

Abstract
A repeat expansion in the C9orf72 gene is the most prevalent genetic cause of frontotemporal dementia (C9‐FTD). Several studies have indicated the involvement of the unfolded protein response (UPR) in C9‐FTD. In human neuropathology, UPR markers are strongly associated with granulovacuolar degeneration (GVD). In this study, we aim to assess the presence of UPR markers together with the presence of dipeptide pathology and GVD in post mortem brain tissue from C9‐FTD cases and neurologically healthy controls. Using immunohistochemistry we assessed the presence of phosphorylated PERK, IRE1α and eIF2α in the frontal cortex, hippocampus and cerebellum of C9‐FTD (n = 18) and control (n = 9) cases. The presence of UPR activation markers was compared with the occurrence of pTDP‐43, p62 and dipeptide repeat (DPR) proteins (poly(GA), ‐(GR) & ‐(GP)) as well as casein kinase 1 delta (CK1δ), a marker for GVD. Increased presence of UPR markers was observed in the hippocampus and cerebellum in C9‐FTD compared to control cases. In the hippocampus, overall levels of pPERK and peIF2α were higher in C9‐FTD, including in granule cells of the dentate gyrus (DG). UPR markers were also observed in granule cells of the cerebellum in C9‐FTD. In addition, increased levels of CK1δ were observed in granule cells in the DG of the hippocampus and granular layer of the cerebellum in C9‐FTD. Double‐labelling experiments indicate a strong association between UPR markers and the presence of dipeptide pathology as well as GVD. We conclude that UPR markers are increased in C9‐FTD and that their presence is associated with dipeptide pathology and GVD. Increased presence of UPR markers and CK1δ in granule cells in the cerebellum and hippocampus could be a unique feature of C9‐FTD.
Keywords: C9orf72, cerebellum, dentate gyrus, granule cells, granulovacuolar degeneration, unfolded protein response
Introduction
Frontotemporal dementia (FTD) is a neurodegenerative disease and the second most common cause of presenile dementia. A repeat expansion mutation in the C9orf72 gene is the most common genetic cause of FTD (C9‐FTD). It is found in 12%–25% of familial FTD patients and 3%–6% of the sporadic FTD patients (8, 17). The repeat expansion in C9orf72 also occurs in more than 30% of familial amyotrophic lateral sclerosis (ALS) patients, providing an explanation for the association between ALS and FTD, which share common neuropathological features (34). C9‐FTD commonly presents with changes in behavior and personality, and/or the dysfunction in the language domain (19). The expanded hexanucleotide repeat (GGGGCC) in the first intron of C9orf72 produces RNA foci that are translated by repeat associated non‐AUG dependent (RAN) translation into five aggregation prone dipeptide repeat (DPR) proteins; poly(GA), poly(GR), poly(GP), poly(PR) and poly(PA) (21, 32, 39). Alongside RNA foci and DPR protein aggregates, C9‐FTD patients display inclusions of TAR DNA‐binding protein 43 (TDP43) in affected brain areas (18, 19, 22, 27). The frontal cortex, hippocampus and cerebellum are the most vulnerable regions to C9‐FTD pathology. While TDP43 pathology can be observed in several FTD subtypes and related diseases, RNA foci and DPR proteins are characteristic for C9‐FTD, and therefore their aggregation has been proposed to play a central role in the neurodegenerative process.
The presence of protein aggregates in the cell can lead to a disturbance in protein homeostasis that results in activation of signaling pathways aimed at counteracting this disturbance. The endoplasmic reticulum (ER), as a major site of protein homeostasis, plays a key role in protein quality control. The presence of misfolded proteins and protein aggregates activates the unfolded protein response (UPR) in order to protect the cell from ER stress and prevent the toxic accumulation of protein aggregates. The UPR is regulated by three signaling cascades involving RNA‐activated protein kinase R (PKR)‐like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol requiring enzyme 1 alpha (IRE1α) (15, 29). Once activated, PERK, ATF6 and IRE1α set off a translationally and transcriptionally regulated signaling network aimed at restoring protein homeostasis in the ER. PERK auto‐phosphorylates and in turn phosphorylates eukaryotic initiation factor 2 alpha (eIF2α), which halts protein synthesis whilst also activating the transcription of proapoptotic factors such as CHOP and the translation of ATF4 (2, 11, 12, 15). In human brain tissue, activation of the UPR has been detected in several neurodegenerative diseases including Alzheimer’s disease (AD), frontotemporal lobar degeneration with tau pathology (FTLD‐tau), Parkinson’s disease (PD) and ALS (for review see (28)). Prominent UPR activation is associated with the accumulation of phosphorylated tau and has been found in AD and FTLD‐tau (14, 23, 31). In addition, UPR activation markers are associated with granulovacuolar degeneration (GVD), characterized by basophilic granules predominantly in the hippocampal neurons in AD (1, 37). Interestingly, it was recently shown that the prevalence of GVD is increased in C9‐FTD (26)
There is accumulating evidence supporting the link between toxic dipeptides, UPR markers and neurodegeneration (7, 25, 38). In human post mortem brain tissue, mRNA levels of UPR proteins, ATF4 and CHOP are significantly increased in the frontal cortex of ALS patients with the C9orf72 repeat expansion compared to sporadic ALS patients (38). In patients with frontotemporal lobar degeneration with TDP43 inclusions, and without the C9orf72 repeat expansion (FTLD‐TDP), UPR activation markers phosphorylated PERK (pPERK) and phosphorylated IRE1α (pIRE1α) were not observed in the frontal cortex and hippocampus (23). To our knowledge there is no data on the localization and levels of UPR markers in C9‐FTD. To this end we set out to study the presence of UPR in C9‐FTD. We assessed the presence of UPR markers, protein aggregates and the presence of GVD in post mortem brain tissue from C9‐FTD cases and neurologically healthy controls.
Materials and methods
Subjects
Brain samples from C9‐FTD (n = 18), FTLD‐TDP cases without a repeat expansion mutation in the C9orf72 (n = 8) and neurologically healthy control donors (n = 9) were obtained from the Netherlands Brain Bank (Amsterdam, the Netherlands), the Alzheimer Centre Rotterdam and department of Neurology, Erasmus Medical Centre (Rotterdam, the Netherlands), the department of pathology, Amsterdam UMC (Amsterdam, the Netherlands), and the Dutch Surveillance Centre for Prion Diseases, Department of Pathology, University Medical Centre Utrecht (Utrecht, the Netherlands) (Table 1: pathological and clinical data of all donors).
Table 1.
Demographic information of donors used in study.
| Case no./neuropathological diagnosis | Age of symptom onset | Age at death | Gender (M/F) | PMD (h:min) | Braak stage (NFT) | Clinical diagnosis/cause of death |
|---|---|---|---|---|---|---|
| 1. C9‐FTD | 51 | 56 | F | 3:00 | n/a | bvFTD |
| 2. C9‐FTD | 54 | 63 | M | 4:45 | 1 | PPA |
| 3. C9‐FTD | 65 | 72 | F | 4:50 | 2 | bvFTD |
| 4. C9‐FTD | 39 | 42 | F | 5:45 | n/a | bvFTD/ALS |
| 5. C9‐FTD | 60 | 68 | F | 6:50 | 2 | bvFTD |
| 6. C9‐FTD | 61 | 64 | F | 9:05 | 1 | bvFTD/ALS |
| 7. C9‐FTD | 69 | 75 | M | 6:25 | 1 | bvFTD |
| 8. C9‐FTD | 52 | 59 | M | 8:00 | 2 | bvFTD/ALS |
| 9. C9‐FTD | 53 | 60 | M | 5:05 | 1 | bvFTD |
| 10. C9‐FTD | 56 | 64 | M | 6:20 | 3 | bvFTD |
| 11. C9‐FTD | 71 | 74 | F | <12 | 2 | bvFTD |
| 12. C9‐FTD | 62 | 70 | F | 4:40 | 3 | bvFTD |
| 13. C9‐FTD | 60 | 67 | F | 6:45 | 3 | bvFTD |
| 14. C9‐FTD | 50 | 64 | M | 4:35 | 2 | bvFTD |
| 15. C9‐FTD | 45 | 52 | F | 8:16 | 1 | bvFTD |
| 16. C9‐FTD | 68 | 75 | F | 5:15 | 2 | bvFTD/ALS |
| 17. C9‐FTD | 40 | 47 | F | <12 | 0 | FTD/CJD |
| 18. C9‐FTD | 44 | 46 | F | <12 | 0 | FTD/CJD |
| 1. Control | n/a | 51 | F | <12 | 0 | Aortic aneurysm |
| 2. Control | n/a | 66 | F | <12 | 0 | Heart failure |
| 3. Control | n/a | 82 | F | <12 | 0 | Heart failure |
| 4. Control | n/a | 53 | M | <12 | 0 | Fatal injury |
| 5. Control | n/a | 70 | M | <12 | 0 | Aspiration‐related |
| 6. Control | n/a | 39 | F | <12 | 0 | Fatal injury |
| 7. Control | n/a | 63 | M | <12 | 0 | Cancer |
| 8. Control | n/a | 65 | M | <12 | 0 | Cancer |
| 9. Control | n/a | 66 | M | <12 | 0 | Heart failure |
| 1. FTLD‐TDP | 55 | 66 | M | 5:20 | 0 | bvFTD |
| 2. FTLD‐TDP | 51 | 67 | F | 4:35 | 2 | Schizoaffective disorder |
| 3. FTLD‐TDP | 58 | 69 | M | 5:20 | 0 | svPPA |
| 4. FTLD‐TDP | 52 | 63 | M | 7:35 | 2 | nfPPA |
| 5. FTLD‐TDP | 56 | 68 | M | 7:00 | 1 | svPPA |
| 6. FTLD‐TDP | 62 | 64 | M | 4:29 | 2 | PPA |
| 7. FTLD‐TDP | 55 | 61 | F | 6:30 | 0 | bvFTD |
| 8. FTLD‐TDP | 57 | 67 | F | 7:15 | 3 | bvFTD |
Abbreviations: F = female; M = male; n/a = not available; NFT = neurofibrillary tangles; PMD, post mortem delay.
All cases were neuropathologically assessed based on histochemical stainings including hematoxylin and eosin, Congo red, Bodian or Gallyas and methenamine silver and immunohistochemical stainings including amyloid‐beta, pTau, 3R‐tau, 4R‐tau, pTDP43, alpha‐synuclein and p62. These stainings were performed on formalin‐fixed paraffin‐embedded (FFPE) brain tissue of multiple brain regions including the frontal cortex, temporal pole, superior parietal lobe, occipital pole, amygdala and the hippocampus. Neuropathological staging of AD (NFTs) was evaluated according to Braak and Braak (4)
Both clinical and neuropathological criteria for the disease were met for FTD donors (16, 20). Neurologically healthy controls were donors who died without history of dementia, psychiatric or neurological diseases. All donors or their next of kin provided written informed consent for brain autopsy and use of tissue and medical records for research purposes. This study has been performed according to the Declaration of Helsinki, and the procedures have been approved by the local ethics committee (Amsterdam UMC, Amsterdam, the Netherlands).
Immunohistochemical procedure
Paraffin embedded formalin‐fixed tissue sections (5 μm thick) were cut from the frontal cortex, hippocampus and cerebellum from the FTD and control donors. After deparaffinization, endogenous peroxidase activity was blocked with 0.3% H2O2 in phosphate buffer saline (PBS; pH 7.4) for 30 min. Sections were then treated in 0.1 M citrate buffer (pH 6.0) heated by autoclave (at 121°C for 10 min) for antigen retrieval. Tissue sections were incubated overnight at room temperature with primary antibodies directed against pTDP43 (1:8000; mouse, clone 11‐9, Cosmo Bio, Japan), p62 (1:1000; mouse, clone 3/P62 LCK LIGAND, BD Transduction Laboratories, San Jose, CA, USA), AT8 (1:800; mouse, clone AT8, Thermo Fisher Scientific, MA, USA), casein kinase 1 delta (CK1δ) (1:100; mouse, clone C‐8, Santa Cruz Biotechnology, Dallas TX, USA), poly(GA) (1:2000; rabbit, Cosmo Bio), poly(GR) (1:8000; rabbit, Cosmo Bio), poly(GP) (1:2000; rabbit, Cosmo Bio), peIF2α (1:500; rabbit, Sigma‐Aldrich, St. Louis, MO, USA), pPERK (1:12 800; rabbit, Santa Cruz Biotechnology) and pIRE1α (1:10 000; rabbit, Novus Biologicals, Centennial, CO, USA). Sections were then washed with PBS and incubated with HRP‐labeled Envision (DAKO, Glostrup, Denmark) for 30 min. Color was developed with 3,3′‐diaminobenzidine (DAB; DAKO). Sections were counterstained with haematoxylin and mounted using Quick D (Klinipath; Duiven, the Netherlands).
Double‐immunofluorescence
For double‐immunofluorescence, sections were incubated overnight with a combination of primary antibodies using varying concentrations; p62 (1:1000; mouse), poly(GA) (1:2000; mouse, Millipore, Burlington, MA, USA), poly(GR) (1:1000; rat, Millipore), pPERK (1:6000; rabbit), pIRE1α (1:10 000; rabbit) and CK1δ (1:400; rabbit, Thermo Fisher Scientific). Subsequently, sections were incubated with fluorescent probed secondary antibodies diluted 1:250 for 1 h; Goat Anti‐Rabbit‐594 (Invitrogen, Carlsbad, CA, USA), Goat Anti‐Rat 488 (Invitrogen) and Goat Anti‐Mouse 488 (Life Technologies, Carlsbad, CA, USA). Auto‐fluorescence was blocked with 0.2% Sudan Black for 5 min at room temperature and then slides were mounted with DAPI Fluoromount G (Southern Biotech, AL, USA).
Semi‐quantitative assessment and statistics
To compare the relative presence of immunoreactivity between control and C9‐FTD cases, we applied a semi‐quantitative assessment. Immunopositive neurons were assessed in at least 5 microscopic visual fields using a ×40 objective (400× magnification) in one tissue section based on the following criteria: 0 = absent, 1 = rare (1–10 positive cells), 2 = moderate density (11–20) and 3 = high density (>21) of immunoreactive neurons per microscopic visual field. Neurons were identified based on morphology and nuclear haematoxylin staining. For the medial frontal gyrus multiple visual fields were randomly selected in non‐curved areas that contained all six cortical layers. In this study the CA1/subiculum and the dentate gyrus (DG) were assessed at the level of the middle hippocampal region. Within the hippocampus, adjoining visual subfields were assessed. CA1/subiculum subfields were distinguished based on cell densities and relative thickness of the layers. For the cerebellum only the granular layer was assessed and multiple visual subfields were randomly selected. Statistical analyses were performed using Graphpad Prism (version 7.0a). Semi‐quantitative scores were compared between C9‐FTD and controls using Mann Whitney U Test, where P‐values <0.05 were considered significant. Correlation assessment was performed using Spearman’s correlation for nonparametric data. A P‐value <0.05 was considered significant.
Results
Assessment of pathology and UPR activation markers in C9‐FTD
Comparing the 18 C9‐FTD and 9 neurologically healthy control cases, we found inclusions positive for p62, pTDP43 and the sense translated DPR proteins: poly(GA), poly(GR) and poly(GP) in the frontal cortex, hippocampus and cerebellum of the C9‐FTD cases (Figure 1). p62 and DPR protein pathology was severe in all regions analysed, while pTDP43 pathology was absent in the cerebellum. Moderate presence of immunoreactivity for phosphorylated tau was present in the hippocampus while it was almost absent in the cerebellum (Table S1).
Figure 1.
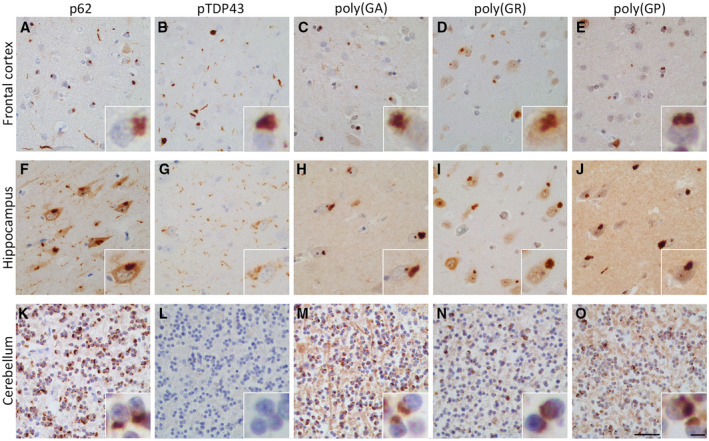
Neuropathology of C9‐FTD in the frontal cortex, hippocampus and cerebellum. Immunohistochemical analysis confirmed C9‐FTD cases in our study exhibited typical disease related pathology. The frontal cortex and hippocampus showed a strong presence of pTDP43, p62 and the DPR proteins poly(GA), poly(GR) and poly(GP) (A–J). In the cerebellum no pTDP43 was seen, however, p62 and the DPR proteins were in abundance (K–O). Cases 14 & 15 were used to illustrate pathology (A–E and F–O respectively). Scale bar represent 50 µm (A–O), and in insert 5 µm.
In the frontal cortex, pPERK, pIRE1α and peIF2α immunoreactivity was observed in C9‐FTD cases (Figure 2A–C), however, when compared to controls they did not reach significance (Figure 2D). Within the C9‐FTD cases no correlation was observed in the frontal cortex between UPR markers and different protein aggregates (p62, pTDP43, Poly(GA), poly(GR) and poly(GP)). In the CA1/subiculum region of the hippocampus a significant increase in pPERK and peIF2α was observed in C9‐FTD compared to controls (P = 0.01 and P = 0.02, respectively), with no difference seen in pIRE1α immunoreactivity (Figure 2E–H). No correlation was observed in this area between UPR markers and different protein aggregates.
Figure 2.
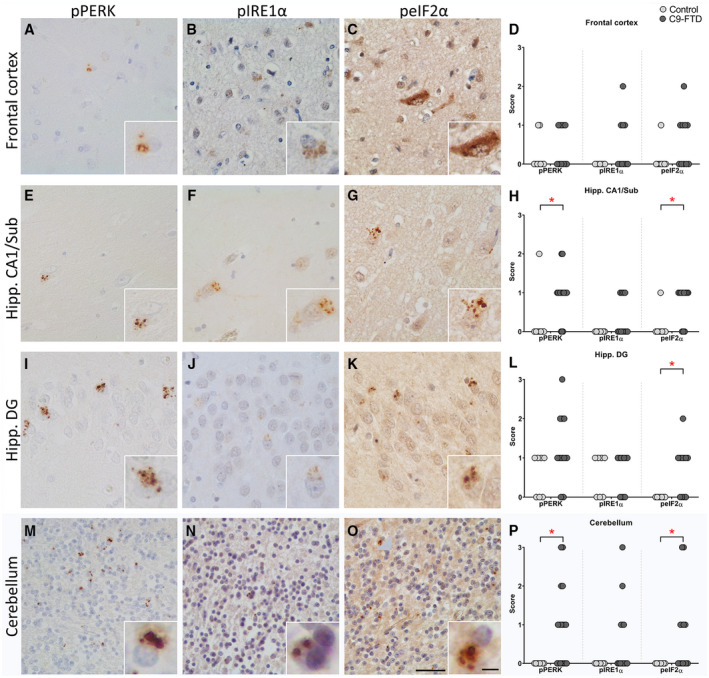
UPR activation markers in the frontal cortex, hippocampus and cerebellum in C9‐FTD and control cases are increased. Immunohistochemistry showing presence of pPERK, pIRE1α and peIF2α in the frontal cortex, CA1/subiculum and DG of the hippocampus and cerebellum in C9‐FTD (A–C, E–G, I–K, M–O). Graphs display UPR marker density scores in the frontal cortex (D), hippocampus CA1/subiculum (H), hippocampus DG (L) and cerebellum granular layer (P), based on the criteria 0 = absent, 1 = rare (1–10 positive cells), 2 = moderate density (11–20) and 3 = high density(>21). To illustrate UPR activation cases 2 (O, J), 6 (G), 9 (B), 11 (C), 14 (A) and 15 (E, F, I, K, M, N) were used. Each dot represents a single cases. Scale bar represent 50 µm, and in insert 5 µm. *P < 0.05.
The hippocampal DG was independently assessed and showed increased levels of pPERK and peIF2α immunoreactivity in granule cells in C9‐FTD, with peIF2α reaching significance when compared to controls (P = 0.01) (Figure 2I–L). Within the C9‐FTD cases no significant correlation was observed in this area between UPR markers and different protein aggregates.
In the C9‐FTD cerebellum, high levels of pPERK, peIF2α and pIRE1α were observed in granule cells in the granular layer, with pPERK and peIF2α reaching significance when compared to controls (P = 0.01 and P = 0.045 respectively) (Figure 2M–P). No pPERK, pIRE1α or peIF2α immunoreactivity was observed in the cerebellum of controls (score = 0, Table S2).
UPR activation markers co‐occur with p62 and poly(GA)
Following the observation that UPR markers were increased in C9‐FTD cerebellum, the co‐ localization of UPR markers with p62 and DPR protein aggregates was assessed (Figure 3). Neurons positive for pPERK and pIRE1α were also positive for p62, however, not all p62 positive neurons were positive for the UPR activation markers. In neurons positive for both UPR markers and p62 the signals did not co‐ localize, showing a different subcellular distribution (Figure 3A,B). Similar findings were observed for poly(GA) inclusions and the UPR activation markers; a subset of cells were positive for both UPR and the DPR protein, however, the majority of poly(GA) inclusion containing neurons were negative for UPR markers (Figure 3C,D). Poly(GR) inclusions and UPR markers were not observed in the same neuron (Figure 3E,F). The small and partial overlap between p62, the DPR protein pathology and UPR markers in the C9‐FTD cerebellum is reflected in the overall assessment; C9‐FTD cases scored 2 or higher for p62 and DPR protein density, whereas the UPR activation marker densities were much lower with only 4 of the 18 patients scoring 2 or higher for UPR markers (Tables S1 and S2).
Figure 3.
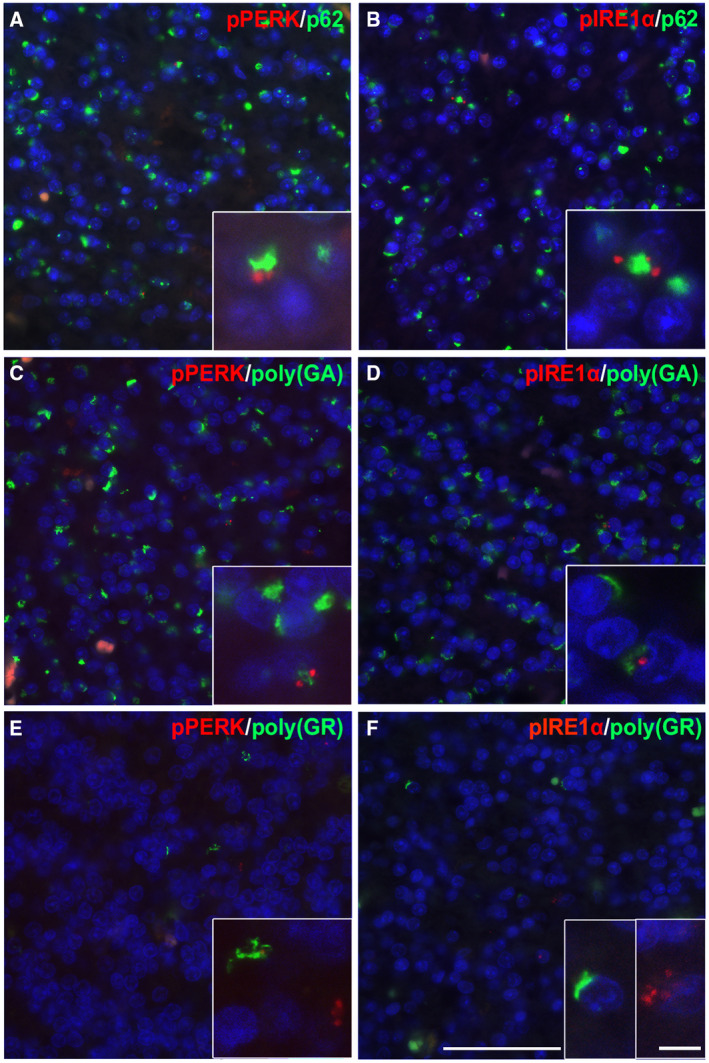
UPR activation markers in granule cells in the C9‐FTD cerebellum. Immunofluorescence staining of pPERK (red) and pIRE1α (red) with p62 (green, A, B), poly(GA) (green, C, D) and poly(GR) (green, E, F) in the granular cell layer of the cerebellum in C9‐FTD. The UPR activation markers (red) were upregulated in a subset of p62 and poly(GA) positive granule cells (green). Though a subset of cells expressed both the DPR proteins and the UPR activation markers, these were not co‐localized and were observed in different regions of the cell. No poly(GR) positive cells were found to be positive for UPR activation markers. Nuclei were stained with DAPI (blue). All images taken from case 2. Scale bar represents 50 µm and in insert 20 µm.
UPR markers are associated with GVD in C9‐FTD hippocampus and cerebellum
Previously, we have shown that UPR markers are associated with GVD in AD (14). Using the GVD marker CK1δ, clear structures resembling GVD could be observed in the granule cells of the DG in the hippocampus of C9‐FTD cases (Figure 4A,B). The granule cells in the granular layer of the cerebellum in C9‐FTD cases also showed a clear presence of GVD (Figure 4C,D). Assessment of CK1δ positive GVD structures in various brain regions showed no differences between control and C9‐FTD cases in the frontal cortex. The presence of CK1δ positive GVD structures was significantly upregulated in C9‐FTD in the hippocampus CA1/subiculum region and the DG, as well as in the cerebellum (P < 0.01, P < 0.01 and P = 0.02 respectively) (Figure 4E). Over all brain regions the densities of GVD correlated well with the presence of UPR markers (Table S2).
Figure 4.
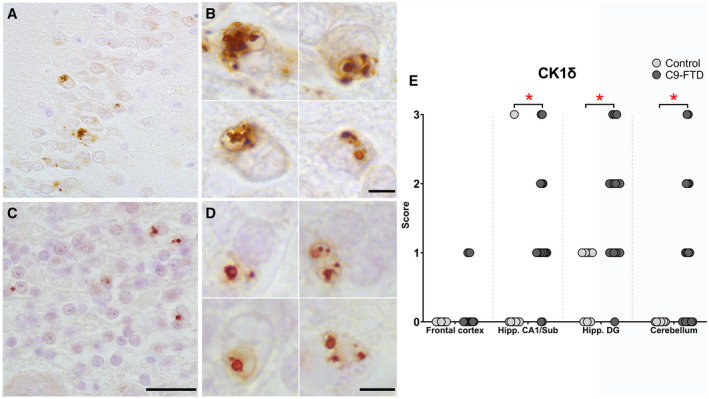
CK1δ positive GVD structures in granule cells of the dentate gyrus in the hippocampus and granular layer in the cerebellum in C9‐FTD. In C9‐FTD, multiple CK1δ positive GVD structures were observed in individual granule cells of the DG in the hippocampus (A, B). Similar CK1δ positive GVD structures were observed in granule cells of the granular layer in the cerebellum (C, D). Occurrence of CK1δ positive GVD structures were not significantly increased in the frontal cortex of C9‐FTD cases. In the hippocampal CA1/Sub and DG, as well as the granular layer of the cerebellum the occurrence of CK1δ positive GVD structures were significantly increased in C9‐FTD patients (e). Nuclei were stained with haematoxylin (blue). Images taken of case 15 (for A, B) and case 2 (C, D). Each dot represents a single case. Scale bars represent 50 µm (A, C) and 5 µm (B, D), *P < 0.05.
Within the DG of C9‐FTD cases CK1δ positive GVD structures significantly correlated with the occurrence of p62 (P = 0.033), poly(GP) (P = 0.045), poly(GR) (P = 0.030), and pPERK (P = 0.003). In addition, double‐immunofluorescence on C9‐FTD hippocampal DG and cerebellum showed co‐ localization of pPERK and pIRE1α with CK1δ (Figure 5A–D). CK1δ was also observed in cells with p62, but these markers did not show co‐ localization and not all cells with p62 inclusions contained CK1δ (Figure 5E,F). In 8 FTLD‐TDP cases without a repeat expansion mutation in the C9orf72 gene no GVD was observed after Haematoxylin and Eosin staining or CK1δ immunohistochemistry.
Figure 5.
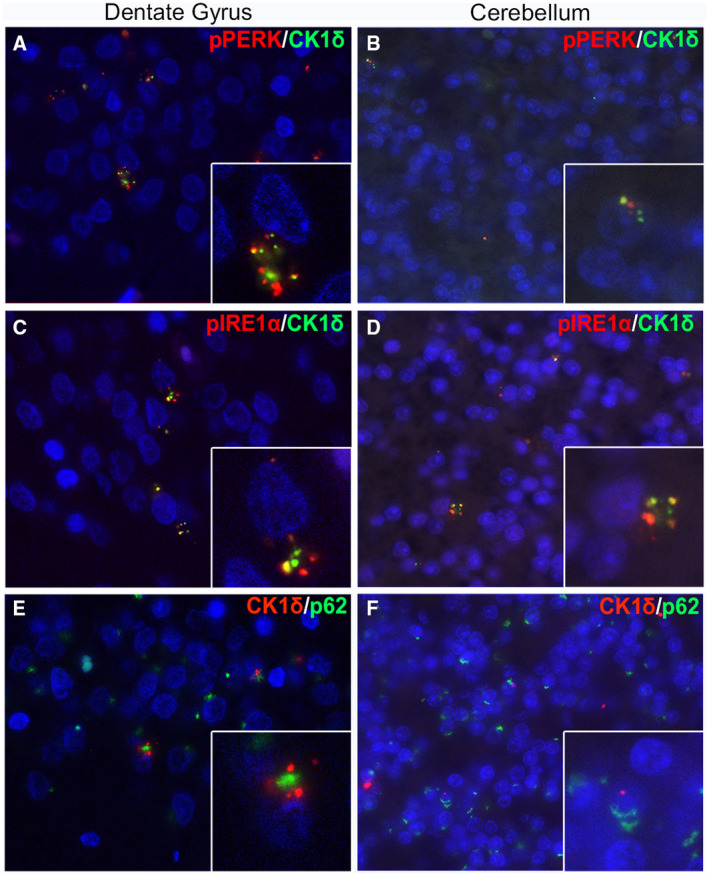
In C9‐FTD, pPERK and pIREα co‐localize with CK1δ in the dentate gyrus and cerebellum. Double‐stainings were performed on the dentate gyrus of the hippocampus and granular cell layer of the cerebellum in C9‐FTD. Double‐staining of pPERK (red) and CK1δ (green) showed co‐localization (yellow) or co‐presence in granule cells (A, B). Similar co‐ localization (yellow) was observed with pIRE1α (red) and CK1δ (green) in granule cells (C, D). P62 (green) did not show co‐localization with CK1δ (red), although most granule cells show co‐presence (E, F). Nuclei were stained with DAPI (blue). Images taken of cases 2 (B, D), 9 (F) and 15 (A, C, E). Scale bars represent 50 µm and in insert 20 µm.
Discussion
The UPR has been suggested to be a common pathomechanism in neurodegenerative diseases where accumulation of misfolded protein aggregates is a main pathological event. In the present study, we have shown that the UPR activation markers, pPERK, pIRE1α and peIF2α are increased in C9‐FTD compared to control cases. This increase was most pronounced in granular cells in the hippocampal DG and the granular layer of the cerebellum. Similar to previous observations in AD hippocampus, we found the UPR markers to be associated with the occurrence of GVD using CK1δ as marker for this specific type of pathology.
In this study, we found an increased presence of UPR markers in C9‐FTD in close association with the presence of DPR proteins. These results are in agreement with the accumulating evidence that UPR activation is involved in the pathogenesis of C9‐FTD/ALS. Transcriptomic analysis of C9‐ALS brain tissue revealed upregulation of ER stress and UPR related genes compared with brain tissue derived from sporadic ALS patients (25). Increased levels of the ER chaperone GRP78/BiP is also observed in C9orf72 iPSC‐derived motor neurons compared with control‐iPSC‐derived motor neurons (7). Recently, it was shown that RAN translation is regulated via phosphorylation of eIF2α, which indicates that activation of the UPR via the PERK pathway results in increased generation of DPR proteins (6, 10). Moreover, inhibition of the peIF2α signaling pathway suppresses RAN translation and formation of DPR proteins (6). In this study, we observed that DPR proteins in C9‐FTD are more abundant than UPR markers, favoring the hypothesis that UPR activation occurs downstream of DPR protein accumulation.
In C9‐FTD cerebellum, UPR markers co‐occurred with poly(GA) and not with poly(GR) DPR proteins. Interestingly, presence of the DPR protein poly(GA) in primary neurons leads to increased levels of UPR stress markers (38). This favors a strong linkage between the presence of poly(GA) and UPR activation in neurons. DPR proteins and UPR activation markers did not co‐ localize at the subcellular level. This corroborates with the observation that increased poly(GA) protein levels outside the ER‐Golgi compartment lead to an increase in pPERK and other UPR markers in primary neurons (38). In addition, UPR activation markers also co‐occur but do not show a subcellular co‐ localization with intracellular pTau and alpha‐synuclein accumulations in AD and Parkinson’s disease, respectively (13, 14). We found that whilst positivity for UPR activation markers is dependent on the presence of poly(GA) aggregates, the majority of aggregated poly(GA) in neurons are negative for UPR markers in the C9‐FTD cerebellum.
To our knowledge this is the first report showing the presence of UPR markers and GVD in the cerebellum. GVD primarily occurs in the pyramidal neurons of the hippocampus and is increased in dementia, compared with age‐matched controls. The occurrence of GVD is most extensively reported in AD, but has also been described in Pick’s disease and Progressive Supranuclear Palsy (28, 30, 37). In addition, GVD is strongly associated with (early) signs of tau pathology as well as TDP43 pathology (14, 33). The granules that characterize GVD most likely correspond to a special type of autophagosome or disturbed endolysosomal process (24, 36). Recently, it was reported that GVD is more prevalent in the hippocampal CA2 region in FTLD/ALS cases with C9orf72 mutation compared with sporadic FTLD/ALS cases and control cases (26). The same study also reported GVD‐like structures in granule cells in the hippocampal DG. The current study supports the increased presence of GVD in C9‐FTD hippocampal regions, including the DG. Using haematoxylin–eosin stainings, Riku et al did not observe GVD in the cerebellum of C9‐FTD/ALS donors (26). Also in the present study we were unable to localize granular cells with GVD granules in the cerebellum using a haematoxylin‐eosin staining. Since the granular layer of the cerebellum is dense with nuclei, it is plausible the GVD structures could be overlooked without performing an immunohistochemical analysis using a specific marker for GVD. In this study, we found CK1δ positive GVD structures in the cerebellum in almost all C9‐FTD donors. The selective vulnerability of granule cells for GVD could be a unique hallmark for C9‐FTD.
In the frontal cortex peIF2α is clearly present in the cytoplasm, next to its presence in granules. Besides PERK, eIF2α is also phosphorylated by different stress‐related kinases, that is, PKR (protein kinase double‐stranded RNA‐dependent), GCN2 (general control non‐derepressible‐2), and HRI (heme‐regulated inhibitor) (9). The presence of a cytoplasmic peIF2α staining could be related to activity of PKR, GCN2 and HRI. In addition, we find no significant increase in levels of CK1δ positive GVD structures in the frontal cortex, despite the presence of protein aggregates and neurodegeneration in C9‐FTD. In the present study, 3 out 14 C9‐FTD cases show presence of CK1δ positive GVD structures in the frontal cortex (Table S2). This frequency is comparable with the earlier study by Riku and colleagues, showing that approximately 15% of C9‐FTD/ALS cases had occurrence of GVD in the frontal cortex based on haematoxylin‐eosin staining or pTDP43 immunostaining (26). Previously, we have shown that prion disease, characterised by substantial accumulation of PrPsc and neurodegeneration in the frontal cortex, is not associated with an increase in CK1δ positive neurons (35). In general, the frontal cortex appears to be less affected by GVD compared with other brain regions like the hippocampal area.
In our study, UPR markers and GVD showed a pronounced presence in the C9‐FTD cerebellum. Patients with the C9orf72 mutation do not show classical cerebellar dysfunction symptoms, such as ataxia, which presents with incoordination of balance, gait, extremity and eye movements, as well as dysarthria. It is surprising that the presence of pathology in the cerebellum in C9‐FTD cases cannot be directly linked to the clinical signs of C9‐FTD. In addition, at the neuroimaging level C9‐FTD patients have been found to have substantially more atrophy in the cerebellum compared to sporadic FTD patients and age‐matched healthy controls (3, 5). This paradox highlights how little is known of the functions or the level of resilience of the cerebellum.
In conclusion, we show that UPR activation markers are increased in C9‐FTD, most prominently in granule cells in the hippocampal DG and granular layer of the cerebellum. In C9‐FTD we observed the presence of GVD in the granule cells of the DG as well as the cerebellum, a region normally unaffected by GVD. The prominent presence of UPR markers and GVD in the cerebellum indicates that both can occur independent from TDP43 and Tau pathology. Whether the presence of the UPR or GVD in granule cells in the DG of the hippocampus and cerebellum is causally involved in the neurodegenerative process in C9‐FTD or that they occur merely as a downstream consequence due to the excessive presence of DPR pathology needs to be resolved in future studies. The presence of UPR markers and GVD in granule cells in the DG and the cerebellum could be a unique feature of C9‐FTD.
Competing interests
The authors have no competing interests.
Author contributions
AAD and JJMH designed the study. PGP, IVD, AAD and JJMH coordinated the study and were responsible for writing the manuscript. PGP, IVD, TM and SM performed the experiments and analyzed the data. LHM, WS, JCVS and JMR participated in writing the manuscript, made intellectual contributions and participated in discussions. JMR, JCVS and LHM were responsible for clinical assessment, autopsy material and neuropathological evaluation. All authors read and approved the final manuscript.
Supporting information
Table S1. FTD neuropathology scores of donors used in study.
Table S2. UPR markers and CK1δ immunoreactivity in the cerebellum, hippocampus and frontal cortex.
Acknowledgments
We thank all brain donors and their caregivers for brain donation, the Netherlands Brain Bank and Michiel Kooreman for logistics and help in selecting brain tissue samples. This study was partly supported by a Memorabel ZonMW fellowship grant to AAD (#733050507).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Ball MJ, Lo P (1977) Granulovacuolar degeneration in the ageing brain and in dementia. J Neuropathol Exp Neurol 36:474–487. [DOI] [PubMed] [Google Scholar]
- 2. Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C et al (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24:7469–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bocchetta M, Cardoso MJ, Cash DM, Ourselin S, Warren JD, Rohrer JD (2016) Patterns of regional cerebellar atrophy in genetic frontotemporal dementia. Neuroimage Clin 11:287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 5. Cash DM, Bocchetta M, Thomas DL, Dick KM, van Swieten JC, Borroni B et al (2018) Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging 62:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F et al (2018) C9ORF72 GGGGCC repeat‐associated non‐AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat Commun 9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H et al (2016) C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell‐derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34:2063–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci 70:3493–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ et al (2017) RAN translation at C9orf72‐associated repeat expansions is selectively enhanced by the integrated stress response. Nat Commun 8:2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J et al (2013) ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature 397:271–274. [DOI] [PubMed] [Google Scholar]
- 13. Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W (2007) Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun 354:707–711. [DOI] [PubMed] [Google Scholar]
- 14. Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol 174:1241–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kadowaki H, Nishitoh H (2013) Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes (Basel) 4:306–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E et al (2011) A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol 122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Majounie E, Renton AE, Mok K, Dopper EGP, Waite A, Rollinson S et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol 11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS et al (2013) Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 1:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mann DMA, Snowden JS (2017) Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype. Brain Pathol 27:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 21. Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. [DOI] [PubMed] [Google Scholar]
- 22. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide‐repeat proteins in FTLD/ALS. Science 339:1335–1338. [DOI] [PubMed] [Google Scholar]
- 23. Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ (2012) The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 226:693–702. [DOI] [PubMed] [Google Scholar]
- 24. Okamoto K, Hirai S, Iizuka T, Yanagisawa T, Watanabe M (1991) Reexamination of granulovacuolar degeneration. Acta Neuropathol 82:340–345. [DOI] [PubMed] [Google Scholar]
- 25. Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ et al (2015) Distinct brain transcriptome profiles in C9orf72‐associated and sporadic ALS. Nat Neurosci 18:1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Riku Y, Duyckaerts C, Boluda S, Plu I, Le Ber I, Millecamps S et al (2019) Increased prevalence of granulovacuolar degeneration in C9orf72 mutation. Acta Neuropathol 138:783–793. [DOI] [PubMed] [Google Scholar]
- 27. Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S et al (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 14:291–301. [DOI] [PubMed] [Google Scholar]
- 28. Scheper W, Hoozemans JJ (2015) The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol 130:315–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74:739–789. [DOI] [PubMed] [Google Scholar]
- 30. Schwab C, DeMaggio AJ, Ghoshal N, Binder LI, Kuret J, McGeer PL (2000) Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol Aging 21:503–510. [DOI] [PubMed] [Google Scholar]
- 31. Stutzbach LD, Xie SX, Naj AC, Albin R, Gilman S, Lee VM et al (2013) The unfolded protein response is activated in disease‐affected brain regions in progressive supranuclear palsy and Alzheimer's disease. Acta Neuropathol Commun 1:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor JP (2013) Neuroscience. RNA that gets RAN in neurodegeneration. Science 339:1282–1283. [DOI] [PubMed] [Google Scholar]
- 33. Thal DR, Del Tredici K, Ludolph AC, Hoozemans JJ, Rozemuller AJ, Braak H, Knippschild U (2011) Stages of granulovacuolar degeneration: their relation to Alzheimer's disease and chronic stress response. Acta Neuropathol 122:577–589. [DOI] [PubMed] [Google Scholar]
- 34. van Blitterswijk M, DeJesus‐Hernandez M, Rademakers R(2012) How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol 25:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wiersma VI, van Hecke W, Scheper W, van Osch MA, Hermsen WJ, Rozemuller AJ, Hoozemans JJ (2016) Activation of the unfolded protein response and granulovacuolar degeneration are not common features of human prion pathology. Acta Neuropathol Commun 4:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wiersma VI, van Ziel AM, Vazquez‐Sanchez S, Nolle A, Berenjeno‐Correa E, Bonaterra‐Pastra A et al (2019) Granulovacuolar degeneration bodies are neuron‐selective lysosomal structures induced by intracellular tau pathology. Acta Neuropathol 138:943–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu M, Shibayama H, Kobayashi H, Yamada K, Ishihara R, Zhao P et al (1992) Granulovacuolar degeneration in the hippocampal cortex of aging and demented patients–a quantitative study. Acta Neuropathol 85:1–9. [DOI] [PubMed] [Google Scholar]
- 38. Zhang YJ, Jansen‐West K, Xu YF, Gendron TF, Bieniek KF, Lin WL et al (2014) Aggregation‐prone c9FTD/ALS poly(GA) RAN‐translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol 128:505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zu T, Liu Y, Banez‐Coronel M, Reid T, Pletnikova O, Lewis J et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A 110:E4968–E4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. FTD neuropathology scores of donors used in study.
Table S2. UPR markers and CK1δ immunoreactivity in the cerebellum, hippocampus and frontal cortex.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.