

Abstract
Pulmonary arterial hypertension (PAH) in congenital cardiac shunts can be reversed by hemodynamic unloading (HU) through shunt closure. However, this reversibility potential is lost beyond a certain point in time. The reason why PAH becomes irreversible is unknown. In this study, we used MCT+shunt-induced PAH in rats to identify a dichotomous reversibility response to HU, similar to the human situation. We compared vascular profiles of reversible and irreversible PAH using RNA sequencing. Cumulatively, we report that loss of reversibility is associated with a switch from a proliferative to a senescent vascular phenotype and confirmed markers of senescence in human PAH-CHD tissue. In vitro, we showed that human pulmonary endothelial cells of patients with PAH are more vulnerable to senescence than controls in response to shear stress and confirmed that the senolytic ABT263 induces apoptosis in senescent, but not in normal, endothelial cells. To support the concept that vascular cell senescence is causal to the irreversible nature of end-stage PAH, we targeted senescence using ABT263 and induced reversal of the hemodynamic and structural changes associated with severe PAH refractory to HU. The factors that drive the transition from a reversible to irreversible pulmonary vascular phenotype could also explain the irreversible nature of other PAH etiologies and provide new leads for pharmacological reversal of end-stage PAH.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive vasculopathy of the pulmonary arteries that has no cure. The disease is characterized by a process of occlusive arteriolar remodeling that causes increased pulmonary vascular resistance culminating in symptoms of right ventricular (RV) failure (1). Contemporary vasodilator therapies may improve pulmonary hemodynamics and decelerate progression by alleviating RV afterload but cannot reverse the arteriopathy (2).
PAH associated with congenital heart disease (CHD) with a left-to-right shunt has a distinct reversible stage (3) during which shunt closure [hemodynamic unloading (HU)] restores normal pulmonary blood flow and reverses the vasculopathy. However, there is a point beyond which the potential benefit of HU is lost, and pathologic remodeling and the elevation in pulmonary vascular resistance persist or progress (3). These observations have led to the recommendation that CHD associated with PAH (PAH-CHD) should be repaired in early infancy. However, not all children with PAH-CHD are referred for a surgical intervention in a timely manner, and for many of those, even for some infants, it has been difficult to clearly distinguish those with irreversible from reversible PAH on the basis of hemodynamic assessment (4). The specific factors that drive the transition from reversible to irreversible PAH-CHD could also explain the irreversible nature of other PAH etiologies, potentially providing new leads for pharmacological reversal of end-stage PAH (3).
Multiple preclinical studies have thus far confirmed that HU can reverse vascular changes associated with PAH (5–7). Our study design was based on previous work, in which pulmonary hypertension (PH) was induced in rats by the pyrrolizidine alkaloid monocrotaline (MCT; 60 mg/kg) and HU was carried out 28 days after MCT injection by transplanting the left lung with MCT-PH into a normotensive recipient (5). The latter study showed that the vascular changes induced by MCT at 28 days, medial hypertrophy and endothelial cell (EC) proliferation, are reversed by HU after 14 and 28 days.
Here, we used an established PAH rat model that combines an MCT (60 mg/kg) injection with an aortocaval shunt, which doubles pulmonary blood flow (8, 9), leading to a neointimal-type of vascular remodeling that is similar to human PAH. HU was carried out at different time points during the progression of PAH (14, 21, and 28 days after MCT injection) by transplanting the left lung with PAH into a normotensive recipient without a shunt (Fig. 1A). In partial contrast with aforementioned preclinical research (5–7), but similar to clinical observations in PAH-CHD (3, 10), we found that HU could reverse the early, but not the late, stages of MCT+shunt PAH. This enabled us to characterize transcriptomic and vascular expression profiles that distinguish a reversible from an irreversible response to HU. RNA sequencing (RNA-seq) indicated that a pattern of cellular senescence was associated with HU irreversibility of PAH. By targeting senescence using the senolytic ABT263, we reversed MCT+shunt-induced PAH that was refractory to HU.
Fig. 1. Study design and hemodynamic and vascular morphologic evaluation.
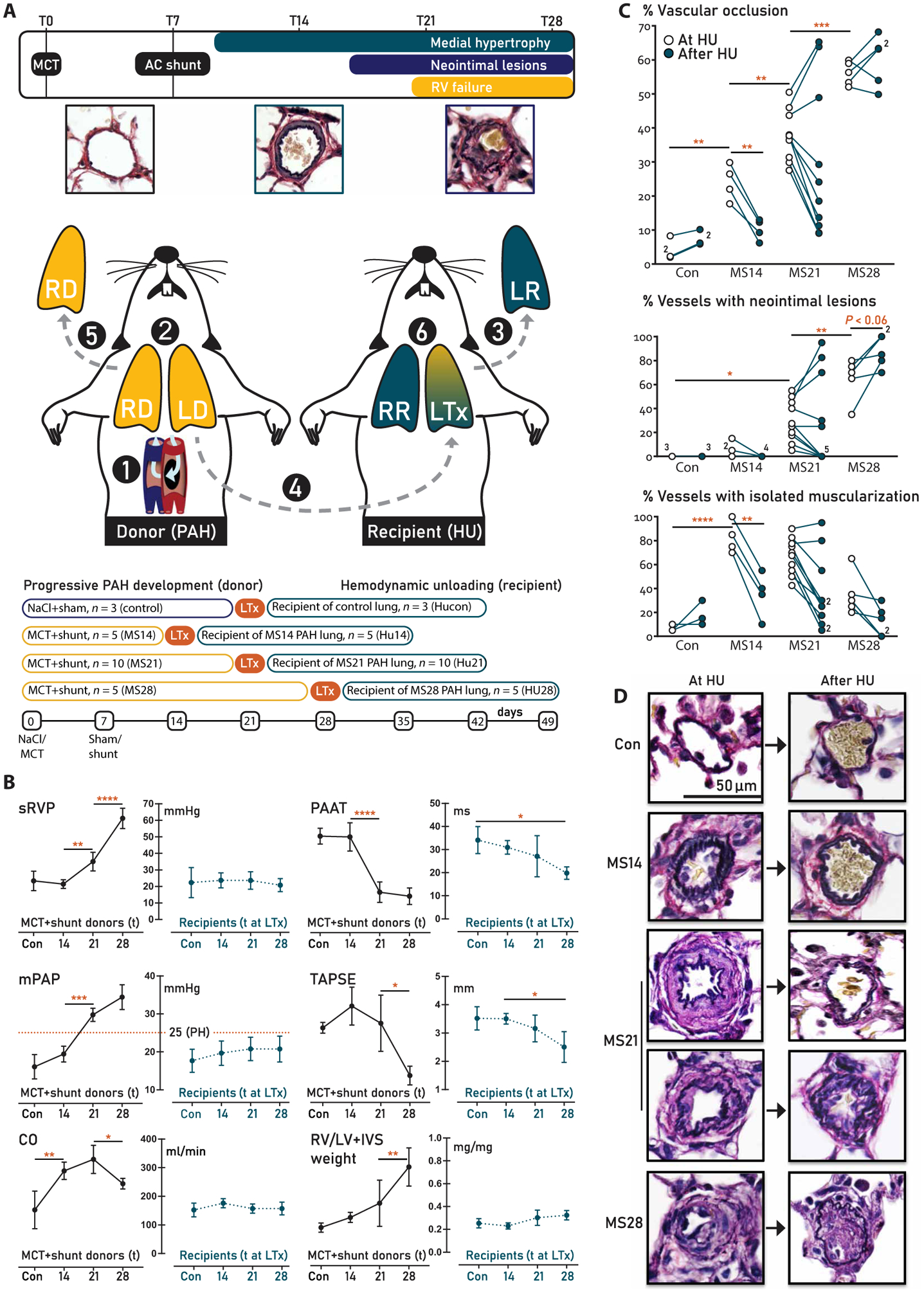
(A) Experimental design. Step 1: PAH was induced by an injection of monocrotaline [MCT; 60 mg/kg, day 0 (T0)] and creation of an aortocaval (AC) shunt, which increases cardiac output (CO) twofold [see (B)]. Step 2: Donor rats developed early, advanced, and end-stage PAH after 14, 21, and 28 days, respectively. Step 3: The left lung of a normotensive, healthy recipient rat was explanted (LR). Step 4: To mimic hemodynamic unloading (HU), the left donor lung (LD) with different PAH stages was transplanted into the recipient. Step 5: The right donor lung (RD) was explanted and used to assess the PAH stage at HU. The donor rat was sacrificed. Step 6: 21 days after transplantation, the recipient rat was sacrificed. The transplanted lung (LTx) was used to assess the effect of HU of lungs with the different stages of PAH. RV, right ventricular; RR, right lung of recipient rat. (B) Hemodynamic evaluation. The variables measured in the donor rat at the time point of HU are indicated in black; those in the recipient 21 days after HU are indicated in teal. N = 5 per PAH group, n = 3 for controls. sRVP, systolic RV pressure; mPAP, mean pulmonary artery pressure; TAPSE, tricuspid annular plain systolic excursion; PAAT, pulmonary artery acceleration time; RV/LV+IVS, RV to left ventricular plus septal weight ratio. The red dotted line in mPAP panels indicates pulmonary hypertension. (C) Quantification of vascular remodeling and (D) representative examples of Verhoeff staining. Left: Representative examples of the intra-acinar vessels at the different time points of PAH progression, showing progressive medial hypertrophy (MS14 and MS21) and the development of a neointima (MS21 and MS28). Right: Representative examples of the effect of HU after 21 days, showing normalization of the MS14 and some MS21 stages, and progressive remodeling, leading to acellular neointimal fibrosis of the other MS21 and all MS28 PAH stages. Data are reported as means ± SD. Statistics by one-way ANOVA with Bonferroni correction or Kruskal-Wallis with Dunn’s correction. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
RESULTS
MCT+shunt-induced PAH and HU in rats
According to a previously published protocol (9), MCT+shunt-induced PAH was created in 35 adult male syngeneic Lewis rats (200 g) by a subcutaneous injection of MCT (60 mg/kg, T0), followed by aortocaval shunt surgery 7 days later (8, 9). We used three time points of PAH progression previously defined (11) to establish the following groups: MCT+shunt day 14 (MS14) (n = 10), MS21 (n = 15), and MS28 (n = 10). Six rats were injected with saline and served as sham-operated controls. For full hemodynamic characterization of these time points, five PAH rats per group and three controls were evaluated by echocardiography (echo) and right heart catheterization before sacrifice (9). The other MS14, MS21, and MS28 rats (n = 5, n = 10, and n = 5, respectively) and controls (n = 3) served as donors for a unilateral orthotopic left lung transplantation (LTx) (12) into a syngeneic recipient rat without a shunt (Fig. 1A). Recipient rats are referred to as hemodynamically unloaded (HU) by their transplanted lung disease stage at LTx: HU14 (n = 5), HU21 (n = 10), HU28 (n = 5), and HUcon (n = 3).
First, an echo was performed in the donor rats. Then, the left donor lung with PAH was transplanted into a healthy recipient Lewis rat (250 g). The middle lobe of the right donor lung was used to assess baseline vascular morphology at HU. One recipient rat of the HU14 group was sacrificed 1 week after LTx because of surgical complications. This rat and its donor were excluded from all analyses. All other recipients fully recovered within 2 days after LTx. Twenty-one days after LTx, all recipients underwent hemodynamic evaluation by echo and heart catheterization, followed by sacrifice. The left transplanted lung was used to assess the effect of HU on vascular morphology.
HU reverses early but not late stages of MCT+shunt-induced PAH
MCT+shunt progressively increased systolic RV pressure (sRVP) and mean pulmonary artery pressure (mPAP). Pulmonary artery acceleration time (PAAT) was decreased at MS21 and MS28, also indicating pulmonary hypertension (Fig. 1B). At MS14, cardiac output (CO) and tricuspid annular plane systolic excursion (TAPSE) were increased compared to controls, indicating adequate ventricular adaptation to volume overload. CO decreased after MS21, which, combined with the increased sRVP and mPAP, reflects elevated pulmonary vascular resistance. TAPSE also decreased after MS21, reflecting RV dysfunction. The relative RV weight and liver wet-dry ratio progressively increased during PAH progression, indicating RV hypertrophy and decompensation. Severe dyspnea, fatigue, pleural edema, and ascites were observed in 60% of the rats at day 28. All recipient rats (without a shunt and with a left lung with different PAH stages) had normal sRVP, mPAP, and CO at sacrifice, 21 days after HU. PAAT and TAPSE were decreased and relative RV weight was increased at HU28 versus HU14, which might reflect residual pulmonary hypertension (Fig. 1B).
In control donors, most intra-acinar vessels were nonmuscularized. LTx did not affect the normal aspect of the vascular morphology. In MS14 donors, vascular occlusion was increased compared to control, most intra-acinar vessels (83 ± 13%) were muscularized, but neointimal lesions were scarce (5 ± 7%). HU at day 14 reversed vascular occlusion in the transplanted lung, which was associated with a decreased percentage of muscularized vessels (Fig. 1, C and D). In MS21 donors, vascular occlusion further increased compared to MS14. Vessels (67 ± 15%) showed isolated muscularization, and in 30 ± 17%, a neointima was observed. The effect of HU at day 21 was dichotomous. In 7 of 10 HU21 recipients, vascular occlusion and the percentage of vessels with muscularization or a neointima decreased, indicating reversal in this subgroup. Conversely, in the other 3 of 10 HU21 recipients, vascular occlusion and the percentage of vessels with a neointima increased, indicating progression of PAH. In MS28 donors, vascular occlusion and the percentage of vessels with a neointima further increased compared to MS21. The percentage of vessels with isolated muscularization decreased compared to MS21, which can be attributed to internal elastic laminae disintegration and lack of a separate, discernible media layer. HU at this stage did not reverse vascular remodeling: Rather, the percentage of vessels with a neointima further increased (Fig. 1, C and D). In conclusion, HU led to partial reversal or complete normalization in all MS14 and 7 of 10 MS21 donors. HU did not lead to reversal in all MS28 and 3 of 10 MS21 donors.
Vascular profiles associated with the reversibility of MCT+shunt-induced PAH
To study profiles associated with PAH reversibility, rats were reassigned on the basis of response to HU, leading to the following groups: normal controls (n = 5), reversible PAH at HU (right donor lung, n = 11), irreversible PAH at HU (right donor lung, n = 8), reversed PAH 21 days after HU (transplanted lung, n = 11), and nonreversed/progressed PAH after HU (transplanted lung, n = 8). Next-generation RNA-seq was performed on all donor right lung tissue. One irreversible PAH sample was excluded after quality control. Principal components analysis (Fig. 2A) showed differential clustering of reversible versus irreversible PAH in component 1 and clustering of controls versus both PAH stages in component 2. At cutoffs of >1 log2 fold change (LogFC) and q < 0.05, a total of 751 genes were differentially expressed, of which 82 were increased, and 9 were decreased in reversible PAH only; 150 were increased and 298 were decreased in irreversible PAH only; and 181 were increased and 31 were decreased in both PAH stages compared to control (Fig. 2B). Pathways and biological processes associated with reversible PAH included those related to proliferation and DNA repair. Pathways and biological processes associated with irreversible PAH included those related to immunologic responses, tissue remodeling, aging, and cell survival-associated pathways such as Rap1, PI3k-Akt, and HIF1 signaling (Fig. 2C). Pathways related to inflammatory responses, chemokine signaling, AGE-RAGE, and p53 pathways were observed in reversible and irreversible stages of PAH.
Fig. 2. RNA-seq analysis and cellular deconvolution of rat lung tissue.
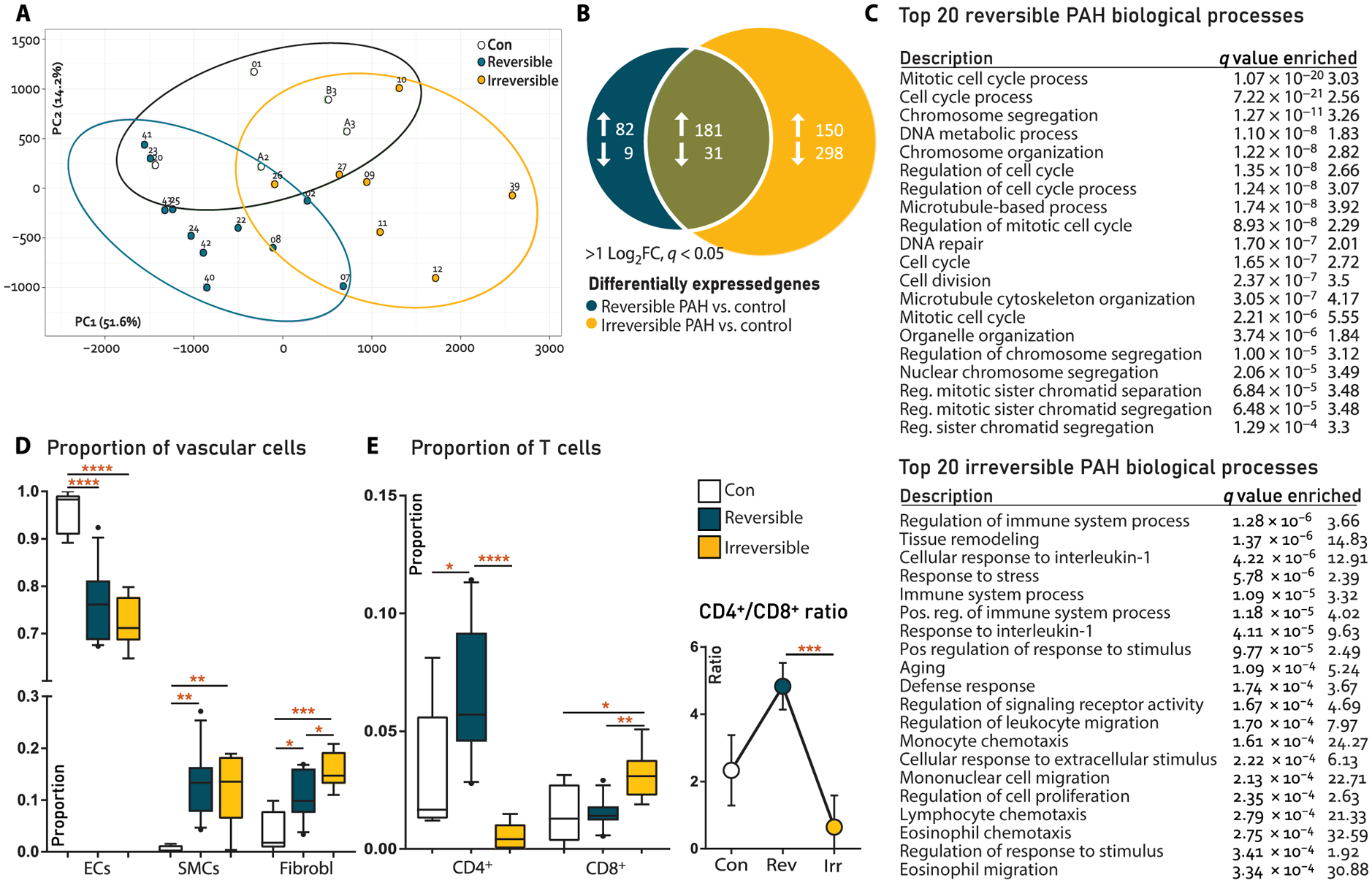
(A) Principal components analysis showing clustering of reversible versus irreversible PAH in component 1. (B) Venn diagram of differentially expressed genes (DEGs) at q < 0.01 and >2log2FC cutoffs. (C) List of 20 most enriched biological processes associated to reversible or irreversible PAH only (pathway analysis). (D) Deconvolution for vascular cells, analyzing endothelial cell (EC) versus smooth muscle cell (SMC) and fibroblast (Fibrobl) proportions. Con, N = 5; reversible, N = 11; irreversible, N = 7. (E) Deconvolution for immune cells, showing CD4+ T cells versus CD8+ T cells (left) and CD4+/CD8+ T cell ratios (right). Con, N = 5; reversible, N = 11; irreversible, N = 7. Statistical analyses (A to C) performed on raw reads using pathway analysis algorithm in R. Cellular deconvolution analyses (D and E) performed on raw reads using the support vector regression algorithm in R; also, see cellular deconvolution (Materials and Methods). Data are reported as means ± SD. Statistical differences in (D and E) were assessed by one-way ANOVA with Bonferroni correction. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Using cellular deconvolution, a method to assess the proportion of specific cells in mixed-cell tissue (13), we found a decrease in the EC proportion in reversible PAH versus control, which further decreased in irreversible PAH, indicating EC death. The smooth muscle cell (SMC) proportion was increased in both PAH stages versus control, indicating neomuscularization and medial hypertrophy. The proportion of fibroblasts was increased in reversible PAH and increased further in irreversible PAH, indicating progressive fibrosis (Fig. 2D). Deconvolution for immune cells showed an increased CD4+/CD8+ T cell ratio in reversible PAH, which inverted in irreversible PAH (Fig. 2E). CD4+/CD8+ inversion is associated with HIV infection, autoimmunity, and immunosenescence (14, 15). There were no differences in proportions of other immune cells.
Pathway analysis indicated that reversible PAH is characterized by increased proliferation and DNA repair. Cellular deconvolution additionally indicated increased endothelial apoptosis in reversible PAH. The differentially expressed genes (DEGs; q < 0.05; >1.5log2FC) associated to these processes (cell proliferation, GO:0008283; apoptotic processes, GO:0006915; DNA repair, GO:0006281) are visualized in Fig. 3 (A to C). The expression of BMPR2, a hallmark gene of PAH, was down-regulated upon transition toward irreversible PAH (Fig. 3D). We confirmed and quantified the roles of proliferation, apoptosis, and DNA damage by immunohistochemistry (Fig. 3, E and F).
Fig. 3. Vascular profiles associated to the reversibility of PAH in MCT+shunt rats.
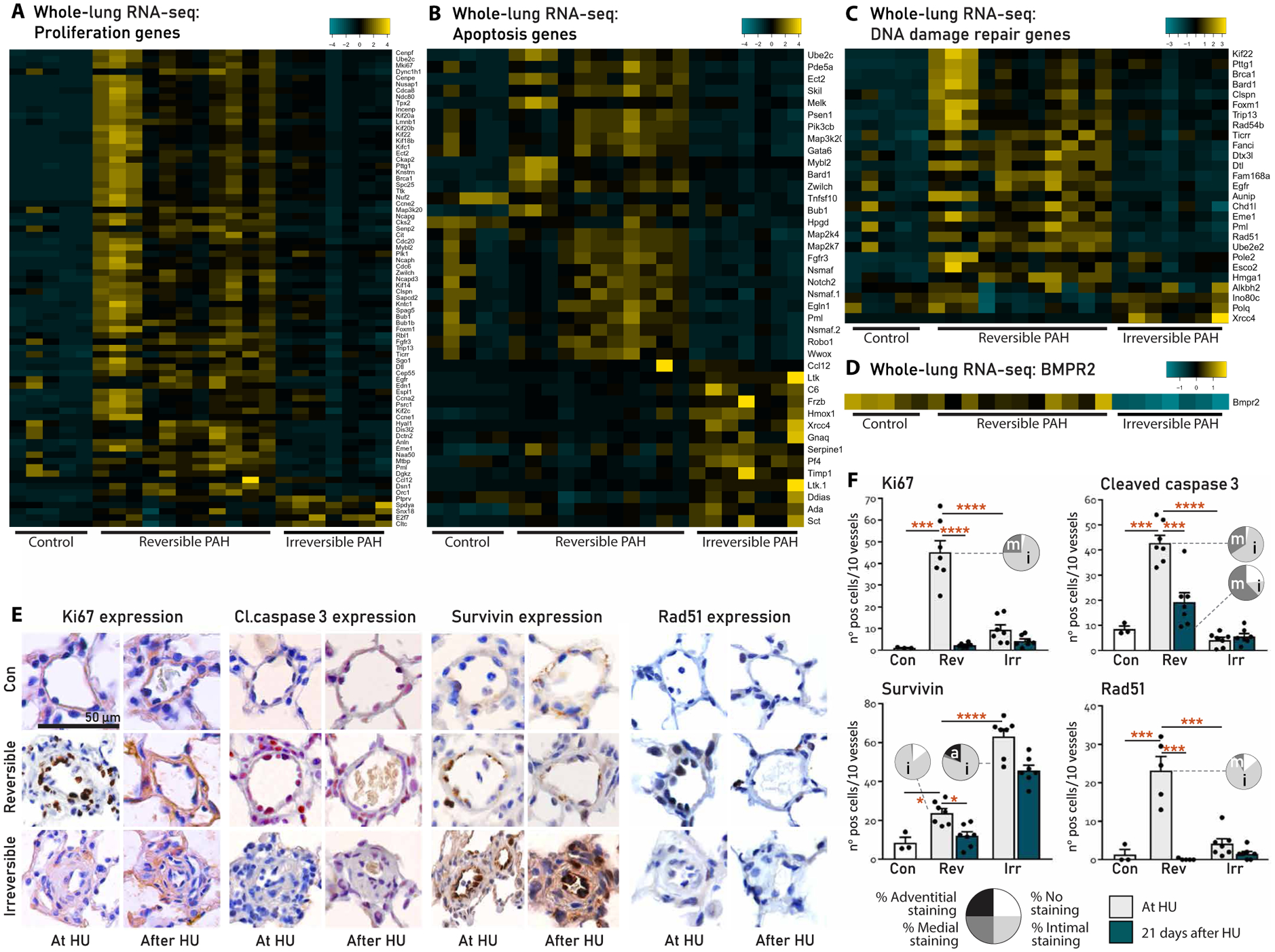
(A to D) Functional categorization of DEGs at q < 0.01 and >1log2FC cutoffs in lungs from controls and rats with reversible or irreversible PAH, made using the heatmap algorithm in R. (E) Representative immunohistochemistry of the intra-acinar vessels stained for Ki67, cleaved caspase 3, survivin, and Rad51 in lungs from controls and rats with reversible PAH and irreversible PAH at HU and 21 days after HU. (F) Quantification of vascular expression from (E). Black bars represent the expression at HU; green bars represent the expression 21 days after HU. Vascular localization of expression per group is represented in the pie charts [white, no expression; light gray, intima (i); dark gray, media (m); black, perivascular/adventitia (a)]. Con, n = 3; reversible, n = 7; irreversible, n = 7. Data are reported as means ± SD. Statistics by one-way ANOVA with Bonferroni correction or Kruskal-Wallis with Dunn’s correction. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
The expression of Ki67 (proliferation) and cleaved caspase 3 (apoptosis) was increased in the endothelium of reversible PAH compared to control and irreversible PAH, reflecting high EC turnover. Survivin, a protein associated with apoptosis resistance, was increased in irreversible versus reversible PAH. Survivin expression was also located in the endothelium. Rad51, the top DEG for DNA repair, was increased in reversible PAH compared to control and irreversible PAH. Nuclear Rad51 foci were present in the endothelium of reversible PAH and absent in irreversible PAH.
In reversible PAH, all proteins that were induced during PAH development were normalized by HU, except for cleaved caspase 3, which was decreased but remained higher than controls. Cleaved caspase 3 was located predominantly in the media (Fig. 3, E and F), possibly reflecting regression of medial muscularization. In irreversible PAH, HU did not affect the expression of the proteins that were assessed.
This transition from a hyperproliferative, proapoptotic phenotype to a growth-arrested, apoptosis-resistant, and proinflammatory milieu in the context of DNA damage suggested a role for cellular senescence in the irreversibility of PAH. DEGs associated with cellular senescence (GO:0090398) and the senescence-associated secretory phenotype (SASP) (16, 17) were up-regulated in irreversible PAH (Fig. 4, A and B). We confirmed this by immunohistochemistry for the senescence markers p16ink4A and p21cip1 and SASP markers matrix metalloproteinase 2 (MMP2) and interleukin-6 (IL6), which were all increased in irreversible compared to reversible PAH (Fig. 4, C and D). p21 was located in the ECs and adventitia. p16 was mainly located in the adventitia. MMP2 was located in the endothelium and the adventitia. IL6 was mainly located in the adventitia and showed diffuse staining in the media/neointima. In addition, we assessed the expression of tumor necrosis factor–α (TNFα), as both vascular remodeling in PAH and EC senescence are known to be driven by TNFα (18–20). TNFα was increased in the endothelium of reversible PAH compared to control. Expression further increased in irreversible PAH where it was located in the neointima and adventitia (Fig. 4, C and D).
Fig. 4. Vascular profiles associated to the irreversibility of PAH.
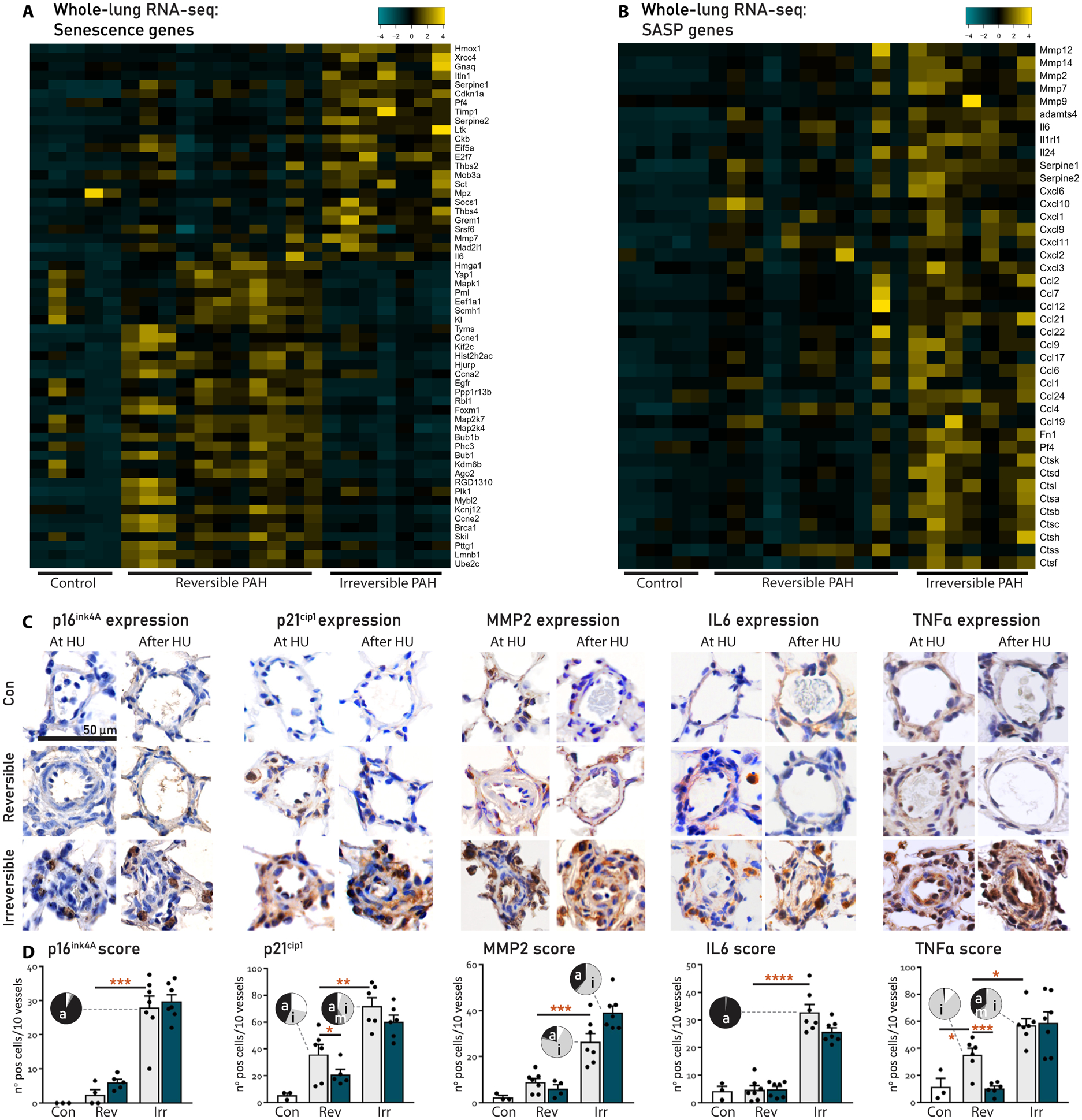
(A and B) Functional categorization of DEGs at q < 0.01 and >1log2FC cutoffs in lung tissue from controls and rats with reversible or irreversible PAH, made using the heatmap algorithm in R. SASP, senescence-associated secretory phenotype. (C) Representative immunohistochemistry of the intra-acinar vessels stained for expression of p16Ink4A, p21cip1, MMP2, IL6, and TNFα in lungs of controls and rats with reversible PAH and irreversible PAH at HU and 21 days after HU. (D) Quantification of vascular expression from (C). Black bars represent the expression at HU; green bars represent the expression 21 days after HU. Vascular localization of expression per group is represented in the pie charts [white, no expression; light gray, intima (i); dark gray, media (m); black, perivascular/adventitia (a)]. Con, n = 3; reversible, n = 7; irreversible, n = 7. Data are reported as means ± SD. Statistics by one-way ANOVA with Bonferroni correction or Kruskal-Wallis with Dunn’s correction. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
A similar senescent vascular profile in human irreversible PAH-CHD
The vascular profile of irreversible shunt-induced PAH in rats was corroborated in human lung explant tissue from patients with irreversible PAH-CHD versus controls (Fig. 5). Patient 1 is a 16-year-old female with repaired ventricular septal defect (VSD; postoperative PAH-CHD); patient 2 is a 30-year-old male with unrepaired atrial septal defect (ASD; Eisenmenger PAH-CHD). Ki67 and cleaved caspase 3 were not present in the vessel wall in controls nor in patients. Rad51 was not observed in controls, but Rad51 foci were present in the luminal cells of mild lesions in PAH (Fig. 5) and in more severe lesions. Survivin was largely absent in controls but stained intensely in the luminal cells of severe lesions in PAH. In patients with PAH, p21 was expressed in plexiform lesions lining the plexus channels. Whereas p16 localized predominantly in the luminal cells and in the adventitia. Vascular p21 and p16 expression was not observed in controls.
Fig. 5. A senescent vascular profile in human irreversible PAH-CHD.
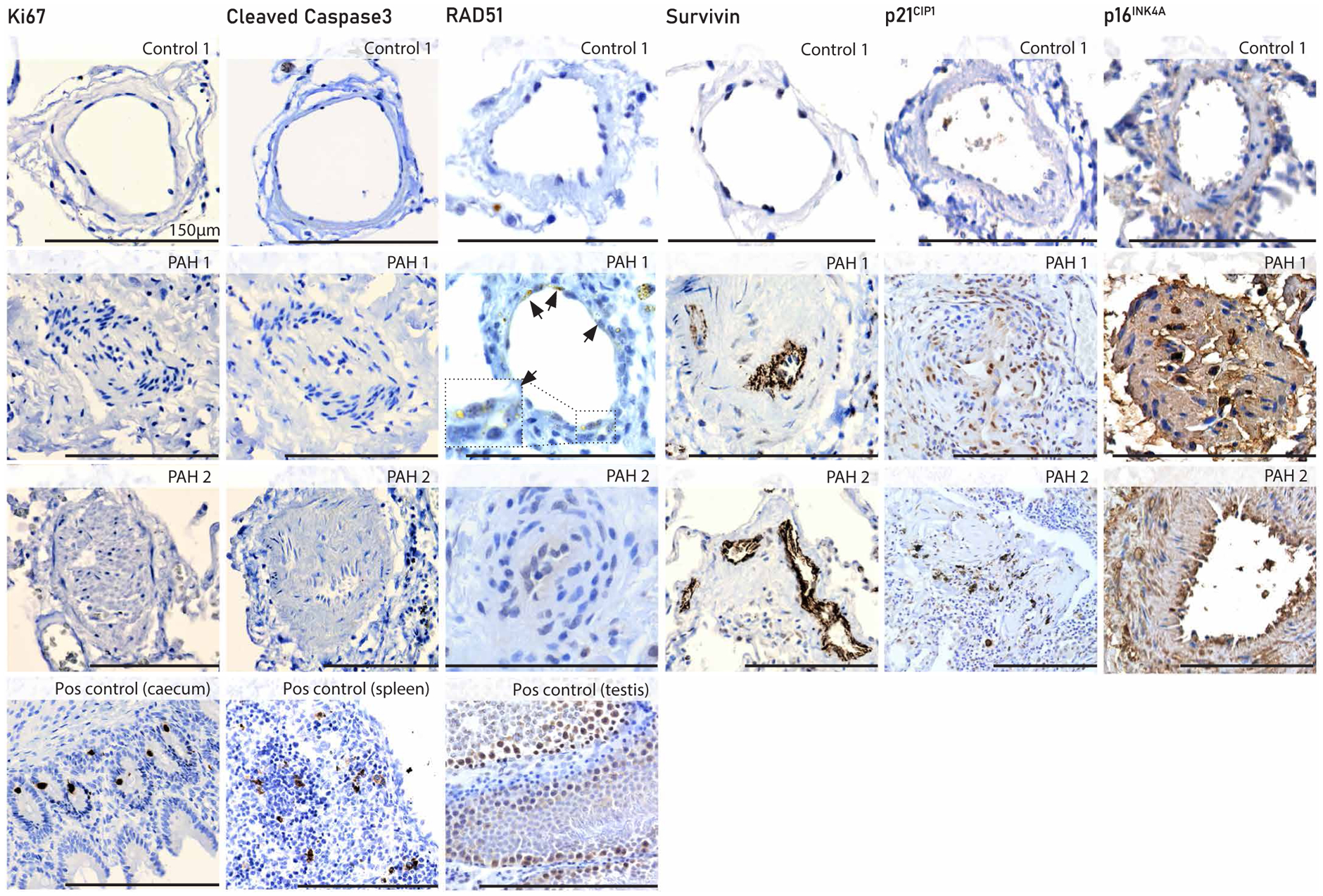
Immunohistochemistry of Ki67, cleaved caspase 3, Rad51, survivin, p21CIP1, and p16INK4A expression in human pulmonary arteries (<150 μm) in explant tissue of a patient without PAH (control 1), a pediatric patient with end-stage PAH-CHD (PAH 1), and an adult patient with end-stage PAH-CHD (PAH 2). Positive (Pos) controls (human tissue) were added when no vascular expression was expected. Arrowheads in Rad51 denote positive Rad51 nuclear foci. The inset is shown at higher magnification.
Shear stress induces DNA damage in PAH ECs and a phenotypic switch in SMCs
To investigate the effect of disturbed blood flow on the possible development of senescence in PAH, we cultured pulmonary microvascular ECs (pMVECs), isolated from patients with idiopathic PAH (n = 5) or control patients (n = 5), under static conditions or low shear stress (2.5 dyne/cm2) and high shear stress (HSS; 15 dyne/cm2). In static conditions, BMPR2 expression was twice as low in isolated PAH pMVECs compared to control cells (Fig. 6A). Staining for γH2A.X, a marker for DNA double-strand breaks, showed increased pMVEC DNA damage under HSS in control and even more in PAH cells compared to static conditions (Fig. 6B). In PAH pMVECs, HSS increased p16 and PAI1 mRNA. These markers were largely unaffected by shear stress in control cells (Fig. 6C), whereas KLF2, a positive control for shear stress, was increased by shear stress in both PAH and control pMVECs. These data indicated that disturbed blood flow can induce DNA damage and p16 expression, possibly leading to senescence, and that PAH pMVECs are more vulnerable to this than control cells.
Fig. 6. Prosenescent effects of shear stress on human pulmonary microvascular ECs.
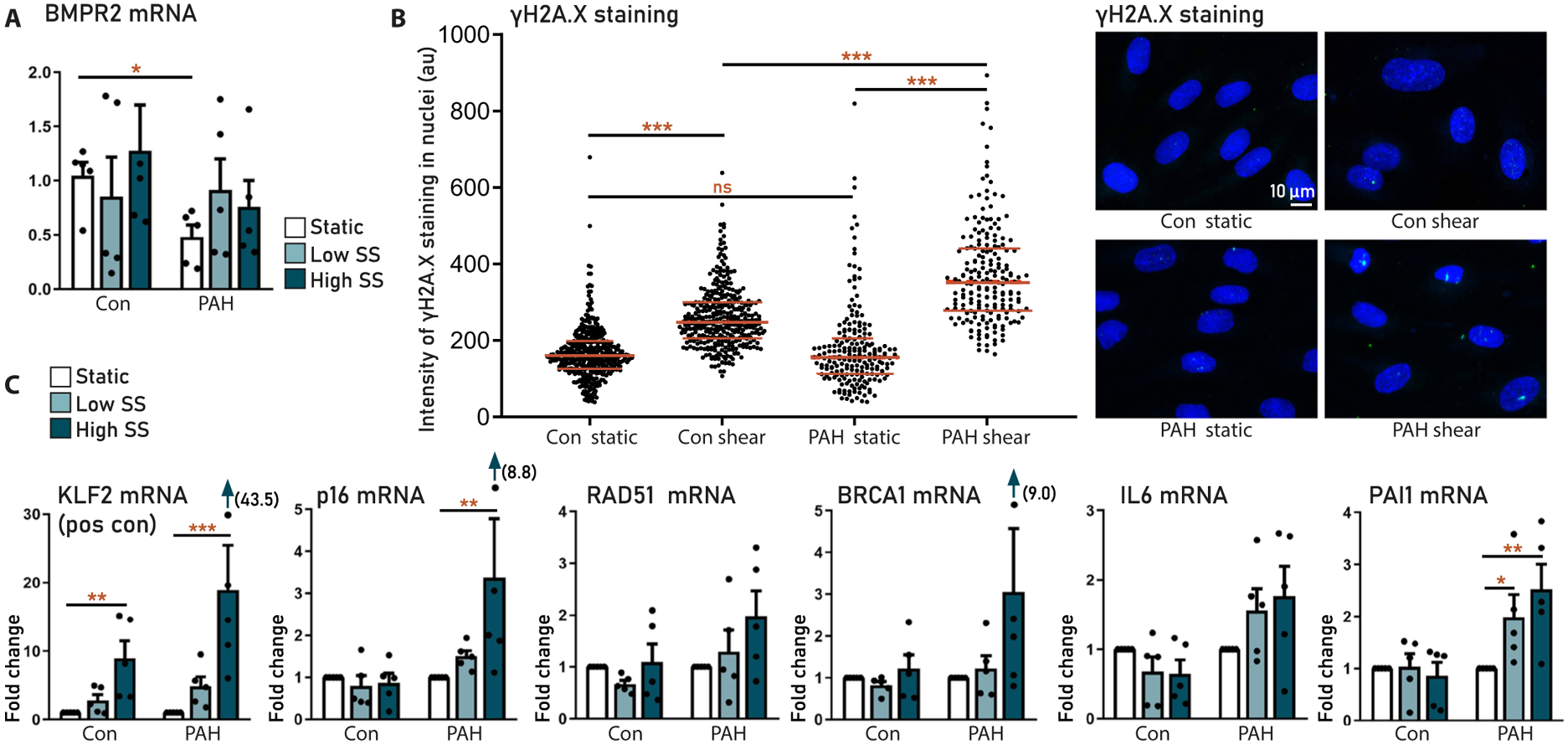
Pulmonary microvascular ECs (pMVECs) isolated from patients with idiopathic PAH (PAH, n = 5) and controls (con, n = 5). (A) BMPR2 expression in control and PAH MVECs in static and low shear stress (2.5 dyne/cm2) and HSS (15 dyne/cm2) conditions. (B) Quantification (left) and representative images (right) of DNA damage (γH2A.X staining) in control and PAH MVECs cultured under static and shear stress conditions. (C) Effect of shear stress on the expression of senescence and DNA damage markers in control and PAH MVECs. Data are reported as means ± SD. Statistics by two-sided t test or Kruskal-Wallis test. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We further aimed to assess the possible effect of the SASP of senescent pMVECs on SMCs. To this end, we first induced senescence in pMVECs from controls (n = 3) and patients with PAH (n = 3) by (i) 10-gray (Gy) γ-irradiation and (ii) a 10-day stimulation with 10 μM TNFα (19). Sham-irradiated, unstimulated cells served as a control. After 4 and 11 days, respectively, irradiation and TNFα both increased p16, p21, and IL6 mRNA and caused the cells to become multinucleated, flat, and enlarged, indicating senescence (Fig. 7A). Cell medium preconditioned by senescent versus nonsenescent pMVECs was then used to culture naïve pulmonary vascular SMCs. We observed that the conditioned medium from senescent pMVECs increased proliferation of SMCs (Fig. 7B). IL6 mRNA was also increased in SMCs cultured with SASP medium, whereas CNN1, a marker of SMC contractile function, was decreased (Fig. 7C). These data indicated a role for the endothelial SASP in SMC function and vascular remodeling.
Fig. 7. Effects of the human MVEC SASP on SMCs and targeted apoptosis of senescent MVECs.
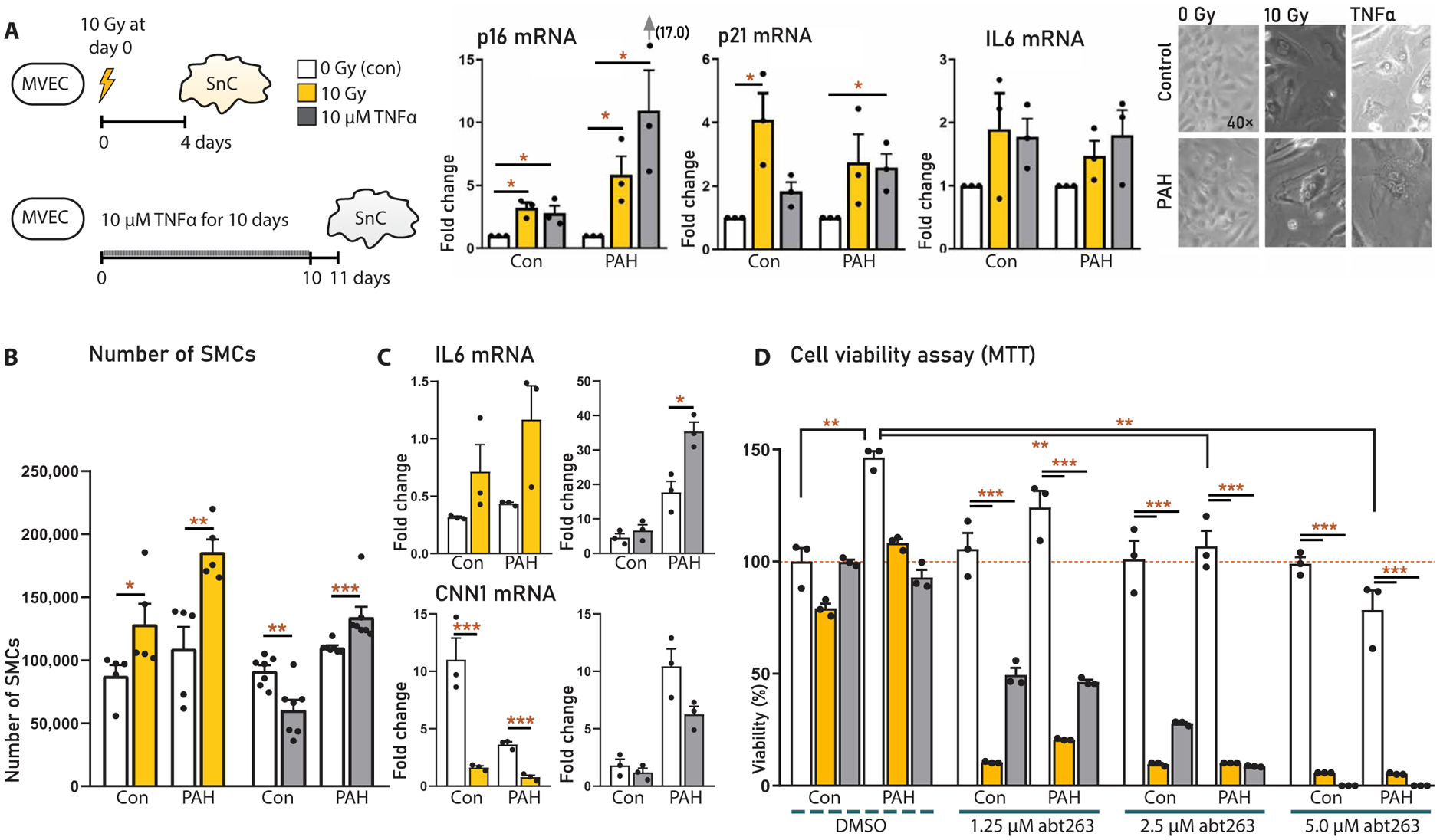
pMVECs isolated from idiopathic PAH and control patients. (A) Experimental schematic (left), mRNA expression analysis (middle), and microscopy (right) of human PAH and control pulmonary MVEC senescence, induced by irradiation or TNFα. Experiments were repeated three times. (B) Proliferation of naïve SMCs cultured with medium preconditioned by senescent or nonsenescent control or PAH MVECs. Experiments were repeated five to seven times. (C) IL6 and CNN1 expression in SMCs from (B) assessed by PCR. Experiments were repeated three times. (D) MTS assay of MVEC viability with or without ABT263. MVECs were isolated from controls and PAH, with or without irradiation or TNFα-induced senescence; experiments were repeated three times. Data are reported as means ± SD. Statistics by two-sided t test or Kruskal-Wallis test. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
As proof of the concept that cellular senescence is causally involved in the transition toward irreversible PAH, we investigated specific elimination of senescent cells using the senolytic agent ABT263. ABT263 treatment specifically decreased cell viability in the senescent control and PAH pMVECs, whereas viability remained intact in the sham-irradiated pMVECs (Fig. 7D). ABT263 treatment (2.5 μM) also normalized the hyperproliferative phenotype in sham-irradiated PAH pMVECs, which was 1.5× higher than controls at baseline. These data indicate that ABT263 both effectively eliminates senescent cells and allows for normal but not hyper-proliferation of pMVECs.
Reversal of shunt-induced PAH by targeted apoptosis of senescent cells
To confirm the selectivity of ABT263 for senescent cells in vitro and that cellular senescence is causally involved in the transition toward irreversible experimental end-stage MCT+shunt-PAH, we first performed a dose-response study in rat ECs. Senescence was induced in rat ECs using 10-Gy γ-irradiation as described above. Irradiation increased p21 and IL6 mRNA; decreased proliferative potential [laminB1 and 5-ethynyl-2′-deoxyuridine (EdU) staining]; and caused multinucleation, cellular flattening, and enlargement (Fig. 8A). p16 was not detected in these cells. Four days after irradiation, treatment with ABT263 (1.25, 2.5, and 5 μM for 24 hours) selectively decreased cell viability up to 20% in the senescent cells, whereas viability remained intact in control ECs (Fig. 8B).
Fig. 8. Targeted senolysis by ABT263 reverses established experimental PAH.
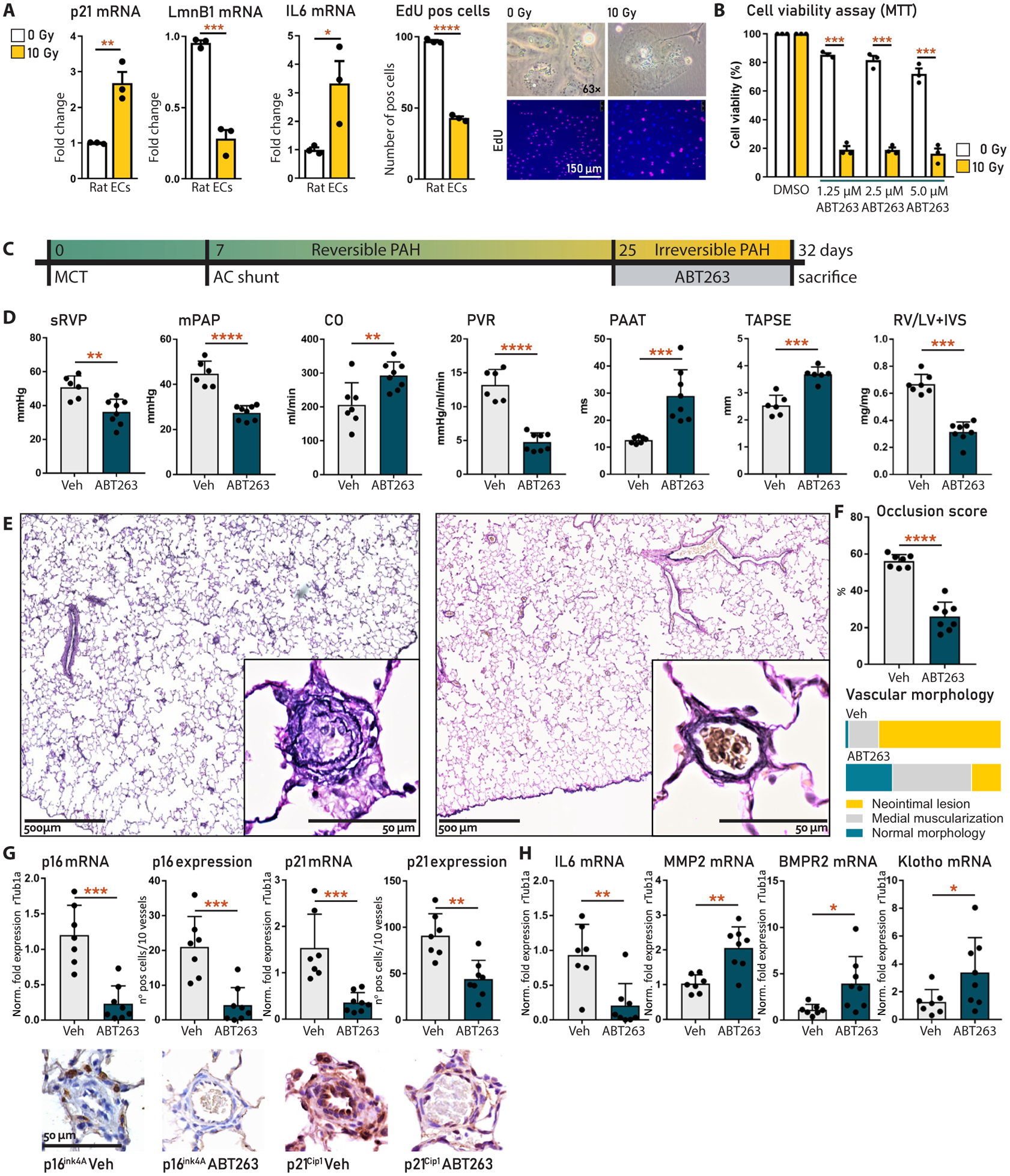
(A) Characterization of rat EC senescence induced by irradiation, measured by mRNA expression (left) and microscopic phenotype (right) including EdU. Experiments were repeated three times (B) MTS assay of rat EC viability with or without irradiation-induced senescence, treated with or without ABT263. Experiments were repeated three times (C) Experimental design. ABT263 (10 mg/kg) or Vehicle (5% DMSO and +4% Tween in PBS) was administered to MCT+shunt rats (n = 10 per group) daily via an intraperitoneal injection (10 mg/kg) from days 25 to 32. (D) Hemodynamic parameters in rats treated with vehicle (n = 7) or ABT263 (n = 8). (E) Verhoeff staining of remodeled vessels in ABT263-treated rat lung (right) versus vehicle-treated lung (left). (F) Semiquantification of (C). (G) Vascular p16 and p21 mRNA and protein expression in ABT263- and vehicle-treated rat lungs. Representative immunohistochemistry images are shown below. (H) Vascular IL6, MMP2, BMPR2, and Klotho mRNA in ABT263- and vehicle-treated rat lungs. Data are reported as means ± SD. Statistics by one-way ANOVA with Bonferroni correction or Kruskal-Wallis with Dunn’s correction. Relevant significant differences are indicated with a black bar and asterisk. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To investigate whether ABT263 can reverse MCT+shunt PAH in vivo, we induced MCT+shunt PAH in 20 Wistar rats and administered daily intraperitoneal injections with ABT263 (10 mg/kg; n = 10) or vehicle [n = 10; 5% dimethyl sulfoxide (DMSO) and +3.5% Tween 20] from days 25 to 32, an advanced-stage PAH that we characterized as HU irreversible (Fig. 8C). The shunts in these rats remained functional during treatment, driving disease progression during the treatment period. One rat died postoperatively because of a leaking shunt. In both groups, two rats died of severe right heart failure that occurred before or 1 day after treatment initiation at day 25 (indicating severe end-stage PAH) and were therefore excluded from all analyses. Dyspnea, pleural edema, and ascites occurred in five of seven vehicle-treated rats versus zero of eight in the ABT263 group. sRVP, mPAP, pulmonary vascular resistance, and relative RV weight were decreased, and CO, PAAT, and TAPSE were increased by ABT263 treatment (P < 0.05) (Fig. 8D), indicating disease regression. ABT263 significantly decreased luminal occlusion, reduced the percentage of vessels with a neointima, and increased those with a normal morphology (P < 0.05), indicating reversed remodeling or potential vascular regeneration (Fig. 8, E and F). ABT263 treatment decreased p16 and p21 mRNA, the number of p16-positive adventitial cells, and the number of p21- positive endothelial and adventitial cells, indicating clearance of senescent cells (Fig. 8G). The SASP marker IL6 was also decreased by ABT263. MMP2 mRNA, however, was increased by ABT263. In this context of regressing PAH, this may indicate a physiological role for MMP2, which is also implied in vascular development or neovascularization (21). We therefore also assessed BMPR2 and Klotho, both associated with vascular regeneration, which were also increased by ABT263 treatment (Fig. 8H). Together, these data show that ABT263 reduces the senescence burden in vivo, which is asso ciated with reversed vascular remodeling, evidence for a proregenerative milieu, and improved pulmonary hemodynamics.
DISCUSSION
Using experimental shunt-induced PAH, here, we showed that HU can reverse pulmonary vascular remodeling but not beyond a certain point of no return. The dichotomous response to HU enabled us to identify and compare vascular profiles of reversible and irreversible PAH using RNA-seq. We found that loss of reversibility is associated with a switch from a proliferative to a senescent vascular phenotype. To support the concept that vascular cell senescence is causal to the irreversible nature of end-stage PAH, we targeted senescence using ABT263 and induced reversal of the hemodynamic and structural changes associated with severe PAH refractory to HU. This class of drugs may represent a new line of treatment for advanced PAH.
The pulmonary vascular histomorphology associated with the (ir)reversibility of PAH-CHD has been described extensively in clinical studies (22). Several decades ago, it was shown in patients with open-shunt PAH-CHD that HU by banding of the main pulmonary artery can reverse structural changes like medial hypertrophy and intimal hyperplasia. Acellular neointimal fibrosis, degradation of the elastic laminae, and the formation of plexiform lesions were associated to disease progression (10). Several preclinical studies have since then confirmed that medial hypertrophy can be reversed by HU. In rats with PH induced by MCT and right, middle, and lower lobectomy, both medial hypertrophy and increased EC proliferation were normalized by HU through transplantation into a normotensive recipient at 28 days after MCT injection (5). Note that pulmonary blood flow in latter study is increased because about one-third of lung tissue is removed but not to the same magnitude as in our shunted rats, in which pulmonary blood flow is increased two- to threefold, explaining the difference in PAH severity between MCT+pneumonectomy versus MCT+shunt in rats. Pigs with an aortocaval shunt that was closed after 5 weeks show a similar pattern of reversed medial hypertrophy and normalized EC proliferation (7). These two studies performed HU in a stage of PH wherein neointimal lesions are not yet present. A recent study in the Sugen 5416+hypoxia rat model, however, showed that HU by left pulmonary artery banding (PAB) at 5 weeks can also reverse established neointimal lesions (6). Our study describes both a reversible and an irreversible stage of vascular remodeling in a PAH model that has features of neointimal vascular remodeling that do not change after HU and may progress, consistent with a severe rise in PAH. These observations suggest that the reversibility of PAH is governed not only by the presence but also by the molecular characteristics of neointimal lesions.
We described that the progression from reversible to irreversible PAH with HU coincides with three distinct changes in the pulmonary vasculature that are consistent with senescence: (i) a switch from a proliferative and/or regenerative state into growth arrest; (ii) a shift from proapoptosis to apoptosis resistance, as also previously described in human PAH-CHD (23); and (iii) a marked increase in proinflammatory cytokines accompanied by inversion of the CD4+/CD8+ T cell ratio in the lung. Furthermore, we observed an initial increase followed by a substantial reduction in genes associated with DNA damage repair. We confirmed senescence by demonstrating increased pulmonary vascular expression of p16ink4A and p21Cip1 both in rats and in human end-stage PAH-CHD. The vascular expression pattern of p16 in PAH-CHD is consistent with previous observations in idiopathic PAH (24). In vitro, we showed that pulmonary MVECs isolated from patients with PAH are more prone to develop senescence via increased sensitivity to DNA damage in response to HSS compared to control cells. We hypothesize that this effect may be due to decreased expression of BMPR2 in PAH MVECs, which was recently shown to compromise genomic integrity of ECs (25). We also found that the SASP of senescent MVECs stimulates proliferation and causes a phenotypical switch of SMCs in culture, providing insight into mechanisms of vascular remodeling beyond the EC. Moreover, we showed that low-dose treatment with the senolytic ABT263 selectively induces apoptosis in senescent MVECs in vitro and reversed advanced vascular remodeling and shunt-induced PAH in rats. Combined, these data serve as proof of the concept that cellular senescence contributes critically to the transition from reversible to irreversible PAH. After this transition has occurred, we hypothesize that the SASP may exacerbate the end-stage vascular sclerotic phenotype by promoting medial hypertrophy, suppressing vascular regeneration, maintaining inflammation, promoting fibrosis, and degrading elastin (fig. S1).
A central role for cellular senescence in the pathology of various degenerative vascular and pulmonary pathologies has become apparent in recent years. In atherosclerosis, senescent ECs and SMCs were shown to accumulate in plaques, which typically occur in areas where flow is disturbed (26). Senescent ECs in plaques produce less nitric oxide and prostacyclin and develop a proinflammatory SASP, leading to vascular dysfunction, constriction, and sclerosis (27). Clearance of senescent cells improved vascular function in atherosclerotic mice (28). Senescence was also described in degenerative pulmonary diseases such as idiopathic pulmonary fibrosis (IPF) (29) or chronic obstructive pulmonary disease (30), both of which are also associated with vascular remodeling and pulmonary hypertension (31). Accumulating evidence, including this study, indicates that DNA damage and insufficient DNA repair are early and central phenomena in PAH as well (32). Many triggers linked to the development of PAH are also genotoxic, such as treatment with dasatinib, chronic inflammation, or oxidative stress. Moreover, it was shown recently that reduced BMPR2 signaling, a critical feature of all PAH etiologies and a genetic cause, impairs normal DNA damage repair mediated by BRCA1 and BRCA2 (33) and Rad51 (25). DNA damage was also shown to cause BMPR2 loss itself, which suggests a vicious cycle that could account for DNA damage accumulation (33). These data are consistent with the decrease in BRCA1/2 and Rad51, as well as in BMPR2 that we observed upon transition from reversible to irreversible PAH in vivo, and the increased DNA damage in PAH MVECs with halved BMPR2 expression upon shear stress in vitro. Excessive DNA damage can eventually lead to either apoptotic cell death, reflected here in the decreased proportion of ECs in reversible PAH, or survival of cells with genomic abnormalities, causing dysfunction, hyperproliferation, or, lastly, senescence (17). Disturbed blood flow can further regulate the development of EC senescence via p53, a context-dependent mediator for cell fate, that decides between apoptosis or senescence (26). The presence of increased TNFα in this context appears to shift this balance toward senescence (26). The heightened expression of neointimal TNFα observed in this study indicates that this process may also play a role in the induction of vascular cell senescence in PAH.
In MCT+shunt rats, senescence did not appear to be limited to the ECs, as we also observed senescence markers in adventitial cells, likely fibroblasts or immune cells. The role of fibroblast senescence is well described, for example, in IPF (29) and systemic sclerosis (34), pathologies also both associated with PAH. Immunosenescence, induced by aging or oxidative stress, is characterized by chronic low-grade inflammation (35) and a reduced CD4/CD8 ratio (36). These hallmarks are also consistent with our observations in the transition toward irreversible PAH. In macrophages, p16 expression is associated with proliferation and differentiation toward the proinflammatory M1 macrophage (37), leading to increased IL6 secretion. Senolytic drugs can eliminate these p16-positive macrophages (38) and senescent vascular cells, indicating a possible dual mode of action to reverse vascular remodeling. Moreover, senolytics such as ABT263 or the forkhead box protein O4 peptide Foxo4-DRI have shown to promote tissue regeneration after clearance of senescent cells (39). The increased expression of BMPR2, Klotho, and MMP2 after ABT263 treatment in this study suggests that regeneration of lung vasculature occurs after clearance of senescent cells. This aspect could additionally benefit patients with PAH in whom vascular loss or the inability to recruit vessels during exercise contributes to the severity of the disease. Given a favorable safety profile, these agents may have a place in the future treatment of PAH-CHD and, perhaps, other forms of PAH as well. ABT263 has already been evaluated in multiple phase 2 clinical trials for different malignancies (small-cell lung cancer, NCT00445198). Senescence is associated with a distinct secretome that could be investigated as a peripheral blood biomarker to identify PAH that is refractory to HU and select patients that may respond to senolytics.
A limitation of this study is that the individual contribution of either MCT or the shunt to the development of irreversible PAH cannot be discerned. Single hit models such as MCT-only, shunt-only, or hypoxia-only models, however, are characterized only by medial hypertrophy-type vascular remodeling and lack the neointimal lesions reminiscent of PAH. All of these models are deemed reversible and, therefore, not suitable to investigate the (ir)reversibility of severe, neointimal PAH (40–42). It is likely that the combination of hits (MCT and shunt, Sugen 5416, and hypoxia) causes the occurrence of neointimal lesions. Likewise, in human PAH-CHD, it is also the combination of increased flow and increased pressure (as in a VSD) that leads to irreversible PAH within 2 years, whereas increased flow alone (as in an ASD) causes irreversible PAH in only 20% of patients after more than 30 years (3). A second limitation is that LTx, used here as a surrogate for HU, may also contribute to the reversibility of vascular lesions because it places a diseased lung into a healthy, noninflammatory environment. Still, it is apparent from the rat groups that were not reversible by LTx (which also showed the most inflammation), that this effect is not leading. The fact that MCT+pneumonectomy is reversible by LTx at day 28 (5), while MCT+shunt at day 28 is not, does make it plausible that volume loading/offloading is important, as the degree of pulmonary overflow (1.5-fold versus 2- to 3-fold increase) is the only variable that is changed between these setups. Human and animal studies that use methods of HU without transplantation, such as shunt closure or PAB, also support the fact that pulmonary vascular lesions can be reversed just by reducing pulmonary blood flow (6, 7, 10). Unfortunately, shunt closure is not technically feasible in this model nor is performing PAB surgery during advanced PAH (due to RV dysfunction) (6). Another question that remains uncertain is in which cell type senescence is first induced, or which senescent cell type contributes primarily to irreversibility in MCT+shunt-PAH. We chose to focus on the ECs because of their sensitivity to disturbed flow, but senescence in tissue usually involves multiple cell types as was also observed in the pulmonary vasculature of rats with irreversible PAH in this study.
Here, using HU, we identified a reversible and an irreversible stage of vascular remodeling in experimental shunt-induced PAH. We report that loss of PAH reversibility by HU is associated with a senescent vascular phenotype. Targeted senolysis reversed vascular remodeling and improved pulmonary hemodynamics in the end stage of experimental shunt-induced PAH. This serves as proof of the concept that senescence contributes critically to the transition of reversible to irreversible disease and indicates senolytic class drugs as a new line of treatment for end-stage PAH.
MATERIALS AND METHODS
Study design
The goal of this study was to identify histological and molecular characteristics related to the reversibility of severe PAH and to elucidate the mechanisms associated to the transition from reversible to irreversible disease. The study design was based on the clinical observation that PAH in congenital cardiac shunts can be reversed by HU through shunt closure, but this potential is lost beyond a certain point of no return. To reproduce this clinical phenomenon, we used an established shunt-associated rat model for severe PAH that combines an MCT injection (60 mg/kg) with an aortocaval shunt, which doubles pulmonary blood flow and leads to a neointimal type of vascular remodeling. HU was carried out at different time points during the progression of PAH (14, 21, and 28 days after MCT injection) by transplanting the left lung with PAH into a normotensive recipient without a shunt. Similar to clinical observations in PAH-CHD, we found that HU could reverse the early, but not the late, stages of MCT+shunt PAH. We could then characterize transcriptomic and vascular expression profiles that distinguish a reversible from an irreversible response to HU using RNA-seq and immunohistochemistry, which indicated that a pattern of cellular senescence was associated with HU irreversibility of PAH. In vitro, we further investigated the role of cellular senescence in vascular remodeling and evaluated the senolytic drug ABT263 in its selectivity to induce apoptosis in senescent versus healthy pMVECs. Last, to support the concept that vascular cell senescence is causal to the irreversible nature of end-stage PAH in vivo, we treated rats with advanced MCT+shunt PAH with ABT263 and induced reversal of the hemodynamic and structural changes associated with severe PAH refractory to HU.
All experimental protocols were approved by the Dutch National, and Institutional Animal Care And Use Committee of the University of Groningen, Netherlands and were conducted according to published guidelines for animal and translational research in PAH (43). Prospectively selected primary and secondary end points for all animal studies were vascular occlusion score and mPAP, respectively. Animal experiments were terminated at the defined time point or when severe dyspnea developed, as assessed twice a day by ABCDE criteria defined previously (11). Sample sizes were based on previous research in our laboratories, reporting occlusion scores around 60 ± 10% in end stage PAH and an effect size of ±20-point reduction in vascular occlusion (associated with hemodynamic improvement). Rats were randomly assigned to each experimental group and were housed randomly in group cages to ensure blinding for the researcher when conducting the in vivo analyses. Ex vivo, all samples were renumbered randomly for further blinding. The MS21/HU21 group (n = 5) was replicated once. All in vitro experiments were replicated three times.
MCT+shunt PAH in rats
MCT+shunt-associated PAH was induced in adults male rats (200 to 250 g) by a subcutaneous injection (60 mg/kg) of MCT at T0 (Sigma-Aldrich), followed by aortocaval shunt surgery 7 days later, described in detail previously (9). Control rats were injected with saline and sham-operated (NaCl+sham). Syngeneic Lewis rats (Envigo) were used for the transplantation experiments and Wistar rats (Charles River Laboratories) for the ABT263 experiment. MCT+shunt rats are referred to as MCT+shunt followed by the time point of PAH progression in days after MCT.
HU by single LTx
Lewis rats in which PAH was induced by MCT+shunt up to the predefined time points of PAH progression (MS14, MS21, and MS28) served as donors for a syngeneic unilateral orthotopic LTx (12) into a healthy recipient Lewis rat without a shunt (Fig. 1A). NaCl+sham donors were used as control. Recipient rats are referred to as hemodynamically unloaded (HU) by their disease stage at LTx. Recipient rats (250 to 300 g) were anesthetized using 3% isoflurane, intubated, and ventilated mechanically at 21% O2. The left lung was explanted via a lateral thoracotomy. Simultaneously, the donor rats were intubated and anesthetized using 3% isoflurane. After echo, a sternal thoracotomy was performed. The lungs were heparinized and flushed with cold saline via a cannula in the RV. The left donor lung was wrapped in foil with 4°C saline, explanted, and transplanted into the recipient. Cold ischemic time to reperfusion was 30 to 40 min. The lungs were reinflated using positive pressure ventilation. The donor rat was sacrificed after left lung explantation. The lower and middle lobes of the right donor lung were infused with 4% paraformaldehyde and paraffin-embedded for histomorphology to assess baseline disease stage at HU. The upper right lobe was snap-frozen for RNA extraction. The heart and liver of the donor were harvested for evaluation of PAH-associated sequelae. Twenty-one days after LTx, all recipients underwent hemodynamic evaluation by echo and right heart catheterization, followed by sacrifice. The left transplanted lung was infused with 4% paraformaldehyde and paraffin-embedded for histomorphology to assess the effect of HU.
Hemodynamic evaluation
Hemodynamic evaluation was performed using echo and closed-chest right heart catheterization, using a standardized protocol described previously (9, 11).
In vivo intervention in MCT+shunt PAH with ABT263
Twenty Wistar rats were acquired from Charles River Laboratories (France). PAH was induced by MCT+shunt. From day 25 to day 32, daily intraperitoneal injections were given with either vehicle [5% DMSO and +4% Tween in phosphate-buffered saline (PBS)] or ABT263 (10 mg/kg) in vehicle (n = 10 per group). Rats were sacrificed at day 32 or in pairs upon development of severe dyspnea. Hemodynamic and histopathologic evaluation were carried out as described above.
Human PAH lung tissue
Sections from lung explant tissue of patients with end-stage PAH-CHD and controls were acquired from the Biobank at the Pulmonary Vascular Research Institute at Stanford University, Palo Alto, CA, USA. Patient characteristics are listed in table S1. Immunohistochemistry was carried out for Ki67 (1:75), cleaved caspase 3 (1:150), Rad51 (1:100), survivin (1:50), p21 (1:100), and p16 (1:100). Species-specific G coupled to peroxidase were used as secondary or tertiary antibody and then stained by diaminobenzidine. Slides were scanned on ×40 magnification and used for qualitative analysis.
Real-time polymerase chain reaction
Total RNA was extracted from MVECs using NucleoSpin RNA II (Macherey-Nagel, Düren) and from rat lung tissue using TriPure (Roche), and cDNA was synthesized using iScript (Bio-Rad). Real-time quantitative polymerase chain reaction (qPCR) was performed with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and run in triplicate on a QuantStudio 7 Flex Real-time PCR System (Applied Biosystems). Analysis of qPCR results was done using the ΔΔCt method. Glyceraldehyde-3-phosphate dehydrogenase and rTUB1 were used as internal controls. Amplification specificity was controlled by postamplification melting curve analysis. In addition, qPCR products were electrophoresed on 2% agarose gels (w/v) to confirm the presence of a single band of expected size. Primer sequences are listed in table S2.
Statistical analyses
Statistical analyses were performed using SPSS. For all variables, normality was assessed using the Shapiro-Wilk test. α < 0.05 was considered significant. To assess differences between time points within the pre- or post-HU groups, we used one-way analysis of variance (ANOVA) with Bonferroni correction for parametric and Kruskal-Wallis for nonparametric variables. We used a two-sided T test or Mann-Whitney U test to assess the effects of HU on lung morphology or the effect of ABT263 versus vehicle. Transcriptomic data were analyzed using R. All data are reported as means ± SD. Individual data for animal experiments are provided in data file S1.
Supplementary Material
Acknowledgments:
We acknowledge M. Weij and A. Smit for performing the aortocaval shunt surgery and hemodynamic measurements, R. Szulcek and X. Pan of the VU Medical Center in Amsterdam for optimizing the human MVEC cell culture, E. Mavrogiannis (UMCG) for laboratory assistance in the ABT263 in vivo experiment, and T. Guimaraes Ferraz (LUMC) for work in the human MVEC ABT263 in vitro experiment.
Funding: This work was supported by the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organization for Health Research and Development, the Royal Netherlands Academy of Sciences (CVON-Phaedra 2012-08), and the Sebald Fund and the Ter Meulen Fund (Royal Netherlands Academy of Sciences). B.B. and R.M.F.B. are also supported by the Dutch Heart Foundation (NHS2013-T091, Cobra3). M.R. is supported by the Dunlevie Professorship. M.R. and P.K. are supported by the Integrative Omics grant and the NIH NHLBI RO.
Footnotes
Competing interests: The UMCG, which uses R.A.D.B. has received research grants and/or fees from AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche. R.A.D.B. received speaker fees from Abbott, AstraZeneca, Novartis, and Roche, all not relevant to this study. M. Demaria is a founder, advisor, and shareholder of Cleara Biotech, which is not relevant to this study. All other co-authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. RNA-seq data was submitted to GEO-NCBI (GEO accession number GSE149899).
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/12/554/eaaw4974/DC1
Materials and Methods
Fig. S1. Working model for the role of cellular senescence in vascular remodeling in PAH.
Fig. S2. Matrix of vascular cell type–specific genes.
Table S1. Characteristics of human PAH-CHD.
Table S2. Human and rat primer sequences for PCR.
Data file S1. Individual data tables for all animal experiments.
References (44–48)
REFERENCES AND NOTES
- 1.Rabinovitch M, Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest 122, 4306–4313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zijlstra WM, Douwes JM, Rosenzweig EB, Schokker S, Krishnan U, Roofthooft MT, Miller-Reed K, Hillege HL, Ivy DD, Berger RM, Survival differences in pediatric pulmonary arterial hypertension: Clues to a better understanding of outcome and optimal treatment strategies. J. Am. Coll. Cardiol 63, 2159–2169 (2014). [DOI] [PubMed] [Google Scholar]
- 3.van der Feen DE, Bartelds B, de Boer RA, Berger RM, Pulmonary arterial hypertension in congenital heart disease: Translational opportunities to study the reversibilityof pulmonary vascular disease. Eur. Heart J 38, 2034–2041 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Ploegstra M-J, Zijlstra WMH, Douwes JM, Hillege HL, Berger RM, Prognostic factors in pediatric pulmonary arterial hypertension: A systematic review and meta-analysis. Int. J. Cardiol 184, 198–207 (2015). [DOI] [PubMed] [Google Scholar]
- 5.O’Blenes SB, Fischer S, McIntyre B, Keshavjee S, Rabinovitch M, Hemodynamic unloading leads to regression of pulmonary vascular disease in rats. J. Thorac. Cardiovasc. Surg 121, 279–289 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Abe K, Shinoda M, Tanaka M, Kuwabara Y, Yoshida K, Hirooka Y, McMurtry IF, Oka M, Sunagawa K, Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc. Res 111, 16–25 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Mercier O, Sage E, de Perrot M, Tu L, Marcos E, Decante B, Baudet B, Herve P, Dartevelle P, Eddahibi S, Fadel E, Regression of flow-induced pulmonary arterial vasculopathy after flow correction in piglets. J. Thorac. Cardiovasc. Surg 137, 1538–1546 (2009). [DOI] [PubMed] [Google Scholar]
- 8.van Albada ME, Schoemaker RG, Kemna MS, Cromme-Dijkhuis AH, van Veghel R, Berger RM, The role of increased pulmonary blood flow in pulmonary arterial hypertension. Eur. Respir. J 26, 487–493 (2005). [DOI] [PubMed] [Google Scholar]
- 9.van der Feen DE, Weij M, Smit-van Oosten A, Jorna LM, Hagdorn QA, Bartelds B, Berger RM, Shunt surgery, right heart catheterization, and vascular morphometry in a rat model for flow-induced pulmonary arterial hypertension. J. Vis. Exp 2017, 55065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagenvoort CA, Wagenvoort N, Draulans-Noë Y, Reversibility of plexogenic pulmonary arteriopathy following banding of the pulmonary artery. J. Thorac. Cardiovasc. Surg 87, 876–886 (1984). [PubMed] [Google Scholar]
- 11.Dickinson MG, Kowalski PS, Bartelds B, Borgdorff MA, van der Feen D, Sietsma H, Molema G, Kamps JAM, Berger RM, A critical role for Egr-1 during vascular remodelling in pulmonary arterial hypertension. Cardiovasc. Res 103, 573–584 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Bullard AG, Petersen A, Wildevuur CR, Prop J, Crapo JD, Lung transplantation in the rat. A morphometric analysis of the gas exchange region. Transplantation 52, 443–449 (1991). [DOI] [PubMed] [Google Scholar]
- 13.Vallania F, Tam A, Lofgren S, Schaffert S, Azad TD, Bongen E, Alsup M, Alonso M, Davis M, Engleman E, Khatri P, Leveraging heterogeneity across multiple data sets increases accuracy of cell-mixture deconvolution and reduces biological and technical biases. bioRxiv 206466 [Preprint]. 2017. 10.1101/206466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller GC, Gottlieb MG, Correa BL, Filho IG, Moresco RN, Bauer ME, The inverted CD4:CD8 ratio is associated with gender-related changes in oxidative stress during aging. Cell. Immunol 296, 149–154 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Sainz T, Serrano-Villar S, Díaz L, Tome MIG, Gurbindo MD, de José MI, Mellado MJ, Ramos JT, Zamora J, Moreno S, Muñoz-Fernández MA, The CD4/CD8 ratio as a marker T-cell activation, senescence and activation/exhaustion in treated HIV-infected children and young adults. AIDS 27, 1513–1516 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Coppé J-P, Desprez P-Y, Krtolica A, Campisi J, The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Childs BG, Durik M, Baker DJ, van Deursen JM, Cellular senescence in agingand age-related disease: From mechanisms to therapy. Nat. Med 21, 1424–1435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hurst LA, Dunmore BJ, Long L, Crosby A, Al-Lamki R, Deighton J, Southwood M, Yang X, Nikolic MZ, Herrera B, Inman GJ, Bradley JR, Rana AA, Upton PD, Morrell NW, TNF α drives pulmonary arterial hypertension by suppressing the BMP type-II receptor and altering NOTCH signalling. Nat. Commun 8, 14079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, Stuppner H, Lipovac M, Uhrin P, Breuss JM, Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci. Rep 7, 39501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beyne-Rauzy O, Recher C, Dastugue N, Demur C, Pottier G, Laurent G, Sabatier L, Mansat-De Mas V, Tumor necrosis factor alpha induces senescence and chromosomal instability in human leukemic cells. Oncogene 23, 7507–7516 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Rundhaug JE, Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med 9, 267–285 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagenvoort CA, Open lung biopsies in congenital heart disease for evaluationof pulmonary vascular disease. Predictive value with regard to corrective operability. Histopathology 9, 417–436 (1985). [DOI] [PubMed] [Google Scholar]
- 23.Lévy M, Maurey C, Celermajer DS, Vouhé PR, Danel C, Bonnet D, Israël-Biet D, Impaired apoptosis of pulmonary endothelial cells is associated with intimal proliferation and irreversibility of pulmonary hypertension in congenital heart disease. J. Am. Coll. Cardiol 49, 803–810 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF, The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med 178, 558–564 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vattulainen-Collanus S, Southwood M, Yang XD, Moore S, Ghatpande P, Morrell NW, Lagna G, Hata A, Bone morphogenetic protein signaling is required for RAD51-mediated maintenance of genome integrity in vascular endothelial cells. Commun. Biol 1, 149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, White A, Biswas S, Khamis R, Chong CK, Cheung W-M, Sherwin SJ, Bennett MR, Gil J, Mason JC, Haskard DO, Evans PC, Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler. Thromb. Vasc. Biol 34, 985–995 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Minamino T, Komuro I, Vascular cell senescence: Contribution to atherosclerosis. Circ. Res 100, 15–26 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, Miller JD, Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK, Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun 8, 14532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, Rideau D, Parpaleix A, Huang J, Amsellem V, Vienney N, Validire P, Maitre B, Attwe A, Lukas C, Vindrieux D, Boczkowski J, Derumeaux G, Pende M, Bernard D, Meiners S, Adnot S, mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 3, e93203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Feen DE, Berger RM, Bartelds B, Converging paths of pulmonary arterial hypertension and cellular senescence. Am. J. Respir. Cell Mol. Biol 61, 1–20 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils-Bonnet S, Tremblay È, Couture C, Michelakis ED, Provencher S, Bonnet S, Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129, 786–797 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Li M, Vattulainen S, Aho J, Orcholski M, Rojas V, Yuan K, Helenius M, Taimen P, Myllykangas S, De Jesus Perez V, Koskenvuo JW, Alastalo T-P, Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol 50, 1118–1128 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Piera-Velazquez S, Jimenez SA, Role of cellular senescence and NOX4-mediated oxidative stress in systemic sclerosis pathogenesis. Curr. Rheumatol. Rep 17, 473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu HT, Park S, Shin E-C, Lee W-W, T cell senescence and cardiovascular diseases. Clin. Exp. Med 16, 257–263 (2016). [DOI] [PubMed] [Google Scholar]
- 36.van den Hoogen LL, Sims GP, van Roon JAG, Dorothea R, Fritsch-Stork E, Aging and systemic lupus erythematosus—Immunosenescence and beyond. Curr. Aging Sci 8, 158–177 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Frescas D, Hall BM, Strom E, Virtuoso LP, Gupta M, Gleiberman AS, Rydkina E, Balan V, Vujcic S, Chernova OB, Gudkov AV, Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle 16, 1526–1533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kakkola L, Denisova OV, Tynell J, Viiliäinen J, Ysenbaert T, Matos RC, Nagaraj A, Öhman T, Kuivanen S, Paavilainen H, Feng L, Yadav B, Julkunen I, Vapalahti O, Hukkanen V, Stenman J, Aittokallio T, Verschuren EW, Ojala PM, Nyman T, Saelens X, Dzeyk K, Kainov DE, Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 4, e742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IJcken WF, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, Hoeijmakers JHJ, Campisi J, de Keizer PLJ, Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruiter G, de Man FS, Schalij I, Sairras S, Grünberg K, Westerhof N, van der Laarse WJ, Vonk-Noordegraaf A, Reversibility of the monocrotaline pulmonary hypertension rat model. Eur. Respir. J 42, 553–556 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Rungatscher A, Linardi D, Milani E, Ucci G, Nicolato E, Merigo F, Salvetti B, Mazzucco A, Luciani GB, Faggian G, Chronic overcirculation-induced pulmonary arterial hypertension in aorto-caval shunt. Microvasc. Res 94, 73–79 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF, Animal modelsof pulmonary arterial hypertension: the hope for etiological discoveryand pharmacological cure. Am. J. Physiol. Cell. Mol. Physiol 297, L1013–L1032 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Provencher S, Archer SL, Ramirez FD, Hibbert B, Paulin R, Boucherat O, Lacasse Y, Bonnet S, Standards and methodological rigor in pulmonary arterial hypertension preclinical and translational research. Circ. Res 122, 1021–1032 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Zhu N, Welch CL, Wang J, Allen PM, Gonzaga-Jauregui C, Ma L, King AK, Krishnan U, Rosenzweig EB, Ivy DD, Austin ED, Hamid R, Pauciulo MW, Lutz KA, Nichols WC, Reid JG, Overton JD, Baras A, Dewey FE, Shen Y, Chung WK, Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 10, 56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mencke R, Hillebrands J-L; NIGRAM Consortium, The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res. Rev 35, 124–146 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF, Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. 19, 1178–1180 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Herranz N, Gil J, Mechanisms and functions of cellular senescence. J. Clin. Invest 128, 1238–1246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szulcek R, Happé CM, Rol N, Fontijn RD, Dickhoff C, Hartemink KJ, Grünberg K, Tu L, Timens W, Nossent GD, Paul MA, Leyen TA, Horrevoets AJ, de Man FS, Guignabert C, Yu PB, Vonk-Noordegraaf A, van Nieuw Amerongen GP, Bogaard HJ, Delayed microvascular shear adaptation in pulmonary arterial hypertension. roleof platelet endothelial cell adhesion molecule-1 cleavage. Am. J. Respir. Crit. Care Med 193, 1410–1420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.