

Abstract
Transient-receptor-potential cation channel, subfamily M, member 3 (TRPM3) serves as a polymodal calcium sensor in diverse mammalian cell-types. Mutation of the human TRPM3 gene (TRPM3) has been linked with inherited forms of early-onset cataract with or without other eye abnormalities. Here we have characterized the ocular phenotypes of germline ‘knock-in’ mice that harbor a human cataract-associated isoleucine-to-methionine mutation (p.I65M) in TRPM3 (Trpm3-mutant) compared with germline ‘knock-out’ mice that functionally lack TRPM3 (Trpm3-null). Despite strong expression of Trpm3 in lens epithelial cells, neither heterozygous (Trpm3+/−) nor homozygous (Trpm3−/−) Trpm3-null mice developed cataract; however, the latter exhibited a mild impairment of lens growth. By contrast, homozygous Trpm3-M/M mutants developed severe, progressive, anterior pyramid-like cataract with microphthalmia, whereas, heterozygous Trpm3-I/M and hemizygous Trpm3-M/- mutants developed anterior pyramidal cataract with delayed onset and progression - consistent with a semi-dominant lens phenotype. Histochemical staining revealed abnormal accumulation of calcium phosphate-like deposits and collagen fibrils in Trpm3-mutant lenses and immunoblotting detected increased αII-spectrin cleavage products consistent with calpain hyper-activation. Immunofluorescent confocal microscopy of Trpm3-M/M mutant lenses revealed fiber cell membrane degeneration that was accompanied by accumulation of alpha-smooth muscle actin positive (α-SMA+ve) myofibroblast-like cells and macrosialin positive (CD68+ve) macrophage-like cells. Collectively, our mouse model data support an ocular disease association for TRPM3 in humans and suggest that (1) Trpm3 deficiency impaired lens growth but not lens transparency and (2) Trpm3 dysfunction resulted in progressive lens degeneration and calcification coupled with pro-fibrotic (α-SMA+ve) and immune (CD68+ve) cell responses.
Keywords: TRP-channel, lens, cataract, calcium, myofibroblast, macrophage
1. Introduction
The melastatin sub-family of transient receptor potential cation channels comprises eight members (TRPM1–8) that share sequence homology with the founder member TRPM1 (melastatin 1) - a diagnostic and prognostic marker for primary cutaneous metastatic melanoma and a causative gene for night-blindness [1, 2]. TRPM3 (melastatin 2), which is the most closely related melastatin channel to TRPM1 (~57% amino acid identity), was first isolated from human kidney and mouse brain [3–6] and has been shown to function as a spontaneous calcium ion (Ca2+) channel with permeability to other cations influenced by alternative splicing [5–7]. Like voltage-gated ion channels, TRPM3-channels share a transmembrane topology of tetramers around a central pore that can be activated by several physico-chemical stimuli including hypotonic extracellular stress, the neuro-steroid pregnenolone sulfate, heat, and cell-membrane phosphoinositol phosphates [4, 8–11]. TRPM3-channels are widely expressed in neuronal (e.g., dorsal root ganglia) and non-neuronal (e.g., epithelia) mammalian tissues and have been implicated in critical physiological functions including insulin secretion [8], noxious heat detection [12, 13], vasoconstriction [14], vasodilation [15], and inflammatory pain [16].
Genetic variants of the human TRPM3 gene (TRPM3) have been associated with several systemic conditions or diseases. Non-coding TRPM3 variants have been tentatively associated with longevity, elevated low-density lipoproteins and triglycerides, systemic sclerosis, aspirin-exacerbated respiratory disease, chronic fatigue syndrome and thyroid nodules [17–22]. Rare deletions involving coding exons of TRPM3 have been documented in Kabuki syndrome and in autism [23, 24]. Recently, de novo coding variants in TRPM3 have been functionally implicated in developmental delay and intellectual disability that was variably associated with epilepsy, hypotonia, joint dislocations, and oculomotor disorders [25–28]. By contrast, a non-coding TRPM3 gene variant found in the whippet breed of racing dogs has been strongly associated with increased racing speed [29].
In addition to systemic phenotypes, TRPM3 gene variants have been associated with eye abnormalities and disease. In mice, disruption of the TRPM3 gene (Trpm3) resulted in an attenuated pupillary response to light [30]. By contrast, in humans coding mutations in TRPM3 have been linked with inherited forms of early-onset or pediatric cataract with or without glaucoma and anterior eye abnormalities [31–33]. Here, we compare and contrast the ocular phenotype of germline knock-in mice harboring a human cataract-linked coding mutation (p.Ile65Met, p.I65M) in TRPM3 (Trpm3-mutant) with that of germline knock-out mice functionally lacking TRPM3 (Trpm3-null).
2. Materials and Methods
2.1. Gene-targeted mice
Trpm3-mutant mice were generated by clustered regularly interspersed short palindromic repeats and CRISPR-associated protein 9 (CRISPR/Cas9) gene-editing technology in our Genome Engineering and iPSC Center (geic.wustl.edu). Briefly, guide RNAs (gRNAs) were designed flanking the target-site and selected based on minimum off-target sites (in silico) and distance from target-site. A donor single-stranded oligo-deoxynucleotide (ssODN) was designed to introduce a missense transversion (c.195T>G) into exon-4 of Trpm3 to replicate the disease mutation found in humans [31]. Sequences of the gRNAs and ssODN were as follows: Trpm3.g2, 5′-tgtttcaggctcagaaatccngg; Trpm3.g6, 5′-ataaaatgctctttcaatccngg; Trpm3.ssODN, 5’-atg ggg gtc ttt ggt gct cgg tat gat gtg aac aca ttc tct ttt ata aaa tgc tct ttc Cat cca aga ctt ctg agc ctg aaa caa aac gag aga gag aga aaa aaa gat gaa tat aaa ttt taa atc t-3’. The gRNAs and ssODN were validated following nucleofection of mouse Nura2a cells using the Cel-1 endonuclease mismatch detection assay [34] to measure non-homologous end joining frequency (% NHEJ) and then by next-generation sequencing (MiSeq, Illumina) to measure homology directed repair frequency (% HDR), respectively. Germline-targeted knock-in mice were generated using standard protocols in our Mouse Embryo Stem (ES) Cell Core facility (https://mescc.wustl.edu/). Briefly, gRNAs, donor oligo, and nuclease expression vector were injected at the blastocyst stage of B6JxCBA hybrid mice then implanted into pseudo-pregnant foster females. Offspring were screened by PCR amplification and Sanger sequencing to identify correctly edited founders (F0) that were subsequently maintained on a predominantly C57BL6/J genetic background (B6J, Jackson Laboratory, JAX, Bar Harbor, ME, USA), in part, to rescue a deletion mutation in the gene coding for lens beaded filament structural protein-2 (Bfsp2, CP49) that is carried by certain mouse strains (e.g., 129) [35–37].
Heterozygous Trpm3-null (Trpm3+/−) mice (International Strain Designation B6;129S5-Trpm3tm1Lex/Ori, Common strain name LEXKO-380) were obtained from the European Mouse Mutant Archive (EMMA; www.emmanet.org) as cryopreserved embryos (EMMA ID - EM:02333), and re-derived using standard thawing and implantation techniques. Pigmented founders were subsequently maintained on a predominantly B6J background as for Trpm3-mutant mice above, whereas, albino founders were maintained on a congenic albino B6J background (B6(Cg)-Tyrc−2J/J, JAX).
Mice were humanely killed according to American Veterinary Medical Association (AVMA) guidelines and the eyes removed. Whole eyes or dissected lenses bathed in phosphate buffered saline (PBS, #P4417–100TAB, MilliporeSigma, Burlington, MA, USA) were imaged under a dissecting microscope (Stemi 2000; Zeiss, Thornwood, NY, USA) fitted with a digital camera (Spot Insight, Sterling Heights, MI, USA). All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University in compliance with the Institute for Laboratory Animal Research (ILAR) guidelines.
2.2. Polymerase chain reaction (PCR) amplification and sequencing
For mouse genotyping, genomic DNA was isolated from ear biopsy and PCR-amplified using gene-specific primers (Table 1, Supplemental Fig. 1) as described [38)]. For, reverse-transcript (RT)-PCR, total RNA was isolated from lens pairs, stored (−20°C) in RNALater stabilizing solution (ThermoFisher Scientific, Waltham, MA, USA), using the RNeasy Plus Micro kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA (~1 μg) was reverse transcribed using the iScript cDNA synthesis kit then PCR-amplified in iQ SYBR Green Supermix with allele-specific primers (Table 1) using an iQ5 Real-time PCR cycler running iQ5 software (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Sanger sequencing was performed using fluorescent dideoxy-terminator chemistry as described [39]. Briefly, genomic DNA (2.5 ng/μl, 10 μl reactions) or cDNA (~10% of RT reaction) was PCR-amplified (35 cycles) in a GeneAmp 9700 thermal cycler (Applied Biosystems, Grand Island, NY) using Top Taq mastermix kit (Qiagen) and gene-specific primers (Table 1). Resulting PCR amplicons were enzyme-purified with ExoSAP-IT (USB Corporation, Cleveland, OH, USA) and direct cycle-sequenced in both directions with M13 forward or reverse sequencing primers and the BigDye Terminator v3.1 kit (Applied Biosystems) followed by capillary electrophoresis on a 3130xl Genetic Analyzer running Sequence Analysis (v.6.0) software (Applied Biosystems) and Chromas (v2.6) software (Technelysium, Tewantin, Queensland, Australia).
Table 1.
Primer sequences used for PCR-amplification and Sanger sequencing of Trpm3.
| Primer name | Location | Strand | Sequence (5’ - 3’) | Amplicon (bp) | Target |
|---|---|---|---|---|---|
| Mm Trpm3 Ex4F* | Intron 3 | Sense | tgtaaaacgacggccagtACATGATGTTGCTGCCTAGC | 250 | Exon 4 |
| Wt Trpm3 I65 F | Exon 4 | Sense | CAGGCTCAGAAATCCTGGATT | 193 | I65 allele |
| Mt Trpm3 M65 F | Exon 4 | Sense | CAGGCTCAGAAGTCTTGGATG | 193 | M65 allele |
| Mm Trpm3 Ex4R* | Intron 4 | Antisense | caggaaacagctatgaccGGTTCCATTTCAGTGTGTGG | 250/193 | Exon 4 |
| Lexko380–466F | Exon 21 | Sense | AGCTCCTGGACCAGTCCTAC | 466 | Exon 21_wild-type |
| Neo3a | β-geo | Sense | GCAGCGCATCGCCTTCTATC | 396 | β-geo_LexKO380 |
| Lexko380–6 | Intron 21 | Antisense | CCTCAAGACTCCCAATCCAAG | 466/396 | Exon 21_LexKO380 |
| Trpm3 RT20F | Exon 20 | Sense | GGAGAGGACGGAAGACAAC | 551 | Exon 21_LexKO380 (RNA) |
| Trpm3 RT22R | Exon 22 | Antisense | GAACTCCAAGCTGAGAATTGAAG | Exon 21_LexKO380 (RNA) |
M13 ‘tail’ sequencing primer (18-mer) shown in lowercase font
2.3. β-galactosidase reporter staining
β-galactosidase (β-gal) activity of the lacZ reporter gene was detected in situ using the X-Gal Staining Assay Kit (Genlantis, San Diego, CA, USA). Briefly, eyes were fixed in formaldehyde-glutaraldehyde buffer (2 × 1 hr, 20°C) then incubated with X-gal staining solution (2 hr, 20°C).
Stained eyes were subjected to standard formaldehyde-fixed paraffin-embedding (FFPE) and serial sectioning (4 μm) with a microtome (RM2255, Leica Microsystems, Buffalo Grove, IL, USA) then dewaxed (xylene/alcohol series), counterstained (5 min, 20°C) with Nuclear Fast Red solution (0.1% wt/vol in 5% wt/vol aluminium sulfate in distilled water, MilliporeSigma), mounted (Cytoseal XYL media, ThermoFisher), and imaged with an upright BX61 microscope fitted with a digital camera (Olympus, Center Valley, PA, USA).
2.4. Histochemistry
Enucleated eyes were fixed (16–24 hr., 4°C) in 4% paraformaldehyde (#15710, Electron Microscopy Sciences, EMS, Hatfield, PA, USA) diluted in PBS unless otherwise stated below then subjected to standard FFPE and serial sectioning (4 μm, saggital plane) with a microtome (RM2255, Leica). Dewaxed and hydrated sections (xylene/alcohol/water series) were stained as below, then alcohol-dehydrated, xylene-cleared, mounted (Cytoseal XYL) and imaged with a BX61 microscope (Olympus). For hematoxylin and eosin (H&E) staining of nuclei and cytoplasm, respectively, FFPE sections were treated with Harris hematoxylin (pH 2.3, VWR, Radnor, PA, USA) for 5 min and then with eosin alcohol (pH 4.1, VWR) for 10 sec −1 min.
For staining of mineralization, we used the von Kossa precipitation reaction in which calcium cations (Ca2+) in calcium salts (e.g., calcium phosphate) are replaced under acidic conditions with silver ions that upon photochemical (UV) reduction form dark-brown or black metallic silver deposits [40, 41]. Briefly, FFPE sections were incubated in silver nitrate solution (1% wt/vol in distilled water, MilliporeSigma) and exposed to bright sunlight (1–2 hr), washed (5 min, 20°C) in sodium thiosulfate solution (5% wt/vol in distilled water, MilliporeSigma) to remove un-reacted silver, then counterstained (5 min, 20°C) with Nuclear Fast Red solution (EMS). For calcification, we stained with the anthraquinone dye alizarin red S that reacts with calcium ions to form a bright orange or red chelate [40, 42]. Briefly, eyes were fixed in 10% neutral buffered formalin (ThermoFisher) and FFPE sections stained (1–5 min, 20°C) with alizarin red S solution (2% wt/vol sodium alizarin sulphonate in distilled water, ACROS Organics/ThermoFisher) adjusted to pH 4.1~4.3 with ammonium hydroxide solution (10% wt/vol in distilled water). Collagen staining was performed using the Picrosirius Red Stain Kit (Polysciences Inc., Warrington, PA, USA) according to the manufacturer’s protocol. Picrosirius red is a linear anionic dye with six acidic sulfonate groups that can react with the basic cationic groups along collagen fibrils [43, 44]. Briefly, FFPE sections were stained with Harris hematoxylin (8 min), rinsed (H2O), placed for 2 min in Solution A (phosphomolybdic acid), then for 1 hr in Solution B (0.1% Sirius red F3B - C.I. 35782 - in saturated aqueous picric acid), acid-rinsed (2 min) in Solution C (0.1 N HCL), and ethanol-rinsed (70%, 45 sec).
2.5. Immunoblot analysis
Immunoblot analysis of αII-spectrin cleavage products was performed essentially as described [38, 45]. Briefly, dissected lenses preserved (−20° C) in RNAlater stabilizing solution (Invitrogen/ThermoFisher) were re-suspended in PBS (50 ul per lens pair) containing protease inhibitor (Halt, ThermoFisher) using a Bullet Blender (Next Advance, Troy, NY, USA) tissue homogenizer (2 min, setting 8) then centrifuged (10,000 × g, 5 min) to pellet cell-nuclei. Post-nuclear lysate was mixed with an equal volume of Laemmli buffer (Bio-Rad) and soluble protein concentration was measured (480 nm) with the non-interfering assay (G-Bioscience, St. Louis, MO, USA). Lens soluble proteins (~ 25 ug/lane) were adjusted to 5 mM DTT (100 mM stock, Sigma), denatured (95° C, 5 min), separated by SDS-PAGE (4–12% gradient gels, 1 × 3-[N-morpholino]propanesulfonic acid/MOPS NuPage running buffer (150 V, 2 hr, Novex/Invitrogen), then blotted (30 V, 2 hr, NuPage transfer buffer + 10% methanol) onto nitrocellulose (Novex) using an XCell SureLock Mini-Cell electrophoresis system and XCell II blot module (Novex/ThermoFisher). Air-dried blots were blocked (30–60 min, Odyssey Block (OB) solution, Li-Cor, Lincoln, NE, USA) then sequentially incubated (16 hr, 4° C, OB solution + 0.1% Tween 20, OBT) with primary antibodies to α-spectrin (1:1000, MAB1622, MilliporeSigma) and β-actin (1:5000, #3700, Cell Signaling Technology, CST, Danvers, MA, USA) followed by secondary antibody (1:25,000, goat-anti-mouse IRD680LT in OBT, Li-Cor), rinsed (4 × 5 min PBST, 1 × 5 min PBS), air-dried (30–60 min), and visualized with an Odyssey Infrared Imaging System (Li-Cor).
2.6. Immunofluorescence confocal microscopy
Enucleated eyes were processed using standard FFPE-section or cryo-section techniques and immuno-localization performed as described (38, 46). Primary antibodies used were, anti-aquaporin 0 (anti-AQP0, AB3071, EMD Millipore, Billerica, MA, USA), anti-alpha smooth muscle actin (α-SMA, ab5694, Abcam, Cambridge, MA, USA), and Alexa Fluor-488 anti-mouse cluster of differentiation 68 antigen (anti-CD68, #137011, Biolegend, San Diego, CA, USA). Briefly, for anti-AQP0 and anti-αSMA localization, eyes were fixed (16–24 hr., 4°C) in 4% paraformaldehyde (#15710, EMS) diluted in PBS and FFPE-sections (4 μm) cut in the sagittal plane using a microtome (RM2255, Leica). For anti-CD68 localization, eyes were fixed as above and cryo-protected by serial incubation (16 hr) in 15% and 30% sucrose/PBS, then embedded in Tissue-Tek O.C.T. compound (#62550–01, EMS) and sagittal cryo-sections (15 μm) cut using a cryostat (Cryotome E, ThermoFisher). Eye sections were permeabilized (0.1% Triton X100/PBS 10 min.), blocked (1 hr., 20°C) in Image-iT FX Signal Enhancer (ThermoFisher) then incubated with primary antibody (16 hr, 4°C). Sections for anti-AQP0 and anti-αSMA labeling were further incubated in Alexa Fluor 488-conjugated secondary antibody (1 hr., 20°C). Cell nuclei were stained (15 min, 20°C) with DAPI (MilliporeSigma). Images were captured with a confocal microscope (FV1000, Olympus) and managed in Photoshop (Adobe Systems, San Jose, CA, USA).
2.7. Statistical analysis
One-way analysis of variance (ANOVA) was used to determine statistical significance (p) ± standard deviation of lens size.
3. RESULTS
3.1. Trpm3-null mice develop transparent lenses of reduced size
To model the effects of TRPM3 deficiency on lens phenotype, we re-derived germline Trpm3-null mice that were generated previously using homologous recombination to disrupt Trpm3 by introducing a plasmid-construct containing a β-galactosidase-(lacZ)-neomycin (β-geo) fusion cassette flanked by a 5′- internal ribosome entry site (IRES) [12] (Fig. 1A). According to the Genome Reference Consortium Mouse Build 38 patch release 6 (GRCm38.p6) curated by the National Center for Biotechnology Information (NCBI) Gene database - mouse (C57BL/6J) Trpm3 (Gene ID: 226025) comprises at least 32 exons spanning ~ 0.86 Mb on murine chromosome 19 (19:22137797–22995410) and undergoes extensive alternative splicing to generate over 30 transcript variants and protein isoforms (https://www.ncbi.nlm.nih.gov/gene/226025) [33]. Genomic PCR amplification and Sanger sequencing confirmed that the β-geo cassette disrupted the boundary between exon-21 and intron-21 of Trpm3 (Fig. 1B). RT-PCR amplification and cDNA sequencing of lens transcripts revealed that insertion of the β-geo cassette resulted in abnormal Trpm3 mRNA transcript(s) characterized by (1) skipping of exon-21, (2) mis-splicing of exon-20 to exon-22 generating a reading frame-shift, and (3) introduction of a premature translation stop-codon (TAA) at the start of exon-22 (p.R700fsX1) (Fig. 1C). Since disruption of Trpm3 occurred at the exon/intron-21 boundary, upstream from the transmembrane ion-transport domain in exons 24–26 (Fig. 1A), all full-length channel isoforms will be disabled consistent with a functionally null allele (Trpm3−).
Figure 1. Gene-targeting strategy forTrpm3-mutant versus Trpm3-null mice.
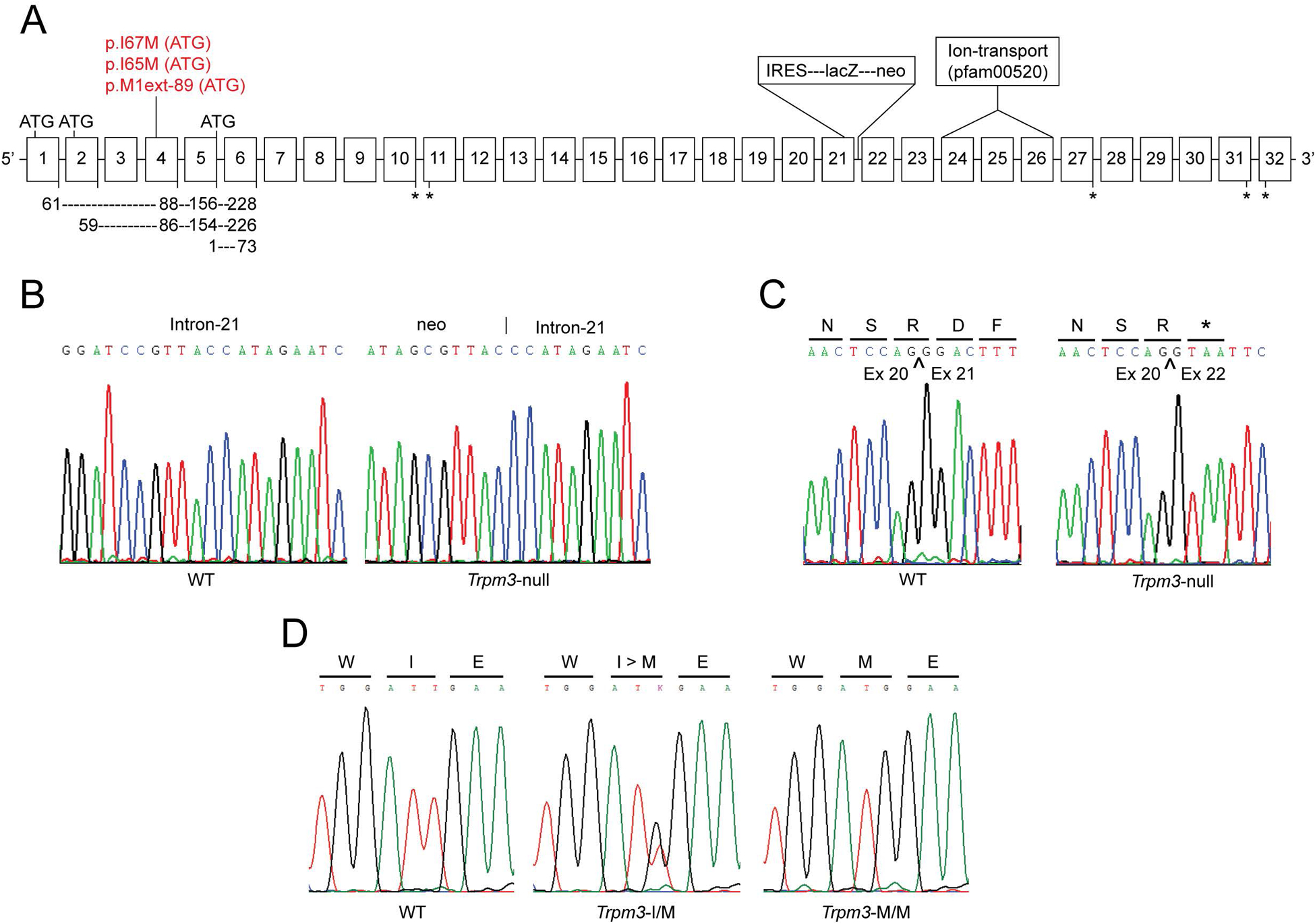
(A) Schematic showing exon organization (boxes 1–32) of Trpm3 and the relative locations of the p.I>M substitution in Trpm3-mutant mice and the β-geo fusion cassette in Trpm3-null mice. ATG denotes alternative translation start-sites in exons 1, 2, 4, and 5. Depending on transcript variant, the I-to-M substitution represents missense changes (p.I65M, p.I67M) or a novel translation start-site (M1ext-89M) shown in red. Exons encoding the transmembrane cation channel domain (pfam00520) are indicated. Asterisks denote alternative translation stop-sites. (B) Genomic DNA sequence showing the novel junction between the 3′-end of the β-geo cassette and intron −21. (C) cDNA sequence derived from RT-PCR amplification of lens RNA from wild-type (WT) and Trpm3-null (Trpm3−/−) mice showing that insertion of the β-geo cassette resulted in a mis-splicing frame-shift and premature termination site (p.R700fsX1). Bars under the single letter amino-acid code indicate the translation (codon) reading-frame. (D) Genomic DNA sequence of exon-4 showing germline knock-in of the p.I>M missense substitution in Trpm3-mutant mice. WT - wild-type (I/I), heterozygote (I/M), and homozygote (M/M).
While most of the re-derived founder (F0) mice were pigmented, several had a white coat with pink eyes and genomic sequencing confirmed that they were homozygous for the tyrosinase classic-2J (Tyrc−2J) albino mutation (c.291G>T, p.R77L) on chromosome 7 that first arose spontaneously (in 1970) in the C57 Black 6 (C57BL/6) inbred strain [47] (Supplemental Fig. 2). Subsequently, pigmented F0 mice were bred with pigmented B6J mice, whereas, albino F0 mice were crossed with congenic B6J albino mice (B6(Cg)-Tyrc−2J/J) to maintain the recessive B6-albino phenotype and enhance visualization of β-galactosidase reporter activity in ordinarily pigmented ocular tissues (see below). Both heterozygous Trpm3-null (Trpm3+/−) and homozygous Trpm3-null (Trpm3−/−) mice (pigmented and albino) were viable and fertile with no obvious physical or systemic abnormalities.
To confirm Trpm3 disruption at the protein translation level, and simultaneously localize Trpm3 expression in the lens, we undertook β-galactosidase (lacZ) reporter staining of eye sections from albino Trpm3−/− mice at post-natal day two (P2). β-galactosidase staining revealed strong expression of Trpm3 across the anterior epithelium of the lens (Fig. 2B). In addition, strong Trpm3 expression extended to the normally pigmented epithelial cell monolayers of the iris, ciliary body, and retina (Fig. 2C, D). These reporter expression data confirm disruption of Trpm3 resulting in loss of protein function particularly in the lens epithelium and pigmented ocular epithelia.
Figure 2. Ocular expression of Trpm3.
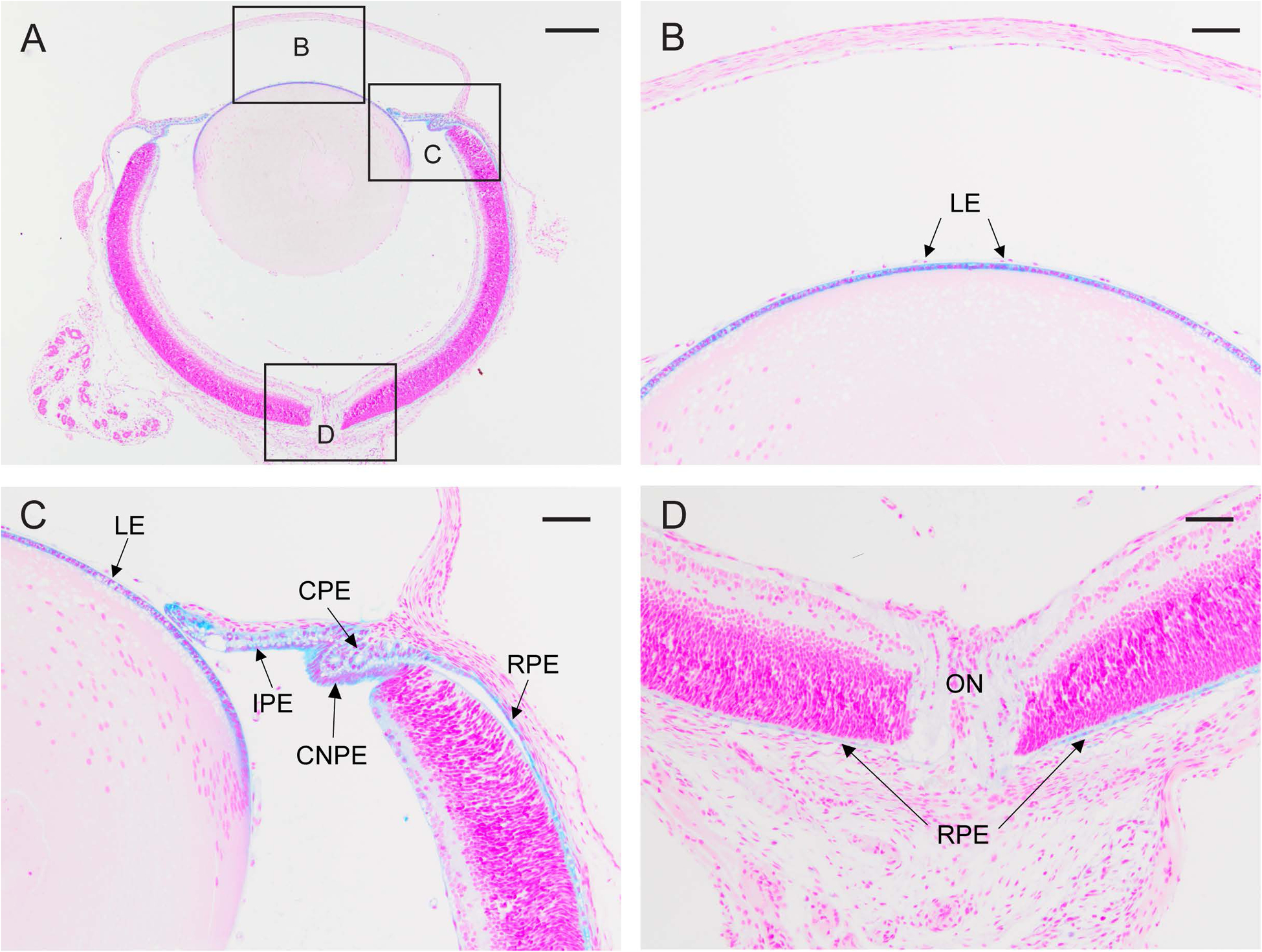
β-galactosidase reporter staining (blue) of the albino Trpm3-null eye (A) at P2 showing strong Trpm3 expression in the lens epithelium (LE) in high-magnification panels B and C, the ciliary (pigmented) epithelium (CPE), ciliary non-pigmented epithelium (CNPE), and iris (pigmented) epithelium (IPE) in panel C, and the retinal (pigmented) epithelium (RPE) in panels C and D. ON, optic nerve (D). A-D counterstained with nuclear fast red. Scale bar, 200 μm (A), 50 μm (B-D).
Enucleated eyes of Tprm3−/− mice were grossly similar to those of wild-type littermates; however, they often appeared slightly smaller than wild-type (Fig. 3A, B). Upon dissection, Trpm3−/− lenses were transparent (Fig. 3C, D); however, they were statistically smaller than wild-type (Fig. 4). Tprm3+/− eyes and lenses were grossly indistinguishable in appearance and size from wild-type (data not shown). Collectively, these observations suggest that absence (but not deficiency) of Trpm3 function impaired overall lens growth without loss of lens transparency.
Figure 3. Ocular phenotype of Trpm3-null mice.
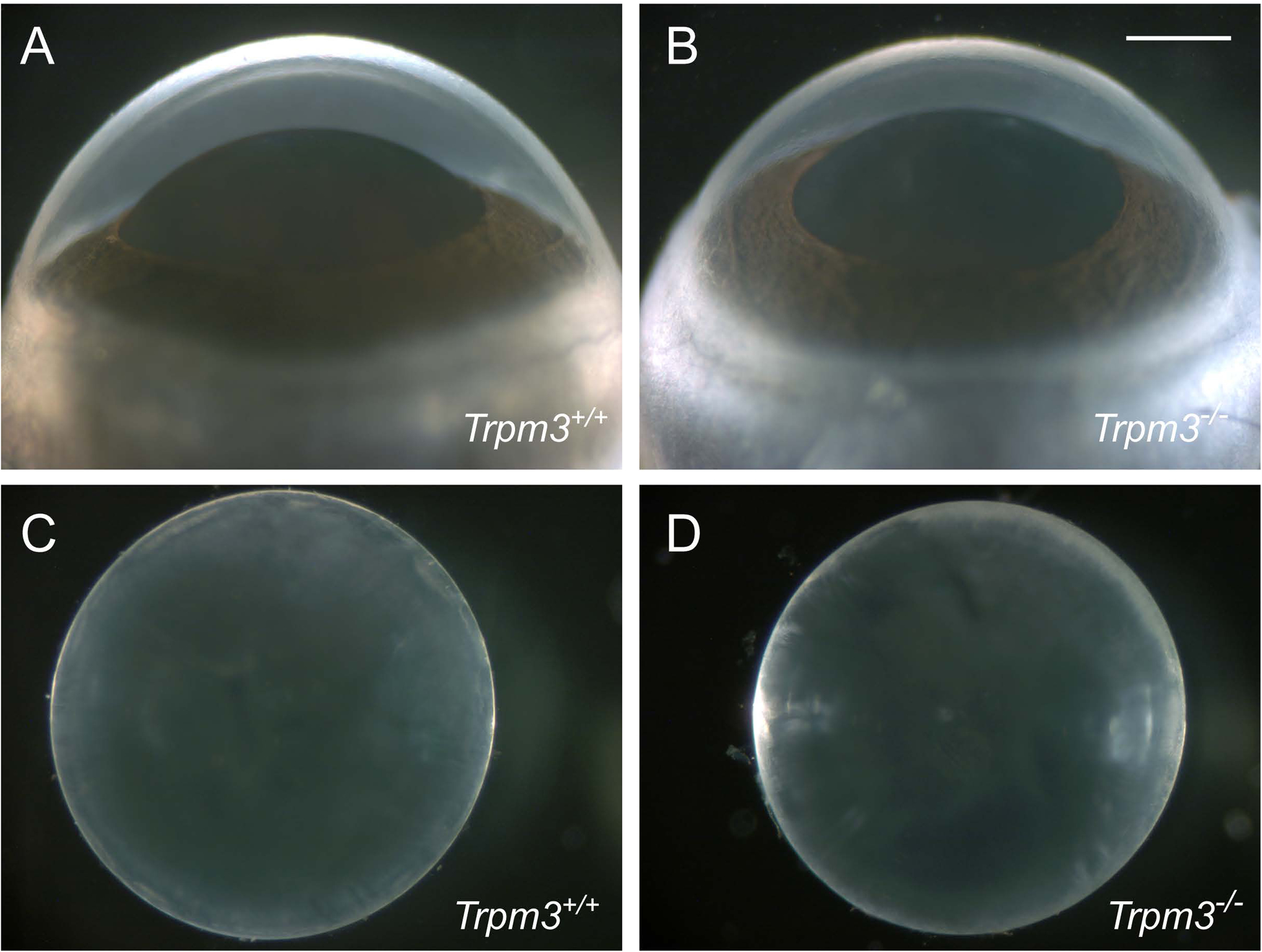
Representative dark-field images of eyes (A, B) and lenses (C, D) from wild-type (A, C) and Trpm3-null mice (B, D) at P28. Scale bar: 500 μm.
Figure 4. Lens size in Trpm3-null and Trpm3-mutant mice.
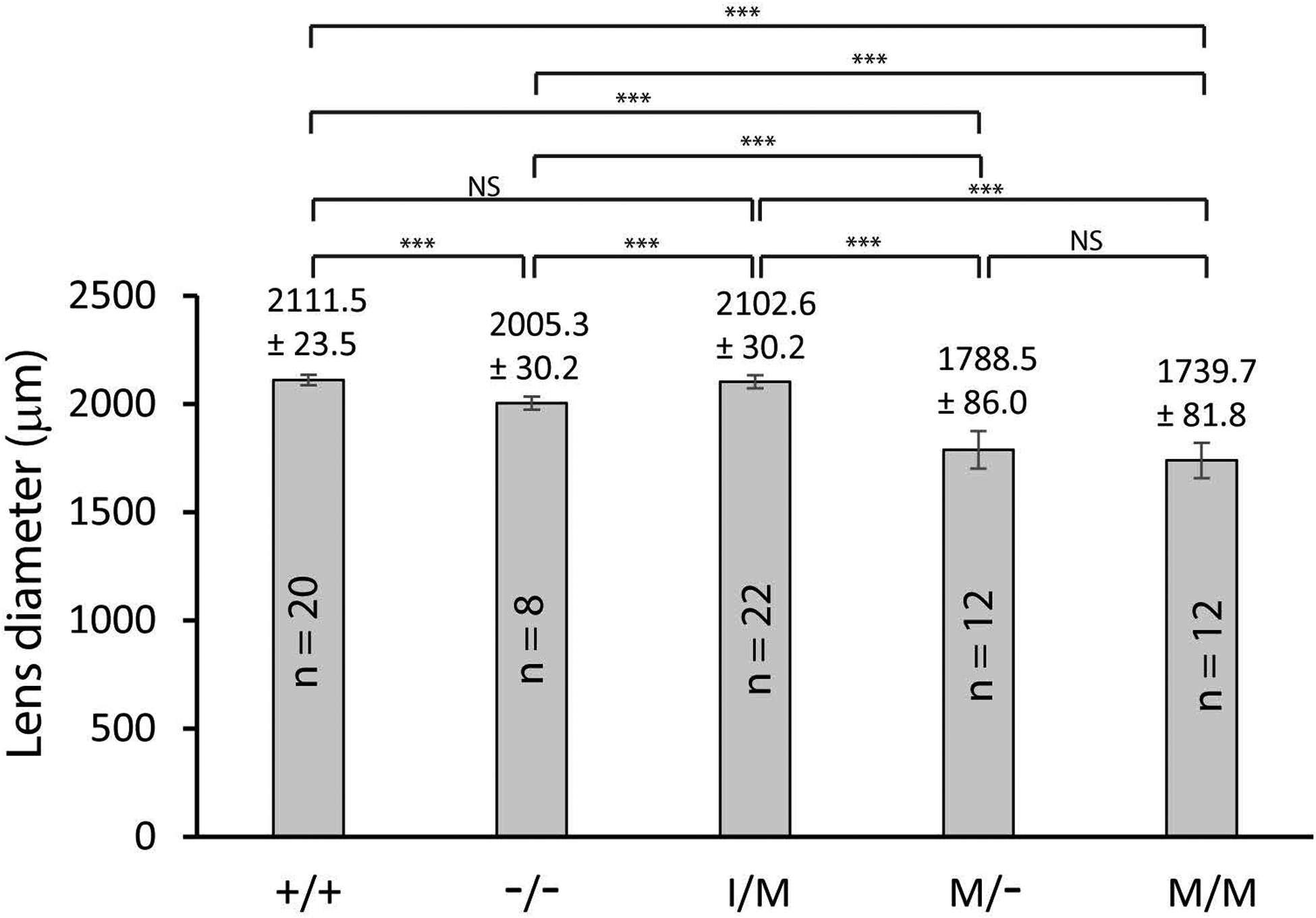
Bar graphs comparing the equatorial diameter of lenses (P28) from wild-type (+/+), Trpm3-null (−/−), and Trpm3-mutant (I/M, M/-, M/M) mice. Error bars represent standard deviation. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. NS, not significant.
3.2. Trpm3-mutant mice develop cataract
To model TRPM3 dysfunction, we used CRISPR-Cas9 gene-editing technology to generate a knock-in mouse mutant that inherits a human cataract-associated mutation (c.195A>G, p.Ile65Met or p.I65M) found in exon-4 of TRPM3 (chromosome 9q) at codon 65 (ATA>ATG) of transcript variant-9 (NM_001007471) coding for isoform-k (NP_001007472) [31, 33]. The equivalent mutation site in mice (c.195T>G, p.I65M) was located in exon-4 of Trpm3 at codon 65 (ATT>ATG) of transcript variant-23 (NM _001362504) that encodes isoform-w (NP_001349433). Genomic PCR-amplification and Sanger sequencing confirmed introduction of the c.195T>G transversion in 14 of 84 (16%) founder (F0) mice. However, four of these mice had additional deleterious ‘near-target’ sequence changes (Supplemental Fig. 3) leaving 10 (12%) correctly edited F0 mice (5 heterozygotes, 5 homozygotes). Subsequent sequencing of F0 offspring confirmed germline transmission of the c.195T>G mutation in exon-4 of Trpm3 (Fig. 1D).
According to the NCBI Gene database (https://www.ncbi.nlm.nih.gov/gene/226025) 29 of 30 Trpm3 RefSeq alternative transcript splice-variants include exon-4 either as a coding exon (20 transcripts) or a non-coding exon (9 transcripts). Trpm3 exons 1 and 2 are coding exons, whereas, exon-3 is non-coding. However, exons 1–3 are mutually exclusive, with 5 transcript variants containing exon-1, 15 variants containing exon-2, 7 variants containing exon-3, and two variants lacking exons 1–3. Translation of mutant transcripts (c.T>G in exon-4) that also contain either exon-1 (61 codons) or exon-2 (59 codons) is predicted to result in the relatively conservative amino-acid substitution of isoleucine (aliphatic) to methionine (sulfur-containing) at codon 67 (p.I67M) or at codon 65 (p.I65M), respectively, at the protein level (Fig. 1A). However, mutant transcripts that include exon-3 and exon-4 as non-coding exons, start translation 267 nucleotides downstream of the c.T>G mutation site (in exon-4) placing an alternative, in-frame, translation start-codon (ATT>ATG) 89 codons upstream from the consensus start-site (p.M1 located at the last codon of exon-5) that extends the amino (N)-terminal domain of at least seven TRPM3 isoforms by 89 novel amino acids (p.M1ext-89) (Fig. 1) [31, 33]. For convenience, however, we have designated mutant mice and lenses heterozygous and homozygous for the human cataract mutation as Trpm3-I/M and Trpm3-M/M, respectively, throughout. Further, we crossed Trpm3-M/M mice with Trpm3−/− mice to generate hemizygous Trpm3-mutant mice and lenses (Trpm3-M/-). Like Trpm3-null mice, Trpm3-mutant mice (I/M, M/-, and M/M) were viable and fertile without gross physical, systemic, or behavioral signs; however, Trpm3-M/M mutants often exhibited small eyes or microphthalmia (Fig. 5G).
Figure 5. Ocular phenotype of Trpm3-mutant mice.
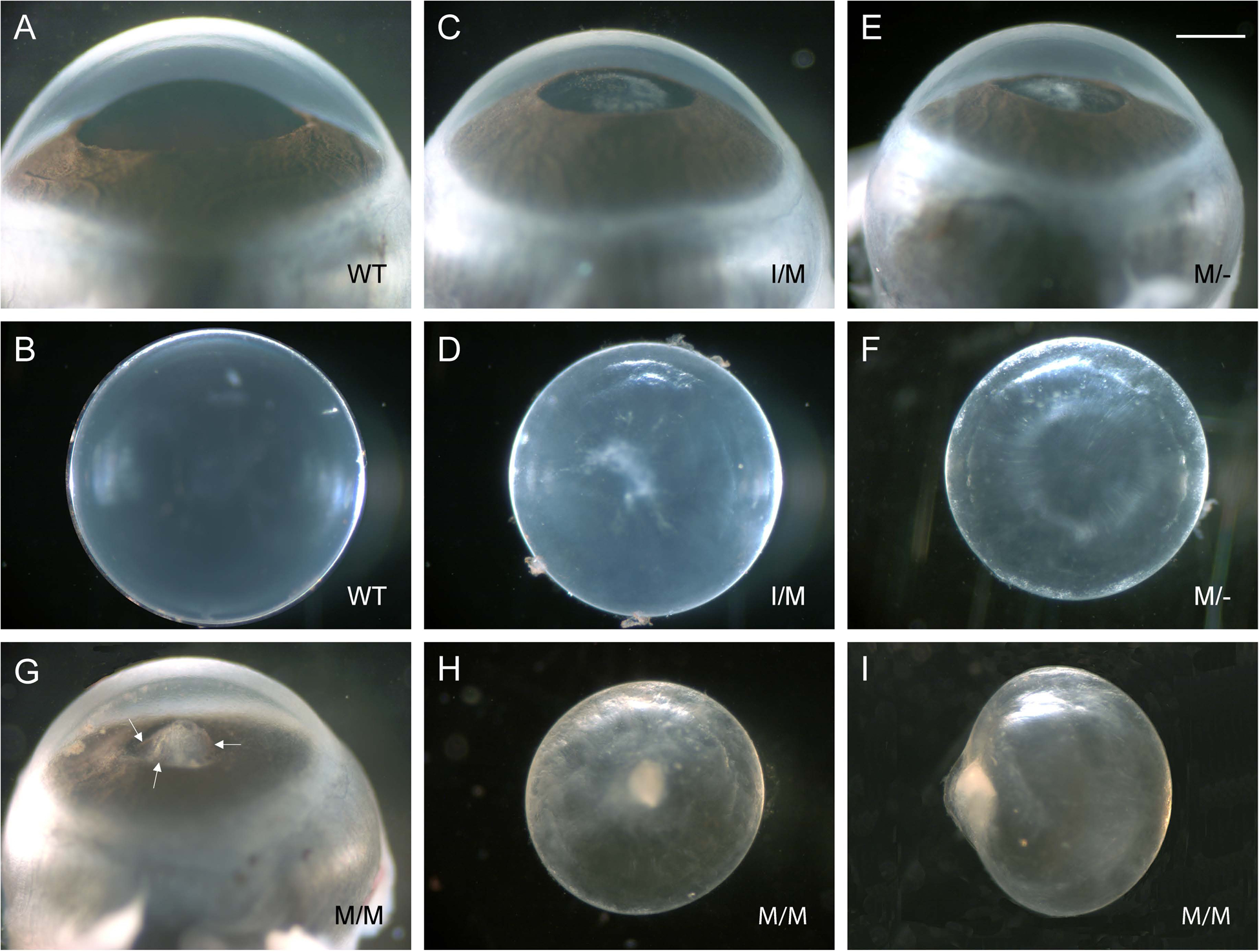
Representative dark-field images of eyes (A, C, E, G) and lenses (B, D, F, H, I) from wild-type (A, B), Trpm3-I/M (C, D), Trpm3-M/- (E, F), and Trpm3-M/M (G-I) mutant mice at P28 showing relatively mild anterior polar cataract in Trpm3-I/M and Trpm3-M/- mutant lenses (C-F) and more severe anterior pyramidal cataract in Trpm3-M/M mutant lenses (G-I). Arrows in panel G point to black-brown flecks of melanin pigment on anterior protrusion of Trpm3-M/M mutant lens. Scale bar: 500 μm.
Upon dissection at wean-age (P21-P28), Trpm3-M/M mutant lenses were small (microphakia) with dense anterior pyramid-shaped or pyramidal cataract that, in vivo, protruded from the anterior of the lens through the iris aperture or pupil into the anterior chamber of the eye, which is filled with aqueous humor (Fig. 5G–I). Often, the anterior pyramidal surface of Trpm3-M/M mutant lenses was punctuated with spots of black/brown pigment suggesting that they were melanin deposits derived from physical contact of the iris pigment epithelium with the protruding lens (Fig. 5G). Similarly, Trpm3-I/M and Trpm3-M/- mutant lenses developed an anterior polar opacification at P21-P28 (Fig. 5C–F) that usually progressed by three months of age into an anterior pyramidal cataract resembling that of Trpm3-M/M mutant lenses (see Fig. 6–8, 10). Whereas Trpm3-I/M mutant lenses were similar in size to wild-type, Trpm3-M/- and Trpm3-M/M mutant lenses were significantly smaller than wild-type – consistent with impaired lens growth (Fig. 4).
Figure 6. Histology of the Trpm3-mutant lens.
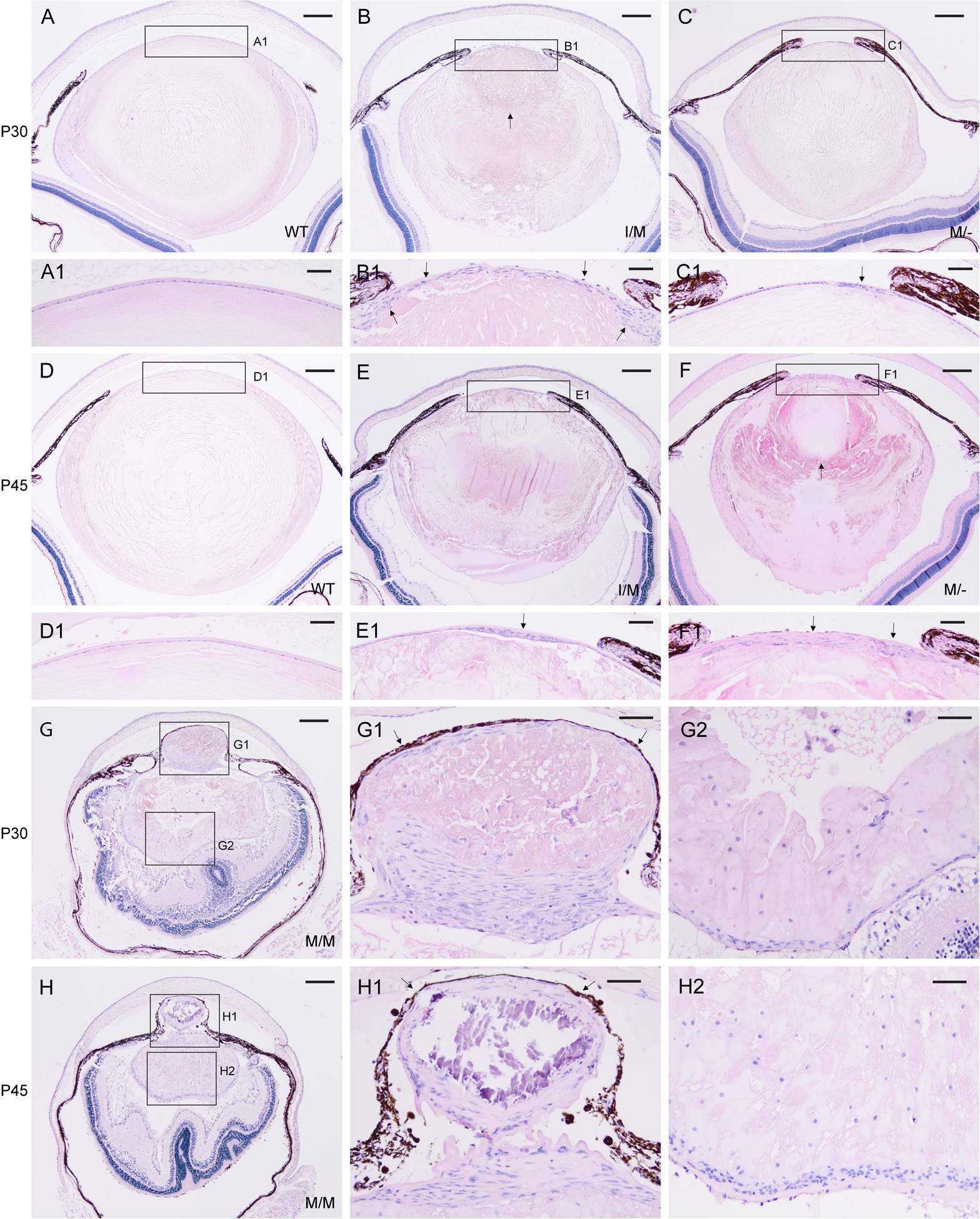
Representative H&E staining of lenses from wild-type (A, D), Trpm3-I/M (B, E), Trpm3-M/- (C, F), and Trpm3-M/M (G, H) mutant mice at P30 (A-C, G) and P45 (D-F, H) showing progressive lens degeneration and accumulation of nucleated cells (B, C, E, F, G, H, G1, G2, H1, H2). Arrows indicate abnormal multi-layering of the lens epithelium (B1, C1, E1, F1), anterior displacement of the lens nucleus (B, F), and iris or melanin pigment adhesion to the anterior lens capsule (G1, H1). In addition to cell nuclei, hematoxylin stained blue/black mineralization-like deposits (H1). Scale bar: 200 μm (A-H), 50 μm (A1–H1, G2, H2).
Histochemical (H&E) staining of FFPE eye sections revealed progressive lens degeneration in Trpm3-I/M, Trpm3-M/-, and Trpm3-M/M mutant mice at P30 and P45 (Fig. 6B, C, E, F, G, H). In addition, lens degeneration was variably accompanied by abnormal multi-layering of the anterior lens epithelium (Fig. 6B1, C1, E1, F1), accumulation of nucleated cells within and around the lens extending from the equator to the posterior pole (Fig. 6G2, H2), anterior displacement of the lens nucleus (Fig. 6F), and partial adhesion of the iris pigmented epithelium (or melanin pigment) to the anterior lens surface (Fig. 6G1, H1). While anterior cataract development was fully penetrant, Trpm3-mutant lenses of each genotype (I/M, M/-, M/M) displayed phenotypic variability in onset, progression, and severity. Taken overall, the Trpm3-mutant cataract was consistent with a semi-dominant lens phenotype associated with deleterious gain-of-function effects on the TRPM3 cation channel.
3.3. Trpm3-mutant lenses accumulate calcium deposits and hyper-activate calpain
In addition to cell nuclei, hematoxylin staining further revealed deep blue-black/purple deposits in the Trpm3-M/M mutant lens cytoplasm with sharply defined edges reminiscent of mineralized or calcified tissue (Fig. 6G, H) [40]. To establish the presence of mineralized or calcified tissue in the lens, we performed two classic histochemical staining methods used to detect bone mineralization. In Trpm3-M/M mutant lenses, von Kossa staining revealed strong, progressive, accumulation of black (silver) deposits particularly at the anterior and posterior poles (Fig. 7D, H), whereas, such staining was weak or subtle in Trpm3-I/M and Trpm3-M/- mutant lenses (Fig. 7B, C, F, G). Alizarin red S produced light-pink staining surrounding the core region or nucleus of wild-type lenses (Fig. 8A, E). In Trpm3-I/M and Trpm3-M/- mutant lenses, alizarin red S produced variegated perinuclear and nuclear staining that became progressively darker with age (P30-P45) extending to the whole lens (Fig. 8B, C, F, G). By contrast, alizarin red S staining of Trpm3-M/M mutant lenses revealed progressive accumulation of dark-red chelate deposits again concentrated at the anterior and posterior poles (Fig. 8D, H).
Figure 7. Mineralization of the Trpm3-mutant lens.
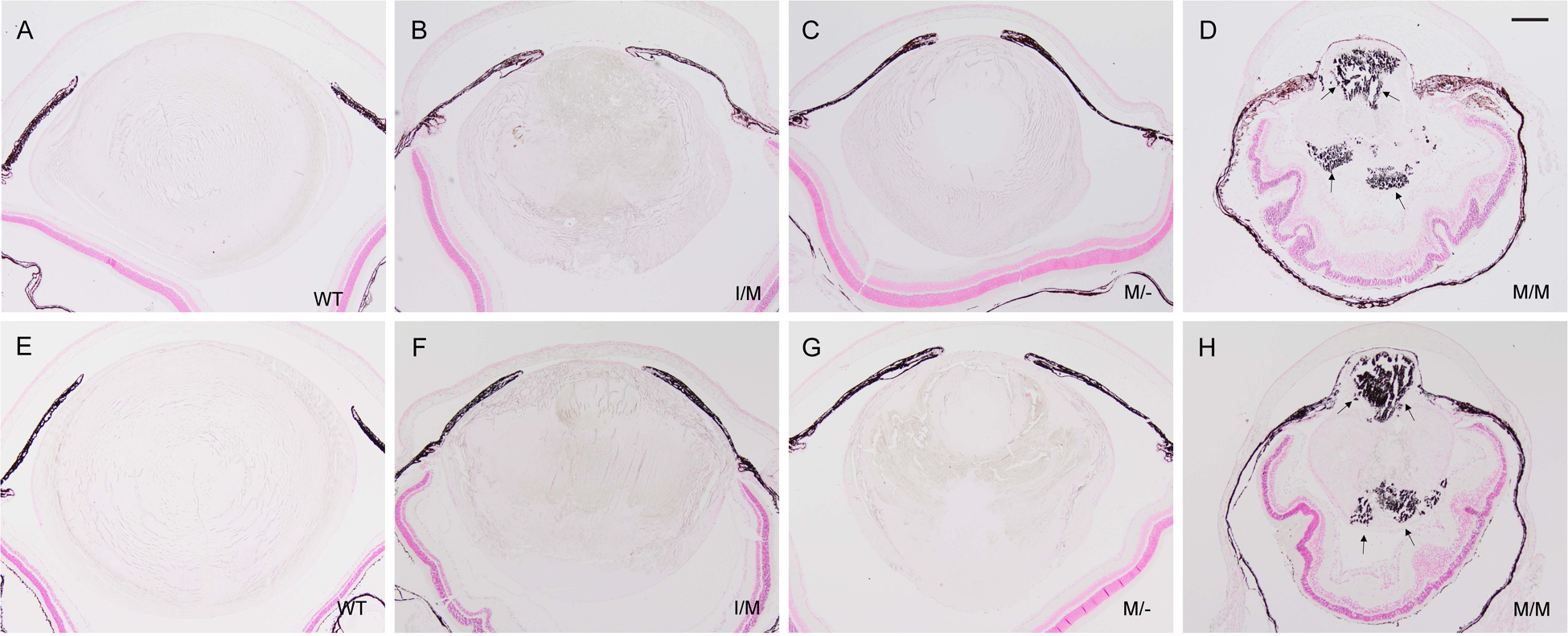
Representative von Kossa (silver) staining of lenses from wild-type (A, E), Trpm3-I/M (B, F), Trpm3-M/- (C, G), and Trpm3-M/M (D, H) mutant mice at P30 (A-D) and P45 (E-H) showing progressive accumulation of calcium phosphate-like (black) deposits (D, H arrows). A-H counterstained with nuclear fast red. Note, the iris anterior to the lens (A-H) and the retinal pigment epithelium posterior to the lens (D, H) appear dark brown/black due to melanin pigmentation. Scale bar: 200 μm.
Figure 8. Calcification of the Trpm3-mutant lens.
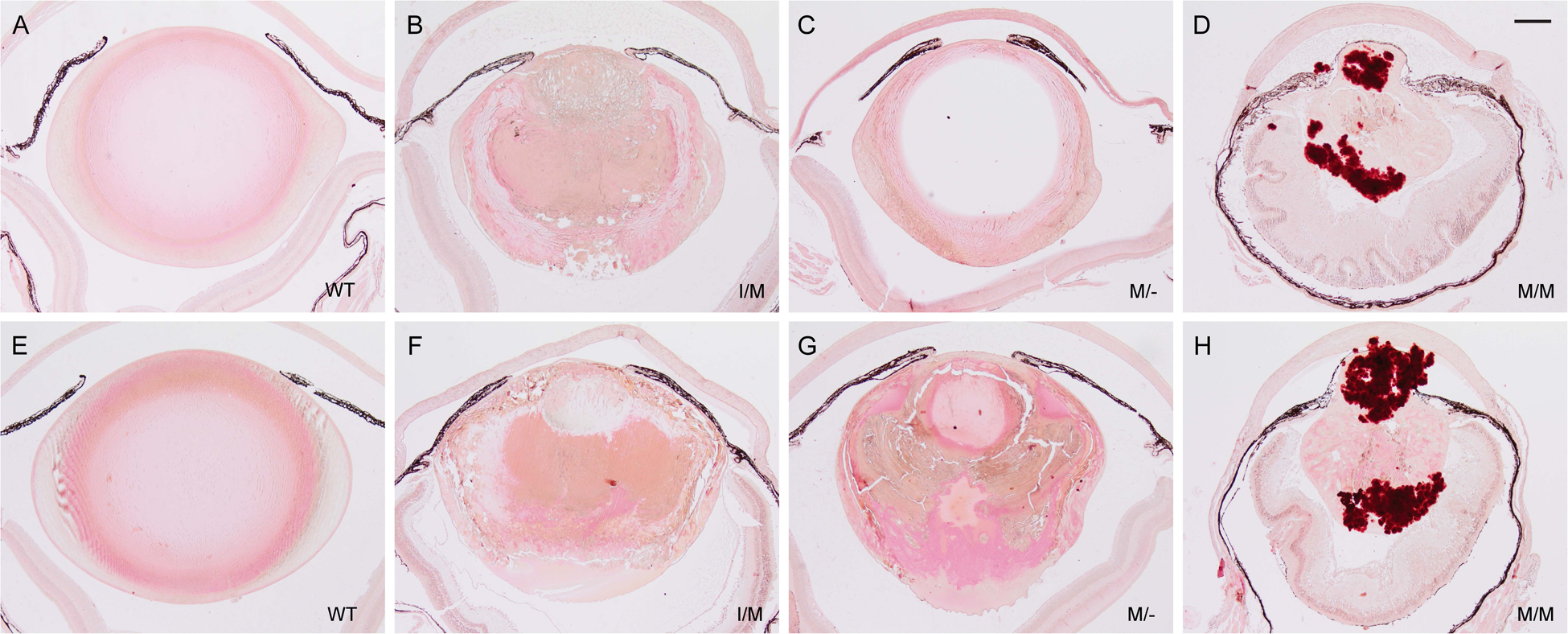
Representative alizarin red S staining of lenses from wild-type (A, E), Trpm3-I/M (B, F), Trpm3-M/- (C, G), and Trpm3-M/M (D, H) mutant mice at P30 (A-D) and P45 (E-H) showing progressive accumulation of calcium (red-orange) deposits (D, H). Scale bar: 200 μm.
Since Ca2+ accumulation was evident in the Trpm3-mutant lenses, we sought to determine if cytosolic calcium-activated cysteine protease, or calpain, activity was elevated. Immunoblot analysis revealed that calpain-specific cleavage of αII-spectrin to a C-terminal fragment of 145 kDa was increased in Trpm3-I/M and Trpm3-MM mutant lenses compared to Trpm3-null and wild-type lenses (Fig. 9). Further, uncleaved αII-spectrin and a calpain-cleavage product of 150 kDa were reduced in Trpm3-M/M mutant lenses compared to Trpm3-I/M mutant, Trpm3-null, and wild-type lenses. Smaller αII-spectrin cleavage products (< 100 kDa) were also more abundant in Trpm3-M/M mutant lenses than in Trpm3-I/M mutant, Trpm3-null and wild-type lenses (Fig. 9). Combined, histochemical staining (von Kossa, alizarin red S) and αII-spectrin proteolysis revealed progressive bone-like mineralization/calcification along with calpain hyper-activation during cataract development in Trpm3-mutant lenses.
Figure 9. Calpain hyper-activation in the Trpm3-mutant lens.
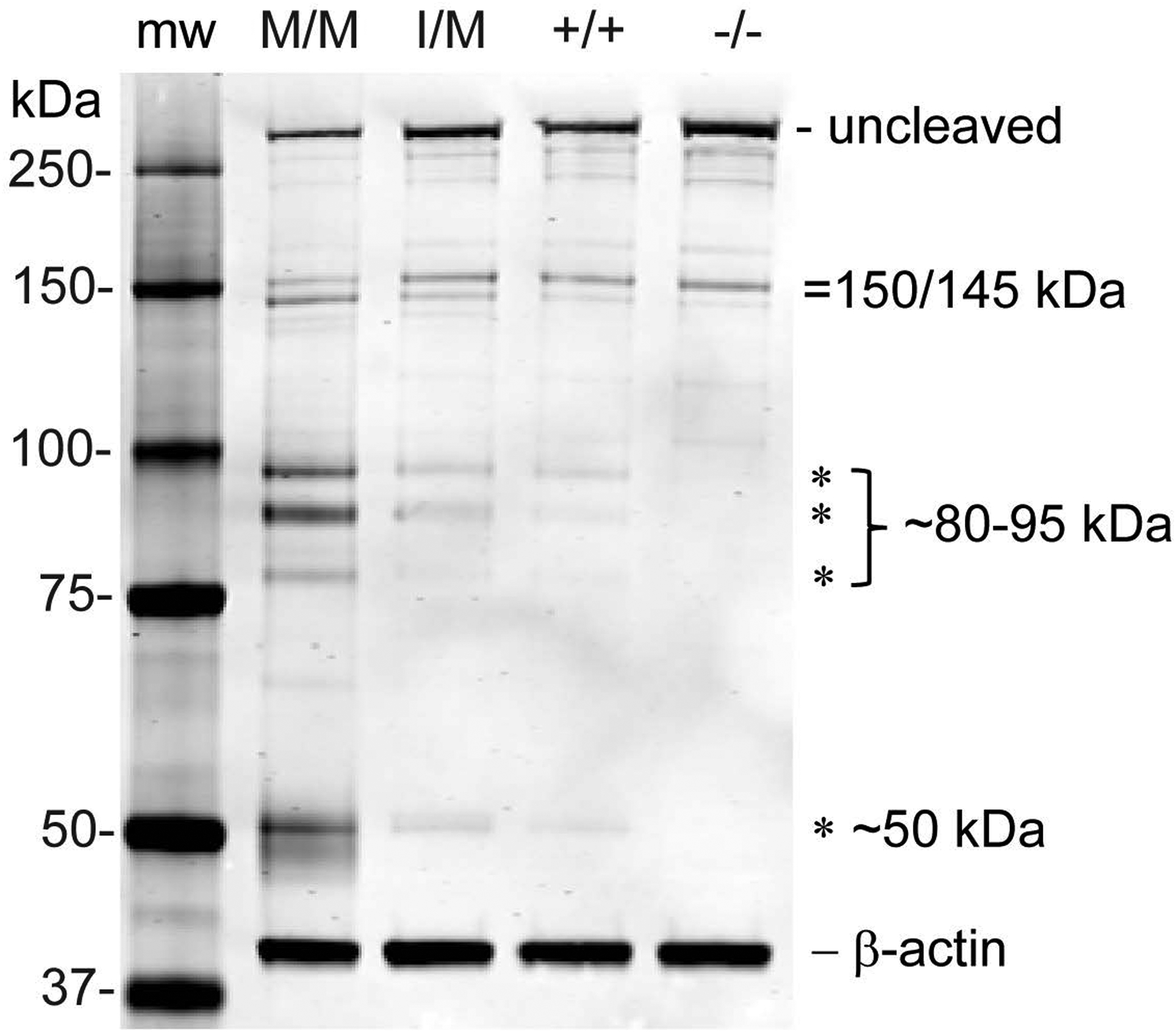
Representative immunoblot analysis of αII-spectrin showing increased caplain cleavage products of 145 kDa, ~80–95 kDa, and ~50 kDa in the Trpm3-M/M and Trpm3-I/M mutant lenses compared with Trpm3-null (−/−) and wild-type (+/+) lenses (P21). Molecular weight markers (mw).
3.4. Trpm3-mutant lenses accumulate collagen fibrils
Since TRP-channels and disturbance of Ca2+ homeostasis have both been associated with tissue fibrosis [48], we performed picrosirius red staining of Trpm3-mutant lenses to visualize changes in collagen distribution compared with wild-type. In adult wild-type lenses, picrosirius red staining outlined the surrounding basement membrane or capsule that is rich in collagen type IV [49]. Staining was strongest across the anterior capsule underlying the lens epithelium compared with the posterior capsule supporting lens fiber cells (Fig. 10A, B) – consistent with increased thickness of the anterior capsule (~10 μm) compared with the posterior capsule (~3 μm) [49]. In addition, faint picrosirius red staining was confined to the outermost (nascent) fiber cells generated at the lens equator region (Fig. 10A). Trpm3-null lenses displayed a similar picrosirius red staining pattern to that observed in wild-type (Fig. 10B). By contrast, in Trpm3-mutant lenses strong picrosirius red staining was localized to the anterior pyramidal cataract region with weaker staining extending into the nuclear region of Trpm3-I/M, Trpm3-M/-, and Trpm3-M/M mutant lenses at P90 (Fig. 10C–H). Further, picrosirius red staining of Trpm3-mutant lenses suggested that the capsule was largely intact in equatorial regions. However, compared to wild-type, the capsule appeared variably thickened and/or folded or wrinkled in the anterior pyramidal cataract regions of Trpm3-I/M, Trpm3-M/-, and Trpm3-M/M mutant lenses (Fig. 10D, F, H) – raising the possibility that it may have been ruptured and subsequently repaired. Overall, the excess collagen deposition particularly within the anterior pyramidal cataract region was consistent with a fibrotic-like response in the Trpm3-mutant lens.
Figure 10. Collagen deposition in the Trpm3-mutant lens.
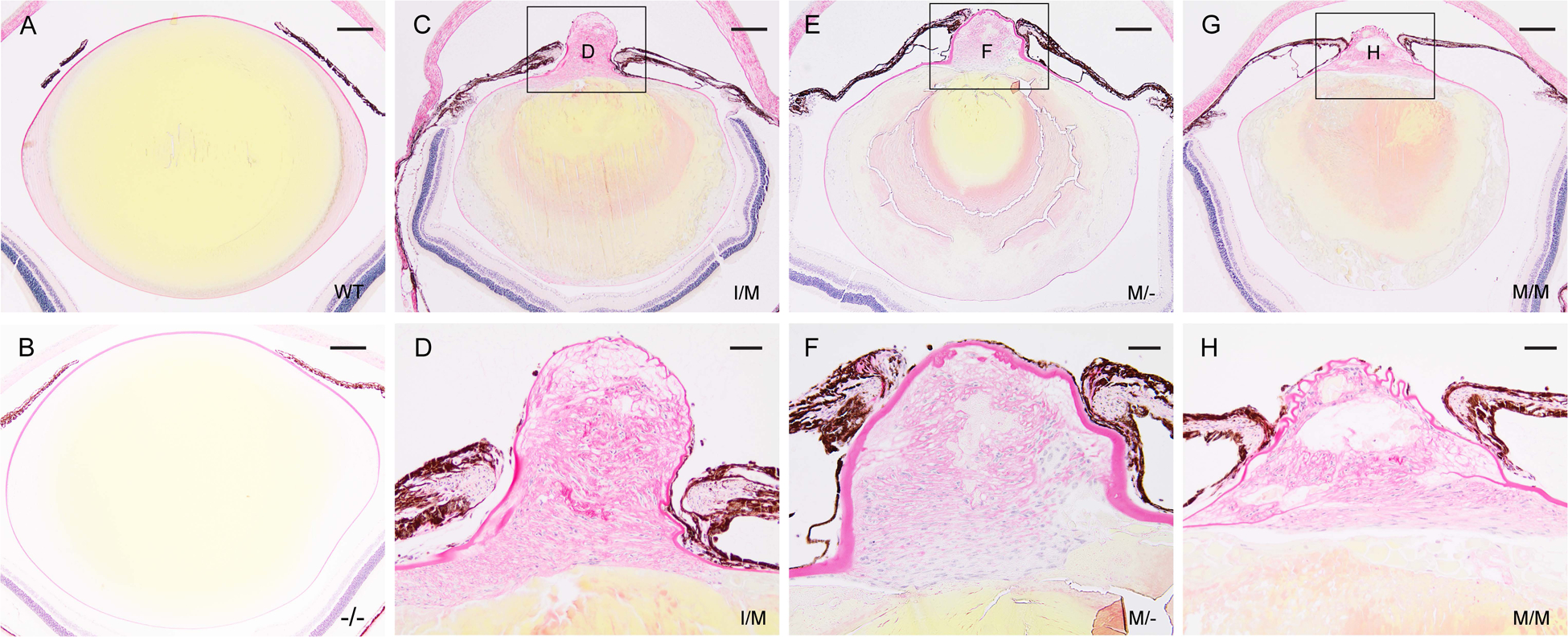
Representative picrosirius red staining of lenses (P90) from wild-type (A), Trpm3-null (B), Trpm3-I/M mutant (C, D), Trpm3-M/- mutant (E, F), and Trpm3-M/M mutant (G, H) mice showing accumulation of collagen-like fibrils or bundles in the anterior pyramidal protrusion and variable thickening or folding of the anterior capsule (C-H). By contrast, picrosirius red staining of wild-type (A) and Trpm3-null (B) lenses was confined to the surrounding capsule and the outermost equatorial fiber cells, whereas, the lens core cytoplasm stained yellow. Scale bar: 200 μm (A-C, E, G), 50 μm (D, F, H).
3.5. Trpm3-mutant lenses accumulate myofibroblast-like and macrophage-like cells
Since H&E histopathology revealed lens fiber cell degeneration and abnormal accumulation of nucleated cells in the Trpm3-M/M mutant lens (Fig. 6G, H), we undertook immuno-localization of specific markers for cell membrane integrity, mesenchymal cells, and immune cells. First, we imaged Trpm3-M/M mutant lens fiber cell organization with antibody to lens major intrinsic protein or aquaporin-0 (AQP0). Anti-AQP0 labeling revealed degeneration of lens fiber cell membranes at P22 – particularly at the anterior pole and along the optical-axis to the posterior pole (Fig. 11B). Second, we imaged Trpm3-mutant and Trpm3-null lenses with antibody to the mesenchymal or myofibroblast marker alpha smooth muscle actin (α-SMA). At three months of age (P90), neither wild-type nor Trpm3-null lenses exhibited α-SMA+ve cells (Fig. 12A–C). By contrast, anti-α-SMA labeling of Trpm3-mutant (I/M, M/-, and M/M) lenses (P90) revealed accumulation of α-SMA+ve cells in several sub-capsular regions including the posterior pole and the anterior pyramidal cataract protrusion (Fig. 12D–F). Notably, the latter region was also rich in collagen fibrils (Fig. 10D, F, H). Finally, we imaged Trpm3-M/M mutant lenses with the macrophage-lineage marker macrosialin or cluster of differentiation 68 antigen (CD68). In contrast to wild-type, anti-CD68 labeling of Trpm3-M/M mutant lenses revealed accumulation of CD68+ve cells, at first around the outside of the lens capsule at P30 (Fig. 13B–D) and then with aging (P90) inside the capsule – particularly at or near the anterior pyramidal cataract protrusion (Fig. 13F–H). Collectively, these immuno-localization data suggest that Trpm3 dysfunction in the lens leads to fiber cell degeneration and abnormal accumulation of nucleated cells including myofibroblast-like (αSMA+ve) and macrophage-like (CD68+ve) cells.
Figure 11. Fiber cell degeneration in the Trpm3-mutant lens.
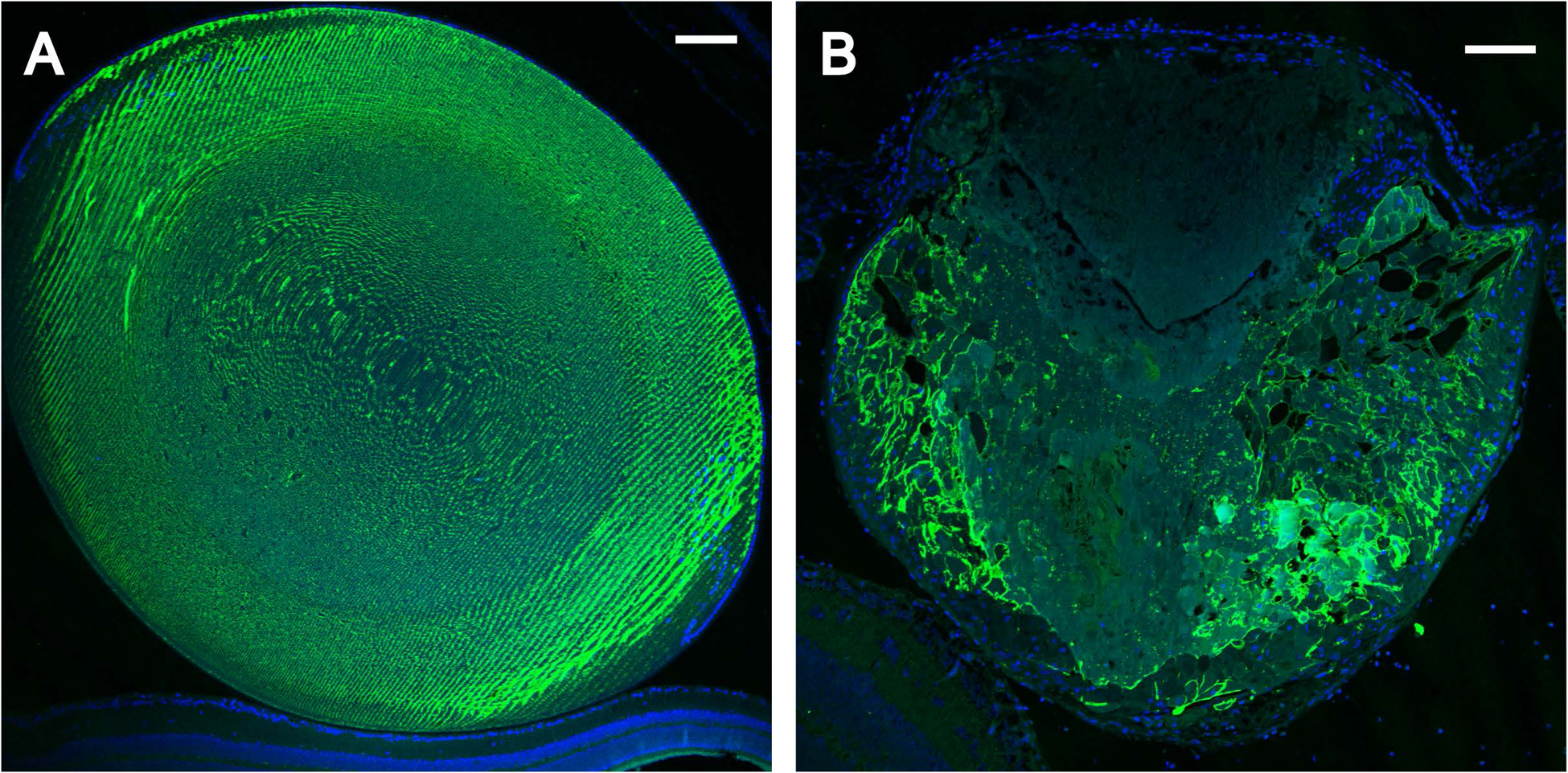
Representative immunofluorescence microscopy images of lenses (P22) from wild-type (A) and Trpm3-M/M mutant (B) mice labelled with anti-AQP0 showing degeneration of fiber cell membranes particularly in the anterior cataract region devoid of AQP0 (B). Cell nuclei were stained with DAPI (blue). Scale bar: 100 μm.
Figure 12. Accumulation of α-SMA+ve myofibroblast-like cells in the Trpm3-mutant lens.
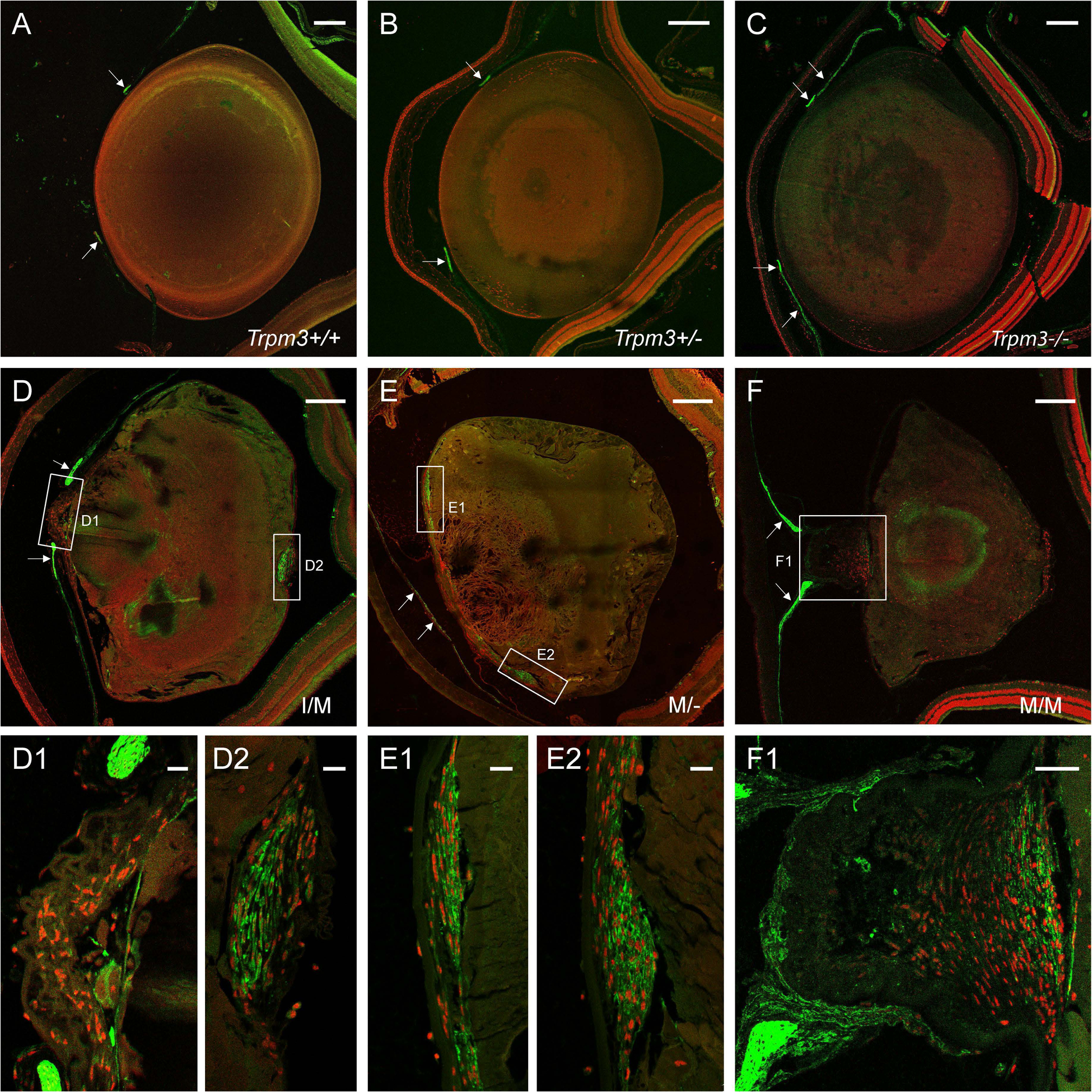
Representative immunofluorescence microscopy images of lenses (P90) from wild-type (A), Trpm3+/− (B), Trpm3−/− (C), Trpm3-I/M mutant (D), Trpm3-M/- mutant (E), and Trpm3-M/M mutant (F) mice showing accumulation of α-SMA+ve cells (green) at the anterior pyramidal cataract region of Trpm3-I/M (D1) and Trpm3-M/- (E1, E2) mutant lenses, the anterior sub-capsular regions of Trpm3-M/M mutant lenses (F1), and the posterior pole of the Trpm3-I/M mutant lens (D2). The α-SMA+ve iris is indicated by arrows in panels A-F. Cell nuclei were stained with DAPI (red). Scale bar: 200 μm (A-F), 20 μm (D1, D2, E1, E2), 50 μm (F1).
Figure 13. Accumulation of CD68+ve macrophage-like cells in the Trpm3-mutant lens.
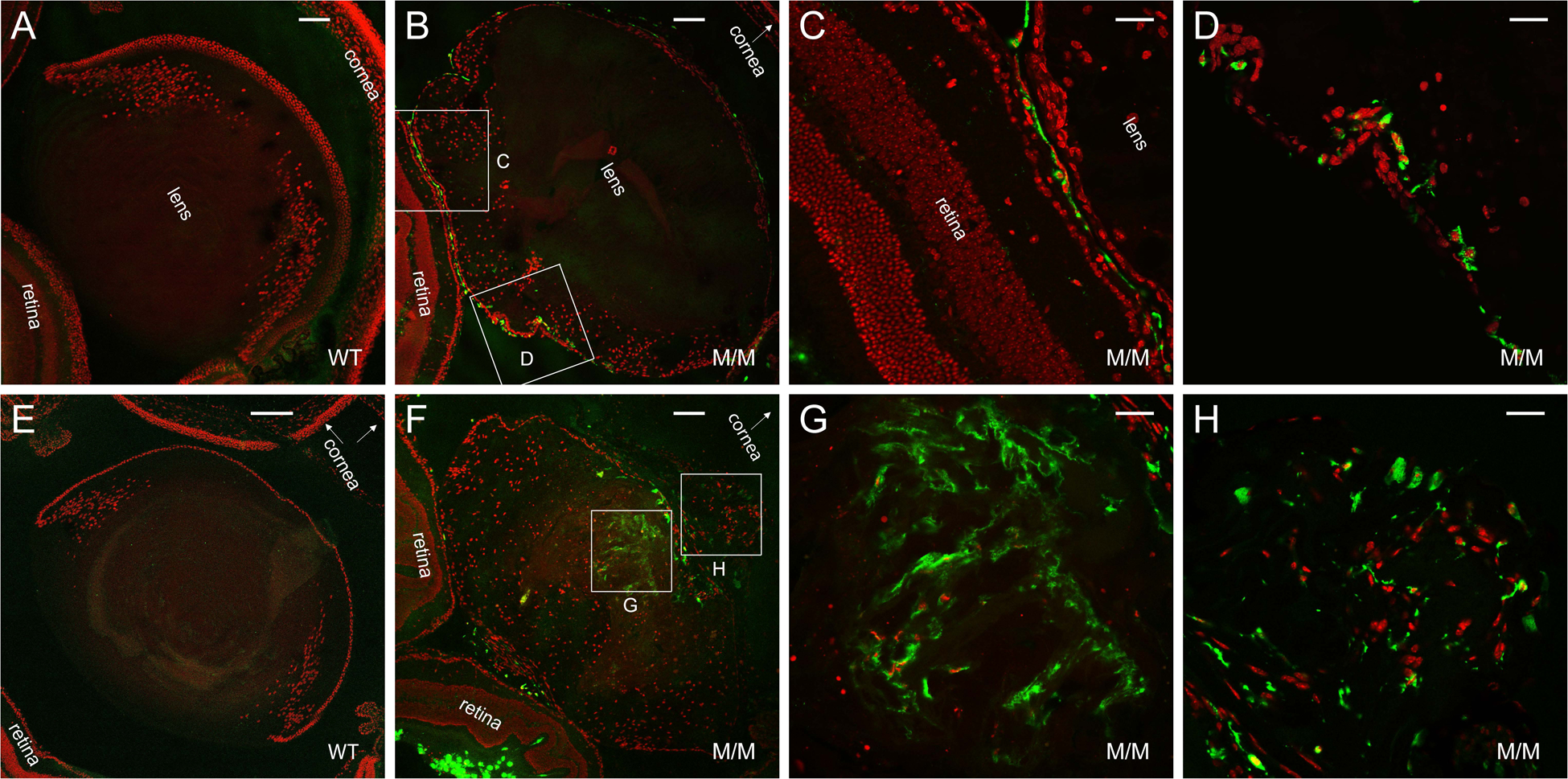
Representative immunofluorescence microscopy images of lenses from wild-type (A, E) and Trpm3-M/M mutant (B-D, F-H) mice at P30 (A-D) and P90 (E-H) showing accumulation of CD68+ve cells (green) around the lens periphery (B-D) then within the lens at or near the anterior pyramidal cataract region (F-H). Cell nuclei were stained with DAPI (red). Scale bar: 100 μm (A, B, F), 200 μm (E), 30 μm (C, D, G, H).
4. Discussion
Mutation of TRPM3 has been linked with rare inherited (autosomal dominant) forms of early-onset cataract with or without other ocular abnormalities in humans [31–33]. In this study, we have shown that homozygous introduction of a human TRPM3 mutation (p.I65M) into the mouse germline (Trpm3-M/M mutant) resulted in a progressive ocular phenotype characterized by a small eye and degenerating lens with an anterior pyramidal cataract accompanied by accumulation of Ca2+ phosphate-like deposits, αII-spectrin cleavage products, α-SMA+ve cells, collagen fibrils, and CD68+ve cells. Similarly, heterozygous Trpm3-I/M and hemizygous Trpm3-M/- mutant mice developed an anterior pyramidal cataract with delayed onset and progression, consistent with a semi-dominant lens phenotype. By contrast, germline loss of Trpm3 function in mice did not result in a cataract phenotype. Collectively, these mouse model data, for the first time, support TRPM3 mutation as a cause of early-onset cataract in humans and suggest that TRPM3-channel dysfunction (but not deficiency) triggered a pathogenic mechanism in the lens involving calcium accumulation and calpain hyper-activation associated with myofibroblast-like (α-SMA+ve) and macrophage-like (CD68+ve) cellular responses.
Despite strong expression of Trpm3 in the lens epithelium, Trpm3-null mice merely developed a mild impairment of lens growth without loss of transparency (Fig. 3). This relatively benign lens phenotype, compared to that of Trpm3-mutant mice (Fig. 5), suggested that TRPM3 loss-of-function may be mitigated, in part, by other TRP-channels expressed in the lens. Candidates for such functional compensation between lens TRP-channels include TRPV1 and TRPV4, which have been shown to participate in maintaining an intracellular hydrostatic pressure gradient within the lens [50]. Besides the lens epithelium Trpm3 was also strongly expressed in ciliary pigmented epithelium, iris pigmented epithelium and retinal pigment epithelium (RPE) (Fig. 2). Although other TRP-channels (e.g., TRPV4) are expressed throughout the eye including the ciliary body and RPE [51–53], we cannot exclude the possibility of physiological deficits in ocular epithelia lacking Trpm3 function. Indeed, Trpm3-null mice have been reported to exhibit an attenuated pupillary response (iris constriction) to bright and dim light conditions [30]. Conceivably, loss of Trpm3 function in the iris pigmented epithelium may contribute to an attenuated pupillary light response (PLR). While visual function testing of Trpm3-null mice elicits a normal electroretinogram [54], absence of Trpm3 expression in the RPE may lead to impaired epithelial barrier function and/or Ca2+ sensing in the sub-retinal space [52]. Similarly, Trpm3 loss-of-function in the ciliary epithelium may affect aqueous humor production. Finally, behavioral studies of Trpm3-null mice have revealed that TRPM3 serves as a thermo-sensitive nociceptor channel involved in the systemic detection of noxious heat [12, 13]. Temperature distribution simulations of the human eye exposed to laser radiation have shown that the temperature of the lens anterior epithelium is 32°C, whereas, the temperature of the RPE at the posterior of the eye is 37°C [55]. However, the role of temperature in regulating ocular TRPM3 is currently unclear. Further studies of Trpm3-null mice may help to elucidate the role(s) of TRPM3-channels in ocular epithelial homeostasis.
In contrast to the mild loss-of-function phenotype of the Trpm3-null lens, the Trpm3-mutant lens developed a progressive, semi-dominant, cataract phenotype suggesting a deleterious gain-of-function mechanism in the lens. The p.I>M mutation in Trpm3 is predicted to affect multiple TRPM3-channel isoforms, either by acting as a missense substitution (p.I65M, p.I67M) with damaging effects on at least 20 isoforms and/or as an N-terminal extension mutation (p.M1ext-89) affecting at least 7 isoforms that initiate translation 89 codons down-stream (Fig. 1) [31, 33]. Recently, missense mutations in TRPM3 associated with intellectual disability and epilepsy in humans, have been shown to over-activate Ca2+ influx and modify gating properties of TRPM3-channels - consistent with a deleterious gain-of-function mechanism [26, 27]. While we have not directly measured channel activity of the TRPM3 p.I>M mutant, our chromogenic histochemistry (von Kossa and alizarin red S staining) and increased αII-spectrin cleavage data (Fig. 7–9) indirectly support abnormal Ca2+ influx via the lens epithelium, accumulation of Ca2+ phosphate-like deposits, and calpain over-activation resulting in lens fiber cell membrane degeneration determined by anti-AQP0 labeling (Fig. 11). At least four active isoforms of calpain (1–3 and 7) are abundantly expressed in the mouse lens [56] and besides their role in remodeling of the membrane cytoskeleton during lens fiber cell differentiation, calpain-mediated proteolysis of lens proteins, including αII-spectrin (α-fodrin), has long been associated with cataract formation in rodents [57, 58].
Besides Ca2+ accumulation and calpain hyper-activation, picrosirius red staining revealed progressive collagen deposition in Trpm3-mutant lenses particularly within the anterior pyramidal region of opacification (Fig. 10). Recently, TRP-channels have emerged as mediators of intracellular calcium signaling dysregulation that leads to fibrotic disease of major vital organs including the lung, heart, and gut [46, 59–61]. Trpm3-M/M mutant lenses also accumulated α-SMA+ve cells within the anterior pyramidal cataract region co-localized with collagen deposition (Fig. 10 and 12). α-SMA is a characteristic marker of contractile myofibroblasts that secrete extracellular matrix components (e.g., fibrillar collagens 1 and 3) and multiple sources of these pro-fibrotic cells have been associated with cataract phenotypes in rodents and humans [62]. First, exposure of the rodent lens epithelium in culture or in vivo to the fibrogenic cytokine transforming growth factor-β (TGFβ) induced transdifferentiation of lens epithelial cells to myofibroblasts resulting in (anterior) sub-capsular cataract [63]. Similarly, in the human capsular bag model of posterior capsular opacification (PCO), which forms after cataract surgery, exposure to TGFβ resulted in increased transdifferentiation of lens epithelial cells to contractile myofibroblasts that cause capsule wrinkling further impairing post-surgical visual acuity [64, 65]. Second, in the mouse lens capsular injury model of anterior sub-capsular cataract (ASC), α-SMA+ve myofibroblasts are believed to be generated by epithelial-to-mesenchymal transition (EMT) type-II that is associated with tissue regeneration and fibrosis [66, 67]. Third, α-SMA+ myofibroblasts associated with severe dysgenesis of mouse lenses that lack N-cadherin have been principally sourced to CD45/β2-integrin+ve immune cells – suggesting that immune surveillance of the lens led to a fibrotic cataract response [68]. Fourth, α-SMA+ve myofibroblasts associated with PCO following mock cataract surgery in rabbits, were primarily sourced from a rare population of myogenic cells that express the skeletal muscle-specific transcription factor, MyoD and the bone morphogenetic protein inhibitor, noggin. These Myo/Nog cells resided in the lens and ciliary body and on the ciliary zonules that suspend the lens in the light-path of the eye [69, 70]. Further immunolocalization studies are underway to determine the origin(s) of α-SMA+ve myofibroblast-like cells in the Trpm3-M/M mutant lens.
Along with α-SMA+ve cells, CD68+ve cells were observed surrounding the Trpm3-M/M mutant lens at P30 and then within the progressively degenerating lens by three months of age (Fig. 13). Although we found no direct signs of lens rupture and fiber cell extrusion, macrophage-like cells may have penetrated breaches in the capsule of Trpm3-MM mutant lenses particularly at the anterior pole. Alternatively, immune cells have been shown to physically traverse (transmigrate) epithelial basement membranes in vivo [71, 72] and recruitment of CD68+ve cells likely via the lens capsule has been reported during severe lens dysgenesis due to lack of N-cadherin [68]. Immune cells, known to populate the vascularized uveal tract of the eye, which includes the iris, ciliary body, and choroid [73], have been proposed to surveille the lens by migrating across the ciliary zonules [68, 74]. Further studies are underway to determine the ocular source and trafficking route of macrophage-like cells found in the Trpm3-MM mutant lens.
In humans segregating the p.I65M mutation in TRPM3 across 5-generations, early-onset cataract was variably associated with a spectrum of ocular abnormalities [31]. While cataract was diagnosed between birth (congenital) and 35 years-of-age, no clinical slit-lamp biomicroscopy images of lens opacities were recorded prior to cataract surgery (age-range 4–40 years) and cataract morphology was not specifically documented. Of those affected with cataract, ~ 60% were also diagnosed with atypical high-tension glaucoma (age-at-diagnosis birth to 57 years) that likely contributed to enucleation surgery in several cases. In addition, certain affected individuals exhibited other structural eye defects including persistent pupillary membrane, retinal detachment, calcium deposits in the cornea, iris abnormalities, anterior segment dysgenesis, and megalocornea. Overall, human TRPM3-related cataract was highly penetrant with variable age-at-onset, whereas, co-existing glaucoma and other structural eye defects were moderately or weakly penetrant, respectively, contributing to inconsistent ocular disease severity and course. These phenotypic observations in humans suggested that TRPM3 dysfunction may extend beyond the lens to exert pathogenic effects in other eye tissues (e.g., iris, ciliary body, and RPE) consistent with the ocular expression pattern of Trpm3 in the mouse (Fig. 2). Our modeling of the human TRPM3-I65M mutation in mice has regenerated an early-onset cataract phenotype - associated with lens calcification – that, while fully penetrant, also displayed variable onset, progression, and severity. Continued studies of the Trpm3-mutant mice reported here will provide new insights regarding the pan-ocular function and dysfunction of TRPM3 cation channels.
Supplementary Material
Acknowledgements
We thank Dr. X. Cui, M. Wallace, and S. Penrose for mouse generation and re-derivation support, B. McMahan and G. Ling for histology support, and anonymous reviewers for insightful comments. Supported by NIH/NEI grants EY028899 (AS) and EY02687 (Core Grant for Vision Research) and an unrestricted grant to the Department of Ophthalmology and Visual Sciences from Research to Prevent Blindness.
Abbreviations:
- TRPM3
Transient receptor potential cation channel, sub-family M, member 3/melastatin-2
- lacZ
β-galactosidase
- β-geo
β-galactosidase-neomycin
- IRES
5′- internal ribosome entry site
- AQP0
aquaporin 0/major intrinsic protein
- α-SMA
alpha-smooth muscle actin
- CD68
cluster of differentiation 68 antigen/macrosialin
- DAPI
4′,6-diamidino-2-phenylindole
Footnotes
Conflict of Interest Statement
There is no conflict of interest in connection with this article.
References
- 1.Irie S, Furukawa T. (2014) Trpm1. Handb Exp Pharmacol 2014;222:387–402. [DOI] [PubMed] [Google Scholar]
- 2.Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog Retin Eye Res 2015;45:58–110. [DOI] [PubMed] [Google Scholar]
- 3.Lee N, Chen J, Sun L, Wu S, Gray KR, Rich A, Huang M, Lin JH, Feder JN, Janovitz EB, Levesque PC, Blanar MA. Expression and characterization of human transient receptor potential melastatin 3 (hTRPM3). J Biol Chem 2003;278:20890–20897. [DOI] [PubMed] [Google Scholar]
- 4.Grimm C, Kraft R, Sauerbruch S, Schultz G, Harteneck C. Molecular and functional characterization of the melastatin-related cation channel TRPM3. J Biol Chem 2003;278:21493–21501. [DOI] [PubMed] [Google Scholar]
- 5.Oberwinkler J, Lis A, Giehl KM, Flockerzi V, Philipp SE. (2005) Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J Biol Chem 2005;280:22540–22548. [DOI] [PubMed] [Google Scholar]
- 6.Oberwinkler J, Philipp SE. Trpm3. Handb Exp Pharmacol 2014;222:427–459. [DOI] [PubMed] [Google Scholar]
- 7.Fruhwald J, Camacho Londono J, Dembla S, Mannebach S, Lis A, Drews A, Wissenbach U, Oberwinkler J, Philipp SE. Alternative splicing of a protein domain indispensable for function of transient receptor potential melastatin 3 (TRPM3) ion channels. J Biol Chem 2012;287:36663–36672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner TF, Loch S, Lambert S, Straub I, Mannebach S, Mathar I, Dufer M, Lis A, Flockerzi V, Philipp SE, Oberwinkler J. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat Cell Biol 2008;10:1421–1430. [DOI] [PubMed] [Google Scholar]
- 9.Held K, Voets T, and Vriens J TRPM3 in temperature sensing and beyond. Temperature (Austin) 2015;2:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toth BI, Konrad M, Ghosh D, Mohr F, Halaszovich CR, Leitner MG, Vriens J, Oberwinkler J, Voets T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J Gen Physiol 2015;146:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badheka D, Borbiro I, Rohacs T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J Gen Physiol 2015;146:65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vriens J, Owsianik G, Hofmann T, Philipp SE, Stab J, Chen X, Benoit M, Xue F, Janssens A, Kerselaers S, Oberwinkler J, Vennekens R, Gudermann T, Nilius B, Voets T. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011;70:482–494. [DOI] [PubMed] [Google Scholar]
- 13.Vandewauw I, De Clercq K, Mulier M, Held K, Pinto S, Van Ranst N. Segal A, Voet T, Vennekens R, Zimmermann K, Vriens J, Voets T. A TRP channel trio mediates acute noxious heat sensing. Nature 2018;555, 662–666. [DOI] [PubMed] [Google Scholar]
- 14.Naylor J, Li J, Milligan CJ, Zeng F, Sukumar P, Hou B, Sedo A, Yuldasheva N, Majeed Y, Beri D, Jiang S, Seymour VA, McKeown L, Kumar B, Harteneck C, O’Regan D, Wheatcroft SB, Kearney MT, Jones C, Porter KE, Beech DJ. Pregnenolone sulphate- and cholesterol-regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circ Res 2010;106:1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alonso-Carbajo L, Alpizar YA, Startek JB, Lopez-Lopez JR, Perez-Garcia MT, Talavera K. Activation of the cation channel TRPM3 in perivascular nerves induces vasodilation of resistance arteries. J Mol Cell Cardiol 2019;129:219–230. [DOI] [PubMed] [Google Scholar]
- 16.Held K, Kichko T, De Clercq K, Klaassen H, Van Bree R, Vanherck JC, Marchand A, Reeh PW, Chaltin P, Voets T, and Vriens J Activation of TRPM3 by a potent synthetic ligand reveals a role in peptide release. Proc Natl Acad Sci U S A 2015;112:E1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yashin AI, Wu D, Arbeev KG, Ukraintseva SV. Joint influence of small-effect genetic variants on human longevity. Aging (Albany NY) 2010;2:612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park SH, Lee JY, Kim S. A methodology for multivariate phenotype-based genome-wide association studies to mine pleiotropic genes. BMC Syst Biol 2011;5 Suppl 2:S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oztuzcu S, Onat AM, Pehlivan Y, Alibaz-Oner F, Donmez S, Cetin GY, Yolbas S, Bozgeyik I, Yilmaz N, Ozgen M, Cagatay Y, Kisacik B, Koca SS, Pamuk ON, Sayarlioglu M, Direskeneli H, Demiryurek AT. Association of TRPM channel gene polymorphisms with systemic sclerosis. In Vivo 2015;29:763–770. [PubMed] [Google Scholar]
- 20.Narayanankutty A, Palma-Lara I, Pavon-Romero G, Perez-Rubio G, Camarena A, Teran LM, Falfan-Valencia R. (2016) Association of TRPM3 Polymorphism (rs10780946) and aspirin-exacerbated respiratory disease (AERD). Lung 2016;194:273–279. [DOI] [PubMed] [Google Scholar]
- 21.Marshall-Gradisnik S, Huth T, Chacko A, Johnston S, Smith P, Staines D. Natural killer cells and single nucleotide polymorphisms of specific ion channels and receptor genes in myalgic encephalomyelitis/chronic fatigue syndrome. Appl Clin Genet 2016;9:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwangbo Y, Lee EK, Son HY, Im SW, Kwak SJ, Yoon JW, Kim MJ, Kim J, Choi HS, Ryu CH, Lee YJ, Kim JI, Cho NH, Park YJ. (2018) Genome-wide association study reveals distinct genetic susceptibility of thyroid nodules from thyroid cancer. J Clin Endocrinol Metab 2018;103:4384–4394. [DOI] [PubMed] [Google Scholar]
- 23.Kuniba H, Yoshiura K, Kondoh T, Ohashi H, Kurosawa K, Tonoki H, Nagai T, Okamoto N, Kato M, Fukushima Y, Kaname T, Naritomi K, Matsumoto T, Moriuchi H, Kishino T, Kinoshita A, Miyake N, Matsumoto N, Niikawa N. Molecular karyotyping in 17 patients and mutation screening in 41 patients with Kabuki syndrome. J Hum Genet 2009;54:304–309. [DOI] [PubMed] [Google Scholar]
- 24.Pagnamenta AT, Holt R, Yusuf M, Pinto D, Wing K, Betancur C, Scherer SW, Volpi EV, Monaco AP. A family with autism and rare copy number variants disrupting the Duchenne/Becker muscular dystrophy gene DMD and TRPM3. J Neurodev Disord 2011;3:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dyment DA, Terhal PA, Rustad CF, Tveten K, Griffith C, Jayakar P, Shinawi M, Ellingwood S, Smith R, van Gassen K, McWalter K, Innes AM., Lines MA. De novo substitutions of TRPM3 cause intellectual disability and epilepsy. Eur J Hum Genet 2019;10:1611–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao S, Yudin Y, Rohacs T. Disease-associated mutations in the human TRPM3 render the channel overactive via two distinct mechanisms. Elife 2020;9:e55634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Hoeymissen E, Held K, Nogueira Freitas AC, Janssens A, Voets T, Vriens J. Gain of channel function and modified gating properties in TRPM3 mutants causing intellectual disability and epilepsy. Elife 2020;9:e57190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Sainte Agathe JM, Van-Gils J, Lasseaux E, Arveiler B, Lacombe D, Pfirrmann C, Raclet V, Gaston L, Plaisant C, Aupy J, Trimouille A. Confirmation and Expansion of the Phenotype Associated with the Recurrent p.Val837Met Variant in TRPM3. Eur J Med Genet 2020;63:103942. [DOI] [PubMed] [Google Scholar]
- 29.Kim J, Williams FJ, Dreger DL, Plassais J, Davis BW, Parker HG, Ostrander EA. Genetic selection of athletic success in sport-hunting dogs. Proc Natl Acad Sci U S A 2018;115:E7212–E7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes S, Pothecary CA, Jagannath A, Foster RG, Hankins MW, Peirson SN. Profound defects in pupillary responses to light in TRPM-channel null mice: a role for TRPM channels in non-image-forming photoreception. Eur J Neurosci 2012;35:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bennett TM, Mackay DS, Siegfried CJ, Shiels A. Mutation of the melastatin-related cation channel, TRPM3, underlies inherited cataract and glaucoma. PLoS One 2014;9:e104000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Leng Y, Han S, Yan L, Lu C, Luo Y, Zhang X, Cao L. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet J Rare Dis 2018;13:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiels A TRPM3_miR-204: a complex locus for eye development and disease. Hum Genomics 2020;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulinski J, Besack D, Oleykowski CA, Godwin AK, Yeung AT. CEL I enzymatic mutation detection assay. Biotechniques 2000;29:44–48. [DOI] [PubMed] [Google Scholar]
- 35.Alizadeh A, Clark J, Seeberger T, Hess J, Blankenship T, FitzGerald PG. Characterization of a mutation in the lens-specific CP49 in the 129 strain of mouse. Invest Ophthalmol Vis Sci 2004;45:884–891. [DOI] [PubMed] [Google Scholar]
- 36.Sandilands A, Wang X, Hutcheson AM, James J, Prescott AR, Wegener A, Pekny M, Gong X, Quinlan RA. Bfsp2 mutation found in mouse 129 strains causes the loss of CP49 and induces vimentin-dependent changes in the lens fibre cell cytoskeleton. Exp Eye Res 2004;78:109–123. [DOI] [PubMed] [Google Scholar]
- 37.Simirskii VN, Lee RS, Wawrousek EF, Duncan MK. Inbred FVB/N mice are mutant at the cp49/Bfsp2 locus and lack beaded filament proteins in the lens. Invest Ophthalmol Vis Sci 2006;47:4931–4934. [DOI] [PubMed] [Google Scholar]
- 38.Zhou Y, Bennett TM, Shiels A. Lens ER-stress response during cataract development in Mip-mutant mice. Biochim Biophys Acta 2016;1862:1433–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mackay DS, Bennett TM, Culican SM, Shiels A. (2014) Exome sequencing identifies novel and recurrent mutations in GJA8 and CRYGD associated with inherited cataract. Hum Genomics 2014;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGee-Russell SM. (1958) Histochemical methods for calcium. J Histochem Cytochem 1958;6:22–42. [DOI] [PubMed] [Google Scholar]
- 41.Bonewald LF, Harris SE, Rosser J, Dallas MR, Dallas SL, Camacho NP, Boyan B, Boskey A. von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation. Calcif Tissue Int 2003;72:537–547. [DOI] [PubMed] [Google Scholar]
- 42.Rigueur D, Lyons KM. Whole-mount skeletal staining. Methods Mol Biol 2014;1130:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Junquiera LC, Junqueira LC, Brentani RR. A simple and sensitive method for the quantitative estimation of collagen. Anal Biochem 1979;94:96–99. [DOI] [PubMed] [Google Scholar]
- 44.Lattouf R, Younes R, Lutomski D, Naaman N, Godeau G, Senni K, Changotade S. Picrosirius red staining: a useful tool to appraise collagen networks in normal and pathological tissues. J Histochem Cytochem 2014;62:751–758. [DOI] [PubMed] [Google Scholar]
- 45.Bennett TM, Zhou Y, Shiels A. Lens transcriptome profile during cataract development in Mip-null mice. Biochem Biophys Res Commun 2016;478:988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou Y, Bennett TM, Shiels A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019;109:16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Fur N, Kelsall SR, Mintz B. Base substitution at different alternative splice donor sites of the tyrosinase gene in murine albinism. Genomics 1996;37:245–248. [DOI] [PubMed] [Google Scholar]
- 48.Janssen LJ, Mukherjee S, Ask K. Calcium Homeostasis and Ionic Mechanisms in Pulmonary Fibroblasts. Am J Respir Cell Mol Biol 2010;53:135–148. [DOI] [PubMed] [Google Scholar]
- 49.Danysh BP, Duncan MK. The lens capsule. Exp Eye Res 2009;88:151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao J, Sun X, White TW, Delamere NA, Mathias RT. Feedback Regulation of Intracellular Hydrostatic Pressure in Surface Cells of the Lens. Biophys J 2015;109:1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alkozi HA, Pintor J. TRPV4 activation triggers the release of melatonin from human non-pigmented ciliary epithelial cells. Exp Eye Res 2015;136:34–37. [DOI] [PubMed] [Google Scholar]
- 52.Zhao PY, Gan G, Peng S, Wang SB, Chen B, Adelman RA, Rizzolo LJ. TRP Channels Localize to Subdomains of the Apical Plasma Membrane in Human Fetal Retinal Pigment Epithelium. Invest Ophthalmol Vis Sci 2015;56:1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jo AO, Lakk M, Frye AM, Phuong TT, Redmon SN, Roberts R, Berkowitz BA, Yarishkin O, Krizaj D. Differential volume regulation and calcium signaling in two ciliary body cell types is subserved by TRPV4 channels. Proc Natl Acad Sci U S A 2016;113:3885–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown RL, Xiong WH, Peters JH, Tekmen-Clark M, Strycharska-Orczyk I, Reed BT, Morgans CW, Duvoisin RM. TRPM3 expression in mouse retina. PLoS One 2015;10:e0117615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mirnezami SA, Rajaei Jafarabadi M, Abrishami M. Temperature distribution simulation of the human eye exposed to laser radiation. J Lasers Med Sci 2013;4:175–181. [PMC free article] [PubMed] [Google Scholar]
- 56.De Maria A, Shi Y, Kumar NM, Bassnett S. Calpain expression and activity during lens fiber cell differentiation. J Biol Chem 2009;284:13542–13550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakamura Y, Fukiage C, Shih M, Ma H, David LL, Azuma M, Shearer TR. Contribution of calpain Lp82-induced proteolysis to experimental cataractogenesis in mice. Invest Ophthalmol Vis Sci 2000;41:1460–1466. [PubMed] [Google Scholar]
- 58.Sakamoto-Mizutani K, Fukiage C, Tamada Y, Azuma M, Shearer TR. Contribution of ubiquitous calpains to cataractogenesis in the spontaneous diabetic WBN/Kob rat. Exp Eye Res 2002;75:611–617. [DOI] [PubMed] [Google Scholar]
- 59.Belvisi MG, Birrell MA. The emerging role of transient receptor potential channels in chronic lung disease. Eur Respir J 2017;50:1601357. [DOI] [PubMed] [Google Scholar]
- 60.Inoue R, Kurahara LH, Hiraishi K. TRP channels in cardiac and intestinal fibrosis. Semin Cell Dev Biol 2019;94:40–49. [DOI] [PubMed] [Google Scholar]
- 61.Grebert C, Becq F, Vandebrouck C. Focus on TRP channels in cystic fibrosis. Cell Calcium 2019;81:29–37. [DOI] [PubMed] [Google Scholar]
- 62.Shu DY, Lovicu FJ. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog Retin Eye Res 2017;60:44–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells Tissues Organs 2005;179:43–55. [DOI] [PubMed] [Google Scholar]
- 64.Wormstone IM, Wang L, Liu CS. Posterior capsule opacification. Exp Eye Res 2009;88:257–269. [DOI] [PubMed] [Google Scholar]
- 65.Eldred JA, Dawes LJ, Wormstone IM. The lens as a model for fibrotic disease. Philos Trans R Soc Lond B Biol Sci 2011;366:1301–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xiao W, Chen X, Li W, Ye S, Wang W, Luo L, Liu Y. Quantitative analysis of injury-induced anterior subcapsular cataract in the mouse: a model of lens epithelial cells proliferation and epithelial-mesenchymal transition. Sci Rep 2015;5:8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shirai K, Tanaka SI, Lovicu FJ, Saika S. The murine lens: A model to investigate in vivo epithelial-mesenchymal transition. Dev Dyn 2018;247:340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Logan CM, Bowen CJ, Menko AS. Induction of immune surveillance of the dysmorphogenic lens. Sci Rep 2017;7:16235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerhart J, Withers C, Gerhart C, Werner L, Mamalis N, Bravo-Nuevo A, Scheinfeld V, FitzGerald P, Getts R, George-Weinstein M. Myo/Nog cells are present in the ciliary processes, on the zonule of Zinn and posterior capsule of the lens following cataract surgery. Exp Eye Res 2018;171:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerhart J, Werner L, Mamalis N, Infanti J, Withers C, Abdalla F, Gerhart C, Bravo-Nuevo A, Gerhart O, Getts L, Rhodes K, Bowers J, Getts R, George-Weinstein M. Depletion of Myo/Nog cells in the lens mitigates posterior capsule opacification in rabbits. Invest Ophthalmol Vis Sci 2019;60:1813–1823. [DOI] [PubMed] [Google Scholar]
- 71.Kelley LC, Lohmer LL, Hagedorn EJ, Sherwood DR. Traversing the basement membrane in vivo: a diversity of strategies. J Cell Biol 2014;204:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van den Berg MCW, MacCarthy-Morrogh L, Carter D, Morris J, Ribeiro Bravo I, Feng Y, Martin P. (2019) Proteolytic and opportunistic breaching of the basement membrane zone by immune cells during tumor initiation. Cell Rep 2019;27:2837–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Forrester JV, Xu H. Good news-bad news: the Yin and Yang of immune privilege in the eye. Front Immunol 2012;3:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeDreu J, Bowen CJ, Logan CM, Pal-Ghosh S, Parlanti P, Stepp MA, Menko AS. An immune response to the avascular lens following wounding of the cornea involves ciliary zonule fibrils. FASEB J 2020;34:9316–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.