

Abstract
Formation of capillary blood vasculature is a critical requirement for native as well as engineered organs and can be induced in vitro by co-culturing endothelial cells with fibroblasts. However, whether these fibroblasts are required only in the initial morphogenesis of endothelial cells or needed throughout is unknown, and the ability to remove these stromal cells after assembly could be useful for clinical translation. In this study, we introduce a technique termed CAMEO (Controlled Apoptosis in Multicellular Tissues for Engineered Organogenesis), whereby fibroblasts are selectively ablated on demand, and utilize it to probe the dispensability of fibroblasts in vascular morphogenesis. The presence of fibroblasts is shown to be necessary only during the first few days of endothelial cell morphogenesis, after which they can be ablated without significantly affecting the structural and functional features of the developed vasculature. Furthermore, we demonstrate the use of CAMEO to vascularize a construct containing primary human hepatocytes that improved tissue function. In conclusion, this study suggests that transient, initial support from fibroblasts is sufficient to drive vascular morphogenesis in engineered tissues, and this strategy of engineering-via-elimination may provide a new general approach for achieving desired functions and cell compositions in engineered organs.
Keywords: neovascularization, vasculogenesis, fibroblast, vascular engineering, organ engineering
1. Introduction
Establishing new capillary beds is critical for supporting the metabolic demands and overall health of tissues and organs. This occurs not only in development, but throughout adult life in both regenerative and disease settings.[1] One mechanism by which such vessel beds form involves the spontaneous assembly of endothelial cells into interconnected networks, and ultimately, perfusable blood vessels.[2,3] Understanding and harnessing this vasculogenesis process for translational applications has been of keen interest.[4,5]
A key observation was that co-culture of endothelial cells with fibroblasts could induce endothelial cells to form vascular networks in vitro with minimal additional growth factors and cytokines.[6] In the presence of fibroblasts, endothelial cells spontaneously connected and developed multicellular structures associated with endothelial cell polarization, lumen formation, tubulogenesis, and vessel maturation following a process reminiscent of vasculogenesis.[7–11] Incorporating this process in microfluidic culture systems, it has more recently been shown that vasculogenic networks can integrate and support flow.[12–16]
The exact mechanism by which fibroblasts support vascular morphogenesis is not well understood, but the benefit is even conserved in vivo as incorporation of fibroblasts with endothelial cells in implantable constructs enhances in vivo vascularization.[17–20] The utility of such stromal populations in supporting cell function extends well beyond effects on endothelial cells, as they have been used in supporting stem cells culture, hepatocyte function, cardiac function, and more.[21–25] Therefore, stromal cells are a convenient, all-in-one population of cells for many co-culture systems. However, it remains unclear whether the stromal cells are needed permanently to support these specialized cell functions, and in particular for endothelial cell vasculogenesis, whether they are required only during the initial morphogenetic events.
To address this question, we developed a technique termed CAMEO (Controlled Apoptosis in Multicellular Tissues for Engineered Organogenesis) which allows for a temporal control over the ablation of a select cell population, such as fibroblasts, within a 3D multicellular tissue after a short period of co-culture. This technique repurposes the inducible caspase-9 (iCasp9) kill-switch gene that was originally developed as a fail-safe for T-cell therapies.[26–28] By combining CAMEO with our microfluidic vasculogenesis model, we demonstrate that transient, initial support from fibroblasts is sufficient to drive functional neovascularization in engineered tissues. We then apply this insight to engineer vascularized, perfusable liver tissues embedded with human primary hepatocytes. Our approach of “engineering-via-elimination” provides new opportunities for not only studying the time dependencies of cell populations for processes such as vasculogenesis as demonstrated here, but also engineering vasculature in multicellular organ-specific tissues where the presence of fibroblasts may be undesirable.
2. Results and Discussion
2.1. Fibroblasts Enhance Self-Assembly of a Perfused Vasculature in Microfluidic Devices
We first set out to establish a system that enables the 3D morphogenesis of an endothelial cell and fibroblast co-culture to result in a self-assembled, perfusable vascular network connected to microfluidic channels. Building on a previously reported device that we originally designed to study angiogenesis in vitro,[29,30] we developed a new iteration that supports vasculogenesis. This new microfluidic device consists of a polydimethylsiloxane (PDMS) silicone-based mold bonded to a glass coverslip that features a central tissue chamber (cells plus extracellular matrix) through which two endothelial cell-lined vessels can be perfused. To achieve this arrangement, briefly, two guides on the side of the PDMS device were designed for the insertion of two parallel needles (300 μm in diameter and 1mm apart) that traverse the central chamber (Figure 1a). Once the needles were inserted, a fibrinogen solution containing human umbilical vein endothelial cells (HUVECs) and growth-arrested human dermal fibroblasts (HDFs) were added into the tissue chamber with thrombin. Once the fibrinogen polymerized into fibrin, the needles were removed to create two hollow, microfluidic channels. These channels were then seeded with additional HUVECs to line the walls in a monolayer. This procedure resulted in the formation of two parallel, endothelialized vessels that could be instantly perfused with media and provide nutrients to the cells in the surrounding ECM.
Figure 1.
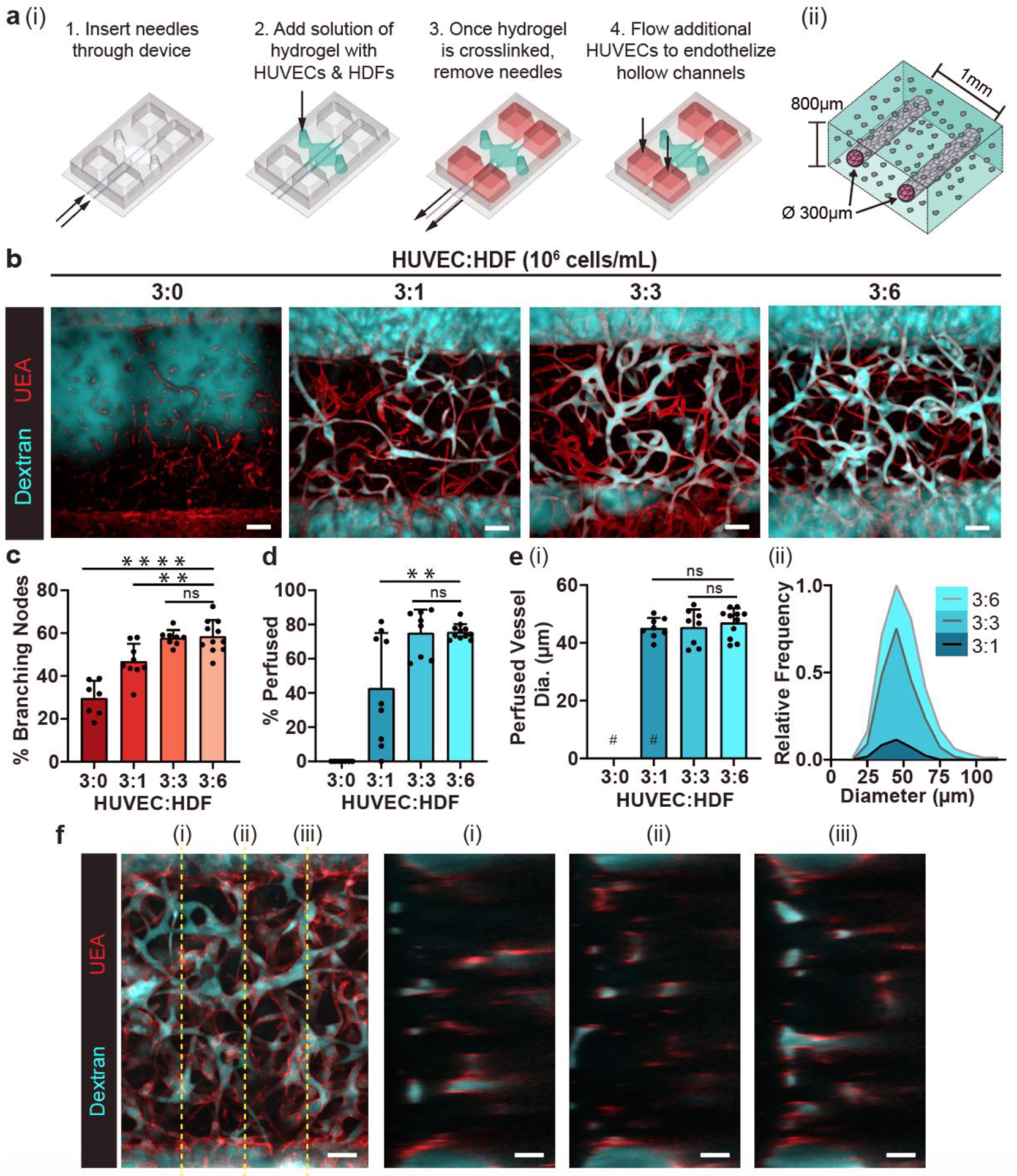
Fibroblasts enhance self-assembly of a perfused vasculature in a 3D microfluidic device. a) Rendering depicting (i) the process of seeding the microfluidic device with a fibrin gel containing HUVECs and HDFs cast around two needle-molded, endothelialized microfluidic channels and (ii) a close-up illustration of the formed tissue inside the microfluidic device. b) Representative max projections of devices fixed on day 7 with varying HUVEC:HDF ratios. Endothelial networks (red) were perfused with 500 kDa dextran (cyan) through one parent channel to visualize connected luminal networks. c) Quantification of the percentage of branching network nodes from total nodes to evaluate network interconnectedness. Each point represents one device. d) Quantification of the percentage of luminal segments that are perfused to the total segments between the channels. e) (i) Quantification of the average diameter of perfused vessel segments for each condition, and (ii) the individual vessels of each ratio condition summed into bins and normalized to the bin with the greatest count. # represents where data points removed due to no vessels being perfused in those devices. f) Multiphoton max projection of a representative 3:6 ratio device. **: p < .01, ****: p < 0.0001, ns = not significant. Scale bars = b) 150 μm and f) 100 μm.
To understand how different densities of fibroblasts would affect the vascular morphogenesis of a given number of endothelial cells, we co-cultured HUVECs (3 million cells/mL) with various concentrations of HDFs (0, 1, 3, and 6 million cells/mL) for 7 days in the fibrin surrounding the needle-molded endothelial channels (Figure 1b). HUVECs alone were able to make branching networks suggestive of functional vasculature (Figure 1c), but when perfused with a fluorophore-conjugated dextran solution (500 kDa) introduced to one of the microfluidic channels, these structures were unable to transport dextran. Only when HDFs were added to the HUVEC culture did we observe highly branched networks that were able to transport the dextran solution through the self-assembled vessels (Figure 1b, c). Furthermore, co-cultures with higher densities of HDFs (3:3 and 3:6 HUVEC:HDF) yielded the highest average percentage of perfused vessels (Figure 1d). While the average diameter of perfused self-assembled vessels (~45 μm) was similar regardless of the density of HDFs (Figure 1e(i)), increasing the density of fibroblasts resulted in a higher density of perfusable vasculature as well as a wider distribution of perfused vessel diameters (Figure 1e(ii)). At the highest concentration of HDFs (3:6 ratio), the engineered tissue was well vascularized all throughout its thickness after the 7-day co-culture (Figure 1f), which prompted us to use this ratio for the rest of the study.
To visualize the process of vasculogenesis in our microfluidic devices, we seeded GFP-labeled HUVECs in the needle-molded microfluidic channels to distinguish them from the non-labeled HUVECs in the bulk fibrin gel. Fixing samples at different times and staining with Ulex Europaeus Agglutinin (UEA) lectin (which labels all HUVECs) allowed us to monitor the process of vascular network formation. As early as day 1, we observed GFP-HUVECs with individual protrusions that started to form intercellular connections (Figure 2a). After 3 days in culture, some HUVECs from the microfluidic channels (GFP-labeled) had migrated into the bulk gel and formed chimeric networks with the (unlabeled) HUVECs in the bulk ECM (Figure 2b). Some of these self-assembled structures became perfusable by day 3, and the percentage of perfused vessels continued to increase over time, achieving ~80% by day 7 (Figure 2c). Interestingly, the average diameters of the perfused vessels in the network remained steady after day 3 (Figure 2d). Together, these data suggest that the vascularization process inside our devices involves both the self-assembly of the bulk HUVECs, and invasion of HUVECs from the microfluidic channels to form a fully interconnected, perfusable vasculature consisting of both the originally templated, larger channels and the self-assembled vasculogenic network.
Figure 2.
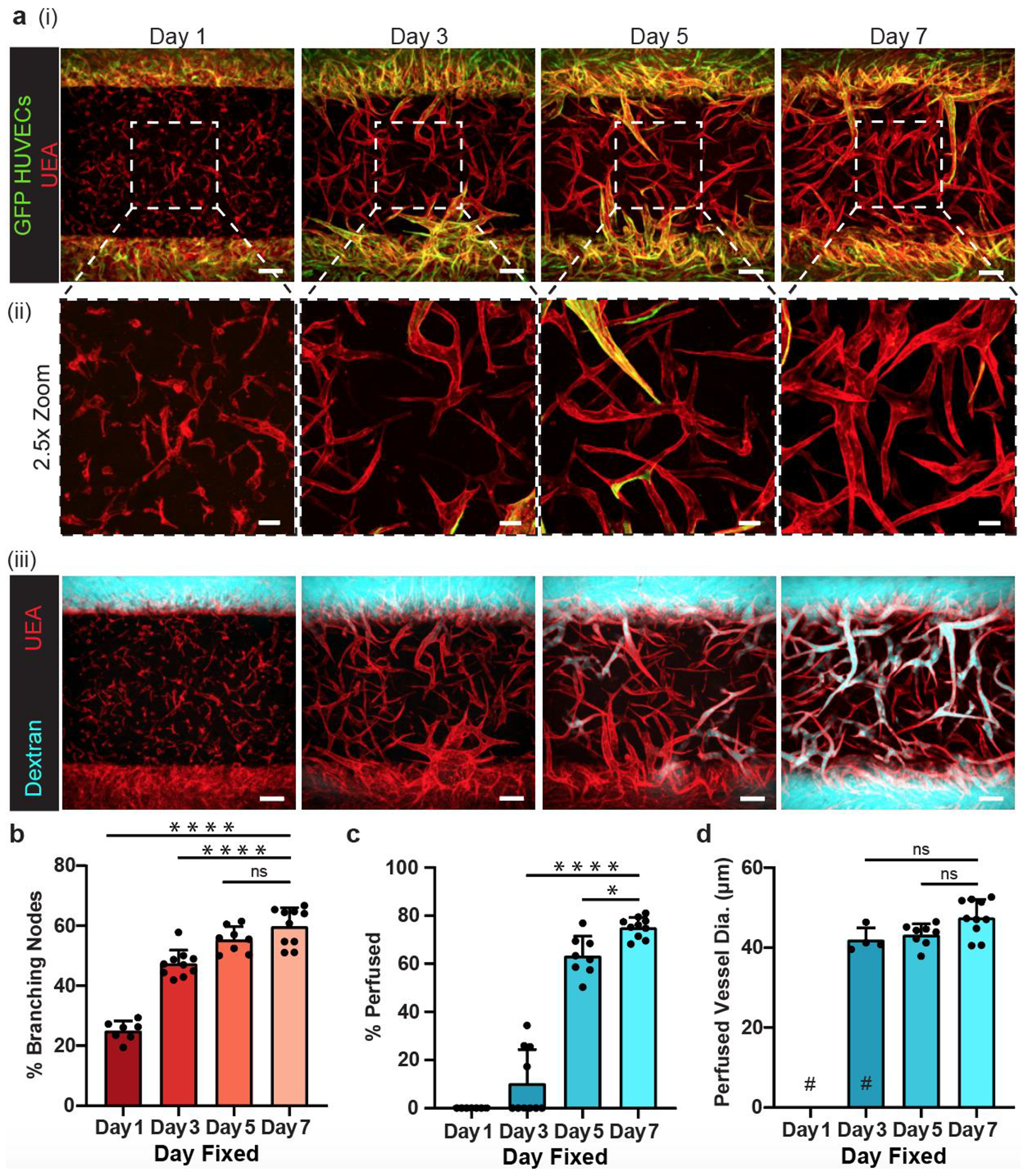
Daily fixations show vasculogenesis formation and functionality. a) (i) Representative images of growth progression from multicellular structures to functionally connected vasculature. GFP-expressing HUVECs (green) were seeded in the needle lumen only and eventually complex with the endothelial cells originating in the bulk gel. (ii) 2.5x zoomed images of the center of the devices. (iii) Same devices shown in (i) with dextran perfusion (cyan) shown along with UEA lectin (red). b) Quantification of percentage of branching nodes, c) percentage of vessels perfused, and d) average diameter of perfused vessels in devices. # represents where data points removed due to no vessels being perfused in those devices. *: p < 0.05, ****: p < 0.0001, ns = not significant. Scale bars a) (i) and (iii) = 150 μm and a) (ii) = 50 μm.
2.2. CAMEO Allows for a Selective Removal of Fibroblasts from Co-Culture
To ablate fibroblasts on demand from the developing co-culture, we utilized a synthetically engineered, inducible caspase-9 (iCasp9) that was originally developed as a safeguard against acute graft-versus-host disease (GVHD) and rare cytokine release syndrome (CRS) induced by T-cell therapies to treat cancer.[27,28] Briefly, iCasp9 is a transgene based on human caspase 9 that features a mutated FKBP12 domain (F36V) that can be dimerized with AP20187, an otherwise inert small molecule chemical inducer of dimerization (CID).[28] Upon dimerization, the iCasp9 proteins become activated, initiating the cascade of caspase-based apoptotic pathways. Since the first description of iCasp9, several clinical studies demonstrated the effectiveness, specificity, and safety of the iCasp9-CID system.[27]
Using this inducible transgene, we established iCasp9-HDFs, and characterized the effectiveness of iCasp9 in these cells. We seeded iCasp9-HDFs on tissue culture plastic, allowed the cells to form a confluent monolayer before adding different concentrations of CID. Even at the lowest concentration of CID used (5 nM), the levels of cellular ATP dropped precipitously within 2–4 hours, suggesting the onset of apoptosis (Figure S1, Supporting Information). In addition, the rapidity of ATP loss increased with increasing CID concentration. Overnight treatment of 10 nM CID resulted in an average of ~96% reduction of iCasp9-HDFs in culture compared to the vehicle control (0.002% ethanol) (Figure 3a, b).
Figure 3.
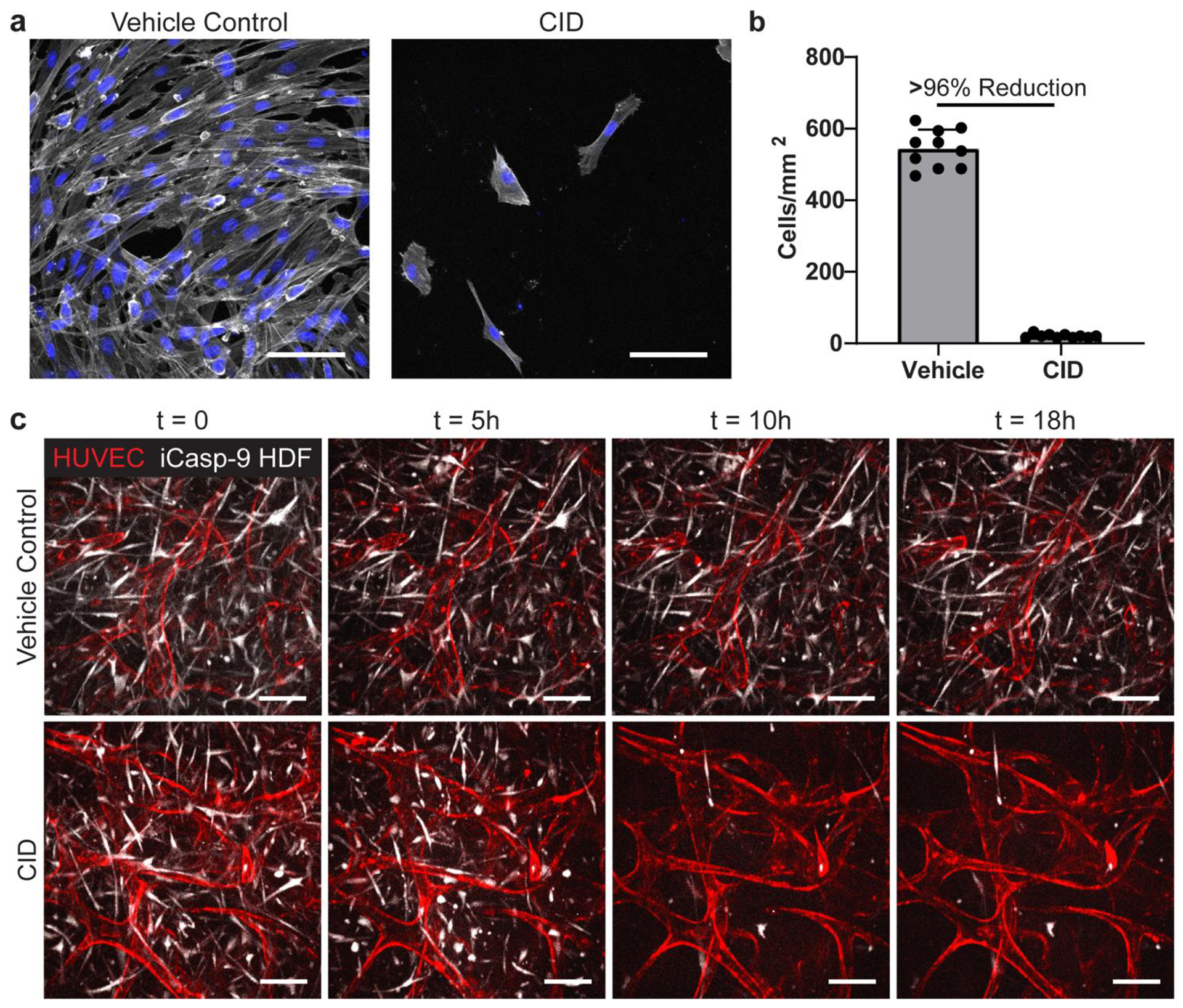
CAMEO allows for a selective removal of fibroblasts from co-culture. a) iCasp-HDFs plated on 2D wells, treated with a vehicle control or CID for 24 hours, fixed, and imaged. Cells stained with phalloidin (white) and DAPI (blue). b) Quantified cell density of vehicle control- and CID-treated cells. c) Time-lapse images of both device conditions in the microfluidic device. Scale bars = 100 μm.
To understand whether the ablation of the fibroblasts could induce an inflammatory response in endothelial cells, we co-cultured the iCasp9-HDFs with endothelial cells, triggered apoptosis with CID, and examined the localization of Nuclear Factor Kappa-light-chain-enhance of activated B cells (NFκB), a TLR-associated marker of the inflammation in HUVECs.[31–33] Whereas the majority of HUVECs treated with 10 ng/mL TNF-α (positive control) showed nuclear localization of NFκB as early as 4 hours and persisted through 48 hours post-treatment, we only observed baseline levels similar to vehicle treatment (~10%) of nuclear NFκB positive HUVECs at 4 hours post-CID treatment and undetectable differences at later time points, suggesting that the HUVECs are not activated by the apoptosis of nearby iCasp9-HDFs (Figure S2, Supporting Information).
To examine the temporal and selective control of the iCasp9-HDFs in a 3D system, we co-cultured mRuby-LifeAct-HUVECs with iCasp9-HDFs (GFP) in our microfluidic platform. After 7 days of co-culture, the cells formed a robust vasculature network as previously observed above, and then we added 10 nM of CID (or the vehicle control) to the media. We observed iCasp9-HDFs starting to contract and round up as early as 1 hour post CID addition and the loss of GFP signal (due to photobleaching and the loss of protein production in apoptotic cells) between 5–10 hours post CID addition (Figure 3c and Movie S1, Supporting Information). Interestingly, the rounding event was preceded by a pulse of contraction of the fibroblasts and the surrounding tissue (Movie S1), consistent with reported caspase-induced ROCK1 activation and ROCK1-mediated actin-myosin contraction.[34,35] Remarkably, we did not observe any immediate, noticeable deterioration of the vascular structure during this massive elimination of HDFs, despite the HUVECs being, in some cases, in direct contact with the dying cells (Figure S3, Supporting Information).
2.3. Transient Co-Culture with Fibroblasts is Sufficient to Support Vasculogenesis.
To understand the dependence of endothelial cells on fibroblasts for vasculogenesis, we co-cultured HUVECs with iCasp9-HDFs in our microfluidic devices and ablated the HDFs with CID at different time points (day 0, 1, 3, and 5) during the 7-day culture period (Figure 4a). Because a small population of HDFs (~4%) remain after CID treatment, we growth-arrested the cells with mitomycin C before co-culture to prevent their ability to proliferate and repopulate the co-culture over this week-long study. When HDFs were ablated within an hour after the completion of the device seeding (day 0), HUVECs were still able to form a network by day 7, but the resulting structure was minimally interconnected (Figure 4b, c), and none of these structures could transport dextran, indicating incomplete vascular morphogenesis similar to HUVEC-only cultures (Figure 4d). Removing HDFs at day 1 resulted in a few perfusable structures by day 7, but most devices still showed little to no perfusion. Removing HDFs at day 3 or day 5 yielded highly interconnected and perfusable endothelial networks that were comparable to the control networks in which HDFs were never ablated. The average diameter of the perfused vessels was comparable as well for all conditions except the day 0-deletion condition, although the distribution of the vessel diameters in the day-1-deletion condition was more dispersed. Similar observations were also made with iCasp9-transduced human lung fibroblasts (iCasp9-HLFs) where the HUVECs were able to self-assemble into perfusable vasculature with only 3 days of co-culture with iCasp9-HLFs (Figure S4, Supporting Information), suggesting that this short-lived dependency on fibroblast support is not specific to HDFs. All together, these data suggested that the presence of fibroblasts during the first 1–3 days of co-culture is critical for the functional vasculogenesis of endothelial cells, but the fibroblasts can be ablated thereafter without structurally affecting the resulting vasculature. With this observation, we termed the technique “Controlled Apoptosis in Multicellular Tissues for Engineered Organogenesis,” or CAMEO.
Figure 4.
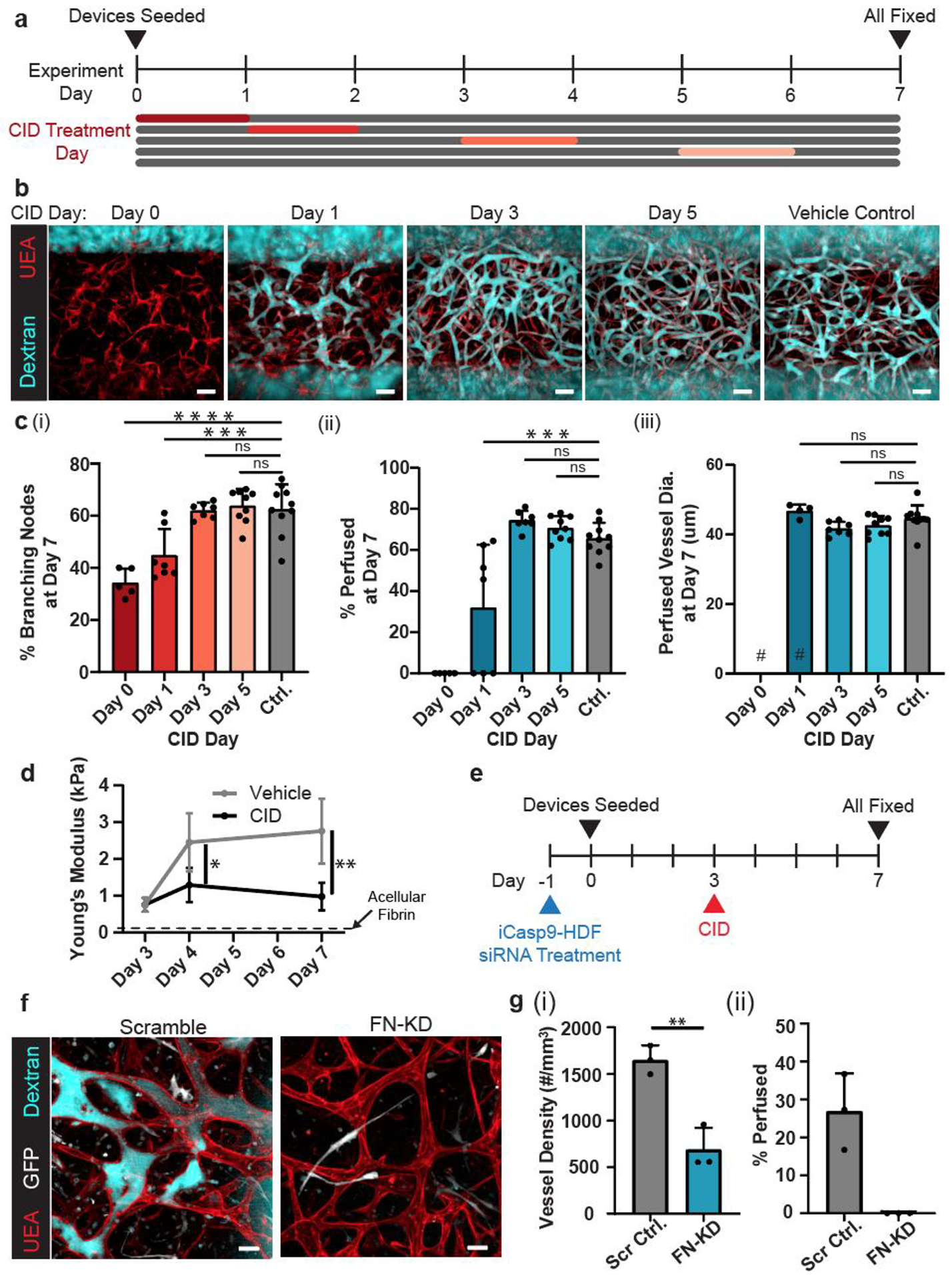
Transient co-culture with fibroblasts is enough to drive vascular morphogenesis. a) Experimental timeline. b) Representative max projections of devices treated with CID or vehicle control on a designated day and fixed on day 7. Devices were stained with UEA lectin (red) and perfused with dextran (cyan). c) Quantification of (i) percentage of branching nodes, (ii) percentage of perfused vessel segments, and (ii) average vessel diameter of perfused vessels. # represents where data points removed due to no vessels being perfused in those devices. d) Young’s modulus of HUVEC-iCasp9-HDF co-culture tissues treated with CID or vehicle at day 3. Dotted line represents the average Young’s modulus of acellular fibrin gel at day 3 (~0.1 kPa). e) Experiment timeline. f) Representative max projection images of dextran-perfusion (cyan), HUVECs (UEA lectin, red) and iCasp9-HDFs and their apoptotic bodies (GFP, white) at day 7. g) Quantification of (i) percentage of structures that are perfused with dextran and (ii) density of the self-assembled network. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, ns = not significant. Scale bars = b) 150 μm, and f) 50 μm.
Given that fibroblasts are important effectors of ECM remodeling, we hypothesized that HDFs may support vascular morphogenesis through their early effects on the ECM. To address this hypothesis, we first characterized the stiffness of the tissues during vascular network formation using nanoindentation and assessed the effect of CAMEO on these mechanical properties. Co-culturing HUVECs and iCasp9-HDFs for 3 days resulted in tissues with a Young’s modulus averaging ~0.75 kPa, which was substantially stiffer than acellular fibrin gels (~0.1 kPa) (Figure 4d). Over the next 4 days (days 3–7), these tissues continued to stiffen, reaching an average Young’s Modulus of ~3 kPa at day 7. In contrast, when we induced apoptosis of iCasp9-HDFs at day 3 through CAMEO, the tissues did not further stiffen (~1 kPa at d7). These data suggest that there is substantial ECM remodeling during culture, and that the changes in ECM are primarily driven by the presence of fibroblasts.
To begin to examine whether HDFs might contribute molecular changes to the ECM during the first days of culture that might support later vascular morphogenesis, we looked to specific ECMs that could be deposited by the fibroblasts. One important matrix protein deposited by fibroblasts during early stages of ECM remodeling is fibronectin, which also has been shown to be important for vascular morphogenesis.[36–38] Therefore, we examined the role of fibronectin in our platform by generating siRNA-mediated fibronectin-knockdown (FN-KD) iCasp9-HDFs and co-culturing them with HUVECs in the microfluidic devices for 3 days before CID treatment (Figure 4e). The density of self-assembled endothelial networks was significantly reduced for HUVECs cocultured with the FN-KD iCasp9-HDFs as compared to scrambled siRNA-treated controls. Furthermore, HUVECs cocultured with FN-KD iCasp9-HDFs failed to generate any perfusable vasculature, whereas those cocultured with control iCasp9-HDFs yielded perfusable vasculature (Figure 4f, g), although to a lower extent than previously seen with wildtype HDFs (Figure 1d) likely due to inherent siRNA cytotoxicity in primary cells. Together, this suggests that the continuation of vascular morphogenesis after the removal of fibroblasts is contingent on provisional ECMs produced by the fibroblasts during the early days of co-culture.
2.4. CAMEO Enhances Function of Vascularized, Engineered Hepatic Tissues.
Given that fibroblasts are only transiently required for supporting the formation of a functional vasculature, we asked whether CAMEO could be used in the vascularization of tissue constructs with organ-specific parenchymal cells without negatively affecting the function of the parenchymal cells. To investigate this, we employed a tri-culture system consisting of HUVECs, iCasp9-HDFs, and primary human hepatocytes in our microfluidic device. After 5 days of tri-culture, the devices were treated with CID to ablate the iCasp9-HDFs, or with the vehicle control. All samples were fixed at day 7 to evaluate vascular perfusion (Figure 5a). In both groups, HUVECs formed perfusable vasculature that permeated the hepatocyte-laden construct (Figure 5b). To evaluate whether the hepatocytes remained functional after CID treatment, levels of albumin and urea, markers for hepatic protein synthesis function and nitrogen metabolism, respectively, were measured in the media. Removal of the iCasp-9 HDFs did not negatively affect the secretory functions of the hepatocytes in our devices (Figures 5c, d); in fact, we observed a statistically higher level of secreted albumin and urea at day 7 in CAMEO devices versus the vehicle control, suggesting potential benefits of removing the fibroblast population in the engineered liver constructs.
Figure 5.
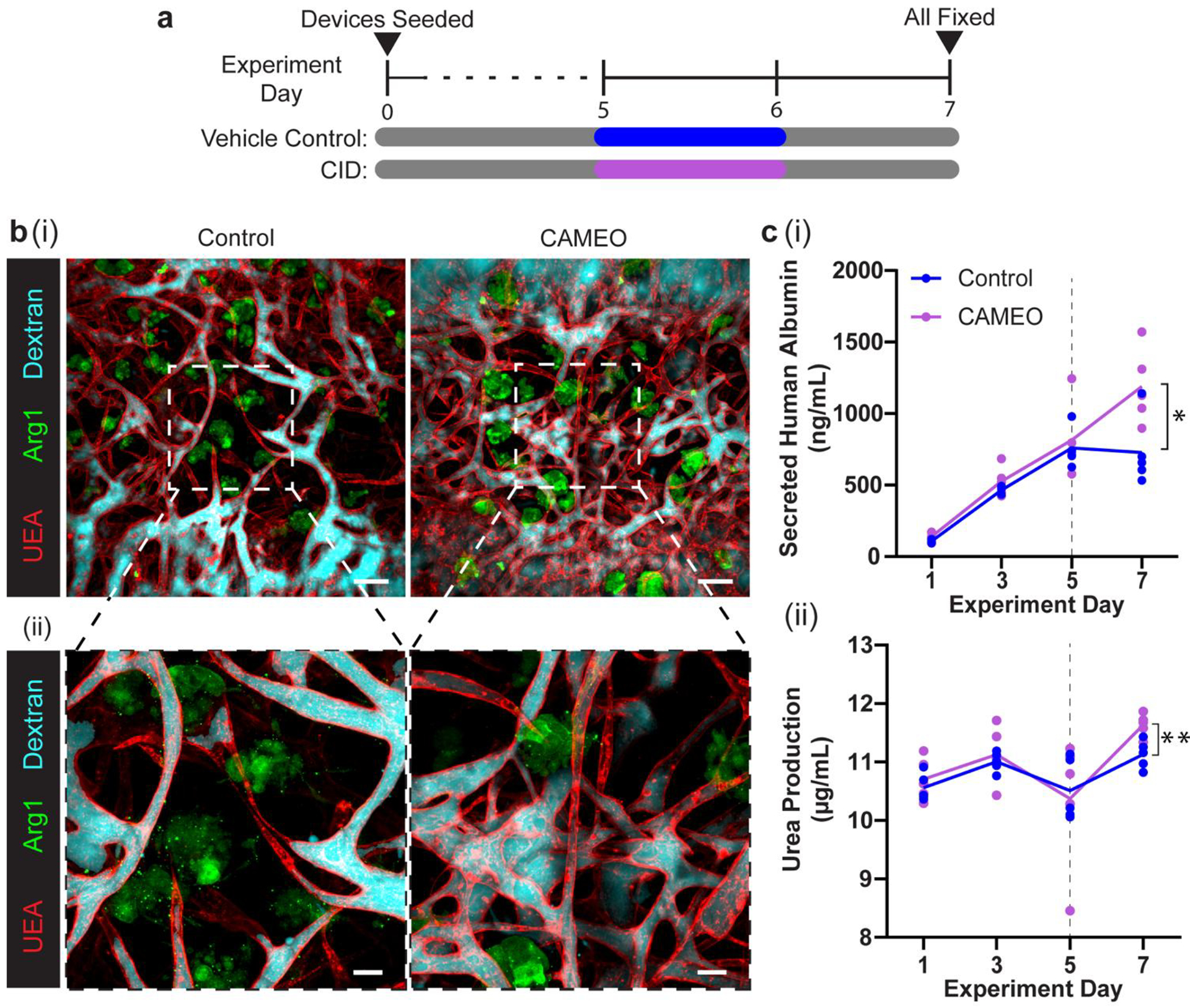
CAMEO enhances function of vascularized engineered hepatic tissues. a) Experimental timeline. b) (i) Representative max projections of dextran-perfused (cyan) vehicle control- and CID-treated devices stained for UEA lectin (red) and human arginase 1 (green), and (ii) 2.5x images. c) (i) Quantification of secreted human albumin and (ii) urea production in each device with mean line shown. Dotted line denotes day vehicle control and CID were dosed for 24 hours. *: p < 0.05, **: p < 0.01. Scale bars = b) (i) 150 μm and b) (ii) 50 μm.
2.5. Discussion
In this study, we explored the dependence of endothelial cells on fibroblasts for forming perfusable vasculature by investigating whether a transient exposure to fibroblasts in a 3D co-culture system is sufficient to drive endothelial cell morphogenesis. Whereas previous studies have shown that fibroblasts contribute to key morphogenetic steps during the formation of endothelial cell networks in vitro,[36] our study suggests that fibroblasts are only required to initiate the process of vascular network formation, which continues to develop in a semi-autonomous fashion even after fibroblasts are ablated. With CAMEO, we demonstrated that self-assembly of HUVECs into perfusable vasculature benefits from the presence of fibroblasts during the first several days of co-culture, but is undisturbed when fibroblasts are ablated as early as 3 days after the co-culture period. Indeed, HUVECs cultured alone formed loose networks that could not mature further into perfusable beds. While transient fibroblast co-culture was sufficient to support endothelial assembly of a perfusable vascular network, additional studies will be required to fully characterize whether later stages of vascular function, such as barrier or anti-coagulation functions, will require additional cell-cell interactions or will remain unaffected.
There are several known mechanisms through which stromal cells can promote vascular morphogenesis. Fibroblasts have been shown to secrete proangiogenic factors in the context of wound healing and tissue regeneration.[39–41] Nonetheless, previous studies have reported that simply transferring conditioned media from stromal cell monocultures to endothelial cells does not induce vascularization to the same extent as seen in co-culture settings. Although some studies have reported success in vascularizing small (100–200 μm thickness) tissues using microfluidic systems with separate, neighboring microcompartments for endothelial cells and fibroblasts,[42,43] a study by Griffith et al. highlighted that the paracrine support of vascular morphogenesis from fibroblasts decreases dramatically with the distance between ECs and fibroblasts in separate co-culture systems,[41] revealing the unique benefits of a directly mixed co-culture for vascularizing larger tissues. Indeed, fibroblasts are known to actively remodel their surrounding ECM via the expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), and to deposit various ECMs such as fibronectin and collagen type III.[36,44–46] Particularly, fibronectin has been described as a provisional matrix in development and wound healing that is important for angiogenesis, vasculogenesis, and vascular remodeling.[47–49] With siRNA knockdown, we demonstrated that the initial fibronectin deposition from fibroblasts to the co-culture matrix is critical for HUVECs to further develop perfusable vascular networks after the ablation of fibroblasts. This suggests that ECM deposition by the fibroblasts in the early days of co-culture may be a critical cue that primes endothelial cells to continue the later stages of vascular morphogenesis without further support from fibroblasts.
Recent advancements in the tissue engineering field have enabled various types of tissues and organoids to be engineered, including brain, kidney, heart, and liver.[23,24,50–52] A main challenge in the field remains in appropriately vascularizing these tissues.[41] While using fibroblast-endothelial cell co-culture methods can contribute to addressing this challenge, fibroblasts are a population of highly heterogenous, uncharacterized cells that could potentially introduce undesirable effects such as tissue contraction, stiffening, and fibrosis.[53–57] Therefore, a vascular engineering technique that relies minimally on these cells may be desirable for clinical translation. Here, we were able to demonstrate the use of CAMEO in generating engineered liver constructs featuring dense, perfusable vasculature and a minimal population of fibroblasts. Notably, the tissues with the fibroblasts ablated at day 5 using CAMEO seemed to have enhanced secretory functions of the human primary hepatocytes compared to tissues with intact fibroblasts at day 7, highlighting unexpected potential benefits of our CAMEO platform. There are many factors that might have contributed to the improved hepatocyte function following fibroblast ablation. First, improved hepatocyte function may have been a result of reduced tissue-stiffening following fibroblast ablation, consistent with other reports that lower matrix stiffness improves primary hepatocyte function.[58–61] Second, it has been previously shown that the interaction between endothelial cells and hepatocytes can promote hepatocyte function, maintenance, and regeneration.[62–65] Removing the fibroblasts from the hepatocyte-HUVEC-HDF tri-culture system could enhance hepatocyte-HUVEC interactions, resulting in increased albumin and urea biosynthesis. Lastly, the high density of fibroblasts in non-ablated cultures may compete with hepatocytes for nutrients. Regardless, CAMEO may prove to be a useful strategy to optimize co-culture conditions for engineered tissues.
Previously, inducible apoptosis transgenes like iCasp9 were developed and primarily only used as a “fail-safe” for cell-based therapies involving T-cells,[27] MSCs,[66] and stem cells.[67] In this study, we repurposed this suicide transgene and demonstrated its utility for studying cell-cell interactions as well as the subsequent impact on the functional vascularization of engineered tissues. CAMEO may offer general utility in applications beyond those demonstrated here. CAMEO could be used in culture settings, for example to eliminate xenogenic feeder-layer fibroblasts that are still widely used to cultivate embryonic, induced, and hematopoietic stem cells.[68] In tissue engineering, it may be useful to eliminate not only stromal populations, but also other cell populations that might have only a transient utility as well. Despite the enormous potential of this approach, further investigation of the induced apoptosis process and the clearing of apoptotic bodies will be needed to accurately evaluate the clinical translatability of CAMEO.
3. Conclusion
In summary, the CAMEO platform enabled by the synthetic kill-switch transgene, iCasp9, allowed for on-demand control over the specific removal of fibroblasts in our endothelial cell-fibroblast vasculogenesis model. Using this system, we demonstrated that only a transient co-culture with fibroblasts is sufficient for the endothelial cells to carry on and complete functional vascular morphogenesis, and then used these insights to generate functionally vascularized liver tissues with a minimal fibroblast population and improved secretory functions. This approach of “engineering-via-elimination” provides not only new opportunities for studying and engineering vasculature for a variety of applications, but more generally, a means to incorporate cell populations into settings where they may have only transient utility.
4. Experimental Section
Cell Preparation:
HUVECs (pooled from 4 donors, Lonza) were cultured in Endothelial Growth Medium-2 (EGM-2, Lonza) and used before P7. HDFs (single donor; Lonza) were cultured in Fibroblast Growth Medium-2 (FGM-2, Lonza) and used before P10. Hepatic aggregates were cultured as described previously.[23,69] Briefly, cryopreserved primary human hepatocytes (Lot ZGF; BioreclamationIVT) were thawed and immediately plated with iCasp9-HDFs in a 1:1 ratio in AggreWells (400 μm pyramidal microwells) and cultured for 3 days in maintenance media containing 10% (v/v) fetal bovine serum (FBS) (Gibco), 1% (v/v) ITS supplement (insulin, transferrin, sodium selenite; BD Biosciences), glucagon (70 ng/mL), dexamethasone (0.04 μg/mL), 0.015 M HEPES, and 1% (v/v) penicillin-streptomycin (Invitrogen) in DMEM with 4.5 g/L glucose (Corning Cellgro).
Lentiviral Transduction:
iCasp9-HDFs and LifeAct-Ruby-HUVECs were generated using pMSCV-F-del Casp9.IRES.GFP (gift from David Spencer, Addgene plasmid #15567) and pLenti.PGK.LifeAct-Ruby.W (gift from Rusty Landsford, Addgene plasmid #51009), respectively. Briefly, individual plasmids were co-transfected into HEK-293T cells with pVSVG, pRSV-REV, and pMDLg/pRRE using the calcium phosphate transfection method. Supernatants containing the assembled viruses were collected after 48 hours and precipitated using PEG-IT (SBI). The concentrated viral pellet was resuspended in PBS and stored in −80°C. HDFs and HUVECs were transduced in growth media overnight with the appropriate lentiviral titers that had been optimized for minimal changes in cell morphology and proliferation. The cells were then washed in PBS the next day and fed with fresh growth media for expansion. For iCasp9-HDFs, GFP+ population (top 30%) was selected via flow-assisted cytometry (FACSMelody, BD Biosciences) where the samples were first gated to exclude debris and cell doublets.
Device Assembly and Cell Seeding:
The molds for the 2-channel microfluidic devices (2 cm × 1 cm × 0.3 cm) were constructed using stereolithography (Proto Labs). Polydimethylsiloxane (PDMS) was cured at a standard mixing ratio overnight at 60°C in the mold, and individual devices were cut and plasma-bonded to glass slides. To enhance ECM bonding to PDMS, the surface inside the tissue chamber of the devices was functionalized with 0.01% poly-L-lysine and 1% glutaraldehyde following plasma-activation and washed overnight in DI water. On the day of seeding, devices were soaked in 70% ethanol (EtOH) and dried. Acupuncture needles (300 μm diameter) (Hwato) were blocked in 0.1% (w/v) bovine serum albumin (BSA) (Sigma) in phosphate buffer saline (PBS) for 45 minutes and inserted through the two needle guides. Devices with needles were UV-sterilized for 15 minutes.
On the day of device seeding, HDFs or iCasp-9 HDFs were growth-arrested with 10 μg/mL mitomycin C in FGM-2 for 2.5 hours and thoroughly washed 5 times in FBM, in order to prevent the small population of HDFs (~4%) that remain after CID treatment from proliferating and repopulating the co-culture. We note that mitomycin C-arrested and untreated HDFs performed similarly in their ability to support vascular morphogenesis, and we used the growth-arrested HDFs for the current study. Both HUVECs and HDFs were lifted from culture plates using TrypLE Express (Gibco), centrifuged at 200 g for 4 minutes, and resuspended to a concentration of 20 million cells/mL in EGM-2. A solution of HUVECs (3 million cells/mL), HDFs or iCasp-9 HDFs (0, 1 million, 3 million, or 6 million cells/mL), fibrinogen (2.5 mg/mL), thrombin (1 U/mL) in EGM-2 was prepared for the bulk hydrogel region of each device. For the devices with hepatocytes, a solution of HUVECs (3 million cells/mL), iCasp-9 HDFs (total of 6 million cells/mL), hepatic aggregates (0.36 million aggregates/mL with about 150 hepatocytes per aggregate), fibrinogen (2.5 mg/mL), thrombin (1 U/mL) was made in a 1:1 media mixture of EGM-2 and hepatocyte maintenance media. After the addition of thrombin, the solution was quickly injected into the tissue chamber, and the devices were repeatedly rotated while the solution crosslinked. Appropriate media was added to each well of the device, and the devices were placed in the incubator (37°C, 5% CO2). After 15 minutes, the needles were carefully ablated from the devices to create 300 μm hollow channels between the wells. Each channel of the device was seeded with additional HUVECs at 2 million cells/mL for at least 5 minutes on each side (top and bottom) in the incubator. Each device received 200 μL of appropriate media daily and cultured on the rocker inside the incubator.
Vessel Network Analysis:
All devices were fixed with 4% PFA for 30 minutes on a rocker and washed with PBS overnight at 4°C. The devices were then blocked in 3% BSA overnight at 4°C. Lectin (UEA DyLight 649, Vector Labs) was diluted in the blocking solution at 1:100 and added to the devices for another 4°C overnight incubation. Devices were then washed overnight with PBS at 4°C and kept in PBS with 0.02% (w/v) sodium azide at 4°C until imaging.
Before imaging, a solution of 500 kDa fluorescein isothiocyanate (FITC)- or tetramethylrhodamine isothiocyanate (TRITC)-conjugated dextran (0.15 mg/mL) was added to one of the microfluidic channels to generate a pressure gradient between the two channels of the device. All device images were then captured by a Leica SP8 confocal microscope (Leica, Wetzlar, Germany) using either a Leica 10x/0.30NA W U-V-I WD-3.60 Water or 25x/0.95NA W VISIR WD-2.50 Water objective and Leica LAS X imaging software. Within experimental runs, the same laser intensities and settings were applied to all samples.
A custom MATLAB script was used to study properties of the vascular network (refer to Supporting Information for the code). The script imports a .tif image stack with a surface marker in one channel, in this case lectin, and the fluorescent dye in another, FITC- or TRITC-dextran. The aspect ratio of each image is adjusted so that each voxel is equally sized in all directions. Since our image resolution is higher in-plane (x- and y-axes) than along the depth (z-axis), this step typically results in a reduction in the number of pixels in the plane and an increase in the number of stack layers. The 3D volume of the cell surface marker channel is then smoothed using a spatial-domain Gaussian kernel to reduce noise in the Lectin channel.
The script relies on user inputs to generate binary images of the complete vascular network from the Lectin signal, and the perfused network according to the FITC-Dextran. First, the user selects a 2D region of interest between the two needle-molded channels on the device, and performs contrast-limited adaptive histogram equalization (CLAHE) on both signals for each z slice.[70] This step is performed to overcome a substantial loss in signal with depth of the volume.
The user then specifies a luminance threshold for each channel and is able to move between different images in the z stack to best capture the vessel structure and perfusion of FITC-Dextran. For the fluorescent dye, the signal is processed using only this specified uniform threshold to capture the regions of perfusion. For the Lectin channel, which exhibits poorer contrast, the user first selects a liberal threshold to segment the 3D volume. The script refines this initial segmentation for each slice using distance regularized level set evolution (DRLSE) with a double well potential (see full code for setup of the iterative segmentation refinement).[71] The DRLSE-segmented slices are merged to form the final volume of the full vessel network. Any holes in the Lectin volume are filled and islands smaller than ~50 μm3 are removed.
From the identified Lectin network, a 3D medial axis skeleton is generated using homotopic thinning.[72,73] This is further converted into a graph with links representing the vessel structures, and nodes to identify branching and terminal points of the vessels.[73] Links are further characterized as perfused and non-perfused vessels based on the overlap of the binarized FITC-Dextran with the vessel network. Links with <25% perfusion are classified as non-perfused links. We report the average length of all links from the Lectin skeleton. The vessel radii are calculated based on the distance between the surface and the medial skeleton of the graph links, and the mean diameters for all vessels, as well as only perfused vessels, are reported. The percentages reported for branching nodes represent the fraction of branching nodes out of all nodes. The user can toggle through the z-stack of the binary channels overlaid on the original images, as well as overlaid on each other to confirm accuracy.
2D Viability and Inflammation Assays:
For the ATP assay, iCasp9-HDFs were plated in monolayer (8,500 cells/well in a 96-well plate). On the next day, cultures were dosed with CID (B/B homodimerizer aka AP20187) (Takara) at concentrations of 5, 50 or 500 nM and assayed for ATP levels using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. For the cell-counting assay, iCasp9-HDFs were first mitomycin-treated for 2.5 hours at 10 μg/mL and washed 3 times with FBM. Then, the cells were plated at 60,000 cells/well in 24-well plates and cultured overnight in the incubator. On the next day, the wells were treated with either 10 nM CID or ethanol vehicle and cultured overnight in the incubator. These wells were then fixed with 4% PFA, washed with PBS, stained with DAPI (1:1000) (Sigma), and stored at 4°C. The nuclei of the cells were imaged with a Nikon TE 200 microscope with a Nikon 20x/0.50NA objective and Spot Imaging 5.3 software. Cell nuclei were counted using CellProfiler to compare cell counts between vehicle and CID treatment.[74] For visualization of the cell bodies, some wells were stained with Alexa Fluor 568 Phalloidin (1:200) (Life Technologies) and imaged with a Leica SP8 confocal microscope (Leica, Wetzlar, Germany) using a Leica 25x/0.95NA W VISIR WD-2.50 Water objective and Leica LAS X imaging software.
For the inflammation assay, 16-well chamber slides (Lab-Tek) were first coated with human fibronectin (Corning) at 10 μg/mL. For CID and vehicle conditions, each well was seeded with 7,000 HUVECs and 14,000 iCasp9-HDFs in EGM-2 media, and the cells were allowed to attach and spread overnight. On the next day, we treated the wells with fresh EGM-2 media containing either CID (10 nM) or vehicle. For the positive control condition, we added 7,000 HUVECs in each well in EGM-2 media, and on the next day, we seeded the wells with fresh EGM-2 media containing human recombinant TNF-α (R&D Systems) at 10 ng/mL. In all conditions, cells were fix at 4, 12, 24, or 48 hours post treatment with 4% PFA. No media was changed between treatment and fixation. The fixed samples were permeabilized with 0.25% Triton X-100 for 10 minutes and blocked with 3% BSA for 1 hour at room temperature. The samples were then stained with rabbit NFκB primary antibody (1:500) (Cell Signaling, D14E12) and UEA lectin (1:500) for 1 hour at room temperature, followed by secondary goat anti-rabbit Alexa Fluor 568 (1:1000) and DAPI (1:1000) staining overnight at 4C. Samples were stored in PBS at 4C until imaging. Samples were imaged by a Leica SP8 confocal microscope (Leica, Wetzlar, Germany) using a 25x/0.95NA W VISIR WD-2.50 Water objective and Leica LAS X imaging software. The percentage of endothelial cells with nuclear NFκB was calculated by manually counting the total number of UEA-positive endothelial cells and the number of UEA-positive and nuclear NFκB -positive endothelial cells.
3D Culture Live Imaging:
Microfluidic devices with LifeAct-Ruby HUVECs and iCasp9-HDFs (GFP) were made and cultured for 7 days as described above. Then, the devices were transferred to a custom-made Petri dish that limits media evaporation. Media was changed to EGM-2 containing OxyFluor (Oxyrase) at a 1:100 dilution. Right before the start of imaging, either CID (10 nM) or vehicle was added to the devices and the petri dish was transferred into the microscope environmental chamber preconditioned to 37°C, 5% CO2, and 100% humidity. A 150 μm stack was imaged in each device by a Yokogawa CSU-10/Zeiss Axiovert 200M inverted spinning-disk microscope using a Zeiss 10x/0.45NA Air objective every 30 minutes for 18 hours. The capture was automated using Metamorph 7.8.9.0 (Molecular Devices).
Nanoindentation Characterization:
Acellular fibrin gels and fibrin gels with HUVECs and iCasp9-HDF at 3:6 HUVEC:HDF ratio were prepared as described above in 2 mm diameter PDMS wells. Nanoindentation characterization was performed using a Piuma Nanoindenter system (Optics 11, Westwood, MA). The spherical indentation probe has a diameter size approximately 100 μm with cantilever spring constant k ~ 0.5 N/m. Samples were immersed in EGM2 medium and measurements were performed at indentation depth of 10 μm and displacement speed of 5μm/s with 4–5 repeats for each condition. The Young’s moduli of these samples were calculated using the built-in Piuma software by fitting force-indentation curves to the Hertzian contact mechanics model, assuming a Poisson ratio of 0.5 for incompressible materials. Optics11 DataViewer software was used for data analysis. Acellular fibrin gels were measured at day 3 and tissues with HUVECs and iCasp9-HDFs were measured at day 3, 4, or 7. For the tissues measured at day 4 or day 7, CID (10 nM) or vehicle was added to the media at day 3.
Co-culture with Fibronectin Knockdown Fibroblasts:
iCasp9-HDFs were plated in 35mm dishes and allowed to expand to ~50% confluence. On the morning of transfection, media was replaced with an antibiotic-free media. At the end of the day, dishes were transfected with 20 pM of either human FN1 siRNA or human scrambled siRNA in a SMARTpool format (Dharmacon). siRNA was diluted in OPTI-MEM (Gibco) media and mixed with Lipofectamine RNAiMAX (Invitrogen) in OPTI-MEM. Mixtures were incubated at room temperature for 5 minutes, after which they were added dropwise to the cell dishes and incubated for 24 hours. On the next day, the cells were washed with FBM 5 times. Microfluidic devices with HUVECs and iCasp9-HDFs (treated with fibronectin or scramble siRNA) in fibrin were prepared as described above at 3:6 HUVEC:HDF ratio and cultured for 7 days in EGM-2. At day 3, all the devices were treated with 10 nM of CID. Devices were fixed at day 7 and imaged for vessel network analysis as described above.
Hepatocyte Functional Assays:
On days 1, 3, 5, and 7, cultured media from the devices was collected and stored at −80°C. Supernatant was thawed to room temperature and assayed for human albumin using an enzyme-linked immunosorbent assay (ELISA) (Bethyl Laboratories) and 3,3’,5,5’-tetramethylbenzidine (TMB, Thermo Fisher), as well as urea using a colorimetric (diacetylmonoxime) assay with acid and heat (Stanbio Labs).
All triculture devices were fixed with 4% PFA for 30 minutes on a rocker and washed with PBS overnight at 4°C. The devices were permeabilized with 0.15% Triton-X for 30 minutes on a rocker and washed with PBS 3 times. The devices were then blocked in 3% BSA overnight at 4°C. Dylight 649 labeled UEA (Vector Laboratories) and primary antibody for rabbit anti-human arginase-1 (Sigma, HPA003595) were diluted in the blocking solution at 1:200 and 1:100 respectively and added to the devices overnight at 4°C. The devices were washed with PBS overnight at 4°C. Goat anti-rabbit Alexa Fluor 568 secondary antibody was diluted in the blocking solution at 1:100 and added to the devices overnight at 4°C. Devices were then washed overnight with PBS overnight at 4°C and kept in PBS with 0.02% (w/v) sodium azide at 4°C until imaging. A FITC-conjugated 500 kDa dextran solution (1.5 mg/mL) was added to the devices as described above right before imaging. All device images were then captured by a Leica SP8 confocal microscope (Leica, Wetzlar, Germany) using either a Leica 10x/0.30NA W U-V-I WD-3.60 Water or 25x/0.95NA W VISIR WD-2.50 Water objective, and Leica LAS X imaging software.
Image Processing:
Max projection images used in figures were assembled and processed using Imaris 9.2.1 (Bitplane). Intensity of stack images was depth compensated by utilizing a built-in autocorrelation correction Matlab plug-in (MathWorks). The dextran channels were smoothed using the Gaussian Filter that used a 3.89 μm filter width in order to make perfused channels more clearly visible in figures (Figure S5, Supporting Information). Thresholding and gamma correction were applied to all images. Both the UEA lectin and TRITC- Dextran were false-colored to red and cyan, respectively. Only unprocessed images were used in the image analysis, explained above. For images containing hepatocytes, false coloring was applied to UEA lectin, FITC Dextran and Arg1 as red, cyan, and green, respectively. We applied uniform volumetric masks that were thresholded to denoise images due to antibody aggregation and fluorophore bleed through. These corrections were applied to the Dextran and Arg1 channels in all hepatocyte images.
Statistical Analysis:
All data are presented as mean ± standard deviation. At least three independent samples were analyzed for quantified sections. All statistical significance was determined using Prism (GraphPad, San Diego, CA). CID-induced cell count reduction in 2D was quantified by a two-tailed Welch’s t-test. Hepatocyte function data (albumin and urea), vascular network metrics for FN-KD experiments, and nanoindentation results were analyzed by unpaired two-tailed t-tests. All other mean analyses were performed using ordinary one-way ANOVA and, where appropriate, followed by Tukey’s honest significance test to evaluate statistical significance (P < 0.05).
Supplementary Material
Acknowledgements
We thank Wilson Wong for guidance and helpful discussions regarding iCasp9 constructs. This work was supported in part by grants from the National Institutes of Health (EB00262, EB08396, UG3 EB017103), the National Science Foundation Cellular Metamaterials Engineering Research Center, the Boston University Biological Design Center, and Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. A.L. acknowledges financial support from the NIH T32 Quantitative Biology and Physiology training grant, and S.S. was supported by the American Heart Association Postdoctoral Fellowship. A.X.C. was supported by the National Science Foundation Graduate Research Fellowship (1122374). S.N.B.is a Howard Hughes Medical Institute Investigator.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
S.N.B. is a director at Vertex, co-founder and consultant at Glympse Bio, consultant for Cristal, Maverick, and Moderna, and receives sponsored research funding from Johnson & Johnson.
Contributor Information
H.-H. Greco Song, Harvard-MIT Program in Health Sciences and Technology, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Alex Lammers, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Subramanian Sundaram, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Logan Rubio, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA.
Amanda X. Chen, Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02142.
Linqing Li, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Jeroen Eyckmans, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA; Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
Sangeeta N. Bhatia, Harvard-MIT Program in Health Sciences and Technology, Institute for Medical Engineering and Science, Massachusetts Institute of Technology, Cambridge, MA 02139, USA Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA; David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 02142; Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA 02142, USA; Howard Hughes Medical Institute, Chevy Chase, MD 20815, USA.
Christopher S. Chen, Biological Design Center, Department of Biomedical Engineering, Boston University, Boston, MA 02215, USA Harvard Wyss Institute for Biologically Inspired Engineering, Boston, MA 02115, USA.
References
- [1].Potente M, Gerhardt H, Carmeliet P, Cell 2011, 146, 873. [DOI] [PubMed] [Google Scholar]
- [2].Luttun A, Carmeliet P, Stem Cells From Basic Res. to Ther 2014, 2, 104. [Google Scholar]
- [3].Herbert SP, Stainier DYR, Nat. Rev. Mol. Cell Biol 2011, 12, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Traore MA, George SC, Tissue Eng. Part B Rev 2017, 23, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Song HHG, Rumma RT, Ozaki CK, Edelman ER, Chen CS, Vascular Tissue Engineering: Progress, Challenges, and Clinical Promise. Cell Stem Cell 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davis GE, Camarillo CW, Exp. Cell Res 1996, 224, 39. [DOI] [PubMed] [Google Scholar]
- [7].Saunders WB, Bohnsack BL, Faske JB, Anthis NJ, Bayless KJ, Hirschi KK, Davis GE, J. Cell Biol 2006, 175, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barry DM, Koo Y, Norden PR, Wylie LA, Xu K, Wichaidit C, Azizoglu DB, Zheng Y, Cobb MH, Davis GE, Cleaver O, Circ. Res 2016, 119, 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Koh W, Mahan RD, Davis GE, J. Cell Sci 2008, 121, 989. [DOI] [PubMed] [Google Scholar]
- [10].Stratman AN, Davis GE, Microsc. Microanal 2012, 18, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kamei M, Brian Saunders W, Bayless KJ, Dye L, Davis GE, Weinstein BM, Nature 2006, 442, 453. [DOI] [PubMed] [Google Scholar]
- [12].Song JW, Munn LL, Proc. Natl. Acad. Sci 2011, 108, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Galie PA, Nguyen D-HT, Choi CK, Cohen DM, Janmey PA, Chen CS, Proc. Natl. Acad. Sci 2014, 111, 7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alonzo LF, Moya ML, Shirure VS, George SC, Lab Chip 2015, 15, 3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kim S, Lee H, Chung M, Jeon NL, Lab Chip 2013, 13, 1489. [DOI] [PubMed] [Google Scholar]
- [16].Hsu Y-H, Moya ML, Abiri P, Hughes CCW, George SC, Lee AP, Lab Chip 2013, 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cheng G, Liao S, Wong HK, Lacorre DA, Di Tomaso E, Au P, Fukumura D, Jain RK, Munn LL, Blood 2011, 118, 4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Samuel R, Daheron L, Liao S, Vardam T, Kamoun WS, Batista A, Buecker C, Schäfer R, Han X, Au P, Scadden DT, Duda DG, Fukumura D, Jain RK, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen X, Aledia AS, Popson SA, Him L, Hughes CCW, George SC, Tissue Eng. Part A 2010, 16, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, Marini R, van Blitterswijk CA, Mulligan RC, D’Amore PA, Langer R, Nat. Biotechnol 2005, 23, 879. [DOI] [PubMed] [Google Scholar]
- [21].Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R, Proc. Natl. Acad. Sci. U. S. A 2002, 99, 4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Abaci HE, Guo Z, Coffman A, Gillette B, Lee WH, Sia SK, Christiano AM, Adv. Healthc. Mater 2016, 5, 1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stevens KR, Scull MA, Ramanan V, Fortin CL, Chaturvedi RR, Knouse KA, Xiao JW, Fung C, Mirabella T, Chen AX, McCue MG, Yang MT, Fleming HE, Chung K, de Jong YP, Chen CS, Rice CM, Bhatia SN, Sci. Transl. Med 2017, 9, eaah5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G, Nature 2018, 556, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, Haghighi A, Homsy J, Hubner N, Church G, Cook SA, Linke WA, Chen CS, Seidman JG, Seidman CE, Science (80-.) 2015, 349, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tey S-K, Dotti G, Rooney CM, Heslop HE, Brenner MK, Biol. Blood Marrow Transplant 2007, 13, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK, Engl N. J. Med 2011, 365, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Straathof KC, Pulè MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM, Blood 2005, 105, 4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nguyen D-HT, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, Chen CS, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Trappmann B, Baker BM, Polacheck WJ, Choi CK, Burdick JA, Chen CS, Nat. Commun 2017, 8, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ramalingam P, Poulos MG, Lazzari E, Gutkin MC, Lopez D, Kloss CC, Crowley MJ, Katsnelson L, Freire AG, Greenblatt MB, Park CY, Butler JM, Nat. Commun 2020, 11, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pober JS, Sessa WC, Nat. Rev. Immunol 2007, 7, 803. [DOI] [PubMed] [Google Scholar]
- [33].Xiao L, Liu Y, Wang N, Am. J. Physiol. Circ. Physiol 2014, 306, H317. [DOI] [PubMed] [Google Scholar]
- [34].Croft DR, Coleman ML, Li S, Robertson D, Sullivan T, Stewart CL, Olson MF, J. Cell Biol 2005, 168, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wickman GR, Julian L, Mardilovich K, Schumacher S, Munro J, Rath N, Al Zander S, Mleczak A, Sumpton D, Morrice N, Bienvenut WV, Olson MF, Cell Death Differ 2013, 20, 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Newman AC, Nakatsu MN, Chou W, Gershon PD, Hughes CCW, Mol. Biol. Cell 2011, 22, 3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Newman AC, Chou W, Welch-Reardon KM, Fong AH, Popson SA, Phan DT, Sandoval DR, Nguyen DP, Gershon PD, Hughes CCW, Arterioscler. Thromb. Vasc. Biol 2013, 33, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou X, Rowe RG, Hiraoka N, George JP, Wirtz D, Mosher DF, Virtanen I, Chernousov MA, Weiss SJ, Genes Dev 2008, 22, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].DiPietro LA, J. Leukoc. Biol 2016, 100, 979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Xiao Y, Liu C, Chen Z, Blatchley MR, Kim D, Zhou J, Xu M, Gerecht S, Fan R, Adv. Biosyst 2019, 3, 1900089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Griffith CK, Miller C, Sainson RCA, Calvert JW, Jeon NL, Hughes CCW, George SC, Tissue Eng 2005, 11, 257. [DOI] [PubMed] [Google Scholar]
- [42].Kim S, Lee H, Chung M, Jeon NL, Lab Chip 2013, 13, 1489. [DOI] [PubMed] [Google Scholar]
- [43].Whisler JA, Chen MB, Kamm RD, Tissue Eng. Part C Methods 2014, 20, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA, Cell 2005, 121, 335. [DOI] [PubMed] [Google Scholar]
- [45].Costa-Almeida R, Soares R, Granja PL, Tissue Eng J. Regen. Med 2018, 12, 240. [DOI] [PubMed] [Google Scholar]
- [46].Hughes CCW, Curr. Opin. Hematol 2008, 15, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Clark RAF, DellaPelle P, Manseau E, Lanigan JM, Dvorak HF, Colvin RB, J. Invest. Dermatol 1982, 79, 269. [DOI] [PubMed] [Google Scholar]
- [48].Van Der Flier A, Badu-Nkansah K, Whittaker CA, Crowley D, Bronson RT, Lacy-Hulbert A, Hynes RO, Development 2010, 137, 2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chiang HY, Korshunov VA, Serour A, Shi F, Sottile J, Arterioscler. Thromb. Vasc. Biol 2009, 29, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, Otani T, Livesey FJ, Knoblich JA, Nat. Biotechnol 2017, 35, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Homan KA, Gupta N, Kroll KT, Kolesky DB, Skylar-Scott M, Miyoshi T, Mau D, Valerius MT, Ferrante T, Bonventre JV, Lewis JA, Morizane R, Nat. Methods 2019, 16, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Clevers H, Cell 2016, 165, 1586. [DOI] [PubMed] [Google Scholar]
- [53].Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M, Trends Immunol 2001, 22, 199. [DOI] [PubMed] [Google Scholar]
- [54].Wynn TA, Ramalingam TR, Nat. Med 2012, 18, 1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jeon JS, Bersini S, Whisler JA, Chen MB, Dubini G, Charest JL, Moretti M, Kamm RD, Integr. Biol 2012, 100, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R, Nat. Med 2013, 19, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lemos DR, Duffield JS, Sci. Transl. Med 2018, 10, 1. [DOI] [PubMed] [Google Scholar]
- [58].Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, Español-Suñer R, Willenbring H, Weaver VM, Chang TT, Hepatology 2016, 64, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Xia T, Zhao R, Liu W, Huang Q, Chen P, Waju YN, Al-ani MK, Lv Y, Yang L, J. Cell. Physiol 2018, 233, 6996. [DOI] [PubMed] [Google Scholar]
- [60].Natarajan V, Berglund EJ, Chen DX, Kidambi S, RSC Adv 2015, 5, 80956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Godoy P, Hengstler JG, Ilkavets I, Meyer C, Bachmann A, Müller A, Tuschl G, Mueller SO, Dooley S, Hepatology 2009, 49, 2031. [DOI] [PubMed] [Google Scholar]
- [62].Géraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, Leibing T, Demory A, Ulbrich F, Diett M, Singh S, Sticht C, Breitkopf-Heinlein K, Richter K, Karppinen SM, Pihlajaniemi T, Arnold B, Rodewald HR, Augustin HG, Schledzewski K, Goerdt S, J. Clin. Invest 2017, 127, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].DeLeve LD, J. Clin. Invest 2013, 123, 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Manavski Y, Abel T, Hu J, Kleinlützum D, Buchholz CJ, Belz C, Augustin HG, Boon RA, Dimmeler S, Proc. Natl. Acad. Sci 2017, 114, 3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ding B-S, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, Mittal V, Kobayashi H, Shido K, Lyden D, Sato TN, Rabbany SY, Rafii S, Nature 2010, 468, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rossignoli F, Grisendi G, Spano C, Golinelli G, Recchia A, Rovesti G, Orsi G, Veronesi E, Horwitz EM, Dominici M, Cancer Gene Ther 2019, 26, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wu C, Hong SG, Winkler T, Spencer DM, Jares A, Ichwan B, Nicolae A, Guo V, Larochelle A, Dunbar CE, Mol. Ther. - Methods Clin. Dev 2014, 1, 14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Richards M, Fong CY, Chan WK, Wong PC, Bongso A, Nat. Biotechnol 2002, 20, 933. [DOI] [PubMed] [Google Scholar]
- [69].Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, Chaturvedi RR, Li CY, Zandstra PW, Chen CS, Bhatia SN, Nat. Commun 2013, 4, 1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zuiderveld K, Heckbert PS, Ed.; Academic Press Professional, Inc.: San Diego, CA, USA, 1994; pp. 474–485. [Google Scholar]
- [71].Li C, Xu C, Gui C, Fox MD, IEEE Trans. image Process 2010, 19, 3243. [DOI] [PubMed] [Google Scholar]
- [72].Lee T-C, Kashyap RL, Chu C-N, CVGIP Graph. Model. Image Process 1994, 56, 462. [Google Scholar]
- [73].Kollmannsberger P, Kerschnitzki M, Repp F, Wagermaier W, Weinkamer R, Fratzl P, New J Phys 2017, 19, 73019. [Google Scholar]
- [74].McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, Wiegraebe W, Singh S, Becker T, Caicedo JC, Carpenter AE, PLOS Biol 2018, 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.