPC2000 (diluted as suggested by IZON user manual) in 0.2 μm‐filtered PBS. Results were evaluated using the IZON Control Suite 3.2 software.
In some experiments, nanoparticle tracking analysis (NTA) was also used. Briefly, measurements were performed on a ZetaView PMX120 instrument (Particle Metrix, Germany). Separated sEV samples were diluted in PBS to achieve the optimal concentration (50–200 particles/frame). Each sample was measured in 11 positions (2 cycles per position) and the acquired video has been used for particle size and concentration analysis. The following camera parameters were used: shutter speed‐100, sensitivity‐80, frame rate‐30. The recordings were performed at 25C.
2.8. Detection of EVs by transmission electron microscopy (TEM)
MC‐derived EVs were examined by TEM using a protocol we described previously (Visnovitz et al., 2019). For phosphotungstic acid contrasting, samples were fixed with 4% glutaraldehyde onto the surface of 300 mesh formvar‐coated grids and contrasted with 2% phosphotungstic acid. For immune electron microscopy, samples were fixed with 4% paraformaldehyde onto formvar‐coated grids and labelled with polyclonal rabbit anti‐CD63 IgG (H‐193, sc‐15363, Santa Cruz Biotechnology) and c‐Kit (Clone 2B8; eBioscience). Polyclonal goat anti‐rabbit IgG/10 nm gold pre‐adsorbed (Abcam) or polyclonal goat anti‐mouse IgG/5 nm gold pre‐adsorbed (Sigma) were used as secondary antibodies. For background contrasting, 2% phosphotungstic acid was used after permanent fixation by 2% glutaraldehyde. Samples were examined by a JEOL 1011 transmission electron microscope.
2.9. Determination of protein content of EV preparations
Protein concentration of EV preparations was determined with the Micro BCA Protein Assay kit (ThermoFisher) according to the instructions of the manufacturer. Briefly, EVs were diluted 5–10 times and were lysed with 0.5% Triton X‐100 (Sigma) and 0.5% SDS (Sigma). Colour was developed for 1 h at 60°C and absorbance was measured at 562 nm by NanoDrop NP‐1000 (ThermoFisher).
2.10. Determination of the lipid content of EVs by the SPV assay
Lipid content of MC‐derived EVs was measured using the SPV assay protocol that we described previously (Visnovitz et al., 2019). In brief, 200 μl of 96% sulphuric acid (Molar Chemicals) was added to 40 μl EVs suspended in PBS. EVs were incubated at 90°C (AccuBlock digital dry batch, Labnet) for 20 min. After cooling down to room temperature, 120 μl of phospho‐vanillin reagent (1 mg/ml vanillin (Sigma) in 17% phosphoric acid (Sigma)) was added. Colour reaction was measured on 96 well plate (ThermoFisher) at 540 nm (plate reader, Multiskan MS, Labsystems).
2.11. Characterization of EVs by flow cytometry
EVs were incubated with the affinity reagents annexin V (APC, PE, FITC, BD Bioscience), lactadherin (Haematologic Technologies, labelled with Cy5 Fast Conjugation Kit, Abcam), PKH67 (FITC, Sigma‐Aldrich) and antibodies including anti‐CD9 Ab (PE, FITC, ThermoFisher), anti‐CD63 (APC, PE, eBioscience) anti‐CD81 (FITC, ThermoFisher), FcɛRI (PE, FITC, eBioscience), CD117 (APC, PE, eBioscience) or appropriate IgG isotype controls (mouse IgG1, IgG2, IgG2b isotype control (BD Bioscience, Biolegend and SONY). LEVs were also stained by propidium iodide, annexin V, lactadherin and caspase‐3/7 (cellEvent Caspase‐3/7 Green Detection Reagent; Invitrogen) to check their apoptotic features. Antibody dilution was selected based on titration for each batch of reagents or as advised by the suppliers. Samples were incubated with antibodies for 1 h at 4°C in the dark in 50 μl 2% EV‐depleted foetal bovine serum (Gibco) in PBS followed by washing steps and dilution to 300 μl prior to measurement. Autofluorescence, fluorescence of the affinity reagents and the fluorochrome‐labelled antibodies in the absence of EVs were also determined, and in the case of large and medium EVs (mEVs), 0.1% Triton X‐100 lysis was used as a control to disrupt EV signals as we described earlier (Gyorgy et al., 2011; Osteikoetxea et al., 2015). Isolated sEVs were coupled to 4 μm aldehyde/sulphate latex beads (Life Technologies) and blocked with 100 mM glycine for 2 h followed by incubation with 1% bovine serum albumin in PBS overnight. In multicolour experiments, unstained, single‐stained, and fluorescence‐minus‐one controls were also used. In the case of cells, mAb binding was analysed by flow cytometry (FACSCalibur; BD Biosciences and a CytoFLEX S V4‐B2‐Y4‐R3 Flow Cytometer; Beckman Coulter) in the presence of DNase to avoid aggregation. FlowJo (Treestar) was used to analyse flow cytometry data. An example for gating prior to analysis of results is demonstrated in Figure S1.
2.12. Characterization of EVs by Western blot
Total protein was extracted from small, medium and lEVs derived from 20 × 106 BMMCs (unstimulated, 100 ng/ml LPS and 0.5 μM A23187 stimulated), by sonication. Protease and phosphatase inhibitor cocktails were added (Sigma‐Aldrich). Protein samples (10–40 μg) and pre‐stained protein markers (26634, Spectra Multicolor Broad Range Protein Ladder) were separated by SDS‐PAGE and blotted onto 0.2 μm Immun‐Blot PVDF membranes (Bio‐Rad). Membranes were stained according to standard Western blot protocol using anti‐TSG101 (T5701, Sigma‐Aldrich), anti‐Alix (ab186429, Sigma‐Aldrich, Dublin, Ireland) and anti‐rabbit Abs (ab205718, Sigma‐Aldrich). Proteins were visualized with a chemiluminescent HRP substrate (32106, Thermo Scientific) and recorded by Gel documentation system (CHEMI Premium, VWR).
2.13. OptiPrep density gradient of purified EV populations and MC conditioned media
A discontinuous iodixanol gradient was used as described by Van Deun et al (2014) with some modifications. Solutions of 5, 10, 20 and 40% iodixanol were prepared and the gradient was formed by layering 1 ml of each solution on top of each other in a 5 ml open top tube. Half ml of conditioned medium was overlaid onto the top of the gradient in the case of PCMCs, while separated EV samples (BMMCs) were ultracentrifuged from bottom up. Gradients were then centrifuged for 18 h at 100,000 × g, at 4°C in an Optima MAX‐XP bench top ultracentrifuge with MLS‐50 rotor (Beckman Coulter Inc.). Gradient fractions of 0.5 ml were collected from the top of the gradient and washed once in PBS (sEVs: 100,000 × g ‐ 3h, mEVs: 12,500 × g ‐ 1.5h, lEVs: 2000 × g – 40 min). In the case of conditioned medium samples, EV populations were separated from all fractions as described above. To estimate the density of fractions, samples were measured by a precision analytical weight and density was calculated.
2.14. Mass spectrometry (MS) characterization of medium size EVs
EV pellets were re‐suspended in 20 μl water. Samples were subjected to repeated freeze‐thaw cycles and digestion as described previously (Turiak, Ozohanics, Marino, Drahos, & Vekey, 2011). The peptide digest was analysed using a Bruker Maxis II Q‐TOF instrument with CaptiveSpray nanoBooster ionization source. Peptides were separated online using a 25 cm Acclaim Pepmap RSLC nano HPLC column on the Dionex Ultimate 3000 Nano LC System. Data were evaluated with Scaffold (Proteome Software, Inc., Portland, OR 97219, USA).
2.15. In vivo studies
BMMCs from GFP mice were incubated for 1h in the presence or absence of LPS (100 ng/ml ) and washed 2 times with PBS to avoid LPS contamination. For diffusion chamber experiments, commercially available EMD Millipore's Diffusion Chamber Kits (PR00 014 01, Merck) were used. The diffusion chambers were assembled with two membrane filters (GSWP01300 or GVWP01300, both having 5 μm pore size) supplied in the kit using a 100% silicone glue (F.BS, Material Chemical Company, Budapest) that contained no antifungal‐ingredients and acetic acid in traces only. Chambers were then dried and sterilized with ultraviolet light. After sterilization, they were washed in sterile PBS with 5% Pen/Strep overnight. 1 million MCs were injected through the opening of the chamber, and the opening was plugged with a piece of the plastic rod supplied in the kit. Empty (mock‐operated) and MC‐containing chambers were then implanted into the peritoneal cavities of C57BL/6 mice for 24 h before removing them surgically (Figure S2). In additional experiments, EVs separated from the conditioned medium of LPS‐stimulated and unstimulated BMMCs were injected intraperitoneally twice in 24 h (at 0 h and at 22th h). For each injection, separated EVs from the conditioned medium of 106 BMMCs were used. To exclude the possibility of LPS contamination, in control experiments mice were also injected intraperitoneally by 100 ng LPS.
After 24 h, mice were sacrificed, and peritoneal cells were collected by peritoneal lavage. Peritoneal cells were counted and assessed by trypan blue staining. MC numbers were determined using Kimura staining by microscope and by flow cytometry using two pan‐MC markers CD117 (c‐Kit) and FcɛRI. Intracellular TNF‐α was also measured by flow cytometry after Brefeldin A treatment (3 μg/ml for 1h). Cells were also examined under a confocal microscope (Leica SP8). Cells retrieved from removed diffusion chambers were submitted to enumeration and viability test using trypan blue staining. Uptake of GFP‐EVs by other cell types were tested by multicolour flow cytometry similarly to what has been described for MCs. Used antibodies were: CD4 (Clone: RM4‐5, Biolegend), CD8 (Clone: 5H10, Invitrogen), CD45R (Clone: RA3‐6B2, Biolegend), CD11b (Clone: M1/70, Biolegend), CD20 (Clone: SA 275A11, Biolegend), FcɛRI (Clone: MAR1, eBioscience) and CD117 (Clone: 2B8, eBioscience). In multicolour experiments, unstained, single stained and Fluorescence Minus One (FMO) controls were also used.
2.16. Statistical analysis
All data were tested for normality prior to statistical analysis by origin 6.1 software (OriginLab Corporation, Northhampton, MA, USA) and Prism (GraphPad Software, Sandiego, CA, USA). For comparisons between two groups, Student's t‐test was used. For comparison of multiple groups, we used One‐way ANOVA. In all tests, P < 0.05 was considered significant.
3. RESULTS
3.1. Cellular uptake of MC‐derived EVs
To test if MCs were able to exchange cytoplasmic and membrane components between each other, we used two different primary cell culture systems: BMMCs and PCMCs derived from wild type or GFP‐expressing C57BL/6 mice (GFP‐MC). Viability, morphology, purity and integrity was checked for every experiment, and cells were only used if viability was above 99%, and purity was above 99% for BMMCs and 95% for PCMCs, while the rate of degranulation was 50 ± 10% (as accepted) (Figure 1).
FIGURE 1.
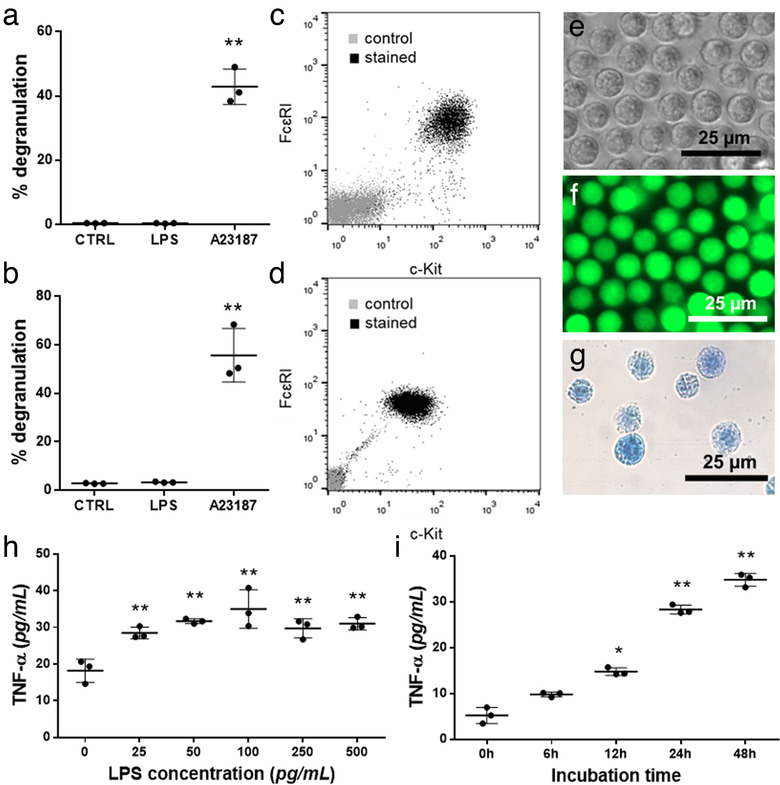
Identification of BMMCs and PCMCs. Mast cell nature was confirmed by showing the degranulation capacity of BMMCs (a) and PCMCs (b) with β‐hexosaminidase assay in the presence of LPS (100 ng/ml ) and A23187 (0.5 μM). Cell purity was assessed by flow cytometry on the 4th week of mast cell differentiation (BMMC, c) or on 10th day of culture (PCMC, d) by flow cytometry, based on FcɛRI and c‐Kit positivity. BMMCs from C57BL/6 mice were studied by phase contrast microscopy (e), while GFP‐BMMCs from C57BL‐GFP mice were analysed by fluorescent microscopy (f). Cellular purity was also checked by Kimura staining (g). The optimal concentration of LPS (h) and incubation time (i) were chosen based on TNF‐α secretion measured by ELISA. Graphs show the mean and SD of three biological replicates, where one point indicates the average of three technical replicates (n = 3, *P ≤ 0.05, **P ≤ 0.01 t test)
When GFP‐MCs and wild type MCs were co‐cultured for 24 h in the presence of (i) PBS, (ii) LPS or (iii) A23187, the fluorescence intensity of wild type MCs increased by 30 ± 23%, 27 ± 17% and 117 ± 88%, respectively (Figure 2a,c). To test if MCs transferred not only cytosolic proteins (GFP) but also membrane components, we also co‐cultured PKH67‐stained and unstained MCs. In the unstained MCs, fluorescence intensity increased by 81 ± 20%, 81 ± 18% and 78 ± 14%, respectively, (Figure 2b,d) upon culture in the presence of PKH67 stained cells. By confocal microscopy we also confirmed in this experimental system that GFP‐MC‐derived EVs were taken up by wild type MCs and are found inside the cell after 24 h (Figure 2e‐h, Supplementary video 1).
FIGURE 2.
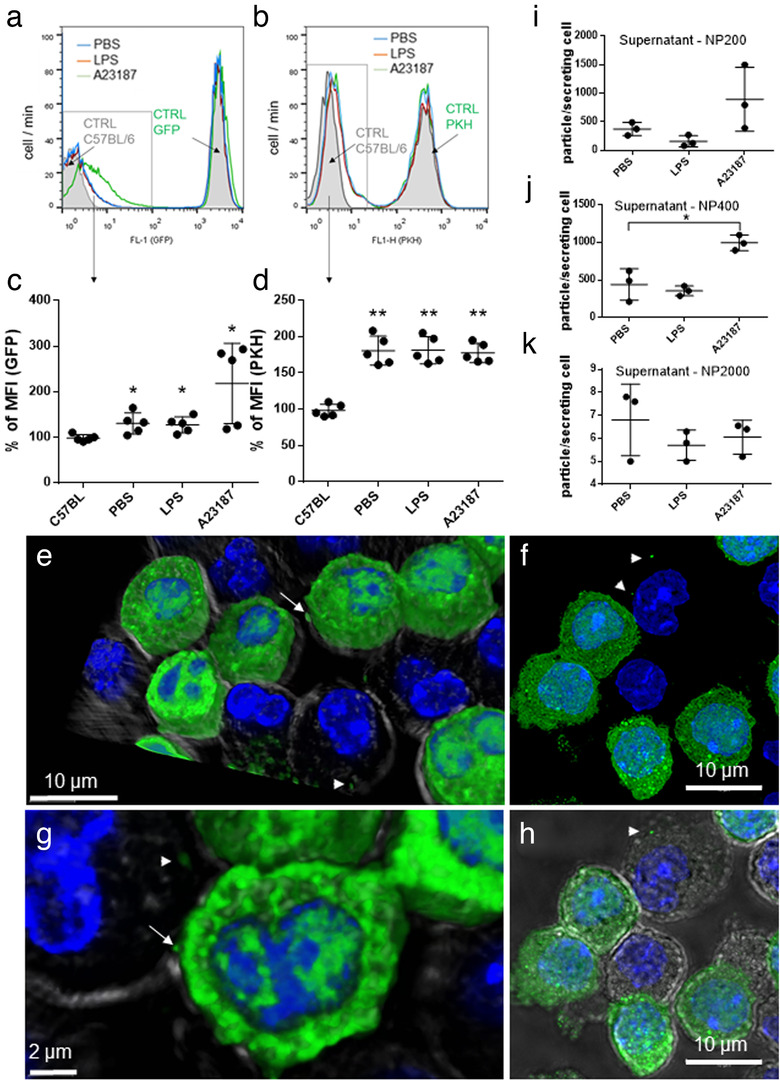
Uptake of MC‐derived EVs by MCs. Non‐fluorescent BMMCs from C57BL/6 mice were co‐cultured with GFP‐BMMCs with cytoplasmic fluorescence, derived from C57BL‐GFP mice (a, c) or PKH‐stained BMMC with fluorescent plasma membrane (b, d) in the presence of PBS or 100 ng/ml LPS. A23187 (0.5 μM) was used as positive control. For negative control, unstained C57BL/6‐derived BMMCs were used (a and b, grey histograms). Scatter plots show the mean and SD of five biological replicates (n = 5), normalized to unstained control. Cell‐free conditioned medium of BMMCs (cultured upon addition of PBS, 100 ng/ml LPS and 0.5 μM A23187) was tested by TRPS with three different pore size membranes (i: NP200 for small EVs, j: NP400 for mEVs and k: NP2000 for lEVs). Graphs show the mean and SD of three independent experiments (n = 3), where one point indicates the average of two technical replicates. e‐h: GFP‐BMMC and wild type BMMCs were co‐cultured for 24 h. Cells were fixed with 2% PFA, nucleus was stained with DAPI and EV‐transfer from GFP‐BMMCs was tested with confocal microscopy (Leica SP8). Cell shapes were visualized by transmitted light. 3D images were generated using LAS X software. Arrows: Releasing EVs, Arrow heads: EVs
3.2. LPS stimulation induces secretion of MC‐derived EVs with similar morphology to those derived from unstimulated MCs
These results raised the possibility that cytoplasm and membrane exchange could take place by EV transfer between cells. Thus, we analysed particles in the conditioned medium of PBS‐, LPS‐ or A23187‐stimulated BMMCs. We found three size‐based particle populations when measuring with qNano (NP200, NP400 and NP2000) including small particles with average diameter of 193 ± 6 nm, medium‐size particles of 317 ± 13 nm and large particles of 1214 ± 25 nm. There was no size difference between particles secreted in the presence of PBS, LPS or A23187 using t test. Concentrations of all three size‐based populations of particles (derived from the same amount of cells) from PBS or LPS‐stimulated MCs showed no significant difference (Figure 2i‐k), while concentration of medium sized particles (measured with membrane NP400) derived from A23187‐stimulated BMMCs (positive control) were slightly higher than those in the PBS and LPS groups (P ≤ 0.05, Figure 2j). As TRPS cannot distinguish membrane coated particles from protein aggregates, this increase may be the result of extracellular granules, as a result of degranulation.
Upon stimulation, MCs secrete pre‐synthetized, stored granules (degranulation) as well as they can release soluble mediators or EVs (Vukman et al., 2017). Therefore, in our case it was crucial to assess if the particles derived from BMMCs or PCMCs were granules or membrane enclosed EVs. To this end, we separated particles with differential centrifugation. We got three particle populations:, small ones after 100,000 × g, medium‐sized ones after 12,500 × g, and large ones after 2,000 × g centrifugation with average diameter of 105 ± 3 nm (Figure 3a), 309 ± 13 nm (Figure 3b) and 1811 ± 59 nm (Figure 3c), respectively. Large particle diameters differed from those measured directly from conditioned medium (P ≤ 0.001, ANOVA), probably because TRPS does not distinguish EVs and protein aggregates.
FIGURE 3.
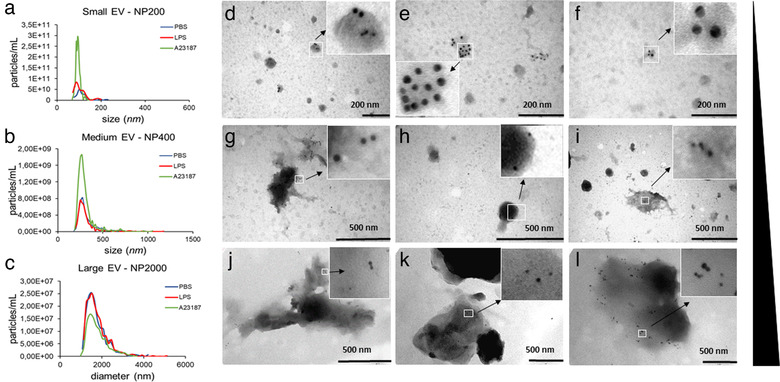
Immune electron microscopic and TRPS analyses of MC‐derived EVs. Particle sizes, size distributions and concentrations of separated large‐, medium size‐ and small‐EVs from the conditioned medium of BMMCs were measured by TRPS using membranes with three different pore sizes (a: NP200 for sEVs, b: NP400 for mEVs and c: NP2000 for lEVs. BMMCs were cultured in the presence of PBS (negative control, blue), LPS (100 ng/ml, red) or A23187 (positive control, green) for 24 h prior to EV separation. Graphs are means of 3 biological replicates with 2‐2 technical replicates (n = 6). EV markers of BMMC‐derived small (d‐f), medium (g‐i) and large (j‐l) EVs were detected by immunoelectron microscopy using nanogold labelling. PBS (unstimulated): 1st column, LPS‐stimulated: 2nd column, and A23187‐stimulated: 3rd column. Gold particles with 10 nm diameter represent CD63 while 5 nm diameter particles indicate c‐Kit
To confirm our TRPS results, we also analysed the separated particles by TEM. Particles from all separations showed the expected EV morphology and their size distribution was close to what has been seen in TRPS except for large structures which collapsed during sample preparation (Figure S3). We also confirmed that these structures positive for CD63 and the MC pan marker c‐Kit by immune‐gold TEM (Figure 3d‐l) therefore, we concluded that they were EVs.
Looking at EV markers by flow cytometry, all three (large, medium and small) size‐based populations from PBS, LPS‐ or A23187‐stimulated BMMCs were positive for CD63, CD9 but only small vesicles were positive for CD81. All samples were positive for fluorochrome‐labelled PKH67 and slightly for lactadherin (5.7 ± 3.2), but only large vesicles showed 9.5 ± 0.5 % positivity for annexin V. Vesicles from GFP‐MC BMMCs were positive for GFP and slightly for lactadherin (Figure 4a, Figure 4b‐table). Results were also confirmed in PCMCs (Figure 4c‐table). While there was no difference between LPS‐stimulated and unstimulated groups, A23187‐induced EVs which showed an increased fluorescent intensity. In order to decide if lEVs were the results of apoptosis or not, besides annexin V, we also incubated these EVs with lactadherin, fluorogenic substrate for activated caspase‐3/7 and propidium iodide. There was no significant difference between unstimulated (3.78 ± 2.10%), LPS‐stimulated (4.97 ± 3.35%) or A23187‐stimulated (4.68 ± 2.52%) mast cell derived lEVs. These results also correlated with viability data of MCs upon stimulation (annexin V‐propidium iodide double negative cells: PBS: 98.27 ± 2.27; LPS: 96.03 ± 2.83; A23187: 9.90 ± 3.10; Figure S4). EVs were also tested for Alix and TSG101, and medium and sEVs were positive for these proteins by Western blot, suggesting their exosome content (Figure 4b,d).
FIGURE 4.
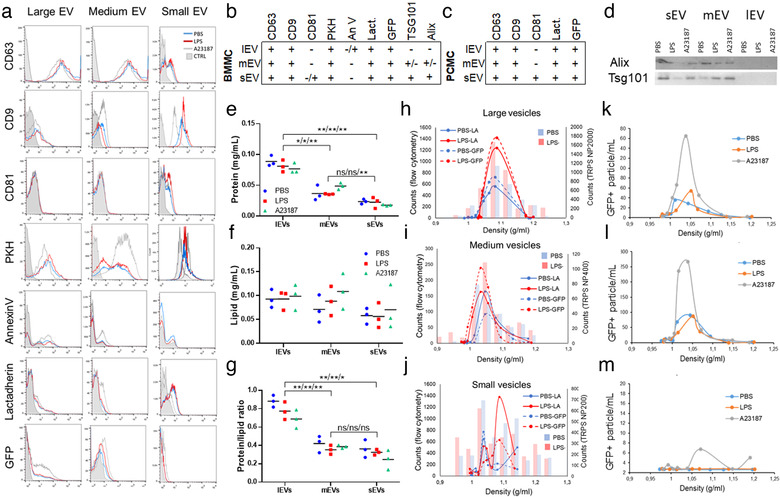
Characterization of mast cell‐derived EV populations. Large, medium and small EVs derived from 100 ng/ml LPS‐stimulated (LPS) or unstimulated (PBS) BMMCs and PCMCs were tested by flow cytometry. A23187 (0.5 μM) was used as positive control. EVs were stained for CD63, CD9 and CD81 or labelled with fluorochrome‐conjugated PKH67, annexin V and lactadherin. GFP expression of EVs derived from C57BL‐GFP mice were also measured. Small EVs were bound onto the surface of aldehyde/sulphate beads (4 μm). Histograms of flow cytometry measurements show single demonstrates of at least three independent experiments with BMMCs (a, n>3). Tables summarize the results on BMMC (b)‐and PCMC (c)‐derived EVs. Exosome markers Alix and TSG101 were detected by Western blot on the 3 × 3 EV populations (d). Protein (e) and lipid (f) content of separated EVs were measured by microBCA protein and SPV lipid assays, respectively. Protein/lipid ratio was also calculated (g). Graphs demonstrate three independent biological replicates (n = 3, *P ≤ 0.05, **P ≤ 0.01, t test). An Optiprep gradient was used to further characterize EVs. Separated small, medium and lEVs from the conditioned medium of BMMCs (from C57BL‐GFP) were ultracentrifuged (<16h, 100,000g, 4°C) bottom up (h‐j), while cell‐free conditioned medium of PCMCs (from C57BL‐GFP) were layered on the top of an Optiprep gradient (k‐m). Particle number was determined by TRPS and their lactadherin binding and positivity for GFP were determined by flow cytometry. Graphs show means of 3 independent experiments (n = 3)
For quality control, we also measured lipid and protein contents and calculated protein/lipid ratios. As expected, the protein/lipid ratio was higher for lEVs (P ≤ 0.01) when compared to mEVs and it was the smallest for sEVs (P ≤ 0.01, t test, Figure 4e‐g).
Density gradient ultracentrifugation was used as a second method for EV isolation. Separated EVs derived from BMMCs were studied by TRPS and flow cytometry. These EVs showed positivity for lactadherin‐binding and for GFP (Figure 4h‐j). Large and medium sized vesicles showed sensitivity to 0.1% of Triton X‐100. When we loaded conditioned medium on the top of a density gradient (PCMC, Figure 4k‐m) or we used bottom to top density gradient ultracentrifugation of separated EVs (BMMCs, Figure 4h‐j), we got similar results. These EVs were all sensitive to 0.1% Triton X‐100 (except for the sEVs) in accordance with our published data (Osteikoetxea et al., 2015).
3.3. MCs take up EVs from the conditioned media of LPS‐stimulated MCs
To confirm that GFP and PKH signal transfer (Figure 2) were due to EV‐mediated interactions, we cultured MCs in the presence of conditioned medium of LPS‐stimulated and non‐stimulated MCs (Figure 5a). If we depleted the conditioned medium in EVs before culture, the relative increase of fluorescent intensity of the acceptor cell was lower (GFP: P < 0.05, t test, Figure 5b, PKH: P < 0.01, t test; Figure 5c,d;). We reconstituted the conditioned medium with EVs or we applied EVs in non‐conditioned medium to acceptor MCs. In these cases, the increase in fluorescent intensity was as high as, or even higher than the EV‐containing cell‐free supernatant (Figure 5b,c). We confirmed the flow cytometry results also with fluorescent and confocal microscopy (Figure 5e‐h).
FIGURE 5.
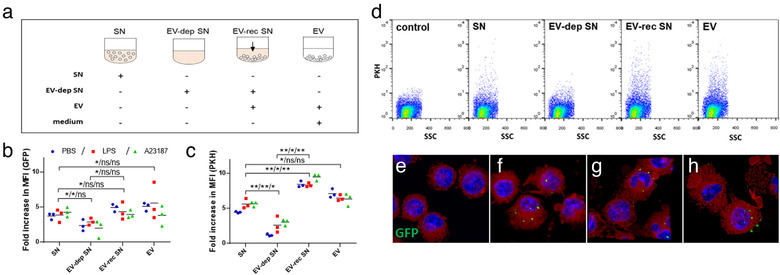
LPS‐stimulated mast cell derived extracellular vesicles are taken up by other mast cells. GFP‐BMMCs (b) and PKH‐stained BMMCs (c, d) were cultured in EV‐free medium in the presence or absence of LPS (100 ng/ml ) for an hour. A23187 (0.5 μM) and PBS were used as positive as negative controls, respectively. EVs were separated from the conditioned medium after 24 h . Unstained BMMCs derived from C57BL/6 mice were cultured in EV‐free medium (control), conditioned medium (SN), EV‐depleted conditioned medium (EV‐dep SN), EV‐reconstructed medium (EV‐rec SN) or separated EVs in fresh medium (a). EV‐uptake was also monitored by confocal microscopy (e‐h: GFP (green) merged with DAPI (blue) and lactadherin (red), e: control BMMCs; f: BMMCs cultured with EVs derived from PBS‐stimulated, g: LPS‐stimulated and h: A23187‐stimulated MCs. The mean of flow cytometric fold increase in MFI values (b, c) normalized to unstained control of three biological replicates (n = 3, *P ≤ 0.05, **P ≤ 0.01 t test) are shown. Flow cytometric plots (d) show single demonstrates of three independent experiments
3.4. LPS stimulated MC‐derived EVs induce TNF‐α secretion in recipient MCs
In the presence of LPS, MCs secrete TNF‐α (Vukman et al., 2012). Here we showed that LPS‐stimulated BMMC‐derived EVs have the same effect on naïve, non‐stimulated MCs (Figure 6a) and this effect was dependent on the concentration of EVs (Figure 6b). If we removed the EVs from the conditioned medium, the TNF‐α secretion was lower (P < 0.001) and if we added EVs back or applied EVs alone, we got the same TNF‐α levels (P = 0.40 and P = 0.89, t test, respectively, Figure 6a). We confirmed our results with more mature PCMC (Figure 6c) as well. Although the drop in TNF‐α secretion was not as spectacular as in the case of BMMCs, it was still significant (P < 0.05, t test). To confirm that our finding was not the result of LPS that adhered onto the surface of EVs, we incubated EVs in LPS containing medium before co‐culture and found no effect on TNF‐α secretion (P = 0.80, Figure 6d). All three EV populations seemed to play a role in the induction of TNF‐α (lEV: P = 0.31, mEV and sEV<0.05, Figure 6e). We also used GW4869 a neutral sphingomyelinase inhibitor suggested to interfere with small EV secretion. The amount of small vesicles decreased (P ≤ 0.05 Figure 6f) in line with published data (Menck et al., 2017). However, there was no effect of GW4869 on TNF‐α secretion (Figure 6g), suggesting that small EVs other than those generated by a ceramide‐related pathway played a role in TNF‐α induction.
FIGURE 6.
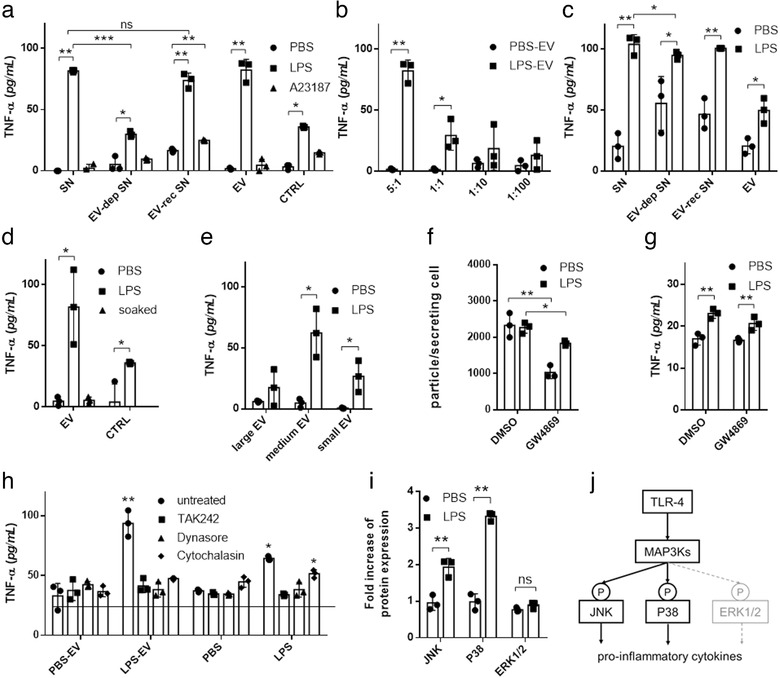
EVs from LPS‐stimulated MCs induce TNF‐α secretion in recipient MCs. BMMCs or PCMCs were cultured in EV‐free medium in the presence or absence of LPS (100 ng/ml ) for an hour. A23187 (0.5 μM) and PBS were used as positive and negative controls, respectively. EVs were separated from the conditioned medium after 24 h. Naïve BMMCs (a) or PCMCs (c) were cultured in EV‐free medium (control) conditioned medium (SN), EV‐depleted conditioned medium (EV‐dep SN), EV‐reconstructed medium (EV‐rec SN) or separated EV containing non‐conditioned medium. For concentration measurements, different ratios of EV donor and acceptor cells were tested (b). Naïve BMMCs were also cultured in the presence of EVs from unstimulated BMMCs incubated in 100 ng/ml LPS for 2 h prior to experiments (d) or large‐, medium size‐ or small‐EVs of stimulated and unstimulated MCs (e). We used GW4869 (10 μM), a neutral sphingomyelinase inhibitor to block small EV generation, 30 min before LPS stimulation of donor cells. As GW4869 was diluted in DMSO, we used DMSO as control. Separated small particles were measured by NTA (f) and were added to naïve cells (same amount as producing cells) after washing for 24 h (g). In inhibition experiments, BMMCs were treated with TAK242 (0.2 μM), dynasore (80 μM) cytochalasin D (10μg/ml ) prior to culture in the presence of separated EVs from conditioned medium of unstimulated (PBS‐EV) or LPS‐stimulated (LPS‐EV) MCs (h). TNF‐α concentration was measured after 24 h with ELISA. Column bars are means of at least three independent experiments (biological replicates, n>3) as the mean and SD of three replicates (n = three, *P ≤ 0.05, **P ≤ 0.01, t test and ANOVA), where one dot indicates the average of three technical replicates. BMMCs were cultured for 24 h in EV‐free complete medium in the presence of isolated EVs derived from LPS‐stimulated (100 ng/ml ) and unstimulated BMMCs. Cells were washed two times lysed and investigated for phosphorylation of ERK1/2, JNK and P38 by RayBio Cell‐based phosphorylation ELISA kits (i). Biological replicates, n≥3, *P ≤ 0.05, **P ≤ 0.01, t test. Schematic picture shows the possible mechanism of MAPK signalling activation (j)
Since it has been demonstrated earlier that EVs induced TNF‐α production in vitro in a TLR4 dependent manner (Couper et al., 2010), we tested if TLR4 binding or EV uptake were involved in MC activation. We used dynasore and cytochalasin D to block endocytosis and EV‐uptake and TAK242 to inhibit TLR4 signalling. We showed that both processes were important in EV‐MC interaction as pre‐treatment of MCs with these inhibitors blocked the effect of LPS‐stimulated MC‐derived EVs on naïve (non‐stimulated) MCs (P < 0.001, ANOVA, Figure 6h).
3.5. MC‐derived EVs activate the MAPK signalling pathway
Since we found that TLR4 activation was crucial in EV‐mediated TNF‐α production, we investigated if the MAPK signalling pathway was activated during this process. MAPK kinase pathway activation due to TLR4 can result in the phosphorylation of three central proteins: ERK1/2, JNK and P38 (Avila & Gonzalez‐Espinosa, 2011). In our experiments, we showed that the JNK and P38 pathways were activated, while the ERK1/2 pathway remained inactive (Figure 6i,j).
3.6. MC‐derived EVs are taken up by MCs and they induce TNF‐α secretion in MCs in vivo in the peritoneal cavity of mice
We wanted to see if EVs exert the same effect in vivo as in vitro, so we implanted GFP‐MC‐containing chambers into the peritoneal cavity of mice. In an in vitro experiment, we confirmed that EVs could be released from the chambers while MCs remained imprisoned in them (Figure 5, Figure 7l). If MCs were stimulated with LPS before implantation, we detected TNF‐α production in peritoneal lavage MCs (P ≤ 0.05, Figure 7a,d), and these MCs contained GFP‐EVs as shown by flow cytometry (P < 0.01, t test, Figure 7b) and confocal microscopy (Figure 7h‐j). For controls, we measured (i) total peritoneal cell numbers, (ii) MC numbers, (iii) cell viability in the peritoneal cavity and iv) viability of cells retrieved from the removed chambers. We did not detect significant difference between numbers of MCs (Figure 7c), total peritoneal cells (Figure 7e) and peritoneal cell viability (Figure 7f) of mice implanted with LPS‐stimulated or un‐stimulated MC‐containing or empty (medium containing) chambers suggesting that EVs did not induce migration or proliferation of peritoneal cells and nor did they escape from the chambers. Live cell retrieval from the chambers was close to 100% (Figure 7g). We also tested if besides MCs, other cell types could also take up GFP‐vesicles released from the implanted chambers. We found an increase in the fluorescence intensity of CD4+ and CD8+ T‐cells, B‐cells and macrophages increased in the peritoneal cavity (Figure 7k). Regarding the cellular composition of the peritoneal cavity, 3.46 ± 2.07% of the peritoneal cells were T‐cells, 24.55 ± 13.86% were B‐cells, 31.30 ± 13.42% were macrophages and 1.94 ± 1.06 were MCs (in line with data in the literature (Meurer et al., 2016; Ray & Dittel, 2010; Vasilyev, Yukhneva, Shurygina, Stukova, & Egorov, 2018)). Out of these cells, 16.48 ± 5.78%, 9.35 ± 0.65%, 12.31 ± 1.81 and 32.59 ± 2.87% of the cells were GFP positive, respectively. Given that this study focuses on MC‐MC communication, in our further experiments we only investigated MCs.
FIGURE 7.
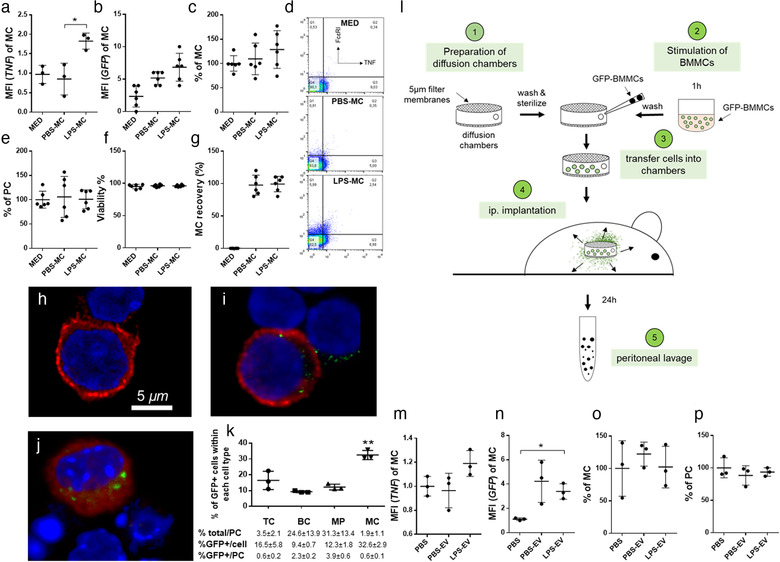
LPS‐stimulated MCs release EVs which spread TNF‐α induction in MCs in vivo in the peritoneal cavity of mice. GFP‐BMMCs were incubated for 1h in the presence or absence of LPS (100 ng/ml ). EMD Millipore's Diffusion Chamber Kits were used with 5 μm pore size membrane filters. Empty (mock‐operated) and MC‐containing (100 ng/ml LPS‐stimulated or unstimulated) chambers were then implanted to the peritoneal cavities of C57BL/6 mice (a‐l). In additional experiments, EVs separated from the conditioned medium of LPS‐stimulated and unstimulated BMMCs were injected i.p. twice in 24 h (n‐p). After 24 h, mice were sacrificed and peritoneal cells were collected by peritoneal lavage. MC numbers (c, o) were determined using Kimura staining by light microscopy and by flow cytometry using the two pan‐MC markers CD117 (c‐Kit) and FcɛRI. Uptake of EVs by MCs was assessed by flow cytometry based on the GFP signal (b, n). Peritoneal cells (PC) were counted (e, p) and their viability (f) was assessed by trypan blue staining. Intracellular TNF‐α (a, m) was also measured by flow cytometry after Brefeldin A treatment (3 μg/ml for 1h). Representative plots of intracellular TNF staining in MCs are shown (d). Cells were examined under a confocal microscope upon staining for FcɛRI expression to identify MC and GFP signal to confirm EV uptake (h: med, i: PBS‐MC, j: LPS‐MC). Cells retrieved from the removed diffusion chambers were subjected to enumeration and viability tests using trypan blue staining (live cell recovery is shown in g). EV‐uptake by different cell types including T cells (TC), B cells (BC), macrophages (MP) and mast cells (MC) was determined as the % of GFP+ cells of each cell type (k). Frequency of different cell types among peritoneal cells (1st row), % of GFP+ cells of each cell type (2nd row) and % of GFP+ cells of a given cell type among all PCs (3rd row) are summarised in the table below figure k. Graphs show the mean and SD of at least 3 independent experiments (biological replicates, n ≥ 3, *P ≤ 0.05, **P ≤ 0.01, t test). Med: medium, MFI: mean fluorescence intensity
To exclude the effect of possible residual LPS, we injected LPS in medium (100 ng/ml ) intraperitoneally to mice. This was the concentration of LPS before the MCs were washed twice prior to implantation. We did not observe a significant increase in the number and intracellular TNF‐α staining of MCs (Figure 6).
We also injected separated BMMC‐EVs in bolus into the peritoneal cavity of mice and we analysed the intracellular TNF‐α staining of FcεRI‐positive MCs. As shown in Figure 7m, repeated bolus injection of EVs slightly increased the TNF‐α content of MCs (P = 0.069, t test) suggesting that EVs may play a role in this activation as MCs were GFP positive (Figure 7n). As in the case of diffusion chamber experiments, we did not find any difference in the total MC or peritoneal cell numbers (Figure 7o,p).
3.7. LPS‐induced EVs have a unique protein content
Although there was no change in the number and morphology of LPS‐induced EVs as compared to PBS controls, we hypothesized that LPS‐induced EVs could have an altered protein content. We tested isolated medium sized EVs with mass spectroscopy and compared mEVs derived from LPS‐stimulated MCs and PBS exposed controls. We identified 424 proteins that were present in both populations, and 12 proteins that were unique to LPS‐mEVs (Figure S7A). Among the shared proteins, the amount of 5 proteins were significantly higher in LPS‐EVs than in PBS EV‐s (P ≤ 0.05). These proteins are listed in SuppTable 1, with their potentially relevant biological functions according to NCBI (https://www.ncbi.nlm.nih.gov/) and UniProt (https://www.uniprot.org/) databases, using Scaffold program. LPS‐mEVs were analysed for their origin and functions in biological processes (Figure S7B,C). We identified 16 proteins with EV localization. Most of the proteins had a biological function (381 from 406 proteins), including role in immune processes (91 proteins), signalling (123 proteins) or response to stimuli (257 proteins) (Figure S7C). We also identified five endogenous TLR4 ligands: HMGB1, heat shock proteins, endoplasmin, myosin S100 A6 presented in MC‐derived EVs (Table S2 (Erridge, 2010, Yu, Wang, & Chen, 2010))
4. DISCUSSION
MCs have been long known for their role in allergic reactions as well in the immune defence against bacteria and parasites. More recently, they have been recognized as orchestrator cells in the immune system being able to regulate many different immune processes by the plethora of their stored and secreted bioactive molecules. The biological significance of MCs is strongly underlined by the new category of pathological conditions collectively termed as ‘mast cell mediator disorders’ (Theoharides, Tsilioni, & Ren, 2019). Furthermore, MC proliferation and accumulation results in the disease ‘mastocytosis’ (Kim et al., 2018, Renke et al., 2019).
MCs are typically localized along epithelia, mucosal membranes and blood vessels. They are key components of innate immunity and play important roles in inflammation. They represent a rich source of the pro‐inflammatory mediator TNF‐α. MC‐derived TNF‐α is required for efficient CD8+ T cell responses to haptens (Dudeck et al., 2015), it promotes the migration and maturation of CD8+ DCs (Suto et al., 2006), distinctly boosts the CD8+ T‐cell‐priming efficiency of CD8+ DCs (Krystel‐Whittemore, Dileepan, & Wood, 2015) and promotes Th17 cell–dependent neutrophil recruitment in ovalbumin‐challenged OTII mice (Nakae, Suto, Berry, & Galli, 2007).
We hypothesized that there was a mechanism by which the relatively low frequency MCs could amplify their effects and extend it in space to an entire network of other MCs. We predicted a mechanism by which MCs may send alarming signals and activate each other in spite of their low frequency to facilitate immune responses. Cytokines and other soluble MC‐derived mediators have been shown to be important in immune cell stimulation in bacterial infections (Mukai, Tsai, Saito, & Galli, 2018). However, the relatively short lifetime of cytokines limit their effects at distances. In contrast, EVs (increasingly recognized players of cell‐to‐cell communication (Vukman et al., 2017)), have been shown to be more stable and may have an effect on MCs in other parts of the body (Kormelink, Mol, de Jong, & Wauben, 2018, Meldolesi, 2018). In the present study, we investigated MC‐derived EVs for their ability to induce a TNF‐α response in other MCs.
Instead of using cell lines, here we analysed the functional consequences of EV release by primary BMMCs and PCMCs in mice. These primary MCs are more reliable in vitro models than cell lines as the latter may lack some important MC receptors (e.g. FcƐRI is absent from the MHC mast cell line). Furthermore, MC lines may show characteristics of other cell types as well (e.g. share molecular features with basophils as observed in the case of RBL‐2H3 cells). While BMMCs show higher purity, they are not as mature as PCMCs. However, these latter cells also have limitations due to their low yield (Yu et al., 2018).
First, we had to confirm that EVs (rather than membrane‐free granules or soluble mediators) mediate communication between MCs. Although in the literature, the distinction between secreted MC granules and EVs is often not obvious (Wernersson & Pejler, 2014), MC degranulation can be clearly distinguished from MC‐derived EV release (Kormelink et al., 2016).
By using immunogold electron microscopy, we confirmed the simultaneous presence of c‐Kit and CD63 on the isolated small, medium and large particles, which thus, proved to be MC‐derived EV populations. By using flow cytometry, we defined their surface markers. In addition, we characterized the EV preparations by their total protein and lipid content and their floatation densities. Finally, we showed that MC‐derived EVs were sensitive to detergent lysis, which further proved their vesicular nature.
Thus, we provided evidence that EVs were secreted in vitro by primary MCs. Furthermore, we demonstrated that the secreted EVs were transferred from MCs to MCs and induced TNF‐α expression in the recipient resting BMMCs or PCMCs.
To gain an insight into the biogenesis of the MC‐derived EVs, first we assessed apoptosis of the releasing MCs to rule out the possibility that the observed EVs were apoptotic vesicles (Poon et al., 2019). Next, we assessed the presence of TSG101, Alix, CD63 and CD81 in association with our EVs (Bobrie, Colombo, Krumeich, Raposo, & Théry, 2012, Kowal et al., 2016). Detection of the above proteins on EVs strongly suggested an endosomal origin of some of our MC‐derived EVs. Finally, we used the neutral sphingomyelinase inhibitor GW4869 to block small EV production (Mariadelva Catalano & O'Driscoll, 2019). While GW4869 reduced the number of LPS‐induced small EVs, it did not interfere with EV transfer and subsequent MC activation. This suggests that EVs other than those generated by a ceramide‐related pathway, might have played a role in the EV‐mediated transfer of MC‐MC pro‐inflammatory response.
Thus, our data presented in this report, show that EVs may also play an important role in the antibacterial defence (Piliponsky, Acharya, & Shubin, 2019) in addition to the de novo produced MC mediators which are well known for being important players in the defence mechanism against bacteria (Theoharides, Kempuraj, Tagen, Conti, & Kalogeromitros, 2007).
The significance of the above in vitro observed, EV‐mediated TNF‐α induction could be best evaluated by addressing the question whether it could be observed also in vivo. To this end, we implanted a disposable device with porous filter walls into the peritoneal cavity of mice (Nishimura, Uchiyama, Yagi, & Hashimoto, 1985). The implanted chambers contained primary cells, which could be retrieved after the removal of the device from mice with ∼ 98% viability and close the original cell number after 24 h. Using this implantable devices we showed that MC‐derived EVs were taken up by MCs, macrophages, T‐cells and B‐cells in line with published data (Vukman et al., 2017). Unexpectedly, we found that MCs were the main acceptor cells of MC‐derived EVs, despite their small cell number (Figure 7h). These results correlate with what is known about MCs that is although they are present in low numbers in tissues, they play crucial role for example during bacterial infections (Zimmermann et al., 2019) or sepsis (Echtenacher, Männel, & Hültner, 1996; Piliponsky et al., 2019)).
Importantly, EVs released through the pores of the implanted chamber were not only taken up by peritoneal cells, but they also induced significant TNF‐α expression by peritoneal MCs and were detectable in the cytoplasm of peritoneal lavage MCs by fluorescent microscopy. In an attempt to reproduce these data, we also injected mice intraperitoneally with bolus injections of separated EVs from conditioned media of GFP‐MCs. We found a significant increase in the fluorescence of wild type MCs in the peritoneal lavage and a close to significant (P = 0.069, t test) increase in TNF‐α production. Clearly, the implantable diffusion chamber enabled us to investigate the continuous in vivo release of EVs. The strength of this approach is clearly demonstrated by the fact that only repeated injections of the separated EVs could recapitulate the effect observed in the case of MCs releasing EVs continuously from the diffusion chamber.
In this study, we also investigated the mechanisms of the cellular uptake of MC‐derived EVs and the induction of the activation of MCs by EVs. During bacterial infection, TLR4 is one of the key receptors for innate immune cell activation. In the case of MCs, it is known that TLR4 plays a central role in recognition of LPS and bacteria (Supajatura et al., 2002; Vukman et al., 2013 and Supajatura et al., 2001)). Here we show that MC‐derived EVs induce TNF‐α production through TLR4, and we identified two MAPK pathways activated during this process. In addition, given that LPS is a known inducer of oxidative stress in MCs (Aidoo et al., 2019), and that membrane lipids are defining components of EVs, we hypothesize that oxidized phospholipids of the EV membrane could have also played a role in TLR4 activation (similarly to what has been described previously (Manček‐Keber et al., 2015)).
Importantly, we found that internalization of EVs by MCs played a key role in MC activation. In the case of bacteria, it has been reported that their uptake by MCs is a critical factor in the induction of immune responses (cytokine secretion, antigen presentation, and migration) (Mielcarek et al., 2001; Trivedi et al., 2013). Similarly to bacteria, EVs are also taken up by MCs, while soluble mediators bind to cell surface receptors. TNFα‐induction by naïve MCs exposed to soluble LPS was lower or equal with what we found upon exposure of MCs to LPS stimulated MC‐derived EVs. Internalized EVs have been shown to transfer molecules to other cells by which they induce activation (Ekström, Valadi, Sjöstrand, Bossios, & Eldh, 2012). Interestingly, Shelke et al demonstrated that when TGF‐β was taken up in association with EVs, it had a prolonged effect on acceptor MCs (Shelke et al., 2019). In the present study, we found that EVs from stimulated MCs contained several proteins with known roles in immune responses (e.g. heat shock protein 70 kDa, H‐2 class histocompatibility antigen and tyrosine‐protein kinase Fgr) which were not detected in EVs from unstimulated MCs. We also identified some proteins (such as HMGB1, heat shock proteins, endoplasmin, myosin, S100 A6 protein) which have been suggested to play a role in endogenous ligand (self‐molecule)‐induced activation of TLR 4 (Erridge, 2010, Yu et al., 2010). Given the lack of published evidence for the intracellular/intravesicular presence and signalling through TLR4, we hypothesize that in our experiments, MC‐derived EV‐associated TLR4 ligands induced signals via cell surface TLR4.
In summary, our data presented here prove that bacterial LPS‐sensing MCs release EVs that in turn, induce TNF‐α in resting MCs. EV communication between members of the MC network may thus, play an important role in spreading and escalating pro‐inflammatory responses to immune stimuli. Our data may provide an explanation how the relatively rare and often enigmatic tissue resident MCs can play key roles in diseases such as autoimmune arthritis, inflammatory bowel disease or Crohn's disease. Of note, there are other examples for EVs extending the cellular responses of a relatively small population of cells to other ones (e.g. dendritic cells extend their antigen presenting capacity to non‐primed other dendritic cells (Thery et al., 2002)). Taken together, the mechanism described in the present report, identifies a novel aspect how MCs of the innate immune system act as master cells in inflammation and immune responses.
In this study, we only investigated the role of EVs in MC‐MC communication. However, the MC‐derived EVs may also have other exciting effects on different other immune and non‐immune recipient cells. The in vivo device introduced in this study may provide a feasible system to investigate many other EV‐mediated interactions beyond those described in this paper.
CONFLICTS OF INTEREST
The authors do not have financial interest to declare.
Supporting information
Supporting Information S1
Supporting Video S1.
ACKNOWLEDGEMENTS
This work was funded by the Hungarian Scientific Research Fund (OTKA PD112085, 111958 and 120237), the National Heart Program (NVKP_16‐1‐2016‐0017), VEKOP‐2.3.2‐16‐2017‐000002, VEKOP2.3.3‐15‐2017‐00016, H2020‐MSCA‐ITN‐2017‐722148 TRAIN EV and Higher Education Excellence Program FIKP, Therapeutic thematic programme). This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under grant agreement No 739593. The first author was also supported by János Bolyai Research Fellowship of the Hungarian Academy of Sciences.
Vukman KV, Ferencz A, Fehér D, et al. An implanted device enables in vivo monitoring of extracellular vesicle‐mediated spread of pro‐inflammatory mast cell response in mice. J Extracell Vesicles. 2020;10:e12023 10.1002/jev2.12023
REFERENCES
- Aidoo, D. B. , Obiri, D. D. , Osafo, N. , Antwi, A. O. , Essel, L. B. , Duduyemi, B. M. , & Ekor, M. (2019). Allergic airway‐induced hypersensitivity is attenuated by bergapten in murine models of inflammation. Advances in Pharmacological Sciences, 2019, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Nedawi, K. , Szemraj, J. , & Cierniewski, C. S. (2005). Mast cell‐derived exosomes activate endothelial cells to secrete plasminogen activator inhibitor type 1, Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 1744–1749. [DOI] [PubMed] [Google Scholar]
- Amanda Carroll‐Portillo, A. , Z., Surviladze , Cambi, A. , Lidke, D. S. , & Wilson, B. S. (2012). Mast cell synapses and exosomes: Membrane contacts for information exchange, Frontiers in Immunology, 15(3), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila, M. , & Gonzalez‐Espinosa, C. (2011). Signaling through Toll‐like receptor 4 and mast cell‐dependent innate immunity responses, Iubmb Life, 63(10), 873–880. [DOI] [PubMed] [Google Scholar]
- Bobrie, A. , Colombo, M. , Krumeich, S. , Raposo, G. , & Théry, C. (2012). Diverse subpopulations of vesicles secreted by different intracellular mechanisms are present in exosome preparations obtained by differential ultracentrifugation, Journal of Extracellular Vesicles, 1, 18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couper, K. N. , Barnes, T.., Hafalla, J. C. , Combes, V. , Ryffel, B. , Secher, T., … de Souza, J. B. , & (2010). Parasite‐derived plasma microparticles contribute significantly to malaria infection‐induced inflammation through potent macrophage stimulation, Plos Pathogens, 6, e1000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudeck, J. , Ghouse, S. M. , Lehmann, C. H. , Hoppe, A. , Schubert, N. , Nedospasov, S. A. , … Dudeck, A. (2015). Mast‐cell‐derived TNF amplifies CD8(+) dendritic cell functionality and CD8(+) T cell priming, Cell Reports, 13, 399–411. [DOI] [PubMed] [Google Scholar]
- Echtenacher, B. , Männel, D. N. , & Hültner, L. (1996). Critical protective role of mast cells in a model of acute septic peritonitis, Nature, 381(6577), 75–77. [DOI] [PubMed] [Google Scholar]
- Ekström, K. , Valadi, H. , Sjöstrand, M. , Bossios, A. , & Eldh, Lötvall J. (2012). Characterization of mRNA and microRNA in human mast cell‐derived exosomes and their transfer to other mast cells and blood CD34 progenitor cells, Journal of Extracellular Vesicles 18389, 1, 10, 3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrom, K. , Valadi, H. , Sjostrand, M. , Malmhall, C. , Bossios, A. , Eldh, M. , & Lotvall, J. (2012). Characterization of mRNA and microRNA in human mast cell‐derived exosomes and their transfer to other mast cells and blood CD34 progenitor cells, Journal of Extracellular Vesicles, 18389, 1, 18389‐01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldh, M. , Ekstrom, K. , Valadi, H. , Sjostrand, M. , Olsson, B. , Jernas, M. , & Lotvall, J. (2010). Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA, Plos One, 5, e15353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge, C. (2010). Endogenous ligands of TLR2 and TLR4: Agonists or assistants? Journal of Leukocyte Biology, 87(6), 989–999. [DOI] [PubMed] [Google Scholar]
- Gyorgy, B. , Modos, K. , Pallinger, E. , Paloczi, K. , Pasztoi, M. , Misjak, P. , … Buzas, E. I. (2011). Detection and isolation of cell‐derived microparticles are compromised by protein complexes resulting from shared biophysical parameters, Blood, 117, e39‐e48. [DOI] [PubMed] [Google Scholar]
- Hugle, T. , Hogan, V. , White, K. E. , & van Laar, J. M. (2011). Mast cells are a source of transforming growth factor beta in systemic sclerosis, Arthritis and Rheumatism, 63, 795–799. [DOI] [PubMed] [Google Scholar]
- Kim, D‐K. , Cho, Y‐E. , Komarow, H. D. , Bandara, G. , Song, B‐J. , Olivera, A. , & Metcalfe, D. D. (2018). Mastocytosis‐derived extracellular vesicles exhibit a mast cell signature, transfer KIT to stellate cells, and promote their activation, Proceedings of the National Academy of Sciences of the United States of America, 115(45), E10692–E10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, I. , Moritani, Y. , & Tanizaki, Y. (1973). Basophils in bronchial asthma with reference to reagin‐type allergy, Clinical Allergy, 3, 195–202. [DOI] [PubMed] [Google Scholar]
- Kormelink, T. G. , Arkesteijn, G. J. A. , van de Lest, C. H. A. , Geerts, W. J. C. , Goerdayal, S. S. , Altelaar, M. A. F. , … Wauben, M. H. M. (2016). Mast cell degranulation is accompanied by the release of a selective subset of extracellular vesicles that contain mast cell–specific proteases, Journal of Immunology, 197(8), 3382–3392. [DOI] [PubMed] [Google Scholar]
- Kormelink, T. G. , Mol, S. , de Jong, E. C. , & Wauben, M. H. M. (2018). The role of extracellular vesicles when innate meets adaptive, Seminars in Immunopathology, 40, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Arras, G. , Colombo, M. , Jouve, M. , Morath, J. P. , Primdal‐Bengtson, B. , … Théry, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes, Proceedings of the National Academy of Sciences of the United States of America, 113(8), E968‐E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystel‐Whittemore, M. , Dileepan, K. N. , & Wood, J. G. (2015). Mast cell: A multi‐functional master cell, Frontiers in Immunology, 6, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manček‐Keber, M. , Frank‐Bertoncelj, M. , Hafner‐Bratkovič, I. , Smole, A. , Zorko, M. , Pirher, N. , … Jerala, R. (2015). Toll‐like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles, Science Signaling, 8(381), ra60. [DOI] [PubMed] [Google Scholar]
- Mariadelva Catalano, M. , & O'Driscoll, L. (2019). Inhibiting extracellular vesicles formation and release: A review of EV inhibitors, Journal of Extracellular Vesicles, 9(1), 1703244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, J. S. , King, C. A. , & McCurdy, J. D. (2003). Mast cell cytokine and chemokine responses to bacterial and viral infection, Current Pharmaceutical Design, 9, 11–24. [DOI] [PubMed] [Google Scholar]
- Meldolesi, J. (2018). Exosomes and ectosomes in intercellular communication Current Biology, 28(8), R435–R444. [DOI] [PubMed] [Google Scholar]
- Menck, K. , Sönmezer, C. , Worst, T. S. , Schulz, M. , Dihazi, G. H. , Streit, F. , … Gross, J. C. (2017). Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane, Journal of Extracellular Vesicles, 6(1), 1378056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurer, S. K. , Neß, M. , Weiskirchen, S. , Kim, P. , Tag, C. G. , Kauffmann, M. , … Weiskirchen, R. (2016). Isolation of mature (peritoneum‐derived) mast cells and immature (bone marrow‐derived) mast cell precursors from mice, Plos One, 11, e0158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielcarek, N. , Hornquist, E. H. , Johansson, B. R. , Locht, C. , Abraham, S. N. , & Holmgren, J. (2001). Interaction of Bordetella pertussis with mast cells, modulation of cytokine secretion by pertussis toxin, Cellular Microbiology, 3, 181–188. [DOI] [PubMed] [Google Scholar]
- Mion, F. , D'Inca, F. , Danelli, L. , Toffoletto, B. , Guarnotta, C. , Frossi, B. , … Pucillo, C. E. (2014). Mast cells control the expansion and differentiation of IL‐10‐competent B cells, Journal of Immunology, 193, 4568–4579. [DOI] [PubMed] [Google Scholar]
- Mukai, K. , Tsai, M. , Saito, H. , & Galli, S. J. (2018). Mast cells as sources of cytokines, chemokines, and growth factors, Immunological Reviews, 282(1), 121–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae, S. , Suto, H. , Berry, G. J. , & Galli, S. J. (2007). Mast cell‐derived TNF can promote Th17 cell‐dependent neutrophil recruitment in ovalbumin‐challenged OTII mice, Blood, 109, 3640–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae, S. , Suto, H. , Iikura, M. , Kakurai, M. , Sedgwick, J. D. , Tsai, M. , & Galli, S. J. (2006). Mast cells enhance T cell activation: Importance of mast cell costimulatory molecules and secreted TNF, Journal of Immunology, 176, 2238–2248. [DOI] [PubMed] [Google Scholar]
- Nishimura, T. , Uchiyama, Y. , Yagi, H. , & Hashimoto, Y. (1985). Use of Millipore diffusion chambers to assay in vivo IL‐2 activity Journal of Immunological Methods, 78(2), 239–245. [DOI] [PubMed] [Google Scholar]
- Osteikoetxea, X. , Balogh, A. , Szabo‐Taylor, K. , Nemeth, A. , Szabo, T. G. , Paloczi, K. , … Buzas, E. I. (2015). Improved characterization of EV preparations based on protein to lipid ratio and lipid properties, Plos One, 10, e0121184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osteikoetxea, X. , Sodar, B. , Nemeth, A. , Szabo‐Taylor, K. , Paloczi, K. , Vukman, K. V. , … Buzas, E. I. (2015). Differential detergent sensitivity of extracellular vesicle subpopulations, Organic & Biomolecular Chemistry, 13, 9775–9782. [DOI] [PubMed] [Google Scholar]
- Piliponsky, A. M. , Acharya, M. , & Shubin, N. J. (2019). Mast Cells in Viral, Bacterial, and Fungal Infection Immunity, International Journal of Molecular Sciences, 20(12), 2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon, I. K. H. , Parkes, M. A. F. , Jiang, L. , Atkin‐Smith, G. K. , Tixeira, R. , Gregory, C. D. , … Baxter, A. A. (2019). Moving beyond size and phosphatidylserine exposure: Evidence for a diversity of apoptotic cell‐derived extracellular vesicles in vitro, Jl Extracell Vesicles, 8(1), 1608786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, A. , & Dittel, B. N. (2010). Isolation of mouse peritoneal cavity cells, Journal of Visualized Experiments: JoVE, 28(35), 1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renke, J. , Kędzierska‐Mieszkowska, S. , Lange, M. , Nedoszytko, B. , Wasilewska, E. , Liberek, A. , … Lipińska, B. (2019). Mast cells in mastocytosis and allergy ‐ Important player in metabolic and immunological homeostasis. Advances in Medical Sciences, 64(1), 124–130. [DOI] [PubMed] [Google Scholar]
- Shelke, G. V. , Yin, Y. , Jang, S. C. , Lässer, C. , Wennmalm, S. , Hoffmann, H. J. , … Lötvall, J. (2019). Endosomal signalling via exosome surface TGFβ‐1, Journal of Extracellular Vesicles, 8(1), 1650458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skokos, D. , Botros, H. G. , Demeure, C. , Morin, J. , Peronet, R. , Birkenmeier, G. , … Mecheri, S. (2003). Mast cell‐derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo, Journal of Immunology, 170, 3037–3045. [DOI] [PubMed] [Google Scholar]
- Skokos, D. , Le Panse, S. , Villa, I. , Rousselle, J. C. , Peronet, R. , David, B. , … Mecheri, S. (2001). Mast cell‐dependent B and T lymphocyte activation is mediated by the secretion of immunologically active exosomes, Journal of Immunology, 166, 868–876. [DOI] [PubMed] [Google Scholar]
- Skokos, D. , Le Panse, S. , Villa, I. , Rousselle, J. C. , Peronet, R. , Namane, A. , … Mecheri, S. (2001). Nonspecific B and T cell‐stimulatory activity mediated by mast cells is associated with exosomes, International Archives of Allergy and Immunology, 124, 133–136. [DOI] [PubMed] [Google Scholar]
- Stassen, M. , Hartmann, A. K. , Delgado, S. J. , Dehmel, S. , & Braun, A. (2019). Mast cells within cellular networks, Journal of Allergy and Clinical Immunology, 144, S46‐S54. [DOI] [PubMed] [Google Scholar]
- Supajatura, V. , Ushio, H. , Nakao, A. , Okumura, K. , Ra, C. , & Ogawa, H. (2001). Protective roles of mast cells against enterobacterial infection are mediated by Toll‐like receptor 4, Journal of Immunology, 167(4), 2250–2256. [DOI] [PubMed] [Google Scholar]
- Supajatura, V. , Ushio, H. , Nakao, A. , Akira, S. , Okumura, K. , Ra, C. , & Ogawa, H. (2002). Differential responses of mast cell Toll‐like receptors 2 and 4 in allergy and innate immunity, Journal of Clinical Investigation, 109, 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto, H. , Nakae, S. , Kakurai, M. , Sedgwick, J. D. , Tsai, M. , & Galli, S. J. (2006). Mast cell‐associated TNF promotes dendritic cell migration, Journal of Immunology, 176, 4102–4112. [DOI] [PubMed] [Google Scholar]
- Theoharides, T. C. , Tsilioni, I. , & Ren, H. (2019). Recent advances in our understanding of mast cell activation ‐ or should it be mast cell mediator disorders? Expert Review of Clinical Immunology, 15, 639–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theoharides, T. C. , Kempuraj, D. , Tagen, M. , Conti, P. , & Kalogeromitros, D. (2007). Differential release of mast cell mediators and the pathogenesis of inflammation, Immunological Reviews, 217(1), 65–78. [DOI] [PubMed] [Google Scholar]
- Thery, C. , Duban, L. , Segura, E. , Veron, P. , Lantz, O. , & Amigorena, S. (2002). Indirect activation of naive CD4+ T cells by dendritic cell‐derived exosomes, Nature Immunology, 3, 1156–1162. [DOI] [PubMed] [Google Scholar]
- Trivedi, N. H. , Guentzel, M. N. , Rodriguez, A. R. , Yu, J‐J. , Forsthuber, T. G. , & Arulanandam, B. P. (2013). Mast cells: Multitalented facilitators of protection against bacterial pathogens, Expert Review of Clinical Immunology, 9(2), 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turiak, L. , Ozohanics, O. , Marino, F. , Drahos, L. , & Vekey, K. (2011). Digestion protocol for small protein amounts for nano‐HPLC‐MS(MS) analysis, Journal of Proteomics, 74, 942–947. [DOI] [PubMed] [Google Scholar]
- Valadi, H. , Ekstrom, K. , Bossios, A. , Sjostrand, M. , Lee, J. J. , & Lotvall, J. O. (2007). Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells, Nature Cell Biology, 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Van Deun, J. , Mestdagh, P. , Sormunen, R. , Cocquyt, V. , Vermaelen, K. , Vandesompele, J. , … Hendrix, A. (2014). The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling, Journal of Extracellular Vesicles, 3, 24858‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev, K. A. , Yukhneva, M. A. , Shurygina, A. S. , Stukova, M. A. , & Egorov, A. Y. (2018). Enhancement of the immunogenicity of influenza A virus by the inhibition of immunosuppressive function of NS1 protein, MIR Journal, 5(1), 48–58. [Google Scholar]
- Visnovitz, T. , Osteikoetxea, X. , Sodar, B. W. , Mihaly, J. , Lorincz, P. , Vukman, K. V. , … Buzas, E. I. (2019). An improved 96 well plate format lipid quantification assay for standardisation of experiments with extracellular vesicles, Journal of Extracellular Vesicles, 8, 1565263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukman, K. V. , Adams, P. N. , Metz, M. , Maurer, M. , & O'Neill, S. M. (2013). Fasciola hepatica tegumental coat impairs mast cells' ability to drive Th1 immune responses, Journal of Immunology, 190, 2873–2879. [DOI] [PubMed] [Google Scholar]
- Vukman, K. V. , Forsonits, A. , Oszvald, A. , Toth, E. A. , & Buzas, E. I. (2017). Mast cell secretome: Soluble and vesicular components, Seminars in Cell & Developmental Biology, 67, 65–73. [DOI] [PubMed] [Google Scholar]
- Vukman, K. V. , Ravida, A. , Aldridge, A. M. , & O'Neill, S. M. (2013). Mannose receptor and macrophage galactose‐type lectin are involved in Bordetella pertussis mast cell interaction, Journal of Leukocyte Biology, 94, 439–448. [DOI] [PubMed] [Google Scholar]
- Vukman, K. V. , Visnovitz, T. , Adams, P. N. , Metz, M. , Maurer, M. , & O'Neill, S. M. (2012). Mast cells cultured from IL‐3‐treated mice show impaired responses to bacterial antigen stimulation, Inflammation Research, 61, 79–85. [DOI] [PubMed] [Google Scholar]
- Wernersson, S. , & Pejler, G. (2014). Mast cell secretory granules: Armed for battle, Nature Reviews Immunology, 14, 478–494. [DOI] [PubMed] [Google Scholar]
- Wierzbicki, M. , & Brzezinska‐Blaszczyk, E. (2009). Diverse effects of bacterial cell wall components on mast cell degranulation, cysteinyl leukotriene generation and migration, Microbiology and Immunology, 53, 694–703. [DOI] [PubMed] [Google Scholar]
- Yu, L. , Wang, L. , & Chen, S. (2010). Endogenous toll‐like receptor ligands and their biological significance, Journal of Cellular and Molecular Medicine, 14(11), 2592–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, T. , He, Z. , Yang, M. , Song, J. , Ma, C. , Ma, S. , … Li, J. (2018). The development of methods for primary mast cells in vitro and ex vivo: An historical review, Experimental Cell Research, 369(2), 179–186. [DOI] [PubMed] [Google Scholar]
- Zimmermann, C. , Troeltzsch, D. , Giménez‐Rivera, V. A. , Galli, S. J. , Metz, M. , Maurer, M. , & Siebenhaar, F. (2019). Mast cells are critical for controlling the bacterial burden and the healing of infected wounds, Proceedings of the National Academy of Sciences of the United States of America, 116(41), 20500–20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Supporting Video S1.