

Abstract
Lemborexant, a dual orexin receptor antagonist, is approved for the treatment of insomnia and is under investigation for treating other sleep disorders. Here we summarize pharmacokinetic, pharmacodynamic, and safety data from 3 randomized, double‐blind, placebo‐controlled phase 1 studies: single ascending doses in healthy adults (Study 001; 1‐200 mg; N = 64), multiple ascending doses in healthy and elderly adults (Study 002; 2.5‐75 mg; N = 55), and multiple doses in healthy white and Japanese adults (Study 003; 2.5‐25 mg; N = 32). Lemborexant exposure increased with increasing dose. The time to maximum concentration ranged from approximately 1 to 3 hours for the 5‐ and 10‐mg doses. The mean effective half‐life was 17 hours for lemborexant 5 mg and 19 hours for lemborexant 10 mg. The plasma concentration at 9 hours postdose was 27% of the maximum concentration following multiple dosing with lemborexant 10 mg. There were no clinically relevant effects on next‐morning residual sleepiness (Karolinska Sleepiness Scale, Digital Symbol Substitution Test, Psychomotor Vigilance Test) for doses through 10 mg/day, indicating no effect of residual plasma concentrations on next‐day residual effects. Lemborexant was well tolerated across the doses tested. There were no clinically relevant effects of age, sex, or race on lemborexant pharmacokinetics, pharmacodynamics, or safety. These results suggest that lemborexant at doses through 25 mg provides an overall pharmacokinetic, pharmacodynamic, and safety profile suitable for obtaining the target pharmacologic effect supporting treatment of insomnia while minimizing residual effects during wake time.
Keywords: insomnia, lemborexant, orexin receptor antagonists, pharmacodynamics, pharmacokinetics, safety
The neuropeptides orexin A and B play a key role in regulating the sleep‐wake cycle. 1 Specifically, orexins promote wakefulness/central arousal by binding to the G protein–coupled orexin‐1 and orexin‐2 receptors. 2 Evidence, primarily from animal studies, has shown that there is diurnal variation in orexin levels corresponding with activity; higher levels occur during active/wakeful periods, and lower levels occur during inactive/sleep periods. 3 The importance of orexins for sleep‐wake regulation is further emphasized by the findings that orexin deficiency is associated with narcolepsy in humans and other mammals 4 and that orexin levels are elevated in individuals with insomnia. 5 The diurnal pharmacologic activity of orexin allows for the treatment of insomnia via a distinct mechanism of action (as opposed to receptor agonist–type agents such as benzodiazepines or nonbenzodiazepine GABAergic drugs).
Lemborexant is a dual orexin receptor antagonist that was recently approved, at doses of 5 and 10 mg, 6 for the treatment of insomnia and is under investigation for the treatment of other sleep disorders. In addition to results from phase 2 and 3 randomized, controlled trials showing lemborexant to be efficacious and well tolerated for the treatment of insomnia, 6 , 7 , 8 various preclinical and clinical studies have been undertaken during the development of lemborexant. Preclinical studies have determined the structure of lemborexant and associated metabolites (M4, M9, and M10) in addition to the in silico, in vitro, and in vivo characteristics of lemborexant. 9 , 10 , 11 , 12 M10 is the major circulating metabolite of lemborexant; however, findings from contribution ratio analysis of the sleep‐promoting effect in humans, evaluation of cerebrospinal fluid/unbound plasma concentration using animal models, and the fact that M10 is a P‐gp substrate indicate that the contribution of M10 to the pharmacologic activity of lemborexant is low and of no clinical importance (Eisai Inc, unpublished data). In vitro studies have demonstrated that lemborexant is predominantly metabolized via cytochrome P450 (CYP)3A (Eisai Inc, unpublished data). As expected, subsequent phase 1, open‐label, drug‐drug interaction studies have revealed that coadministration of lemborexant 10 mg with the strong CYP3A inducer rifampin markedly decreased (by over 90%) both lemborexant maximum drug concentration (Cmax) and area under the concentration‐time curve (AUC) (Eisai Inc, unpublished data). These studies also revealed that coadministration of lemborexant 10 mg with the strong CYP3A inhibitor itraconazole or the moderate CYP3A inhibitor fluconazole increased lemborexant AUC by approximately 4‐fold and Cmax by 1.5‐fold (Eisai Inc, unpublished data). As a consequence, the concomitant use of lemborexant with strong or moderate CYP3A inducers and strong or moderate CYP3A inhibitors should be avoided (the maximum recommended dose of lemborexant with weak CYP3A inhibitors is 5 mg). Clinical pharmacokinetic (PK) data for lemborexant have not previously been published.
Here we summarize PK, pharmacodynamic (PD), and safety data from 3 phase 1 studies of lemborexant, including single ascending doses in healthy adults, multiple ascending doses in healthy adults and elderly subjects, and multiple doses in healthy white and Japanese adults.
Methods
All study protocols were approved by an institutional review board (New England Institutional Review Board, Newton, Massachusetts [Study 001]; IntegReview Independent Review Board, Austin, Texas [Study 002 and Study 003]), and the studies were conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable local regulations. The studies were registered at ClinicalTrials.gov (see Study Population section). All subjects provided written informed consent. The study sites were Clinilabs Inc (New York, New York) and Community Research (Cincinnati, Ohio) for Study 001; California Clinical Trials Medical Group (Glendale and Culver City, California) for Study 002; and California Clinical Trials Medical Group (Glendale, California) for Study 003.
Study Population
Inclusion Criteria
In Study E2006‐A001‐001 (Study 001; NCT01463098), subjects were healthy men or women >18‐64 years of age with a body mass index (BMI) >18 to <30 kg/m2, a habitual time in bed >7 hours, a self‐reported sleep onset latency of ≤30 minutes, and typical total sleep ≥7 hours. In Study E2006‐A001‐002 (Study 002; NCT01673451), subjects were healthy men or women ≥18‐55 years (adult) or ≥65‐80 years (elderly) with a BMI >18 to ≤32 kg/m2, a habitual time in bed ranging from 7.5 to 9 hours, and a self‐reported sleep onset latency ≤30 minutes. In Study E2006‐A001‐003 (Study 003; NCT02039089), subjects were healthy Japanese or white men or women ≥20 to ≤55 years with a BMI ≥18 to <28 kg/m2, a habitual time in bed >7 hours, a self‐reported sleep onset latency ≤30 minutes, and total sleep time ≥7 hours.
Exclusion Criteria
In all 3 studies the main exclusion criteria were a history of significant medical, neurologic, or serious psychiatric illness, a previous diagnosis or current evidence of a sleep disorder, shift work within 2 weeks before screening, having traveled across ≥3 time zones in the 7 days before screening, or a history of gastrointestinal surgery.
Study Design and Treatment Protocol
Study 001
Study 001 was a randomized, double‐blind, placebo‐controlled, single‐ascending‐dose study conducted at 2 sites in the United States. Subjects were stratified by sex and randomized to lemborexant sequential dose‐escalation groups (1, 2.5, 5, 10, 25, 50, 100, or 200 mg) or to placebo. For each group, subjects (n = 8) were randomized, using computer‐generated randomization, in a 1:3 ratio to placebo and lemborexant. All study drugs were administered as single oral doses in the morning under fasting conditions. Subjects were allowed to sleep for 8 hours each day starting at the subject's median habitual bedtime ± 30 minutes.
Study 001 also included a separate component in which PD parameters were assessed in subjects with primary insomnia following single oral dosing of lemborexant, zolpidem immediate release, or placebo. The results of this component of the study will be reported separately.
Study 002
Study 002 was a randomized, double‐blind, placebo‐controlled, multiple‐ascending‐dose study conducted at a single site in the United States. Adult subjects were randomized to lemborexant sequential dose‐escalation groups (2.5, 5, 10, 25, 50, or 75 mg) or to placebo. For each adult group, subjects (n = 8) were randomized, using computer‐generated randomization, in a 1:3 ratio to placebo and lemborexant. In a separate group, elderly subjects (n = 8) were randomized (1:3 ratio) to placebo or lemborexant 25 mg. All study drugs were administered once daily, 30 minutes before bedtime, for 14 consecutive days.
Study 003
Study 003 was a randomized, double‐blind, placebo‐controlled, multiple‐ascending‐dose study conducted at a single site in the United States. Japanese subjects (following a Japanese lifestyle) were randomized to lemborexant sequential dose‐escalation groups (2.5, 10, or 25 mg) or to placebo. For each group, subjects (n = 8) were randomized, using computer‐generated randomization, in a 1:3 ratio to placebo and lemborexant. White subjects (n = 8) were randomized (1:3 ratio) to placebo or lemborexant 10 mg. All study drugs were administered once daily, 30 minutes before bedtime, for 14 consecutive days.
Pharmacokinetic Assessments
Study 001
Blood samples for the assessment of plasma lemborexant concentrations were collected (using sodium heparin as an anticoagulant) before dosing and at predetermined intervals after dosing up to 240 hours. In addition, urine samples for the assessment of urine lemborexant concentrations were collected before dosing and at predetermined intervals after dosing up to 120 hours.
Studies 002 and 003
Blood samples for the assessment of plasma lemborexant concentrations in Study 002 and Study 003, and lemborexant metabolites as well in Study 003, were collected (using sodium heparin as an anticoagulant) before dosing from day 1 through day 14 and at predetermined intervals after dosing on days 1 and 14 (up to 324 hours after the day‐14 dose).
Plasma and urine concentrations of lemborexant were determined at Medpace Bioanalytical Laboratories (Cincinnati, Ohio) using a validated liquid chromatography coupled with tandem mass spectrometry (LC‐MS/MS) method. The method was initially developed to quantitate only the parent lemborexant by protein precipitation using methanol from 100 µL of human sodium heparin plasma or urine before LC‐MS/MS analysis. Deuterated E2006‐d3 was used as the internal standard. Chromatographic separation was achieved on a HyperClone (Phenomenex, Torrance, California), 3 µm DBS, C18, LC‐MS column, 50 × 2 mm with a gradient mobile phase. The mass spectrometer, SCIEX API‐5500 (SCIEX, Redwood City, California), was operated in positive electrospray ionization mode with multiple reaction monitoring of the precursor‐to‐product ion pairs for lemborexant (m/z 411.0 → 287.1) and E2006‐d3 (m/z 414.1 → 290.1). Samples were quantified using peak area ratios. In plasma the interday precision (coefficient of variation) was ≤7.0%, and accuracy was within 6.5% of nominal, whereas the intraday precision (coefficient of variation) was ≤10.4%, and accuracy was within 9.8% of nominal. In urine the inter‐ and intraday precision and accuracy were within the acceptance criteria of ±20% at the lower limit of quantification and ±15% at other quality‐control levels. The lower limit of quantitation was 0.100 ng/mL in plasma or urine, with the calibration curve ranging from 0.100 to 100 ng/mL. The plasma or urine quality‐control samples were stable for up to 94 days at −20°C and −70°C. The plasma method was used to quantitate lemborexant in plasma samples collected in Studies 001 and 002; the urine method was used to quantitate lemborexant in urine samples collected in Study 001.
Exploratory metabolite work on plasma samples from Study 002 revealed 3 metabolites of interest, the hydroxylated M9 and the N‐oxidized M4 and M10. Further details about these metabolites, including assay development, will be reported separately.
Noncompartmental single‐dose PK parameters calculated for lemborexant included Cmax, time to reach maximum (peak) concentration following drug administration (tmax), AUC from time 0 to 24 hours (AUC0‐24h), AUC from time 0 to time of last measurable concentration, AUC from time 0 to infinity, and terminal elimination half‐life (t½). Multiple‐dose PK parameters calculated for lemborexant included trough plasma concentrations, AUC during the dosing interval (AUC0‐τ), accumulation ratio based on Cmax and AUC(0‐τ), and effective half‐life (t½,eff). t½,eff is calculated from drug concentration‐time data obtained after steady‐state dosing (AUCss) and thereby accounts for accumulation of the drug (observed accumulation index = AUCss/AUCday1). 13 , 14 Because lemborexant displays a multicompartment distribution, 15 t½,eff was evaluated as a potentially more clinically appropriate measure than t½, which does not necessarily account for drug accumulation and disposition.
All PK analyses were performed using Phoenix WinNonlin, version 6.2 (Certara, LP, Princeton, New Jersey).
Pharmacodynamic Assessments
Study 001
Subjects were administered the Karolinska Sleepiness Scale (KSS), 16 a Digital Symbol Substitution Test (DSST), 17 and Psychomotor Vigilance Test (PVT) 18 30 minutes before dosing on day 1, then every 2 hours after dosing for 12 hours, and on days 2‐6 starting 30 minutes after lights‐on.
Subjects were asked to rate their sleepiness on the KSS, 16 a 9‐point verbally anchored scale. The verbal anchors and corresponding scores were “extremely alert” (score = 1), “very alert” (2), “alert” (3), “somewhat alert” (4), “neither alert nor sleepy” (5), “some signs of sleepiness” (6), “sleepy but no difficulty remaining awake” (7), “sleepy—some effort to keep awake” (8), and “extremely sleepy—fighting sleep” (9). The key outcome parameter for the KSS was the score from 1 to 9 (least to most sleepy).
The DSST used was from the Repeatable Battery for the Assessment of Neuropsychological Status. 19 This task assesses attention, perceptual speed, motor speed, visual scanning, and memory. Subjects were asked to match symbols to digits (1‐9); a code provided at the top of the page indicated the digit to which a given symbol should be matched. The key outcome parameter was the number of correct matches in 90 seconds (higher scores indicate better performance).
The PVT assesses behavioral alertness by measuring subjects’ response times to visual stimuli over a 10‐minute period. 18 The key outcome parameter was the number of lapses >500 ms (fewer lapses indicate better performance).
Study 002
Subjects were administered the KSS and PVT 15 minutes before habitual bedtime on day −1, 15 minutes before and after dosing on days 1‐14, and 15 minutes, 1, 2, 4, 8, and 12 hours after habitual wake time on days 1‐15.
Study 003
Subjects were administered the KSS and PVT 15 minutes before habitual bedtime on day −1, 15 minutes before and after dosing on days 1‐14, and 15 minutes and 1 and 2 hours after habitual wake time on days 1‐15.
Safety Assessments
In all 3 studies safety assessments included recording of treatment‐emergent adverse events (TEAEs) and serious adverse events (SAEs) per the Medical Dictionary for Regulatory Activities (Version 15 [Study 001], 16.0 [Study 002], or 16.1 [Study 003]), hematology and blood chemistry, urinalysis, vital signs, ECGs including continuous 24‐hour Holter monitoring, and physical examinations. Suicidality was also assessed using the Columbia‐Suicide Severity Rating Scale. 20 The maximum tolerated dose (MTD) was assessed in Studies 001 and 002.
Statistical Analyses
In each study 8 subjects per group were considered adequate to evaluate the preliminary PK, PD, effects of age (adult versus elderly) and race (Japanese versus white), and general safety of lemborexant.
In all studies the PK and PD analysis sets comprised subjects who had sufficient data to derive at least 1 PK and PD parameter, respectively, whereas the safety analysis set comprised subjects who had received study drug and then had at least 1 postdose safety assessment. All results are summarized using descriptive statistics.
Results
Disposition
Study 001
Sixty‐four subjects were randomized; 14 of 16 (87.5%) placebo‐treated subjects and 46 of 48 (95.8%) lemborexant‐treated subjects completed the study. Reasons for discontinuation included subject choice (placebo n = 2, lemborexant n = 1) and lost to follow‐up (lemborexant n = 1).
Study 002
Fifty‐five subjects were randomized. Of these, 13 of 14 (92.9%) placebo‐treated subjects and 38 of 41 (92.7%) lemborexant‐treated subjects completed the study. Reasons for discontinuation included subject choice (placebo n = 1, lemborexant n = 2) and adverse event (AE; lemborexant n = 1 [see Safety section for further details about this AE]).
Study 003
Thirty‐two subjects were randomized; all subjects completed the study.
Demographic and Baseline Characteristics
In all studies, demographic and baseline characteristics were similar between treatment groups (Supplemental Tables S1‐S3).
Pharmacokinetics
Study 001
Lemborexant was generally quantifiable up to 24‐72 hours postdose in the 1‐ to 5‐mg dose groups, up to 120‐240 hours postdose in the 10‐ to 200‐mg dose groups, and exhibited multiexponential decline (Figure 1). Lemborexant plasma concentrations declined approximately 4‐fold from the time of Cmax to 9 hours postdose. Exposure at 9 hours postdose following multiple daily doses of lemborexant 10 mg was 27% of the Cmax, and AUC0‐9h was <60% of AUC0‐24h.
Figure 1.
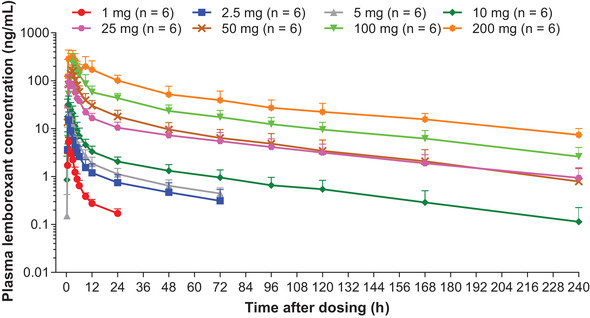
Mean (SD) plasma lemborexant concentration‐time curve for healthy adult subjects who received single doses of lemborexant in Study 001.
PK parameters for Study 001 are summarized in Table 1. Lemborexant was absorbed rapidly following single 1‐ to 10‐mg doses, with a median tmax of approximately 1 hour. Cmax increased with increase in dose across dose groups, median tmax tended to slightly increase with dose, consistent with a dose‐related change in absorption rate with increasing the dose above 10 mg. AUC values also increased with increase in dose. Less than 1% of the administered dose was recovered as unchanged drug in urine across all dose groups.
Table 1.
Study 001: Pharmacokinetic Parameters After Single Doses of Lemborexant in Healthy Adult Subjects
| Lemborexant | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | 1 mg(n = 6) | 2.5 mg(n = 6) | 5 mg(n = 6) | 10 mg(n = 6) | 25 mg(n = 6) | 50 mg(n = 6) | 100 mg(n = 6) | 200 mg(n = 6) |
| Cmax, ng/mL | ||||||||
| Geometric mean (CV) | 5.2 (27.1) | 14.9 (43.0) | 22.3 (19.1) | 32.0 (57.3) | 107 (20.3) | 161 (32.9) | 242 (47.0) | 429 (12.2) |
| Mean (SD) | 5.3 (1.3) | 15.9 (5.7) | 22.7 (4.4) | 36.0 (18.7) | 108 (22.0) | 168 (48.7) | 264 (128) | 431 (51.1) |
| tmax, median (range), h | 1.0 (1.0‐1.1) | 1.0 (1.0‐3.0) | 1.6 (0.9‐3.0) | 1.0 (0.6‐2.0) | 2.0 (1.0‐3.0) | 2.5 (1.0‐3.1) | 3.0 (3.0‐5.0) | 3.0 (1.0‐9.0) |
| AUC0‐24h, ng•h/mL | ||||||||
| Geometric mean (CV) | 17.0 (17.5) | 53.8 (36.4) | 93.2 (18.9) | 150 (38.9) | 648 (15.4) | 1060 (33.0) | 1850 (33.8) | 3970 (24.8) |
| Mean (SD) | 17.2 (3.1) | 56.8 (21.1) | 94.6 (18.8) | 159 (61.9) | 654 (97.6) | 1110 (321) | 1930 (588) | 4080 (1040) |
| AUC0‐t, ng•h/mL | ||||||||
| Geometric mean (CV) | 18.4 (29.9) | 74.4 (45.9) | 126 (18.7) | 274 (30.4) | 1390 (31.3) | 1960 (38.5) | 4300 (35.1) | 9290 (39.5) |
| Mean (SD) | 19.1 (6.2) | 80.2 (32.2) | 128 (26.5) | 284 (80.7) | 1450 (455) | 2080 (775) | 4490 (1300) | 9840 (3510) |
| AUC0‐inf, ng•h/mL | ||||||||
| Geometric mean (CV) | 19.5 (19.8) | 72.3 (54.3) | 146 (20.9) | 299 (31.7) | 1470 (33.7) | 2020 (40.3) | 4520 (36.8) | 9910 (39.5) |
| Mean (SD) | 19.8 (4.0) | 79.7 (42.0) | 149 (34.3) | 311 (90.1) | 1540 (518) | 2150 (834) | 4740 (1420) | 10 500 (3690) |
| t½, mean (SD), h | 23.6 (21.6) | 31.8 (12.7) | 33.4 (6.0) | 56.8 (12.8) | 64.9 (6.7) | 52.1 (14.0) | 61.0 (9.1) | 60.8 (17.9) |
AUC0‐24h indicates area under the concentration‐time curve from time 0 to time 24 hours; AUC0‐inf, area under the concentration‐time curve from time 0 to infinity; AUC0‐t, area under the concentration‐time curve from time 0 to time of last measurable concentration; Cmax, maximum drug concentration; CV, coefficient of variation; t½, terminal elimination half‐life; tmax, time to reach maximum (peak) concentration following drug administration.
Data are geometric mean (% CV) unless otherwise indicated.
Study 002
Study 002 PK parameters are summarized in Table 2. On both day 1 and day 14, lemborexant was rapidly absorbed following single and multiple doses (2.5‐10 mg). On day 14, tmax was 1.0 hour (range 1.0‐3.0 hours) for lemborexant 5 mg and 1.8 hours (range 1.0‐4.0 hours) for lemborexant 10 mg. Consistent with the results of Study 001, both Cmax and AUC0‐24h increased in approximate proportion to dose on both day 1 and day 14. Post‐Cmax lemborexant concentrations exhibited an apparent multiexponential decline (Figure 2A,C). The accumulation at steady state (approximately 1.8‐ to 4.3‐fold) was overpredicted based on the standard calculation used to estimate accumulation, 1/(1 – exp[–kτ]), where τ = 24 hours. Based on the observed accumulation of lemborexant (calculated from the ratio of steady state and single‐dose exposure on day 1) of approximately 2.5‐fold following multiple daily dosing, the geometric mean t½,eff was 17 hours for 5 mg and 19 hours for 10 mg. Based on lemborexant trough concentrations, steady state was reached by day 7 to day 10 across all dose levels. The PK parameters determined for elderly and adult subjects receiving 25 mg lemborexant were similar (Table 2).
Table 2.
Study 002: Pharmacokinetic Parameters on Days 1 and 14 After Multiple Doses of Lemborexant in Healthy Adult and Elderly Subjects
| Lemborexant | |||||||
|---|---|---|---|---|---|---|---|
| Parameter | 2.5 mg(n = 6) | 5 mg(n = 6) | 10 mg(n = 6) | 25 mg(n = 6) | Elderly 25 mg (n = 5) | 50 mg(n = 6) | 75 mg(n = 6) |
| Cmax, ng/mL | |||||||
| Day 1 | |||||||
| Geometric mean (CV) | 9.5 (39.7) | 18.1 (42.0) | 28.0 (48.3) | 89.4 (26.9) | 72.1 (49.0) | 187 (39.2) | 203 (51.2) |
| Mean (SD) | 10.1 (4.3) | 19.4 (7.9) | 30.4 (13.1) | 92.0 (24.0) | 79.4 (43.1) | 199 (81.2) | 223 (103) |
| Day 14 | |||||||
| Geometric mean (CV) | 14.8 (30.6) | 22.0 (49.1) | 44.8 (35.7) | 102 (32.0) | 120 (41.8) | 218 (16.5) | 401 (35.3) |
| Mean (SD) | 15.4 (4.7) | 24.0 (10.7) | 46.9 (14.5) | 107 (38.9) | 128 (47.0) | 220 (33.5) | 420 (140) |
| tmax, median (range), h | |||||||
| Day 1 | 2.0 (1.5‐4.0) | 1.3 (1.0‐3.0) | 3.3 (1.0‐12.0) | 1.5 (1.0‐5.0) | 3.0 (2.0‐5.0) | 2.0 (1.0‐4.0) | 3.0 (1.0‐5.0) |
| Day 14 | 2.0 (1.0‐3.0) | 1.0 (1.0‐3.0) | 1.8 (1.0‐4.0) | 3.0 (1.5‐5.0) | 2.0 (1.5‐4.0) | 2.0 (2.0‐3.0) | 2.0 (1.0‐2.0) |
| AUC0‐24h, ng•h/mL | |||||||
| Day 1 | |||||||
| Geometric mean (CV) | 57.5 (27.3) | 104 (34.4) | 182 (25.2) | 540 (19.2) | 538 (49.4) | 899 (31.3) | 1230 (23.5) |
| Mean (SD) | 59.4 (17.5) | 108 (34.9) | 187 (47.9) | 549 (104) | 596 (347) | 931 (253) | 1260 (301) |
| Day 14 | |||||||
| Geometric mean (CV) | 116 (30.6) | 169 (49.5) | 321 (52.7) | 1050 (36.0) | 1170 (31.0) | 2220 (28.5) | 3710 (23.5) |
| Mean (SD) | 120 (38.0) | 186 (87.5) | 357 (193) | 1100 (387) | 1210 (335) | 2300 (758) | 3790 (857) |
| t½,eff, h, mean (SD) | 24.7 (7.3) | 18.0 (6.5) | 20.9 (10.7) | 23.0 (10.9) | 29.9 (18.2) | 31.3 (11.6) | 45.3 (23.2) |
| RacCmax | |||||||
| Day 14 | |||||||
| Geometric mean (CV) | 1.6 (22.4) | 1.2 (28.4) | 1.6 (49.4) | 1.2 (43.0) | 1.7 (54.2) | 1.2 (32.1) | 2.3 (41.5) |
| Mean (SD) | 1.6 (0.3) | 1.3 (0.3) | 1.8 (0.8) | 1.3 (0.5) | 1.9 (1.0) | 1.2 (0.4) | 2.4 (0.9) |
| RacAUC0‐24h | |||||||
| Day 14 | |||||||
| Geometric mean (CV) | 2.0 (20.7) | 1.6 (22.9) | 1.8 (29.4) | 1.9 (35.9) | 2.2 (49.1) | 2.5 (27.9) | 3.1 (41.2) |
| Mean (SD) | 2.1 (0.4) | 1.7 (0.4) | 1.8 (0.6) | 2.0 (0.6) | 2.4 (1.1) | 2.5 (0.7) | 3.3 (1.4) |
AUC0‐24h indicates area under the concentration‐time curve from time 0 to time 24 hours; Cmax, maximum drug concentration; CV, coefficient of variation; Rac, accumulation ratio based on Cmax and AUC0‐24h; t½,eff, effective half‐life; tmax, time to reach maximum (peak) concentration following drug administration.
Figure 2.
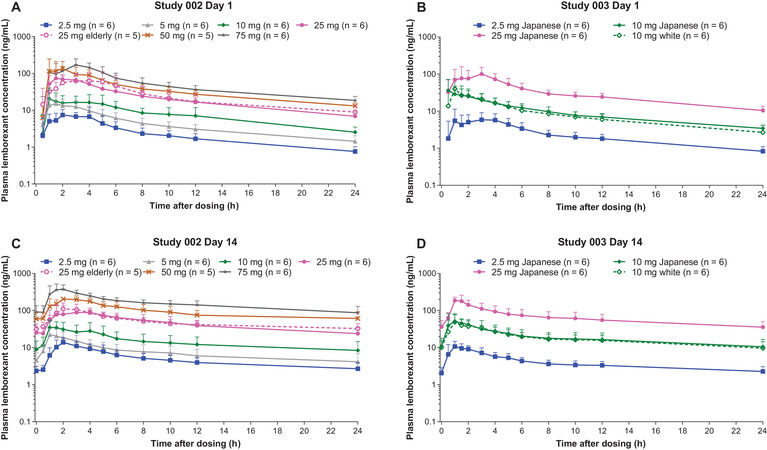
Mean (SD) plasma lemborexant concentration‐time curves for healthy adult and elderly subjects who received lemborexant for 14 days in Study 002 (A,C) and healthy Japanese and white subjects who received lemborexant for 14 days in Study 003 (B,D).
Study 003
PK parameters for Study 003 are summarized in Table 3. Lemborexant was rapidly absorbed following administration of single and multiple doses of lemborexant in both Japanese and white adults, and postdose lemborexant concentrations were characterized in each by a multiexponential decline (Figure 2B,D). There were no differences in PK parameters between the Japanese and white subjects receiving 10‐mg/day doses.
Table 3.
Study 003: Pharmacokinetic Parameters on Days 1 and 14 After Multiple Doses of Lemborexant in Healthy Adult Japanese and White Subjects
| Lemborexant | ||||
|---|---|---|---|---|
| Japanese | White | |||
| Parameter | 2.5 mg (n = 6) | 10 mg (n = 6) | 25 mg (n = 6) | 10 mg (n = 6) |
| Cmax, ng/mL | ||||
| Day 1 | ||||
| Geometric mean (CV) | 10.0 (13.0) | 40.6 (63.5) | 140 (37.8) | 41.9 (54.5) |
| Mean (SD) | 10.0 (1.4) | 46.5 (25.8) | 148 (56.5) | 47.3 (28.1) |
| Day 14 | ||||
| Geometric mean (CV) | 12.8 (27.9) | 64.5 (48.6) | 203 (35.8) | 54.6 (48.3) |
| Mean (SD) | 13.2 (3.4) | 70.2 (30.2) | 213 (66.3) | 59.4 (26.1) |
| tmax, median (range), h | ||||
| Day 1 | 1.5 (1.0‐4.0) | 1.0 (0.5‐6.0) | 2.5 (1.0‐4.0) | 1.0 (1.0‐3.0) |
| Day 14 | 1.3 (0.5‐2.0) | 1.5 (0.5‐2.0) | 1.0 (1.0‐1.5) | 1.3 (1.0‐3.0) |
| AUC0‐24h, ng•h/mL | ||||
| Day 1 | ||||
| Geometric mean (CV) | 53.3 (26.9) | 229 (16.8) | 736 (21.2) | 194 (41.4) |
| Mean (SD) | 54.8 (13.1) | 231 (40.2) | 750 (154) | 208 (83.4) |
| Day 14 | ||||
| Geometric mean (CV) | 93.8 (21.1) | 446 (28.0) | 1480 (35.6) | 385 (55.3) |
| Mean (SD) | 95.6 (21.4) | 459 (110) | 1560 (559) | 431 (226) |
| t½,eff, mean (SD), h | 16.2 (5.5) | 16.7 (5.5) | 20.3 (9.5) | 15.8 (4.0) |
| RacCmax | ||||
| Day 14 | ||||
| Geometric mean (CV) | 1.3 (31.6) | 1.6 (25.1) | 1.5 (46.1) | 1.3 (47.2) |
| Mean (SD) | 1.3 (0.4) | 1.6 (0.4) | 1.6 (0.6) | 1.4 (0.7) |
| RacAUC0‐24h | ||||
| Day 14 | ||||
| Geometric mean (CV) | 1.8 (27.0) | 2.0 (27.2) | 2.0 (25.2) | 2.0 (18.4) |
| Mean (SD) | 1.8 (0.5) | 2.0 (0.5) | 2.1 (0.5) | 2.0 (0.4) |
AUC0‐24h indicates area under the concentration‐time curve from time 0 to time 24 hours; Cmax, maximum drug concentration; CV, coefficient of variation; Rac, accumulation ratio based on Cmax and AUC0‐24h; t½,eff, effective half‐life; tmax, time to reach maximum (peak) concentration following drug administration.
Pharmacodynamics
Study 001
There were dose‐related changes in all measures of sleepiness (PVT, KSS, and DSST) following daytime dosing (Figure 3). PD response on these measures was generally maximal at 2 hours postdose, coinciding, generally, with Cmax. Importantly for lemborexant doses up to 25 mg, there was no evidence of a clinically relevant drug effect beyond 8 hours postdose.
Figure 3.
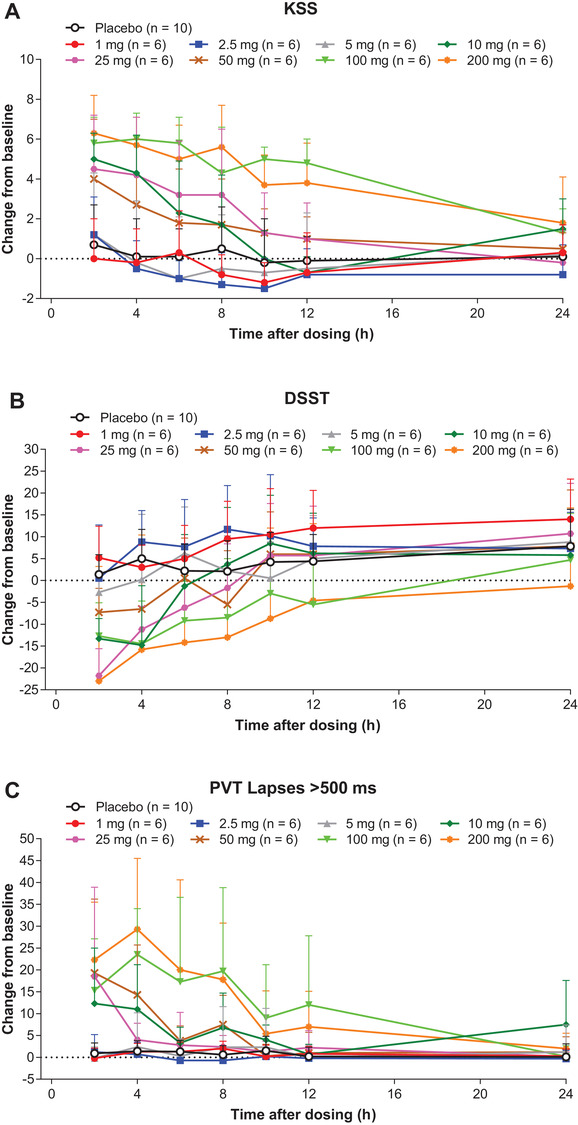
Mean (SD) change from baseline in (A) Karolinska Sleepiness Scale (KSS), (B) Digit Symbol Substitution Test (DSST), and (C) number of lapses >500 ms in Psychomotor Vigilance Test (PVT) in healthy adult subjects who received single doses of lemborexant in Study 001.
There were no apparent differences in any measure of sleepiness between the sexes (data not shown).
Study 002
Measures of next‐morning residual sleepiness (PVT and KSS) were generally similar between the lemborexant 2.5‐ to 25‐mg dose groups and placebo (Figure 4). In the 50‐ and 75‐mg dose groups, which are 5‐ and 7.5‐fold greater than the highest dose (10 mg) currently approved for the treatment of insomnia, there were consistent increases in both measures of residual sleepiness compared with placebo (Figure 4A,B). The magnitude of these increases was generally most pronounced after the first 2‐4 days of treatment rather than the last few days of treatment, suggesting an attenuation of these responses following repeat multiple daily dose administration. The effect of lemborexant on next‐morning residual sleepiness was similar between the elderly and adult lemborexant 25‐mg dose groups (Figure 4A,B).
Figure 4.
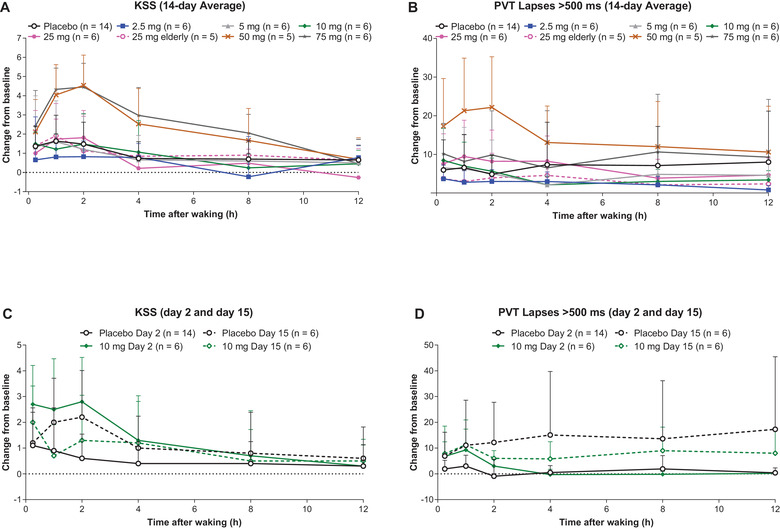
Mean (SD) change from baseline in Karolinska Sleepiness Scale (KSS) and number of lapses >500 ms in Psychomotor Vigilance Test (PVT) over the 14‐day average (A: KSS; B: PVT) and on days 2 and 15 (C: KSS; D: PVT) in healthy adult and elderly subjects who received lemborexant for 14 days in Study 002.
Study 003
There were no consistent or clinically relevant effects of lemborexant 10 mg on next‐morning residual sleepiness (PVT or KSS) in Japanese or white subjects (Supplemental Figure S1). The effect of lemborexant on next‐morning residual sleepiness was similar between the Japanese and white subjects.
Safety
Study 001
Single doses of lemborexant were well tolerated in healthy adults (Table 4A, Supplemental Table S4A). The overall incidence of TEAEs in subjects who received lemborexant was similar to that in subjects receiving placebo (39.6% versus 43.8%). There was no clear dose‐related trend in the prevalence of TEAEs, although few subjects who received the lowest 3 doses of lemborexant reported TEAEs. Further, there were no SAEs, severe TEAEs, or TEAEs leading to discontinuation in any treatment groups. All TEAEs were mild in severity except for 1 event of sleep paralysis of moderate severity reported by a subject who received lemborexant 200 mg, a dose 20‐fold greater than the highest dose (10 mg) approved for the treatment of insomnia. Headache, sleep paralysis, and insomnia were the only TEAEs reported by more than 1 subject in any individual treatment group.
Table 4.
Summary of Adverse Events After Single Doses of Lemborexant in Healthy Adult Subjects, Multiple Doses of Lemborexant in Healthy Adult and Elderly Subjects, and Multiple Doses of Lemborexant in Healthy Adult Japanese and White Subjects
| A: Study 001 | Lemborexant | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Adverse Event, n (%) | Placebo(n = 16) | 1 mg(n = 6) | 2.5 mg(n = 6) | 5 mg(n = 6) | 10 mg(n = 6) | 25 mg(n = 6) | 50 mg(n = 6) | 100 mg(n = 6) | 200 mg(n = 6) |
| TEAEs | 7 (43.8) | 1 (16.7) | 0 | 1 (16.7) | 4 (66.7) | 2 (33.3) | 4 (66.7) | 2 (33.3) | 5 (83.3) |
| Treatment‐related TEAEs | 1 (6.3) | 0 | 0 | 0 | 2 (33.3) | 0 | 3 (50.0) | 2 (33.3) | 4 (66.7) |
| SAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs leading to discontinuation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| B: Study 002 | Lemborexant | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Adverse Event, n (%) | Placebo(n = 16) | 2.5 mg(n = 6) | 5 mg(n = 6) | 10 mg(n = 6) | 25 mg(n = 6) | Elderly 25 mg (n = 5) | 50 mg(n = 6) | 75 mg(n = 6) | |
| TEAEs | 11 (78.6) | 5 (83.3) | 5 (83.3) | 6 (100.0) | 5 (83.3) | 5 (100) | 6 (100) | 6 (100) | |
| Treatment‐related TEAEs | 6 (42.9) | 3 (50.0) | 3 (50.0) | 4 (66.7) | 4 (66.7) | 5 (100) | 6 (100) | 6 (100) | |
| SAEs | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| TEAEs leading to discontinuation | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | |
| Japanese | White | |||||
|---|---|---|---|---|---|---|
| C: Study 003 | Lemborexant | Lemborexant | ||||
| Adverse Event, n (%) | Placebo(n = 6) | 2.5 mg(n = 6) | 10 mg(n = 6) | 25 mg(n = 6) | Placebo (n = 2) | 10 mg (n = 6) |
| TEAEs | 2 (33.3) | 1 (16.7) | 0 | 2 (33.3) | 1 (50.0) | 3 (50.0) |
| Treatment‐related TEAEs | 2 (33.3) | 1 (16.7) | 0 | 2 (33.3) | 0 | 3 (50.0) |
| SAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| TEAEs leading to discontinuation | 0 | 0 | 0 | 0 | 0 | 0 |
SAE indicates serious adverse event; TEAE, treatment‐emergent adverse event.
There were no notable changes from baseline in laboratory values, vital signs, or ECG findings. No suicidal behavior or suicidal ideation was reported during the study.
The MTD of lemborexant was not reached at doses that turned out to be 20‐fold higher than those used in phase 3 studies. Safety signals did not prevent continued testing of ascending doses. However, continued testing of ascending doses was considered unnecessary because the highest dose tested was sufficiently higher (>10‐fold) than the anticipated therapeutic dose range. Therefore, the safety margin achieved was considered sufficient for all further planned clinical testing.
Study 002
Multiple doses of lemborexant were generally well tolerated in healthy adults (Table 4B, Supplemental Table S4B). The overall incidence of TEAEs in subjects who had received lemborexant was higher than that in subjects who had received placebo (92.7% versus 78.6%). There were no SAEs, and most TEAEs were mild in severity. One adult in the lemborexant 25‐mg dose group discontinued the study drug as a result of a TEAE of pyrexia; this TEAE was mild in severity and considered not related to study treatment. The most common TEAE was somnolence, the incidence of which increased with increasing lemborexant dose. Five lemborexant‐treated subjects reported sleep paralysis (25 mg, n = 1; 50 mg, n = 2; 75 mg, n = 2). There were no clinically relevant differences in the incidence of TEAEs between the elderly and adult lemborexant 25‐mg dose groups. Hypoxia was observed in 3 elderly subjects, 1 in the placebo group and 2 in the lemborexant 25‐mg group. All affected subjects were male and had preexisting risk factors for sleep‐disordered breathing, including being male and elderly (1 subject also had a BMI in the overweight range). Polysomnographic screening for the apnea‐hypopnea index had not been conducted to exclude subjects with sleep‐disordered breathing, so it is possible that these 3 subjects had undiagnosed sleep apnea.
There were no notable changes from baseline in laboratory values, vital signs, or ECG findings. As previously reported, 21 concentration‐response analyses of ECG data from Study 002 and Study 003 indicated that lemborexant does not cause QT prolongation, with a wide margin relative to the highest phase 3 dose of 10 mg. No suicidal behavior or suicidal ideation was reported during the study.
The MTD was not reached at doses up to 7.5‐fold higher than those currently approved for the treatment of insomnia.
Study 003
Multiple doses of lemborexant were well tolerated in healthy Japanese and white adults (Table 4C, Supplemental Table S4C). The overall incidence of TEAEs was low across all treatment groups. There were no SAEs, and no TEAE was severe or led to discontinuation in any treatment group. All TEAEs were mild in severity. The most common TEAE was somnolence (all events resolved). There were no clinically relevant differences in the incidence of TEAEs between the Japanese and white subjects receiving 10‐mg/day doses.
There were no clinically relevant laboratory value, vital sign, or ECG findings. No suicidal behavior or suicidal ideation was reported during the study.
Discussion
The phase 1 single‐ and multiple‐ascending‐dose studies described in this report provide important initial clinical pharmacology information about the PK, PD, and safety of lemborexant, including the effects of dose, age, sex, and race. The doses tested represent large multiples (up to 200 mg as a single dose and up to 75 mg as multiple doses for 14 days) of the highest dose (10 mg) currently approved for the treatment of insomnia. At doses up to 25 mg, lemborexant displayed approximately linear PK and had no clinically relevant effect on next‐morning residual sleepiness as measured by PVT, KSS, and DSST. Further, lemborexant was well tolerated and had a favorable safety profile at large margins relative to the highest phase 3 dose (10 mg). There were no clinically relevant effects of age (adult versus elderly), sex, or race/ethnicity (Japanese versus white) on the PK, PD, or safety of lemborexant. In addition, there were no notable differences in PK parameters between daytime (Study 001) and nighttime (Studies 002 and 003) dosing. Together, the findings from these phase 1 studies supported the clinical evaluation of lemborexant as a treatment for insomnia.
The accumulation ratio following multiple dosing calculated from t½ can be overpredicted. As reported elsewhere, lemborexant demonstrates a multiphasic plasma concentration‐time profile. 15 Using the observed data determined after multiple daily dosing to steady state, 13 specifically the AUC accumulation ratio following once‐daily dosing for 14 days, the t½,eff was estimated to be 17 hours for lemborexant 5 mg and 19 hours for lemborexant 10 mg. The t½,eff of lemborexant was consistent between adults and elderly, the sexes, and Japanese and white adults.
An important descriptive PK characteristic of lemborexant at steady state is the approximate 4‐fold reduction in plasma concentrations, from Cmax to 9‐hour concentrations postdose. The exposure at 9 hours postdose, a concentration relevant to residual next‐day effects, following multiple daily doses of 10 mg was 27% of the Cmax, and AUC0‐9h was <60% of AUC0‐24h. These characteristics indicate that a large proportion of lemborexant exposure is eliminated during nighttime, with substantially lower exposure during daytime. Such exposure profiles are likely preferable to minimize residual next‐day effects and other AEs during the day, as observed during the lemborexant clinical program. 6 , 7
Importantly, the wide range of doses assessed in these studies allows for full delineation between doses that show evidence of residual next‐day effects and those that do not show effects. Residual next‐day plasma concentrations did not result in next‐morning residual sleepiness at doses from 2.5 to 25 mg, as evaluated using both objective (PVT and DSST) and subjective (KSS) measures and as demonstrated by the lack of somnolence appearing toward the end of the 14‐day treatment period. The absence of drug effect beyond 8 hours after dosing up to 25 mg/day suggests that lemborexant exposures may fall below the threshold level of orexin receptor occupancy (binding) necessary to block clinically relevant orexin activity during the daytime at doses up to 25 mg. 22 The findings from these phase 1 studies suggest that, at doses up to 25 mg, next‐morning plasma lemborexant concentrations may not exceed the receptor occupancy threshold required for continued sleep promotion in the face of diurnally increasing endogenous orexin concentrations. 3
Lemborexant interacts with the orexin neurotransmitter system as a receptor antagonist. Consequently, this novel mechanism of action enables selection of a candidate drug with a PK profile that supports target pharmacology (competitive orexin receptor binding) at night while minimizing PK exposure/pharmacologic effects during the day. Thus, another important PK/PD finding of these studies is that accumulation of lemborexant in the plasma with multiple dosing did not result in an increasing level of next‐morning residual sleepiness, a finding corroborated in the phase 2 dose‐selection study. 7 The lack of an effect of plasma lemborexant accumulation on next‐morning residual sleepiness may be attributed to the orexin receptor occupancy threshold and the competition with endogenous orexin for receptor sites. Consistent with these PK/PD findings, single and repeated doses of lemborexant (2.5, 5, or 10 mg) had no clinically meaningful residual effect on next‐morning on‐road driving performance (9 hours after bedtime dosing) in a phase 1, randomized controlled study involving healthy adults and elderly subjects. 23
In all 3 studies lemborexant was well tolerated at doses representing large multiples (up to 200 mg as a single dose and up to 75 mg as multiple doses for 14 days) of the highest dose (10 mg) currently approved for the treatment of insomnia and had a favorable safety profile, consistent with the mechanism of action. The safety and tolerability profiles were similar between adults and elderly subjects and between Japanese and white subjects, indicating that no age‐ or race‐based dose adjustments are likely to be needed. There were no SAEs, severe TEAEs, and only 1 TEAE (considered not related to study treatment) that led to discontinuation. Somnolence and, to a lesser extent, sleep paralysis were reported by several subjects, none of whom discontinued the study. These events were more common with supratherapeutic doses (≥25 mg) of lemborexant and are consistent with the known pharmacology of orexin receptor antagonism, 24 findings from previously reported studies of other dual orexin receptor antagonists, 25 , 26 and the phase 2 and 3 studies of lemborexant. 6 , 7 , 8
The phase 1 clinical development program for lemborexant has several notable strengths. These include the testing of a wide range of doses in both single‐ and ascending‐dose studies, the inclusion of both sexes, as well as elderly, Japanese, and white subject groups to evaluate the effects of age, sex, and race on PK, PD, and safety, and the use of multiple measures, both objective and subjective, for the assessment of sleepiness. Limitations of the studies described in this report include those inherent to phase 1 studies in general, such as the number of subjects in each group and the length of treatment and follow‐up.
Conclusions
The findings from phase 1 studies show that, at multiple doses up to 25 mg/day, lemborexant exhibited a linear PK profile and did not exert clinically relevant effects on next‐morning residual sleepiness. Lemborexant also had favorable tolerability and safety profiles at large margins relative to the highest phase 3 dose. Finally, there were no clinically relevant differences in effects attributable to age, sex, or race on the PK, PD, or safety profiles of lemborexant. Overall, the PK, PD, and safety results from these studies suggest that lemborexant at doses up to 25 mg provides a drug profile suitable for pharmacologic effect supporting treatment of insomnia, while exhibiting minimal pharmacologic liability during wake time.
Role of Contributors
All authors participated in the interpretation of study results and in the drafting, critical revision, and approval of the final version of this article. Patricia Murphy, formerly employed by Eisai Inc, contributed to the initial drafting of this manuscript.
Data‐Sharing Statement
Deidentified subject data that underlie the results reported in this article will not be made available, but summary information will be made available on ClinicalTrials.gov following drug approval.
Conflicts of Interest
All authors are employees of Eisai Inc or Eisai Co, Ltd.
Funding
The studies described in this manuscript were funded by Eisai Inc. Medical writing assistance was provided by Luke Carey, PhD, CMPP, and John Bilbruck, PhD, CMPP, of ProScribe—Envision Pharma Group and was funded by Eisai Inc. ProScribe's services complied with international guidelines for Good Publication Practice (GPP3). Eisai Inc was involved in the study design, data collection, data analysis, and preparation of this manuscript.
Supporting information
Figure S1. Mean (SD) change from baseline in Karolinska Sleepiness Scale (KSS) and number of lapses >500 ms in Psychomotor Vigilance Test (PVT) (A, KSS; B, PVT) on days 2 and 15 in healthy Japanese and white subjects who received lemborexant for 14 days in Study 003.
Table S1. Study 001: Demographics and Baseline Characteristics of Healthy Adult Subjects
Table S2. Study 002: Demographics and Baseline Characteristics of Healthy Adult and Elderly Subjects
Table S3. Study 003: Demographics and Baseline Characteristics of Healthy Adult Japanese and White Subjects
Table S4. Summary of Adverse Events After Single Doses of Lemborexant in Healthy Adult Subjects (A: Study 001), Multiple Doses of Lemborexant in Healthy Adult and Elderly Subjects (B: Study 002), and Multiple Doses of Lemborexant in Healthy Adult Japanese and White Subjects (C: Study 003)
Acknowledgments
The authors would like to thank all study subjects as well as study‐site and study‐team personnel.
[The copyright line for this article was changed on 22 June after original online publication]
References
- 1. Mieda M. The roles of orexins in sleep/wake regulation. Neurosci Res. 2017;118:56‐65. [DOI] [PubMed] [Google Scholar]
- 2. Inutsuka A, Yamanaka A. The physiological role of orexin/hypocretin neurons in the regulation of sleep/wakefulness and neuroendocrine functions. Front Endocrinol. 2013;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sakurai T. Orexin deficiency and narcolepsy. Curr Opin Neurobiol. 2013;23(5):760‐766. [DOI] [PubMed] [Google Scholar]
- 5. Tang S, Huang W, Lu S, et al. Increased plasma orexin‐A levels in patients with insomnia disorder are not associated with prepro‐orexin or orexin receptor gene polymorphisms. Peptides. 2017;88:55‐61. [DOI] [PubMed] [Google Scholar]
- 6. Rosenberg R, Murphy P, Zammit G, et al. Comparison of lemborexant with placebo and zolpidem tartrate extended release for the treatment of older adults with insomnia disorder: a phase 3 randomized clinical trial. JAMA Netw Open. 2019;2(12):e1918254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murphy P, Moline M, Mayleben D, et al. Lemborexant, a dual orexin receptor antagonist (DORA) for the treatment of insomnia disorder: results from a Bayesian, adaptive, randomized, double‐blind, placebo‐controlled study. J Clin Sleep Med. 2017;13(11):1289‐1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yardley J, Pinner K, Murphy P, Filippov G, Zammit G, Moline M. Efficacy of lemborexant compared with placebo in adult and elderly subjects with insomnia: results from a phase 3 study (SUNRISE‐2). Presented at: Advances in Sleep and Circadian Science/Sleep Research Society; February 1‐4, 2019; Clearwater Beach, FL.
- 9. Yoshida Y, Naoe Y, Terauchi T, et al. Discovery of (1R,2S)‐2‐{[(2,4‐dimethylpyrimidin‐5‐yl)oxy]methyl}‐2‐(3‐fluorophenyl)‐N‐(5‐fluoropyridin‐2‐yl)cyclopropanecarboxamide (E2006): a potent and efficacious oral orexin receptor antagonist. J Med Chem. 2015;58(11):4648‐4664. [DOI] [PubMed] [Google Scholar]
- 10. Ueno T, Ishida T, Kusano K. Disposition and metabolism of [14C]lemborexant, a novel dual orexin receptor antagonist, in rats and monkeys. Xenobiotica. 2019;46:688‐697. [DOI] [PubMed] [Google Scholar]
- 11. Beuckmann CT, Suzuki M, Ueno T, Nagaoka K, Arai T, Higashiyama H. In vitro and in silico characterization of lemborexant (E2006), a novel dual orexin receptor antagonist. J Pharmacol Exp Ther. 2017;362(2):287‐295. [DOI] [PubMed] [Google Scholar]
- 12. Beuckmann CT, Ueno T, Nakagawa M, Suzuki M, Akasofu S. Preclinical in vivo characterization of lemborexant (E2006), a novel dual orexin receptor antagonist for sleep/wake regulation. Sleep. 2019;42(6):zsz076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boxenbaum H, Battle M. Effective half‐life in clinical pharmacology. J Clin Pharmacol. 1995;35(8):763‐766. [DOI] [PubMed] [Google Scholar]
- 14. Hughes MA, Glass PS, Jacobs JR. Context‐sensitive half‐time in multicompartment pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology. 1992;76(3):334‐341. [DOI] [PubMed] [Google Scholar]
- 15. Lalovic B, Majid O, Landry I, et al. Population pharmacokinetics of lemborexant, a dual orexin receptor antagonist, in healthy adult and elderly subjects and subjects with primary insomnia. Presented at: Annual Meeting of the Population Approach Group in Europe; June 11‐14, 2019; Stockholm, Sweden.
- 16. Akerstedt T, Gillberg M. Subjective and objective sleepiness in the active individual. Int J Neurosci. 1990;52(1‐2):29‐37. [DOI] [PubMed] [Google Scholar]
- 17. Wechsler D. Manual for the Wechsler Adult Intelligence Scale, Revised. New York: Psychological Corporation; 1981. [Google Scholar]
- 18. Lim J, Dinges DF. Sleep deprivation and vigilant attention. Ann N Y Acad Sci. 2008;1129:305‐322. [DOI] [PubMed] [Google Scholar]
- 19. Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol. 1998;20(3):310‐319. [DOI] [PubMed] [Google Scholar]
- 20. Posner K, Brown GK, Stanley B, et al. The Columbia‐Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Murphy PJ, Yasuda S, Nakai K, et al. Concentration‐response modeling of ECG data from early‐phase clinical studies as an alternative clinical and regulatory approach to assessing QT risk—experience from the development program of lemborexant. J Clin Pharmacol. 2017;57(1):96‐104. [DOI] [PubMed] [Google Scholar]
- 22. Gotter AL, Winrow CJ, Brunner J, et al. The duration of sleep promoting efficacy by dual orexin receptor antagonists is dependent upon receptor occupancy threshold. BMC Neurosci. 2013;14:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vermeeren A, Jongen S, Murphy P, et al. On‐the‐road driving performance the morning after bedtime administration of lemborexant in healthy adult and elderly volunteers. Sleep. 2019;42(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brisbare‐Roch C, Dingemanse J, Koberstein R, et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13(2):150‐155. [DOI] [PubMed] [Google Scholar]
- 25. Herring WJ, Connor KM, Ivgy‐May N, et al. Suvorexant in patients with insomnia: results from two 3‐month randomized controlled clinical trials. Biol Psychiatry. 2016;79(2):136‐148. [DOI] [PubMed] [Google Scholar]
- 26. Michelson D, Snyder E, Paradis E, et al. Safety and efficacy of suvorexant during 1‐year treatment of insomnia with subsequent abrupt treatment discontinuation: a phase 3 randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2014;13(5):461‐471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mean (SD) change from baseline in Karolinska Sleepiness Scale (KSS) and number of lapses >500 ms in Psychomotor Vigilance Test (PVT) (A, KSS; B, PVT) on days 2 and 15 in healthy Japanese and white subjects who received lemborexant for 14 days in Study 003.
Table S1. Study 001: Demographics and Baseline Characteristics of Healthy Adult Subjects
Table S2. Study 002: Demographics and Baseline Characteristics of Healthy Adult and Elderly Subjects
Table S3. Study 003: Demographics and Baseline Characteristics of Healthy Adult Japanese and White Subjects
Table S4. Summary of Adverse Events After Single Doses of Lemborexant in Healthy Adult Subjects (A: Study 001), Multiple Doses of Lemborexant in Healthy Adult and Elderly Subjects (B: Study 002), and Multiple Doses of Lemborexant in Healthy Adult Japanese and White Subjects (C: Study 003)