

Abstract
Biliary atresia (BA) is a rare but severe fibroinflammatory disease of the extrahepatic and the intrahepatic bile ducts. Without prompt interventions, BA has fatal outcomes and is the most common indicator for pediatric liver transplantation (LTx). While the mainstay of treatment involves surgically correcting the extrahepatic biliary obstruction via Kasai hepato-portoenterostomy (KHPE), activation of a multitude of biological pathways and yet-to-be-determined etiology in BA continue to foster liver inflammation, cirrhosis and need for LTx. However, important caveats still exist in our understandings of the biliary pathophysiology, the rapidity of liver fibrosis and progression to liver failure, largely due to limited knowledge of the triggers of biliary injury and the inability to accurately model human BA. Although inconclusive, a large body of existing literature points to a potential viral infection in the early peri- or postnatal period as triggers of epithelial injury that perpetuates the downstream biliary disease. Further confounding this issue, are the lack of in-vivo and in-vitro models to efficiently recapitulate the cardinal features of BA, primarily liver fibrosis. To overcome these barriers in BA research, new directions in recent years have enabled (I) identification of additional triggers of biliary injury linked mostly to environmental toxins, (II) development of models to investigate liver fibrogenesis, and (III) translational research using patient-derived organoids. Here, we discuss recent advances that undoubtedly will stimulate future efforts investigating these new and exciting avenues towards mechanistic and drug discovery efforts and disease-preventive measures. The implications of these emerging scientific investigations and disease modeling in severe fibrosing cholangiopathies like BA are enormous and contribute substantially in our understandings of this rare but deadly disease. These findings are also expected to facilitate expeditious identification of translationally targetable pathways and bring us one step closer in treating an infant with BA, a population highly vulnerable to life-long liver related complications.
Keywords: Biliary atresia (BA), neonatal cholestasis, triggers of biliary atresia, mouse models, fibrosis
Introduction
Biliary atresia (BA) is a severe hepatobiliary disease restricted to early infancy that results in fibroinflammatory destruction of the extra- and the intra-hepatic bile ducts, development of rapid cholestasis, cirrhosis and progression to hepatic failure. Although classified as a rare disease with a global incidence of 1:3,000–1:15,000, BA is the most common cause of obstructive cholestasis and accounts for approximately 50% of all pediatric liver transplantations (LTx) performed in the United States (1). If not surgically intervened at early stages of the disease, BA results in rapid deterioration of infant health, failure to thrive, severe complications from liver cirrhosis and death from end-stage liver disease by 2 years of life (2). The only treatment option for long-term survival with native liver is via a surgical procedure called Kasai hepato-portoenterostomy (KHPE), which involves the removal of atretic extrahepatic bile duct (EHBD) and anastomosis using a segment of the small intestine (3). This procedure combined with the recently described MMP7-based diagnostics may facilitate early diagnosis of BA and offer better outcomes (4,5). Even though KHPE facilitates bile drainage and improves liver profiles, about 80% of the infants with BA harbor persistent fibrosis and ductular proliferations inevitably leading to portal hypertension, coagulopathy and liver failure requiring LTx as the most common curative therapy (6).
KHPE is mostly successful only if performed within the first 30–45 days of life (7) with greater benefits of survival with native liver at 2 (58–65%) (8,9) and 15 (34.9%) years of age (10). A recent study using the Healthcare Cost and Utilization Project–National Inpatient Sample (HCUP–NIS) administrative database of 1,243 BA hospitalizations showed that a majority of patients in the US underwent delayed KHPE after 60 days of age greatly increasing healthcare costs for liver cirrhosis related complications (11). The direct health care costs are also higher in patients with BA especially those undergoing LTx, resulting in increased utilization of health services due to portal hypertension, cirrhosis, esophageal varices and gastrointestinal bleedings. For example, the direct healthcare costs for caring a patient with asthma was $3,316 compared to one-year costs of $154,497 for a child with BA post diagnosis (12,13). Although associated with significantly higher costs, LTx improves long-term survival into adulthood albeit with a poor health related quality of life (HRQOL) (14). Clinicians and scientists are therefore not only challenged continually into devising methods to improvise the HRQOL in transplant recipients but also identifying avenues towards early diagnosis for successful KHPE and greater percent of survival with native livers. Pertinent to this discussion, studies also suggest either prenatal or immediate postnatal existence of biliary injury shown either by elevated bile acids or abnormal direct/conjugated bilirubin (DB/CB) levels (15,16). Thus, efforts towards developing non-surgical medical therapies require fundamental knowledge of the etiological agents, that define the evolution of biliary injury, in conjunction with models that recapitulate the fibroinflammation seen in patients with BA. An in-depth understanding of these processes will accelerate efforts to prevent ongoing liver inflammation and fibrosis after KHPE and lower the need for liver transplantations.
In spite of the enormity of experimental and patient-based mechanistic data generated over the past decades, the challenges of early diagnosis and preventing either the inception or progression of BA still remain unmet linked primarily to unknown etiologies and existence of additional pathways of biliary injury (17). In parallel, the rarity of BA and the availability of a single animal model that partially recapitulates the spectrum of extrahepatic biliary disease with no liver fibrosis substantially limits the development of newer treatment approaches (18). In patients with BA, the existence of at least 3 phenotypic patterns: (Group 1: non-syndromic, isolated BA without major malformations, Group 2: isolated BA with at least one malformation as defined by National Birth Defects Prevention Study but without laterality defects, and Group 3: syndromic, with laterality defects), suggest the likelihood of distinct etiologies or risk factors in the early postnatal life (19). In light of these divergences associated with BA, research efforts in the past 5 years have focused particularly towards identifying disease-initiating events and disease modeling with significantly encouraging results. Such efforts have seen a surge in identifying novel triggers linked to environmental factors and translational preclinical studies using biliary disease-specific murine models towards better treatment and preventative measures. Here, we summarize some of the most recent advances and emerging concepts that define the etiology, trigger mechanisms and disease modeling in addressing the pathophysiology of BA and present the following article in accordance with the narrative review checklist (available at http://dx.doi.org/10.21037/dmr-20-97).
Methods
To elucidate recent developments and advances in BA, we performed a thorough review of the literature published from January 2015 to May 2020 by independent searches using NIH National Library of Medicine PubMed, MEDLINE and Web of Science. These publicly available and institutionally accessed databases were used to search the indexed and published articles. To incorporate new research unpublished in standard journals but presented at International meetings and Conferences in this review, we searched for abstracts and proceedings on BA presented at the three major liver-related conferences: the Annual meeting of American Association for the Study of Liver Diseases (AASLD), the European Association for the Study of Liver Diseases (EASL), and the Digestive Disease Week (DDW). Editorials, technical reports and expert opinions were excluded from the literature search.
Triggers of BA: advances and mechanisms
While the exact cause of BA remains unknown, the etiopathogenesis of syndromic or congenital (also referred to as “embryonic”) BA is associated primarily with a variety of structural anomalies at birth (20). Although rare, these congenital abnormalities that occur in ~10% of patients with early onset of BA are associated with splenic malformations, polysplenia, heterotaxy and laterality defects, intestinal malrotation, and situs inversus abdominus (21,22). Distinct from the majority of infants that develop BA perinatally, onset of congenital BA presumably begins in-utero with full phenotypic expression at the time of birth (23). The underlying causality of the embryonic form was linked originally to developmental complications (24) and potential contributions from genetic aberrations linked mostly to defects in early embryonic patterning (25). Patients with other malformations including cardiac anomalies, duodenal and esophageal atresia, annular pancreas, polycystic kidney disease, cleft palate, and gene mutations in epithelial cell ciliary function linked to laterality defects and left-right (LR) patterning defects may present with symptoms similar to ciliopathies (26–28). In this context, several genes involved in maintaining the structural and functional integrity of primary cilia on the apical surface of cholangiocytes have been implicated in biliary diseases including BA (29,30). Evidences from early vertebrate embryogenesis show pivotal functional roles for sensory and motile cilia in establishing LR patterning events (31,32). In cholangiocytes, primary cilia play critical roles not only in sensing extracellular environments of bile flow but also in bile duct maturation, ductular formation (33) and downstream intracellular signaling processes (32). Proposed roles for ciliary dysbiogenesis in syndromic BA is further posited by observations of stunted, abnormally oriented and less abundant cholangiocyte cilia (34–37) and mutations in genes linked to ciliary function including polycystic kidney disease 1 like 1 (PKD1L1) (38), Cripto, FRL-1, Cryptic Family 1 (CFC1) (29,39,40), nodal growth differentiation factor (NODAL), Zic family member 3 (ZIC3) and Left-Right Determination Factor 1 and 2 (LEFTY1, LEFTY 2) (41). Selective reprogramming within the livers of syndromic BA patients with overexpression of the laterality genes SPRY4, ZIC3, SMAD2 and EBAF along with increased expression of five imprinted genes (IGF2, PEG3, PEG10, MEG3, and IPW) has also been noted in genome-wide expression profiling studies (42). A large prospective multicenter study found that BA patients with one or more laterality defects compared to those without any major malformations were significantly younger in age at the time of clinical evaluation (43) suggesting early onset of disease. Furthermore, recent genome-wide association studies (GWAS) in patients with BA (44) have identified variants in three susceptibility genes: glypican (GPC1) adducin 3 (ADD3) (30) and adenosine diphosphate ribosylation factor 6 (ARF6) (45). Absence of each of these genes in zebrafish impedes bile flow and intrahepatic bile duct development further emphasizing the importance of these genetic variants in BA. Despite these evidences, the direct relevance of these findings as “triggers” of Biliary Atresia Splenic Malformation (BASM) and syndromic BA still require validation analyses to firmly establish their functional roles. In general, ciliary dysfunction proximate to gene mutations can be envisaged to underlie pathogenic aspects of syndromic BA.
The triggers of non-syndromic or acquired BA, however, are hypothesized to involve perinatal exposures to noxious stimuli. The most widely accepted hypothesis of initial hepatobiliary injury in acquired BA involves potential viral infection in the early perinatal period, corroborated by extensive experimental evidence from rhesus rotavirus (RRV)-induced BA in newborn mice. The ability of RRV as a trigger of BA-like disease was first demonstrated by Riepenhoff-Talty et al. where oral inoculation of newborn mice by 2 days of age resulted in cholestasis and death by 3 weeks in 50% of mice. Diseased pups showed signs of inflammation and swelling of the EHBDs by day 7 and ballooning and obstruction of the CBD at later stages with no obvious signs of duct atresia typically seen in human BA (46). A later modification by Petersen et al. involving intraperitoneal injection of newborn mice with RRV in the first 24–48 h of life resulted in a more reproducible and demonstrable model of BA (47,48). Similar to restriction of human BA to early newborn life, RRV infection also follows a strict temporal order of disease induction with early inoculations (days 0–3 of life) leading to severe cholestasis and death in >95% of mice and late infections (days 3–7 of life) inducing symptoms in <50% of infected mice with no mortalities (49). Phenotypic analysis showed occurrence of cholestasis between 4th and 11th day post infection with jaundice of non-fur covered skin, acholic stools, bilirubinuria and growth failure (50). Histologically, the model was characterized by a slow evolution to segmental or discontinuous EHBD atresia occurring between 2–3 weeks after intraperitoneal inoculation of RRV (46,51). The process involved an initial injury to the EHBD cholangiocytes by 3 days, exuberant inflammation and intralumenal filling by inflammatory cells by day 7 and a transition to a segmental atresia of the common bile duct (CBD) with occasional dilatations seen in the distal region of the CBD by 2–3 weeks (47). Human infants present with the most commonly observed Type III BA (>90%) that show obliteration of common, hepatic and cystic ducts (CD) and are of the uncorrectable type due to non-anastomosable ducts at porta hepatis (52). However, the RRV model showed discontinuous and “identifiable” atresia with significant variations in severity versus the unresectable and “unidentifiable” CBD in human BA at the time of KHPE (53). The other most important variable is the restriction of the disease model to BALB/c strain that is highly susceptible to RRV infection, with other strains being resistant. Although C57BL/6 mice are highly resistant BA, death in these mice was due to extremely high RRV titers in the brains resulting in neurotropic effects (54). In addition, the immune responses documented in human and BALB/c murine model as fundamental regulators of biliary pathogenesis were also suppressed with substantially lower expressions of hepatic Cxcl1/10/11, CCL2, CD14 and TLR3 and liver T-cell infiltrations (55). It is therefore important to review these findings in the context of a common or global trigger of BA. RRV-based modeling of BA is primarily a strain-specific trigger of BA and may not represent a uniform or a global trigger of BA.
In livers from patients with BA, signatures of multiple viruses including reovirus, rotavirus, Epstein-Barr virus, human papillomavirus and cytomegalovirus (CMV) have been documented and proposed in the etiology of BA (56). The most pertinent studies showed either the presence of CMV DNA in ~60% of BA patients at the time of diagnosis or a CMV-specific T-cell immune response in 56% of patients (19). Hypothesizing CMV as a possible trigger of BA, Zani et al. (57) found infants with CMV IgM+ BA to be more jaundiced with greater degrees of liver inflammation and fibrosis. However, these patients were of older age at the time of KHPE and therefore remains to be ascertained if the age predisposed to secondary viral infections. Nonetheless, any viral infection may play a role in augmenting progressive hepatobiliary injury and fibrosis. Supporting this view, adjuvant antiviral therapy in patients with CMV IgM+ BA significantly improved jaundice clearance. Although survival with native liver increased at 2 years of age, no difference was observed in overall survival (58). CMV DNA was also found in a large number of patients with BA with CMV-pp65 antigenemia distributed in hepatocytes and vascular and biliary duct endothelial cells. In-vitro experiments showed the cytopathic and apoptotic activities of HCMVAD169 in infected human intrahepatic cholangiocyte cells (59). It however remains to be deduced if the viral cytotoxicity is specific to HCMV or any virus with the capacity to infect biliary epithelial cells. Independent of CMV, Rauschenfels et al. (60) investigated expression of Mx protein by immunohistochemistry as a common denominator of viral infection. Although increased Mx protein was found localized to hepatocytes, bile ducts and epithelium, less than half of the biopsies showed expressions for virus DNA/RNA. Similar to previous studies, positivity was limited to older age patients suggesting a secondary phenomenon. Considering the ability of reovirus to induce jaundice, oily fur syndrome, portal lymphoid infiltrates, and sporadic bile duct necrosis in newborn mice, livers from human BA patients were examined for reovirus by RT-PCR, serology and immunohistochemistry with conflicting outcomes. Recently it was shown that reovirus binding to sialic acid co-receptor on ductal epithelial cells is essential for viral infectivity and generation of biliary disease (61). Similar results were obtained in a study reported by Riepenhoff-Talty et al. where 50% of livers from BA patients showed presence of group C rotavirus by RT-PCR (62) while data from Bobo et al. failed to detect group A, B or C rotavirus in patient specimens (63). The inconsistencies in sample collection methodologies and reproducible data, lack of viral sequencing following PCR amplifications and frozen vs. formalin-fixed sampling techniques investigating the presence of virus and/or viral components as causal factors have been inconclusive and suggest a highly likely secondary infection during the course of BA. Some of these viruses also share the key amino acid sequence “SRL” in the RRV Viral Protein 4 (VP4) found to be specific to the rotavirus strains capable of inducing EHBD injury and obstruction in newborn mice, suggesting a common mode of cholangiocyte binding and ductular pathology in human BA (64). However, none of the studies were able to conclusively link the hepatic viral signatures to development of BA and more importantly distinguish between primary and secondarily acquired infections at the time of diagnosis due to altered immune status of the diseased infant.
Relating mouse to human ages in the early postnatal developmental stages, the evolution of the EHBD pathology from inflammation to fibrosis in the RRV model appears to follow a slow transition to atresia. Equating 28 mouse to 180 human days (1 mouse day = 6.43 human days) during early life, a 14-day disease window in mice approximately mirrors a 90-day old infant with BA, a significantly late phase of disease. In infants with BA, most of the diagnosis is performed between 45–60 days of age or earlier that show irreversible fibrosis of the CBD. A distinctive feature of the experimental model is the low degree or lack of liver fibrosis that fails to efficiently recapitulate characteristics of human biliary disease (65). Taken together with the 3 distinct BA phenotypes, these observations suggest unknown identities of the triggering agent likely to be viral and/or other components capable of inducing biliary injury. While this remains speculative and largely unanswered due to paucity of disease-relevant models, identification of the causative factors of BA disease remains a topic of paramount importance.
Independent of the viral infections in the etiology of BA, the recent demonstration of a purified plant toxin, biliatresone, to induce BA-like cholangiocyte cell injury and destruction of bile ducts in zebrafish and newborn mice has significantly raised the likelihood of exposure to environmental toxins in the early perinatal period. Biliatresone, an isoflavonoid biliary toxin, was first shown to cause spontaneous outbreaks of extrahepatic biliary disease resembling BA in Australian neonatal livestock following ingestion of Dysphania species (Dysphania glomulifera and Dysphania littoralis) plants by pregnant dams (66). These incidences which occurred in 1964, 1988, 2007 and 2013 were associated with severe drought resulting in animals grazing on lands typically under water and affected nearly 100% of newborn sheep and in one instance, cows. Although these findings lend credibility to the involvement of an exogenous toxin in BA and may explain the non-mendelian inheritance, twin discordance and genetic abnormalities, human exposures to biliatresone are almost completely unlikely. Conversely, the data strongly suggest that environmental components structurally linked to biliatresone could act as triggers of biliary disease. This prompted us to closely investigate the items that newborn infants are likely to be exposed to or spend most of their times in close proximity with specific emphasis on chemicals capable of mediating biliary toxicity in the immediate postnatal period. Remarkably, several products with significant exposure potential to newborns contained Diphenylmethane diisocyanate (MDI), the most common and extensively used diisocyanate intermediate of the polyurethane chemical (PUC) industry (67). MDI is derived from another commonly used PUC, 4, 4’-Methylenedianiline (MDA), a hepatotoxin known to induce liver injury in multiple rodent studies and responsible for an outbreak of jaundice in the Epping district of Essex (“Epping jaundice”) in early 1965 which affected 84 people (68). In rodents, administration of MDA causes hepatic necroinflammation and fibrosis and portal inflammation, eosinophilic infiltration, cholangitis, cholestasis, and varying degrees of hepatocellular damage in humans. However, the hepatotoxic properties of MDI in mice and human remain unknown. Newborns especially are highly sensitive to toxin exposures via oral, inhalation or dermal routes even to trace quantities of chemical contaminants and volatile organic compounds (VOCs). Varying amounts of residual MDA and MDI have reportedly been found in diapers, transfer of azo dyes in colored prints and textiles due to wet diaper-areas, plastic pacifiers, PU infant crib mattresses, furniture including cribs, nursing chairs and pillows, child safety seats, toys, spandex fibers, PU based spray-foam insulations, carpet underlays/paddings, floor coatings, adhesives, binders, sealants and paints, rubber and plastic products, etc. (69–72). In keeping with a higher incidence of BA in low birth weight and shortened gestational age infants, premature neonates are at a greater PUC exposure risk via contact with several medical devices including PU bags and wraps to prevent heat loss, catheters, feeding tubes, chest drains, limb boards, breathable adhesive films, wound dressings, plasters/band-aids, foam-backed electrodes, adhesive oximetry sensors, suction catheters, forehead pads, infant phototherapy eye protectors etc. (67). In particular, adhesive PU films and wound dressings are in contact for prolonged periods from hours to days providing sufficient time for the PUCs to penetrate the thin neonatal skin (67,73,74). These effects can further be compounded by prolonged sleep patterns of infants (12–13 h/day), increased heat transfer during sleeping, greater inhalation per body mass, higher surface area relative to body weight, poor skin barrier in low birth weight infants, percutaneous respiration, plasticizers in baby products, elevated levels of ROS in newborns and “take-home” exposures (75–78).
Importantly, both MDA and MDI are high production volume (HPV) chemicals with >1 million pounds of production or import into the US. According to the US EPA Toxic Release Inventory (TRI) database, the national aggregate MDA production in 2014 ranged from 100–250 million pounds and 750–1,000 million pounds for MDI. Of greater concern are the total releases and production wastes reaching 1,100–3,600 tonnes/year for MDA and 74–405 tonnes/year for MDI. In addition to trace quantities found in consumer products, the environmental releases and subsequent unknown effects have the potential to predispose susceptible infants to a greater risk of biliary pathogenesis. Taking cues from these studies, we investigated the abilities of the PUCs to induce temporally restricted hepatobiliary pathogenesis. Oral or dermal exposures of newborn mice to small quantities of MDA or MDI rapidly induced symptoms of cholestasis within 72 hours including jaundice, acholic (chalk-colored) stools and bilirubinuria. Collectively, these data establish a probable role for environmental and chemical toxins in the etiopathology of BA.
Interestingly, findings from the chemical BA model appear to superimpose with the concept of Vanishing Bile Duct Syndrome (VBDS), first described in patients undergoing orthotopic LTx (79). VBDS refers to destruction of the intrahepatic bile ducts in the context of drug or toxin induced bile duct damage resulting in chronic cholestasis (80). There are more than 40 drugs that cause VBDS including ciprofloxacin, amoxycillin-clavulanate, ampicillin, chlorpromazine, etc. (81), with instances also found in children treated with trimethoprim-sulfamethoxazole (82). While the classical disease phenotypes characteristic of VBDS exclude EHBD obstruction, the possibility of VBDS affecting the extrahepatic biliary system remains unexplored. Interestingly, factors associated with VBDS share common features with BA including congenital disorders affecting bile duct development, viral infections, immune disorders, and in most cases the need for LTx (83). Indeed, several of the histologic features of acute VBDS correlate with BA including: parenchymal bilirubinostasis, infiltration of portal tracts by mononuclear cells, macrophages and neutrophils, epithelial degeneration, vacuolization and cholangiocyte pyknosis (84). Persistent cholangiocyte injury results in the development of periportal and bridging fibrosis leading eventually to cirrhosis and premature death from liver failure. Overlapping features of VBDS are also observed in other chronic liver diseases including PBC and PSC where biochemical changes persist beyond 3 months (85). Specific to BA, the regenerative capacity of the cholangiocytes lining EHBDs may significantly be lower than the intrahepatic cells rendering them highly susceptible to VBDS. In infants with BA, this may severely limit the ability of EHBDs to re-epithelialize once they undergo fibroinflammatory destruction and are replaced by scar tissue. Although speculative, we believe BA may represent a form of Vanishing Extrahepatic Bile Duct Syndrome (VEBDS). The unique immunological makeup (86), distinct innate immune cellular components (87) and heightened sensitivity and susceptibility to chemicals, toxins and drugs (88,89) may render the newborn to the phenomenon of VEBDS in BA. Supporting this hypothesis, histologic analysis of biliary remnants predominantly showed concentric fibroplasia without inflammation or were inactive with presence of fibrous cords and absent inflammation (90). Strikingly, remnants from the youngest subjects were homogeneously inactive with bland replacement by collagen suggesting significantly earlier onset of extrahepatic biliary injury and atresia. The findings of PUCs to induce complete absence of CBD points to an underlying etiologic trigger that may potentially be extended to other unknown chemicals, environmental toxins, commonly used drugs, viruses, etc.; however, these remain to be further deduced. In common, these initial triggers are expected to stimulate a fibroinflammation-mediated injury of the EHBDs and a continuum to involve IHBDs before and after Kasai procedure. Therefore, recognizing the initial trigger is of prime importance since prolonged or persistent exposures may lead to continued liver damage beyond the initial EHBD pathogenesis and mitigate unwarranted interventional therapies.
Mechanistically, studies performed using in-vitro cholangiocyte spheroids showed that biliatresone mediates cholangiocyte-cell toxicity by decreasing levels of glutathione (GSH) resulting in increased redox stress (91,92). Glutathione is a component of the antioxidant defense mechanism and in the context of our findings, plays an important role in the regulation of complement system. Similarly, transcriptional profiling of liver tissues from biliatresone-exposed zebrafish by Zhao et al. showed alterations in GSH-mediated redox homeostasis suggesting a critical role for GSH in biliatresone-induced hepatocyte injury (93). Using cholangiocyte spheroids, Fried et al. recently demonstrated that decreased cholangiocyte GSH levels increased RhoU and Hey2 expressions and lumenal obstruction substantially altering the NOTCH and Wnt pathways and downregulating Sox17 (94). Similarly, genetic absence of Sox17 in mouse cholangiocyte organoids recapitulates effects similar to biliatresone exposures characterized by loss of apical cell polarity and lumen obstruction (91). Recent work from our group showed enriched glutathione gene signatures in children with BA surviving without LTx for 24 months of age. Using a newly developed mouse model of liver fibrosis, we showed significantly improved histologic scores of liver damage and decreased levels of ALT, GGT and bilirubin in mice treated with N-acetyl-cysteine (NAC) (95). Similarly, markers of glutathione metabolism and oxidative stress were altered in livers of newborn mice exposed to MDA and MDI and correlated with exaggerated liver inflammation and fibrosis. Hepatic expressions of Col1a1, Col1a2, Timp1, Acta2, Vim, Ctgf, Hmox1, Gsta2, Gpx2, Snail1, Bambi, Smurf1, Grem1, Crim1 and Egr1 were significantly increased while expressions of Sod1, Cat and Ddit4 reduced after PUC exposures (96). Interestingly, unpublished data from our laboratory showed increased gene signatures of Hmox1, Gsta2, Gpx2, Sod1 and Ddit4 in EHBDs from mice with experimental BA implying oxidative stress responses and activation of GSH pathways.
In summary, identification of these new triggers of BA are, therefore, expected to stimulate efforts to estimate PUC levels in newborn-related items, limit potential chemical exposures in the early perinatal period and gain deeper understanding of the pathophysiological processes governing duct atresia and complete absence of the CBD.
Disease modeling in BA
Overlapping the discovery of virus-independent triggers of BA, recent work by our group and others has led to the development of new models for investigating BA. Regardless of the clinical variations (embryonic/syndromic or acquired/isolated), infants with BA receive similar medical and surgical care due to limited knowledge of etiopathogenesis. Using microarray data based molecular profiling at diagnosis, we have previously identified gene expression signatures that define inflammation and fibrosis phenotypes of BA (97). Some infants progress rapidly to severe fibrosis at an early age while others show either prominent inflammation, fibrosis or a combination of both. As discussed earlier, modeling the full disease spectrum of BA has been limited to rhesus rotavirus type A (RRV) induced experimental BA where infection of newborn mice results in severe inflammation, obstruction and segmental atresia of the EHBDs with or without CBD dilatations. Although RRV infection produces well-defined symptoms of BA, rodent models using other viruses result in a range of symptoms from none to bilirubinuria and jaundice without full biliary obstruction (98,99). The RRV model is typified by dominant proinflammatory circuits involving IFNγ, TNFα, NK, DC and CD8+ T cells, blocking which ameliorates the disease process (65,100). In contrast, the recently developed PU chemical model offers a unique advantage to explore the rapid fibrogenic responses in the EHBD compared to an inflammation driven obstruction in the RRV model. Exposure to the PU chemicals, MDA and MDI not only induce rapid jaundice, acholic stools and bilirubinuria but also manifest histologically demonstrable epithelial injury, inflammation and complete fibrosis of the CBD indiscernible under the dissecting microscope. These changes are associated with severely abnormal and hypotrophic CD and gallbladder (GB). Chemical exposure of newborn mice resulted in 100% mortality by day 10–12 and revealed a phenotype recapitulating the features of EHBDs in patients with BA (type III; >85%). Importantly, the PUCs reproducibly induce severe cholestasis, EHBD atresia, liver inflammation and 100% mortality in neonatal C57BL/6 mice, a strain highly resistant to RRV-induced BA (only the BALB/c strain is susceptible). This finding for the first time establishes a global model for BA independent of mouse strains facilitating the use of an extensive repertoire of genetically modified mice available exclusively in the C57BL/6 background for mechanistic and preclinical studies. A key distinctive feature of the chemical model from RRV-BA is linked to elevated hepatic expression of Snail1 and Smurf1 in early phase of the disease (96). Snail1 and Smurf1 are both involved in epithelial to mesenchymal transition (EMT), a process implicated in BA liver fibrosis. Hepatocyte-derived Snail1 is shown to play a major role in liver fibrogenesis while Smurf1 regulates TGFβ/PAR6 pathway during EMT and alters cell polarity via dissolution of tight junctions (101). Thus, the chemical model offers additional benefits to investigate pathways relevant to fibroinflammatory pathways in early neonatal liver injuries.
The plant toxin, Biliatresone, that causes selective destruction of the EHBD system has also been used in developing novel models using zebra fish and cholangiocyte spheroids to investigate disease pathogenesis. In particular, this isoflavonoid compound resulted in marked defects of the GB epithelium and extrahepatic biliary system in zebrafish larvae (102). Similar to the extensively studied RRV model of BA and the recently described PUCs, the extrahepatic cholangiocytes appear to be exquisitely sensitive to biliatresone with no change in the architecture of hepatocytes or sinusoidal endothelia (103). Uniquely, the pathophysiology of biliary destruction is restricted to early developmental stages and is associated with a robust innate immune response with progressive accumulation of macrophages and neutrophils around injured EHBDs. The study also showed a reduction in the content of primary cilia and microtubules in primary neonatal mouse extrahepatic cholangiocytes exposed to biliatresone; similar exposures of 3D cholangiocyte spheroids resulted in loss of epithelial monolayer integrity and disruption of apicobasal polarity. Interestingly, treatment of isolated intact EHBDs from neonatal mice with biliatresone resulted in disruption of duct epithelium and patchy lumen obstruction and increased immunostainings for α-SMA, fibronectin EIIIA and type I collagen. As discussed in the preceding section, biliatresone targets GSH, reduced levels of which appears sufficient for inducing cholangiocyte injury (91). Results from these in-vivo and in-vitro models may provide opportunities to explore human-relevant molecules bearing structural similarities and help gain insights into mechanisms of human BA.
In conjunction with severe injury and atresia of the EHBDs, infants with BA show rapid and remarkable progression of liver fibrosis. Even after a successful KHPE, a majority of infants with BA continue to experience enhanced liver fibrosis, periportal duct proliferation, portal hypertension and strictures or dilatations of the intrahepatic bile ducts, with only 13–50% alive with native liver by 2 years of age. While the classical model of extrahepatic BA (eBA) induced within 24 hours of birth has provided important clues to the fibroinflammatory processes operative within the EHBDs, livers from mice with end-stage eBA (day 14 of life) remarkably lack fibrosis and exhibit minimal collagen deposition and marginal portal-portal and/or portal-central bridging fibrosis (18). Besides the early destruction of EHBDs, progressive fibrosis and cirrhosis of the liver in human BA occurs relatively early compared with other neonatal cholestatic diseases, such as Alagille syndrome or A1AT deficiency (104). Studies from our laboratory using a low-dose RRV infection model showed prominent duct proliferation, increased peri-portal and some bridging fibrosis in a small group of mice that fortuitously survive to 3 weeks of age (105). However, the extremely small fraction of such surviving mice precludes the use of this model due to large requirement of newborn mice. A study by Keyzer-Dekker et al. using a small cohort of newborn mice injected with a modified dose of RRV (1×106 vs. 1.5×106) showed focal duct proliferation and moderate CK19 staining at 2 weeks of infection. However, only 12% of the mice showed bridging fibrosis, variable α-SMA and collagen type I staining and were unable to capture the full spectrum of the fibrotic phenotype (106). To overcome the limitations associated with and recapitulate the cardinal features of liver fibrosis, we developed a modified mouse model termed Biliary Atresia Liver Fibrosis (BALF) model, by modulating the time and magnitude of virus exposure (95). Following RRV challenge of 3-day old neonatal mice, portal areas in livers from 3-week old mice post infection showed fibrous septa and portal-portal bridging fibrosis. Analysis using Second Harmonics Generation microscopy showed extensive portal presence of Collagen type I and II fibers. In agreement with the intrahepatic CK7+ duct proliferation seen in >80–90% of patients with BA, livers from BALF mice showed progressive portal triad expansions with reactive ductular proliferations by pan-Cytokeratin staining. In addition, intrahepatic gene expression patterns of Col1a1, Col1a2, Col3a1, Col4a1, Col4a2, Col5a1, Col5a2 and Col6a3, Timp1, Acta2 (α-Sma), Lox and Tgf-β1 were similar to infants with BA (95). More importantly, the long-term survival to 3-weeks was ~80%, a marked improvement that allows translational and interventional studies. Using the BALF model, we recently demonstrated the therapeutic benefits of NAC, a glutathione-related antioxidant. Administration of NAC (150 mg/kg/day) substantially decreased portal inflammation, Sirius red positive areas of fibrosis and α-SMA+ cells. Treatment with NAC also reduced hepatic expression of the fibrosis genes Acta2, Col1a1, Timp1, and Tgfb1 suggesting a potential therapeutic intervention in the development of liver fibrosis in BA (95). Following a similar approach, Mohanty et al. recently developed a liver fibrosis model by challenging newborn mice with an RRV-TUCH rotavirus reassortant, TR(VP2, VP4) (64). Similar to the BALF model, livers from TR(VP2, VP4) challenged mice showed features of hepatobiliary inflammation with Ishak stage 3–5 parenchymal liver fibrosis. The livers also displayed portal-portal and portal-central bridging and the presence of microscopic nodules. TR(VP2, VP4)-infected mice showed long-term survival than the conventional RRV-BA model with an incidence of liver fibrosis in 63% of surviving mice at 4 weeks. These newest advances (Figure 1) have unlocked new opportunities to fuel research into identifying effector immune mechanisms driving fibrosis, develop novel anti-fibrotic therapeutics and drug repurposing efforts.
Figure 1.
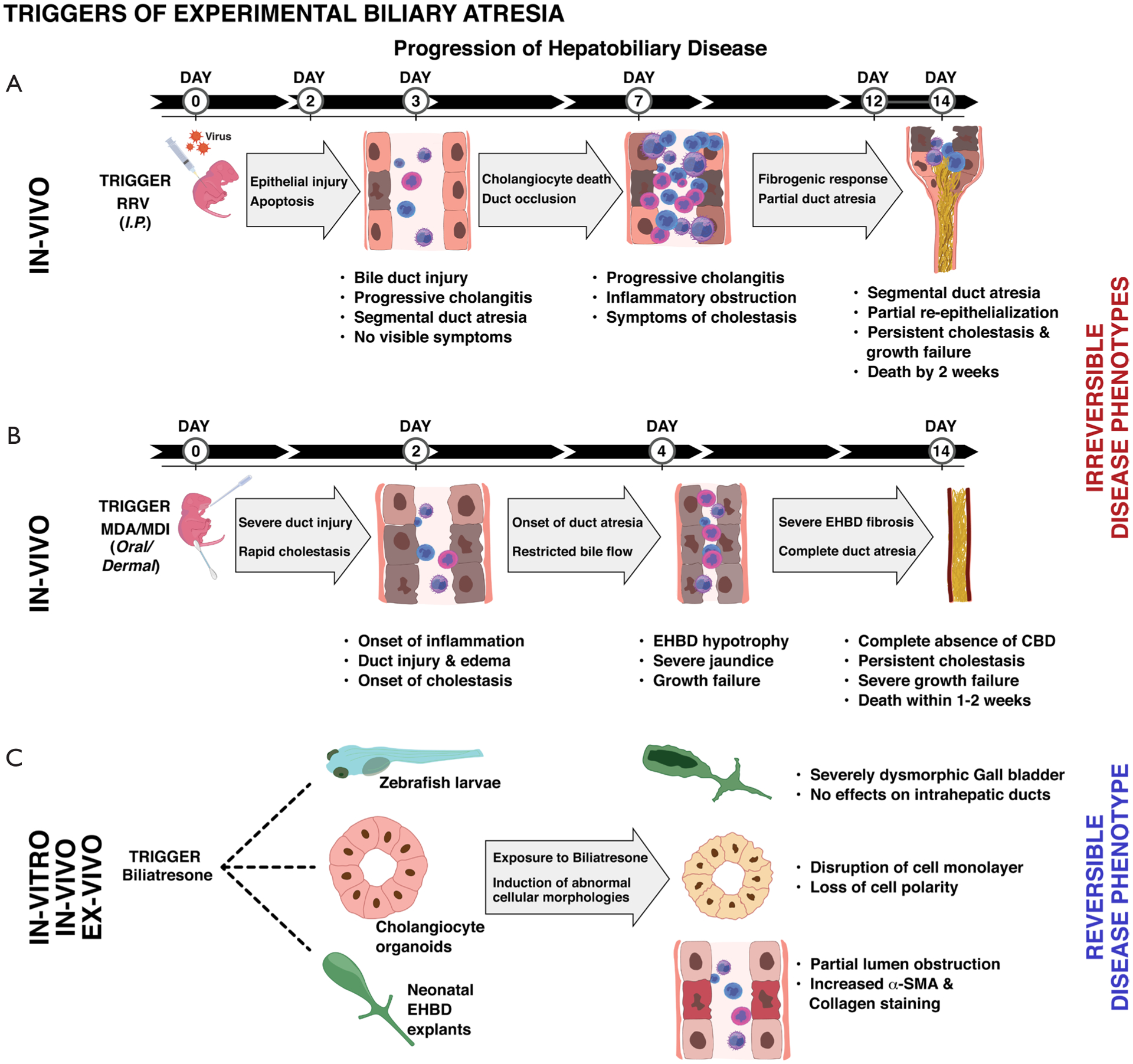
Triggers and outcomes of hepatobiliary pathogenesis in experimental biliary atresia. Flowchart (A) depicts the primary model of experimental BA involving intraperitoneal RRV infection of newborn mice. EHBD manifestations of biliary disease follow a biological continuum from inflammation to segmental atresia and are associated with intrahepatic portal inflammation. (B) Newly identified chemical triggers of BA induce rapid development of severe cholestasis upon oral or dermal exposures. Extrahepatic duct injury is associated with vanishing common bile duct similar to human BA and development of liver fibrosis. Chemical BA model represents a unique approach to investigate very early immunopathogenic mechanisms driving EHBD atresia. (C) depicts recently described in-vivo, in-vitro and ex-vivo activities of the plant toxin, biliatresone, towards cholangiocytes. Biliatresone promotes abnormal cellular pathologies of the extra- but not intrahepatic ducts. BA, Biliary atresia; RRV, rhesus rotavirus; EHBD, extrahepatic bile duct.
Though animal models are an exceptional tool for investigating immunopathogenic underpinnings, differences in murine and human immune makeups may fail to recapitulate the phenotype of human diseases. In-vitro models using human-derived cell lines and iPSC, or somatic cell derived hepatobiliary organoids are rapidly gaining credence as major investigative tools to study pathobiological mechanisms in human BA (Figure 2). Recent body of literature shows that the liver and bile duct derived cell line and organoid approach is gaining popularity especially due to limited in-vitro tools to investigate the pathophysiology of EHBDs. Functionally active cytokeratin-positive cholangiocyte cell lines have been established from rat EHBDs that showed robust expression of Secretin, SSTR2, CFTR, AE2, VEGF-A and NGF and increased secretin-dependent cAMP activity, chloride efflux and proliferation (107). Of note, the inability of in-vivo models to furnish adequate material for investigating biliary cell pathophysiology restricts their usefulness towards diagnostic and research purposes. Combined with the rare incidence of BA, the ability to procure biliary epithelial cells from remnant tissues is considerably restricted. To overcome these limitations, Machhua et al. developed an explant culture method using EHBD tissue from patients with BA and established human biliary epithelial cells that remained viable for >3 months. The long-term culture of these primary cells may therefore provide a cost-efficient model to investigate pathological manifestations of biliary epithelial cells (108). In parallel with the primary cell cultures, recent work has also placed emphasis on the use of induced pluripotent stem cells (iPSCs) and tissue-specific organoids. These approaches represent a new era in investigative biology by providing new models towards understanding role of cholangiocytes in BA pathogenesis and liver regenerative processes (109). Not only is the derivation of duct epithelial cells important, maintenance and propagation of a homogenous population of biliary epithelial cells is also equally critical for multiple downstream applications. To address this, platforms have been developed for the differentiation of human iPSCs to functional cholangiocyte-like cells (CLCs) that recapitulate stages of biliary development. Tian et al. generated disease-specific iPSCs from patients with BA and by CRISPR/Cas9 engineering of healthy iPSCs to lack GPC1 and ADD3. Both lines showed decreased formation of ductal structures, expression of biliary markers (CK7, EpCAM, SOX9, CFTR, etc.) and increased fibrosis markers (a-SMA, Lox2). These iPSCs also expressed YAP and Collagen-1 responsive to pentoxifylline, an anti-fibrogenic drug suggesting their utility in uncovering molecular pathways and developing new therapeutics (110). Sampaziotis et al. developed a method for isolation and propagation of human cholangiocytes from extrahepatic biliary tree to generate extrahepatic cholangiocyte organoids (ECOs) that were transcriptionally and functionally similar to primary cholangiocytes. In-vivo, ECOs showed regenerative capacity, developed bile duct-like structures and on biodegradable scaffolds acquired tissue-like properties to reconstruct and repair GB wall (111). Analogous to ECOs, Shiota et al. recently described a versatile, cost effective, and organ specific preclinical in-vitro EHBD organoid (EHBDO) model generated from adult mouse EHBDs. EHBDOs contain pure epithelial cell populations, express markers of differentiated biliary cells and maintain homogenous morphology through multiple passages (112). These unique characteristics make the EHBDO model suitable for pharmacological testing and biliary cell pathophysiology studies in normal and genetically engineered mice. More recently, Amarachintha et al. generated cholangiocyte organoids (COs) from livers of normal control subjects (N-COs) and patients with BA (BA-COs) as a model system to investigate pathogenic mechanisms of BA. Immunohistochemical and RNASeq analysis showed high KRT19, CFTR, HES1, EPCAM and low ALB, AFP and CYP1A2 expressions by the COs. Epithelial cells from BA-COs recapitulated features of biliary disease with loss of polarity, aberrant expression of ZO-1, F-actin/β-catenin, AQP1, CFTR and SSTR2. Functional assays using Rhodamine123 (R123) showed defective uptake by BA-COs, which did not normalize by addition of Verapamil, a R123 blocker (92). These findings suggest that the BA-COs are a novel model to study fundamental defects specific to BA.
Figure 2.
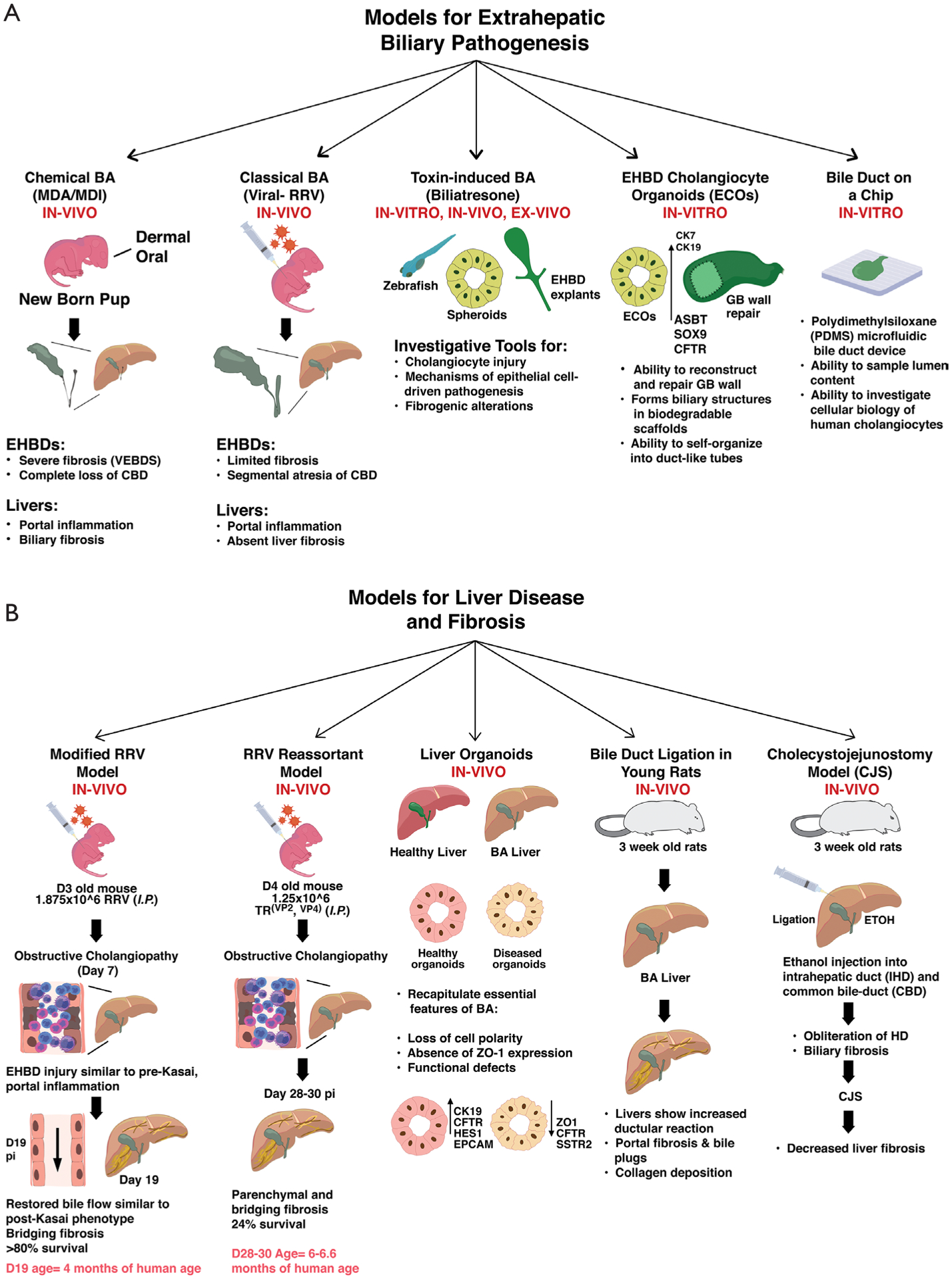
Pre-clinical models for investigating EHBD pathogenesis and liver fibrosis. Panel (A) defines the available models for investigating extrahepatic biliary pathogenesis with specific advantages and outcomes of the individual model systems. Panel (B) shows recent advances in disease modeling for investigating the pathophysiology of liver fibrosis involving in-vivo and in-vitro approaches. These models elucidate features of obstructive cholangiopathies, bridging fibrosis and gene dysregulations associated with liver fibrosis. EHBD, extrahepatic bile duct.
Alongside these advances, a bile duct-on-a-chip has been recently described that efficiently recapitulates the 3-dimensional bile duct architecture and the barrier functions of cholangiocyte layers. Using a microfluidic organ-on-chip technology, Du et al. developed a self-organized cholangiocyte-lined microengineered bile duct with unique biological characteristics of apical-basal polarity, tight junctions and barrier functions. The microfluidic bile duct device consists of a polydimethylsiloxane (PDMS) support containing a mouse cholangiocyte-lined channel through a collagen plug with the apical-basal regions accessible separately through ports. Cholangiocytes form a monolayer within these devices, form a cylindrical tube, express CK19, tight (ZO1) and cell junction (E-cadherin 1) proteins, show polarity and respond to luminal shear flow. The device format offers significant advantage over traditional cholangiocyte spheroids and overcomes the difficulties of sampling lumen contents, accessing apical surface and the inability to control positional spheroid growth in the gel (113). Such a system can prove highly beneficial and indispensable to study cellular biology of human cholangiocytes regionally derived from either livers, GB, or duct remnants from patients with BA.
In addition to these systems, other animal models that could potentially be utilized to study BA pathogenesis have also been investigated. These plausible efforts may significantly benefit future investigations to uncover disease process, enable development of new therapies, drug response, etc. Struecker et al. successfully applied tissue bioengineering principles and performed EHBD replacement by autologous implantation of ex-vivo generated neo-bile ducts in pigs. These ducts were well-tolerated and showed no severe complications, peritonitis and/or biliary leakage, a model that could be applied to BA studies (114). Circumventing the traditional KHPE involving bilioenteric anastomosis via the Roux-en-Y jejunum limb by autologous cholangiocyte derived neo-bile ducts may not only help prevent recurrent episodes of ascending cholangitis or secondary biliary cirrhosis but also facilitate long-term survival with native livers. With recent advancement in surgical techniques, Wang et al. (115) provided a novel rat model for investigating mechanisms and treatments of liver fibrosis in BA after Kasai operation. The model involved cannulating the CHD followed by ligation of the CBD and injecting 95% ethanol into intrahepatic duct (IHD) one-week post-surgery. Without cystojejunostomy, livers showed biliary fibrosis, obliteration of IHD and cystic malformations in CBD with histological findings similar to BA. In this model, cystojejunostomy mirrored Kasai operation and provides a potential model for mechanistic investigations and translational research. A more simplistic surgical model for BA involving bile duct ligation (BDL) in 3-week-old young rats is also reported to study long-term liver functions. Rats that underwent BDL showed significant cholestasis, ductular reaction, portal fibrosis, bile plugs and collagen deposition. While the model fails to capture the severe extrahepatic inflammation and fibrosis characteristic of BA, it may allow investigations of liver fibrosis (116).
Recently, an observational animal model involving Sea Lamprey has also been described. During metamorphosis, the sea lamprey shows developmental BA with complete loss of the entire biliary system and degeneration of bile ducts and GB leading to cholestasis and ductal fibrosis (117). Features similar to human BA including cholangiocyte proliferation, pericholangitis, bile duct obliteration, periportal lymphocytic infiltrations, thickening of the basement membrane and presence of collagen fibrils were observed in this model (118). However, the sea lamprey liver fails to develop liver fibro-necrosis and survives BA and severe cholestasis by adapting to reduce bile salt biosynthesis and metabolizing toxic bile salts to maintain normal plasma levels. Additional studies are required to determine the suitability of this model to explore developmental mechanisms leading up to BA pathogenesis.
Collectively, these emerging and established human and animal in-vivo and in-vitro models (Figure 2) will undoubtedly promote our abilities to investigate pathogenic, developmental, and translational features regulating pathogenesis of BA.
Conclusions
Our review provides a concise summary of the recent discoveries and advances that not only uncover new knowledge of triggers of cellular injury in BA but also advanced techniques of disease modeling. Progressive accumulation of these diverse but unifying theories of disease pathogenesis in the past 5 years have been significant and are expected to propel immediate and future mechanistic, translational and therapeutics discovery efforts to benefit pediatric patients with BA. These data also point to an urgent need of a multidisciplinary approach and collaborative efforts linking immunology, clinical medicine, pathology and the role of environment factors to better understand the disease underpinnings. While this review is focused primarily on BA as a severe prototype of fibrosing cholangiopathy, similar concepts could be applied to PSC, PBC, and other liver diseases. Of significant importance, the capacity of PU chemical intermediates to induce rapid loss of CBD and symptoms of cholestasis in a short window of time and the identification of plant-derived toxins to regulate duct epithelial injury provide alternate dimensions to the long-held hypothesis of viral infection in the etiology of BA. In addition, the diverse origin and structural heterogeneity between PUC and the plant toxins suggest multimodal effectors of cholangiocyte injury and cell death. These findings may be consequential due to the (I) presence of residual chemicals in items that newborns are potentially exposed to and (II) plausible existence of a toxic compound in beets, chard and other consumable plants that co-purifies with biliatresone. More importantly, the existence of multiple EHBD disease phenotypes, molecular representations of inflammatory and fibrosis subtypes at diagnosis, differential response to KHPE, short- and long-term survival with native liver, etc. all point to an involvement of more than a singular trigger of biliary pathogenesis. Further studies are warranted to identify signatures of these chemicals and toxins or their derivatives or structurally similar entities using patient-derived specimens.
Future directions
In keeping with these advances of novel etiopathogenic regulators of biliary disease and a rapid evolution of hitherto unattainable in-vivo murine models defining biological processes of liver fibrosis, our abilities to explore anti-fibrotic therapeutics and drug repurposing efforts to treat liver fibrosis are substantially enhanced. In the context of BA, these findings mark a major milestone since only a minor fraction of infants undergoing KHPE continue to thrive without the need for liver transplantation into adulthood. A greater majority of patients with BA, however, still face the eventuality of needing a liver transplantation either early or late in their lifetime. The progressive nature of the disease and limitations in early diagnosis of the disease further contribute to our inabilities to treat infants with BA before KHPE. To overcome this burden, a detailed interrogation of the cholangiocyte cell biology and ensuing fibroinflammation becomes imperative. In this regard, the recent surge in biliary disease modeling utilizing mouse and patient tissue derived cholangiocyte organoids and the efforts to perfect these techniques for large-scale cell expansions and long-term propagation will enable overcoming the challenges associated with poor access to tissue specimens. Not only have these organoids enabled mechanistic and biological insights but have also been employed in biodegradable scaffolds to generate bioengineered tissue to reconstruct and repair biliary epithelium. These novel developments may also represent a primary step towards developing artificial ducts for treating pediatric and adult cholestatic diseases. Collectively, these high impact scientific data are poised to improve our experimental and therapeutic investigations towards better outcomes in patients with BA.
Acknowledgments
Funding: This work was supported in part by NIH P30 DK078392 (Gene Analysis, Integrative Morphology and Pluripotent Stem Cell and Organoid Cores) of the Digestive Diseases Research Core Center in Cincinnati. We would like to acknowledge the assistance and imaging/graphics support of Mr. Chris Woods, Pathology Research Core. Author contributions: Drafting of the manuscript: AM, UT, RM and PS.
Footnotes
Reporting Checklist: The authors have completed the narrative review checklist. Available at http://dx.doi.org/10.21037/dmr-20-97
Peer Review File: Available at http://dx.doi.org/10.21037/dmr-20-97
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/dmr-20-97).The authors declare no commercial or financial relationships that could be extended as a potential conflict of interest.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
References
- 1.Verkade HJ, Bezerra JA, Davenport M, et al. Biliary atresia and other cholestatic childhood diseases: Advances and future challenges. J Hepatol 2016;65:631–42. [DOI] [PubMed] [Google Scholar]
- 2.Pinto RB, Schneider AC, da Silveira TR. Cirrhosis in children and adolescents: An overview. World J Hepatol 2015;7:392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wildhaber BE. Biliary atresia: 50 years after the first kasai. ISRN Surg 2012;2012:132089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lertudomphonwanit C, Mourya R, Fei L, et al. Large-scale proteomics identifies MMP-7 as a sentinel of epithelial injury and of biliary atresia. Sci Transl Med 2017;9:eaan8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L, Zhou Y, Xu PP, et al. Diagnostic Accuracy of Serum Matrix Metalloproteinase-7 for Biliary Atresia. Hepatology 2018;68:2069–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundaram SS, Mack CL, Feldman AG, et al. Biliary atresia: Indications and timing of liver transplantation and optimization of pretransplant care. Liver Transpl 2017;23:96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sokol RJ, Shepherd RW, Superina R, et al. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology 2007;46:566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saulsbury FT. Identifying the child at risk for repeated abuse. Va Med 1983;110:483–7. [PubMed] [Google Scholar]
- 9.Davenport M, Caponcelli E, Livesey E, et al. Surgical outcome in biliary atresia: etiology affects the influence of age at surgery. Ann Surg 2008;247:694–8. [DOI] [PubMed] [Google Scholar]
- 10.Serinet MO, Wildhaber BE, Broue P, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 2009;123:1280–6. [DOI] [PubMed] [Google Scholar]
- 11.Townsend MR, Jaber A, Abi Nader H, et al. Factors Associated with Timing and Adverse Outcomes in Patients with Biliary Atresia Undergoing Kasai Hepatoportoenterostomy J Pediatr 2018;199:237–42.e2. [DOI] [PubMed] [Google Scholar]
- 12.Boregowda U, Umapathy C, Halim N, et al. Update on the management of gastrointestinal varices. World J Gastrointest Pharmacol Ther 2019;10:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marshall DA, Benchimol EI, MacKenzie A, et al. Direct health-care costs for children diagnosed with genetic diseases are significantly higher than for children with other chronic diseases. Genet Med 2019;21:1049–57. [DOI] [PubMed] [Google Scholar]
- 14.Griva K, Davenport A, Newman SP. Health-related quality of life and long-term survival and graft failure in kidney transplantation: a 12-year follow-up study. Transplantation 2013;95:740–9. [DOI] [PubMed] [Google Scholar]
- 15.Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics 2011;128:e1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noorulla F, Dedon R, Maisels MJ. Association of Early Direct Bilirubin Levels and Biliary Atresia Among Neonates. JAMA Netw Open 2019;2:e1913321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wehrman A, Waisbourd-Zinman O, Wells RG. Recent advances in understanding biliary atresia. F1000Res. 2019;8:F1000 Faculty Rev-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mariotti V, Strazzabosco M, Fabris L, et al. Animal models of biliary injury and altered bile acid metabolism. Biochim Biophys Acta Mol Basis Dis 2018;1864:1254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kilgore A, Mack CL. Update on investigations pertaining to the pathogenesis of biliary atresia. Pediatr Surg Int 2017;33:1233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guttman OR, Roberts EA, Schreiber RA, et al. Biliary atresia with associated structural malformations in Canadian infants. Liver Int 2011;31:1485–93. [DOI] [PubMed] [Google Scholar]
- 21.Chandra RS. Biliary atresia and other structural anomalies in the congenital polysplenia syndrome. J Pediatr 1974;85:649–55. [DOI] [PubMed] [Google Scholar]
- 22.Rasool F, Mirza B. Polysplenia syndrome associated with situs inversus abdominus and type I jejunal atresia. APSP J Case Rep 2011;2:18. [PMC free article] [PubMed] [Google Scholar]
- 23.Makin E, Quaglia A, Kvist N, et al. Congenital biliary atresia: liver injury begins at birth. J Pediatr Surg 2009;44:630–3. [DOI] [PubMed] [Google Scholar]
- 24.Thomson J On Congenital Obliteration of the Bile-Ducts. Trans Edinb Obstet Soc 1892;17:17–49. [PMC free article] [PubMed] [Google Scholar]
- 25.Belmont JW, Mohapatra B, Towbin JA, et al. Molecular genetics of heterotaxy syndromes. Curr Opin Cardiol 2004;19:216–20. [DOI] [PubMed] [Google Scholar]
- 26.Mezina A, Karpen SJ. Genetic contributors and modifiers of biliary atresia. Dig Dis 2015;33:408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girard M, Panasyuk G. Genetics in biliary atresia. Curr Opin Gastroenterol 2019;35:73–81. [DOI] [PubMed] [Google Scholar]
- 28.Stoll C, Morali A, Leheup B, et al. Extrahepatic biliary atresia with laterality sequence anomalies. Genet Couns 2001;12:157–61. [PubMed] [Google Scholar]
- 29.Davit-Spraul A, Baussan C, Hermeziu B, et al. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr 2008;46:111–2. [DOI] [PubMed] [Google Scholar]
- 30.Cui S, Leyva-Vega M, Tsai EA, et al. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 2013;144:1107–15.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng H, Xia H, Deng S. Genetic basis of human left-right asymmetry disorders. Expert Rev Mol Med 2015;16:e19. [DOI] [PubMed] [Google Scholar]
- 32.Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol 2017;9:a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Carvalho E, Ivantes CA, Bezerra JA. Extrahepatic biliary atresia: current concepts and future directions. J Pediatr (Rio J) 2007;83:105–20. [DOI] [PubMed] [Google Scholar]
- 34.Chu AS, Russo PA, Wells RG. Cholangiocyte cilia are abnormal in syndromic and non-syndromic biliary atresia. Mod Pathol 2012;25:751–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teichberg S, Markowitz J, Silverberg M, et al. Abnormal cilia in a child with the polysplenia syndrome and extrahepatic biliary atresia. J Pediatr 1982;100:399–401. [DOI] [PubMed] [Google Scholar]
- 36.Gershoni-Baruch R, Gottfried E, Pery M, et al. Immotile cilia syndrome including polysplenia, situs inversus, and extrahepatic biliary atresia. Am J Med Genet 1989;33:390–3. [DOI] [PubMed] [Google Scholar]
- 37.Karjoo S, Hand NJ, Loarca L, et al. Extrahepatic cholangiocyte cilia are abnormal in biliary atresia. J Pediatr Gastroenterol Nutr 2013;57:96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berauer JP, Mezina AI, Okou DT, et al. Identification of Polycystic Kidney Disease 1 Like 1 Gene Variants in Children With Biliary Atresia Splenic Malformation Syndrome. Hepatology 2019;70:899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacquemin E, Cresteil D, Raynaud N, et al. CFCI gene mutation and biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr 2002;34:326–7. [DOI] [PubMed] [Google Scholar]
- 40.Gonzales E, Davit-Spraul A, Jacquemin E. A Novel CFC1 Mutation in a Family With Heterotaxy and Biliary Atresia Splenic Malformation Syndromes. J Pediatr Gastroenterol Nutr 2020;70:e24–5. [DOI] [PubMed] [Google Scholar]
- 41.Schön P, Tsuchiya K, Lenoir D, et al. Identification, genomic organization, chromosomal mapping and mutation analysis of the human INV gene, the ortholog of a murine gene implicated in left-right axis development and biliary atresia. Hum Genet 2002;110:157–65. [DOI] [PubMed] [Google Scholar]
- 42.Zhang DY, Sabla G, Shivakumar P, et al. Coordinate expression of regulatory genes differentiates embryonic and perinatal forms of biliary atresia. Hepatology 2004;39:954–62. [DOI] [PubMed] [Google Scholar]
- 43.Schwarz KB, Haber BH, Rosenthal P, et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology 2013;58:1724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ningappa M, Min J, Higgs BW, et al. Genome-wide association studies in biliary atresia. Wiley Interdiscip Rev Syst Biol Med 2015;7:267–73. [DOI] [PubMed] [Google Scholar]
- 45.Ningappa M, So J, Glessner J, et al. The Role of ARF6 in Biliary Atresia. PLoS One 2015;10:e0138381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riepenhoff-Talty M, Schaekel K, Clark HF, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res 1993;33:394–9. [DOI] [PubMed] [Google Scholar]
- 47.Petersen C, Biermanns D, Kuske M, et al. New aspects in a murine model for extrahepatic biliary atresia. J Pediatr Surg 1997;32:1190–5. [DOI] [PubMed] [Google Scholar]
- 48.Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. J Hepatol 1998;28:603–7. [DOI] [PubMed] [Google Scholar]
- 49.Mohanty SK, Donnelly B, Bondoc A, et al. Rotavirus replication in the cholangiocyte mediates the temporal dependence of murine biliary atresia. PLoS One 2013;8:e69069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bondoc AJ, Jafri MA, Donnelly B, et al. Prevention of the murine model of biliary atresia after live rotavirus vaccination of dams. J Pediatr Surg 2009;44:1479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bessho K, Bezerra JA. Biliary atresia: will blocking inflammation tame the disease? Annu Rev Med 2011;62:171–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schooler GR, Mavis A. Cystic biliary atresia: A distinct clinical entity that may mimic choledochal cyst. Radiol Case Rep 2018;13:415–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azuma T, Nakamura T, Nakahira M, et al. Pre-operative ultrasonographic diagnosis of biliary atresia--with reference to the presence or absence of the extrahepatic bile duct. Pediatr Surg Int 2003;19:475–7. [DOI] [PubMed] [Google Scholar]
- 54.Petersen C, Madadi-Sanjani O. Role of viruses in biliary atresia: news from mice and men. Innov Surg Sci 2018;3:101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leonhardt J, Kuebler JF, Turowski C, et al. Susceptibility to experimental biliary atresia linked to different hepatic gene expression profiles in two mouse strains. Hepatol Res 2010;40:196–203. [DOI] [PubMed] [Google Scholar]
- 56.Averbukh LD, Wu GY. Evidence for Viral Induction of Biliary Atresia: A Review. J Clin Transl Hepatol 2018;6:410–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zani A, Quaglia A, Hadzic N, et al. Cytomegalovirus-associated biliary atresia: An aetiological and prognostic subgroup. J Pediatr Surg 2015;50:1739–45. [DOI] [PubMed] [Google Scholar]
- 58.Parolini F, Hadzic N, Davenport M. Adjuvant therapy of cytomegalovirus IgM+ve associated biliary atresia: Prima facie evidence of effect. J Pediatr Surg 2019;54:1941–5. [DOI] [PubMed] [Google Scholar]
- 59.Xu Y, Yu J, Zhang R, et al. The perinatal infection of cytomegalovirus is an important etiology for biliary atresia in China. Clin Pediatr (Phila) 2012;51:109–13. [DOI] [PubMed] [Google Scholar]
- 60.Rauschenfels S, Krassmann M, Al-Masri AN, et al. Incidence of hepatotropic viruses in biliary atresia. Eur J Pediatr 2009;168:469–76. [DOI] [PubMed] [Google Scholar]
- 61.Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis 2007;27:233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Riepenhoff-Talty M, Gouvea V, Evans MJ, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infect Dis 1996;174:8–15. [DOI] [PubMed] [Google Scholar]
- 63.Bobo L, Ojeh C, Chiu D, et al. Lack of evidence for rotavirus by polymerase chain reaction/enzyme immunoassay of hepatobiliary samples from children with biliary atresia. Pediatr Res 1997;41:229–34. [DOI] [PubMed] [Google Scholar]
- 64.Mohanty SK, Donnelly B, Lobeck I, et al. The SRL peptide of rhesus rotavirus VP4 protein governs cholangiocyte infection and the murine model of biliary atresia. Hepatology 2017;65:1278–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ortiz-Perez A, Donnelly B, Temple H, et al. Innate Immunity and Pathogenesis of Biliary Atresia. Front Immunol 2020;11:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petersen C, Davenport M. Aetiology of biliary atresia: what is actually known? Orphanet J Rare Dis 2013;8:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Franklin G, Harari H, Ahsan S, et al. Residual Isocyanates in Medical Devices and Products: A Qualitative and Quantitative Assessment. Environ Health Insights 2016;10:175–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salazar-González RA, Zhang X, Doll MA, et al. Role of the human N-acetyltransferase 2 genetic polymorphism in metabolism and genotoxicity of 4, 4’-methylenedianiline. Arch Toxicol 2019;93:2237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krone CA, Klingner TD. Isocyanates, polyurethane and childhood asthma. Pediatr Allergy Immunol 2005;16:368–79. [DOI] [PubMed] [Google Scholar]
- 70.Boor BE, Jarnstrom H, Novoselac A, et al. Infant exposure to emissions of volatile organic compounds from crib mattresses. Environ Sci Technol 2014;48:3541–9. [DOI] [PubMed] [Google Scholar]
- 71.Colareta Ugarte U, Prazad P, Puppala BL, et al. Emission of volatile organic compounds from medical equipment inside neonatal incubators. J Perinatol 2014;34:624–8. [DOI] [PubMed] [Google Scholar]
- 72.Sugaya N, Sato Y, Takahashi M, et al. Analysis of Primary Aromatic Amines Derived from Azo Colorants in Textile Products and Determination of Their Source Colorant. Yakugaku Zasshi 2017;137:95–109. [DOI] [PubMed] [Google Scholar]
- 73.Krone CA, Klingner TD, Ely JT. Polyurethanes and childhood asthma. Med Sci Monit 2003;9:HY39–43. [PubMed] [Google Scholar]
- 74.Van Vliet ED, Reitano EM, Chhabra JS, et al. A review of alternatives to di (2-ethylhexyl) phthalate-containing medical devices in the neonatal intensive care unit. J Perinatol 2011;31:551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liang Y, Xu Y. Emission of phthalates and phthalate alternatives from vinyl flooring and crib mattress covers: the influence of temperature. Environ Sci Technol 2014;48:14228–37. [DOI] [PubMed] [Google Scholar]
- 76.Visscher MO, Adam R, Brink S, et al. Newborn infant skin: physiology, development, and care. Clin Dermatol 2015;33:271–80. [DOI] [PubMed] [Google Scholar]
- 77.Mutinati M, Pantaleo M, Roncetti M, et al. Oxidative stress in neonatology: a review. Reprod Domest Anim 2014;49:7–16. [DOI] [PubMed] [Google Scholar]
- 78.Hyland C, Laribi O. Review of take-home pesticide exposure pathway in children living in agricultural areas. Environ Res 2017;156:559–70. [DOI] [PubMed] [Google Scholar]
- 79.Ludwig J, Wiesner RH, Batts KP, et al. The acute vanishing bile duct syndrome (acute irreversible rejection) after orthotopic liver transplantation. Hepatology 1987;7:476–83. [DOI] [PubMed] [Google Scholar]
- 80.Desmet VJ. Vanishing bile duct syndrome in drug-induced liver disease. J Hepatol 1997;26 Suppl 1:31–5. [DOI] [PubMed] [Google Scholar]
- 81.Konikoff F, Tiomny E, Bujanover Y, et al. An endoscopic study of Campylobacter pylori in an Israeli population. Isr J Med Sci 1989;25:661–3. [PubMed] [Google Scholar]
- 82.Cho HJ, Jwa HJ, Kim KS, et al. Urosodeoxycholic Acid Therapy in a Child with Trimethoprim-Sulfamethoxazole-induced Vanishing Bile Duct Syndrome. Pediatr Gastroenterol Hepatol Nutr 2013;16:273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao Z, Bao L, Yu X, et al. Acute vanishing bile duct syndrome after therapy with cephalosporin, metronidazole, and clotrimazole: A case report. Medicine (Baltimore) 2017;96:e8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li L, Zheng S, Chen Y. Stevens-Johnson syndrome and acute vanishing bile duct syndrome after the use of amoxicillin and naproxen in a child. J Int Med Res 2019;47:4537–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reau NS, Jensen DM. Vanishing bile duct syndrome. Clin Liver Dis 2008;12:203–17, x. [DOI] [PubMed] [Google Scholar]
- 86.Olin A, Henckel E, Chen Y, et al. Stereotypic Immune System Development in Newborn Children. Cell 2018;174:1277–92.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu JC, Khodadadi H, Malik A, et al. Innate Immunity of Neonates and Infants. Front Immunol 2018;9:1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Au WW. Susceptibility of children to environmental toxic substances. Int J Hyg Environ Health 2002;205:501–3. [DOI] [PubMed] [Google Scholar]
- 89.Perlroth NH, Castelo Branco CW. Current knowledge of environmental exposure in children during the sensitive developmental periods. J Pediatr (Rio J) 2017;93:17–27. [DOI] [PubMed] [Google Scholar]
- 90.Bove KE, Thrasher AD, Anders R, et al. Inflammation, Active Fibroplasia, and End-stage Fibrosis in 172 Biliary Atresia Remnants Correlate Poorly With Age at Kasai Portoenterostomy, Visceral Heterotaxy, and Outcome. Am J Surg Pathol 2018;42:1625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Waisbourd-Zinman O, Koh H, Tsai S, et al. The toxin biliatresone causes mouse extrahepatic cholangiocyte damage and fibrosis through decreased glutathione and SOX17. Hepatology 2016;64:880–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amarachintha S, Mourya R, Ayabe H, et al. Patient-Derived Cholangiocyte Organoids Point to Defects in Epithelial Development in Pathogenesis of Biliary Atresia. Hepatology 2019;70:50A. [Google Scholar]
- 93.Zhao X, Lorent K, Wilkins BJ, et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology 2016;64:894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fried S, Har-Zahav A, Wehrman A, et al. Bile Duct Injury in a Toxic Model of Biliary Atresia Is Mediated by Decreased Gsh and Genes in Wnt and Notch Signaling Pathways and Is Applicable to Other Cholangiopathies. Hepatology 2019;70:49A–50A. [Google Scholar]
- 95.Luo Z, Shivakumar P, Mourya R, et al. Gene Expression Signatures Associated With Survival Times of Pediatric Patients With Biliary Atresia Identify Potential Therapeutic Agents. Gastroenterology 2019;157:1138–52.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Navabi N, Shivakumar P. Discovery of Polyurethane Chemicals as New Etiopathogenic Agents in Biliary Atresia. Hepatology 2019;70:1495A–6A.31297851 [Google Scholar]
- 97.Moyer K, Kaimal V, Pacheco C, et al. Staging of biliary atresia at diagnosis by molecular profiling of the liver. Genome Med 2010;2:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Allen SR, Jafri M, Donnelly B, et al. Effect of rotavirus strain on the murine model of biliary atresia. J Virol 2007;81:1671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang W, Donnelly B, Bondoc A, et al. The rhesus rotavirus gene encoding VP4 is a major determinant in the pathogenesis of biliary atresia in newborn mice. J Virol 2011;85:9069–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alvarez F Is biliary atresia an immune mediated disease? J Hepatol 2013;59:648–50. [DOI] [PubMed] [Google Scholar]
- 101.Yu K, Li Q, Shi G, et al. Involvement of epithelial-mesenchymal transition in liver fibrosis. Saudi J Gastroenterol 2018;24:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lorent K, Gong W, Koo KA, et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci Transl Med 2015;7:286ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Loarca L, De Assuncao TM, Jalan-Sakrikar N, et al. Development and characterization of cholangioids from normal and diseased human cholangiocytes as an in vitro model to study primary sclerosing cholangitis. Lab Invest 2017;97:1385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Feldman AG, Sokol RJ. Neonatal Cholestasis. Neoreviews 2013;14: 10.1542/neo.14-2-e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Squires JE, Shivakumar P, Mourya R, et al. Natural killer cells promote long-term hepatobiliary inflammation in a low-dose rotavirus model of experimental biliary atresia. PLoS One 2015;10:e0127191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Keyzer-Dekker CM, Lind RC, Kuebler JF, et al. Liver fibrosis during the development of biliary atresia: Proof of principle in the murine model. J Pediatr Surg 2015;50:1304–9. [DOI] [PubMed] [Google Scholar]
- 107.Venter J, Francis H, Meng F, et al. Development and functional characterization of extrahepatic cholangiocyte lines from normal rats. Dig Liver Dis 2015;47:964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Machhua S, Kumar Y, Prakash Kanojia R, et al. An improved and easy protocol for primary epithelial cell culture from atretic tissue in biliary atresia. Tissue Cell 2019;56:83–9. [DOI] [PubMed] [Google Scholar]
- 109.Guan Y, Xu D, Garfin PM, et al. Human hepatic organoids for the analysis of human genetic diseases. JCI Insight 2017;2:e94954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tian L, Ye Z, Kafka K, et al. Biliary Atresia Relevant Human Induced Pluripotent Stem Cells Recapitulate Key Disease Features in a Dish. J Pediatr Gastroenterol Nutr 2019;68:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sampaziotis F, Justin AW, Tysoe OC, et al. Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids. Nat Med 2017;23:954–63. [DOI] [PubMed] [Google Scholar]
- 112.Shiota J, Zaki NHM, Merchant JL, et al. Generation of Organoids from Mouse Extrahepatic Bile Ducts. J Vis Exp 2019;(146): 10.3791/59544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Du Y, Khandekar G, Llewellyn J, et al. A Bile Duct-on-a-Chip With Organ-Level Functions. Hepatology 2020;71:1350–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Struecker B, Hillebrandt KH, Raschzok N, et al. Implantation of a Tissue-Engineered Neo-Bile Duct in Domestic Pigs. Eur Surg Res 2016;56:61–75. [DOI] [PubMed] [Google Scholar]
- 115.Wang JB, Liu C, Yeh YC, et al. A novel rat model simulating biliary atresia after a Kasai operation. J Invest Surg 2014;27:183–90. [DOI] [PubMed] [Google Scholar]
- 116.Garrido M, Escobar C, Zamora C, et al. Bile duct ligature in young rats: A revisited animal model for biliary atresia. Eur J Histochem 2017;61:2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chung-Davidson YW, Yeh CY, Bussy U, et al. Hsp90 and hepatobiliary transformation during sea lamprey metamorphosis. BMC Dev Biol 2015;15:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chung-Davidson YW, Yeh CY, Li W. The Sea Lamprey as an Etiological Model for Biliary Atresia. Biomed Res Int 2015;2015:832943. [DOI] [PMC free article] [PubMed] [Google Scholar]