

Abstract
Crizotinib and alectinib are anaplastic lymphoma kinase (ALK)‐inhibitors indicated for the treatment of ALK‐positive metastatic non‐small cell lung cancer (NSCLC). At the currently used fixed doses, interindividual variability in exposure is high. The aim of this study was to investigate whether minimum plasma concentrations (Cmin) of crizotinib and alectinib are related to efficacy and toxicity. An observational study was performed, in which ALK‐positive NSCLC patients who were treated with crizotinib and alectinib and from whom pharmacokinetic samples were collected in routine care, were included in the study. Exposure–response analyses were explored using previously proposed Cmin thresholds of 235 ng/mL for crizotinib and 435 ng/mL for alectinib. Forty‐eight crizotinib and 52 alectinib patients were included. For crizotinib, median progression‐free survival (mPFS) was 5.7 vs. 17.4 months for patients with Cmin < 235 ng/mL (48%) and ≥ 235 ng/mL, respectively (P = 0.08). In multivariable analysis, Cmin < 235 ng/mL resulted in a hazard ratio (HR) of 1.79 (95% confidence interval (CI), 0.90–3.59, P = 0.100). In a pooled analysis of all crizotinib patients (not only ALK‐positive, n = 79), the HR was 2.15 (95% CI, 1.21–3.84, P = 0.009). For alectinib, mPFS was 12.6 months vs. not estimable (95% CI, 19.8–not estimable) for patients with Cmin < 435 ng/mL (37%) and ≥ 435 ng/mL, respectively (P = 0.04). Multivariable analysis resulted in an HR of 4.29 (95% CI, 1.33–13.90, P = 0.015). In conclusion, PFS of crizotinib and alectinib treated NSCLC patients is prolonged in patients with Cmin ≥ 235 ng/mL and 435 ng/mL, respectively. Therefore, therapeutic drug monitoring should be part of routine clinical management for these agents.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Crizotinib and alectinib show a high interindividual variability in exposure, while registration studies suggest that exposure might be related to efficacy.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Are the previously proposed efficacy thresholds of minimum plasma concentration (Cmin) ≥ 235 ng/mL for crizotinib and Cmin ≥ 435 ng/mL for alectinib associated with better treatment outcomes in daily clinical practice?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Our results show that patients with a median Cmin above 235 ng/mL and 435 ng/mL for crizotinib and alectinib, respectively, have a prolonged progression‐free survival. Furthermore, a substantial subset of 37–48% of patients does not reach these thresholds.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY AND TRANSLATIONAL SCIENCE?
☑ This subgroup of patients with low exposure and manageable toxicity might benefit from a higher dose of crizotinib and alectinib. Therefore, therapeutic drug monitoring should be part of routine clinical management for these drugs.
Anaplastic lymphoma kinase (ALK) rearrangements are identified as oncogenic drivers in 3–7% of non‐small cell lung cancer (NSCLC) patients. 1 , 2 , 3 Over the past decade, several ALK tyrosine kinase inhibitors have become available and have resulted in significant improvements in overall survival, which is now extended to a median of close to 4 years in the metastatic setting. 4 Crizotinib and alectinib are first and second generation ALK inhibitors, respectively, with alectinib being inherently more effective. 5
Pharmacokinetic exposure to crizotinib and alectinib may be related to toxicity and efficacy. Currently, both crizotinib and alectinib are administered as oral fixed doses of 250 mg twice daily (b.i.d.) and 600 mg b.i.d., respectively, exhibiting a high interindividual variability in pharmacokinetic exposure of 40–45%. 6 , 7 Nevertheless, for both crizotinib and alectinib, no relation between exposure and toxicity could be established based on the currently available data. 6 , 7 However, in the registration study of crizotinib (n = 114) it was shown that patients with a minimum plasma concentration (Cmin) in the lowest quartile (i.e., < 235 ng/mL) had a significantly lower objective response rate and shorter progression‐free survival (PFS) as compared with the upper three quartiles. 6 Similarly, it has been shown that patients treated with alectinib (n = 46) in the lowest tertile of Cmin (i.e., < 435 ng/mL) had less reduction in tumor size compared with the upper two tertiles. 7
Therefore, personalized dosing based on measured drug levels (i.e., therapeutic drug monitoring) may be more rational, as a subgroup of patients might benefit from a higher dose of both crizotinib and alectinib.
However, before deciding whether individualized dosing might be appropriate, exposure–efficacy relationships need to be confirmed in an independent patient cohort. Furthermore, clinical trial populations differ from real‐life patients in several aspects. 8 Therefore, exposure–efficacy analyses should preferably be performed in real‐life patients, instead of in highly selected patients in clinical trials. For this reason, the aim of the observational study reported here was to investigate whether pharmacokinetic exposure to crizotinib and alectinib is related to efficacy and toxicity in a real‐life patient cohort.
METHODS
Patient population and data collection
A retrospective observational cohort study was performed at The Netherlands Cancer Institute, Amsterdam, the Netherlands. Consecutive patients with ALK‐positive NSCLC who were treated with crizotinib or alectinib were included. Pharmacokinetic (PK) samples of these patients were collected as part of routine care. An additional pooled analysis with all patients treated with crizotinib was performed (i.e., ALK‐positive, c‐ros oncogene 1 (ROS1)‐positive and mesenchymal epithelial transition growth factor (cMET) dysregulation). Of these patients, demographic data, prior lines of treatment, crizotinib and alectinib dose, treatment duration, reason for discontinuation, clinically relevant toxicities, and progression‐free survival were retrospectively collected from medical records. Following initiation of treatment, imaging was performed twice every 6 weeks, thereafter every 12 weeks.
Pharmacokinetic exposure
Plasma samples were collected during routine follow‐up visits to the outpatient clinic. Date and time of last drug intake and plasma sampling were recorded in order to calculate the time after dose. Crizotinib and alectinib concentrations were quantified using validated liquid chromatography‐tandem mass spectrometry assays. 9 , 10 As samples were collected at random timepoints during the dosing interval, Cmin was calculated using the following algorithm: 11
in which Cmin is the calculated minimum plasma concentration, Cmeasured is the measured plasma concentration, dosing interval is the time between two consecutive administrations of the drug (i.e., 12 hours for crizotinib and alectinib), TAD is the time after dose (i.e., the time between last intake of the drug and collection of the PK sample), and t 1/2 is the elimination half‐life of the drug (i.e., 42 hours for crizotinib 6 and 32 hours for alectinib 7 ). As crizotinib and alectinib have a longer elimination half‐life than imatinib (i.e., 18 hours 12 ), this algorithm should perform at least similarly to imatinib.
Samples drawn before steady state was reached or more than one t 1/2 after the last dose were excluded from the analysis. The median of all available Cmin levels per patient was taken as a measure of pharmacokinetic exposure.
Exposure–response analyses
Exposure–efficacy analyses were performed using previously proposed thresholds of 235 ng/mL for crizotinib and 435 ng/mL for alectinib. 6 , 7 , 13 PFS of patients with a median Cmin above and below these thresholds was compared using univariable and multivariable Cox regression analyses. A two‐sided P value < 0.05 was considered statistically significant. Statistical analyses were performed using R version 3.3.6 (R Project, Vienna, Austria). 14
Exposure–toxicity analyses were performed by comparing median Cmin between patients with and without clinically relevant toxicities, which was defined as toxicities leading to treatment interruption, dose reduction, or treatment discontinuation.
Median follow‐up time was determined using the reverse Kaplan–Meier method. 15
Ethical regulations
The institutional review board authorized the study on November 15, 2018. The need for written informed consent was waived.
RESULTS
Patient characteristics
In total, 100 consecutive patients were included (48 treated with crizotinib and 52 treated with alectinib) between 2012 and 2019. Baseline characteristics of these patients are provided in Tables 1 and 2 for crizotinib and alectinib, respectively. At the time of data cutoff (April 26, 2019), 11 and 34 patients were still on treatment for crizotinib and alectinib, respectively. For crizotinib, 23 patients (48%) had a median Cmin < 235 ng/mL. For alectinib, 19 patients (37%) had a median Cmin < 435 ng/mL. In general, patients with a low exposure tended to be younger and had a more favorable World Health Organization (WHO) performance status (Tables 1 and 2 ). For alectinib, patients with a low exposure were more often pretreated with ALK inhibitor(s) (i.e., crizotinib and/or ceritinib, Table 2 ). Of the patients who were treated with crizotinib and alectinib sequentially and were included in both data sets (n = 17), seven patients had a low alectinib exposure, of whom five patients also had a low crizotinib exposure. Median follow‐up was 43.6 months (range: 0.8–58.7 months) for crizotinib and 14.4 months (range: 2.2–24.6 months) for alectinib. Baseline characteristics of all patients treated with crizotinib (i.e., ALK‐positive, ROS1‐positive and cMET dysregulation) are provided in Table S1 .
Table 1.
Baseline characteristics of patients treated with crizotinib (n = 48)
| Patient characteristic | Median Cmin < 235 ng/mL (n = 23) | Median Cmin ≥ 235 ng/mL (n = 25) | All patients (n = 48) |
|---|---|---|---|
| Gender, female | 9 (39%) | 14 (56%) | 23 (48%) |
| Age (years) | 48 (21–86) | 60 (25–75) | 53 (21–86) |
| Weight (kg) | 79 (61–126) | 74 (54–96) | 77 (54–126) |
| Tumor stage | |||
| IIIA | 1 (4%) | 0 | 1 (2%) |
| IIIB | 3 (13%) | 2 (8%) | 5 (10%) |
| IV | 19 (83%) | 23 (92%) | 42 (88%) |
| Brain metastases, yes | 3 (13%) | 2 (8%) | 5 (10%) |
| Previous lines of systemic therapy | |||
| 0 | 11 (48%) | 17 (68%) | 28 (58%) |
| 1 | 8 (35%) | 6 (24%) | 14 (29%) |
| ≥ 2 | 4 (17%) | 2 (8%) | 6 (13%) |
| Crizotinib dose a | |||
| 250 mg b.i.d. | 16 (70%) | 21 (84%) | 37 (77%) |
| 200 mg b.i.d. | 3 (13%) | 2 (8%) | 5 (10%) |
| 250 mg q.d. | 3 (13%) | 2 (8%) | 5 (10%) |
| 250 mg q.a.d. | 1 (4%) | 0 | 1 (2%) |
| WHO performance status | |||
| 0 | 16 (70%) | 14 (56%) | 30 (63%) |
| 1 | 7 (30%) | 11 (44%) | 18 (38%) |
Data are expressed as no. (%) or median (range), as appropriate.
b.i.d., twice daily; Cmin, minimum plasma concentration; q.a.d., every other day; q.d., once daily; WHO, World Health Organization.
Lowest dose per patient.
Table 2.
Baseline characteristics of patients treated with alectinib (n = 52)
| Patient characteristic | Median Cmin < 435 ng/mL (n = 19) | Median Cmin ≥ 435 ng/mL (n = 33) | All patients (n = 52) |
|---|---|---|---|
| Gender, female | 8 (42%) | 20 (61%) | 28 (54%) |
| Age (years) | 54 (21–70) | 60 (34–88) | 57 (21–88) |
| Weight (kg) | 76 (54–123) | 79 (49–117) | 78 (49–123) |
| Tumor stage | |||
| IIIA | 0 | 1 (3%) | 1 (2%) |
| IIIB | 0 | 2 (6%) | 2 (4%) |
| IV | 19 (100%) | 30 (91%) | 49 (94%) |
| Brain metastases, yes | 10 (53%) | 10 (30%) | 20 (39%) |
| Previous lines of systemic therapy | |||
| 0 | 3 (16%) | 13 (39%) | 16 (31%) |
| 1 | 8 (42%) | 12 (36%) | 20 (39%) |
| ≥ 2 | 8 (42%) | 8 (24%) | 16 (31%) |
| Prior treatment with ALK inhibitor(s), yes | 16 (84%) | 19 (58%) | 35 (67%) |
| Alectinib dose a | |||
| 600 mg b.i.d. | 16 (84%) | 15 (45%) | 31 (60%) |
| 450 mg b.i.d. | 1 (5%) | 8 (24%) | 9 (17%) |
| 300 mg b.i.d. | 2 (11%) | 10 (30%) | 12 (23%) |
| WHO performance status | |||
| 0 | 11 (58%) | 6 (18%) | 17 (33%) |
| 1 | 8 (42%) | 21 (64%) | 29 (56%) |
| ≥ 2 | 0 | 4 (12%) | 4 (8%) |
| Missing | 0 | 2 (6%) | 2 (4%) |
Data are expressed as no. (%) or median (range), as appropriate.
ALK, anaplastic lymphoma kinase; b.i.d., twice daily; Cmin, minimum plasma concentration; WHO, World Health Organization.
Lowest dose per patient.
Pharmacokinetic measurements
Of the 100 consecutively included patients, a median of three samples per patient (range: 1–15) were available. In total, 376 PK samples were eligible for analysis (235 crizotinib and 141 alectinib). Figure 1 provides an overview of the median Cmin per patient. Median crizotinib Cmin per patient was 244 ng/mL (range: 103–688 ng/mL), with an interindividual and intraindividual variability of 45% and 20%, respectively, at the standard dose of 250 mg b.i.d. Median alectinib Cmin per patient was 517 ng/mL (range: 141–1944 ng/mL), with an interindividual and intraindividual variability of 57% and 27%, respectively, at the standard dose of 600 mg b.i.d.
Figure 1.
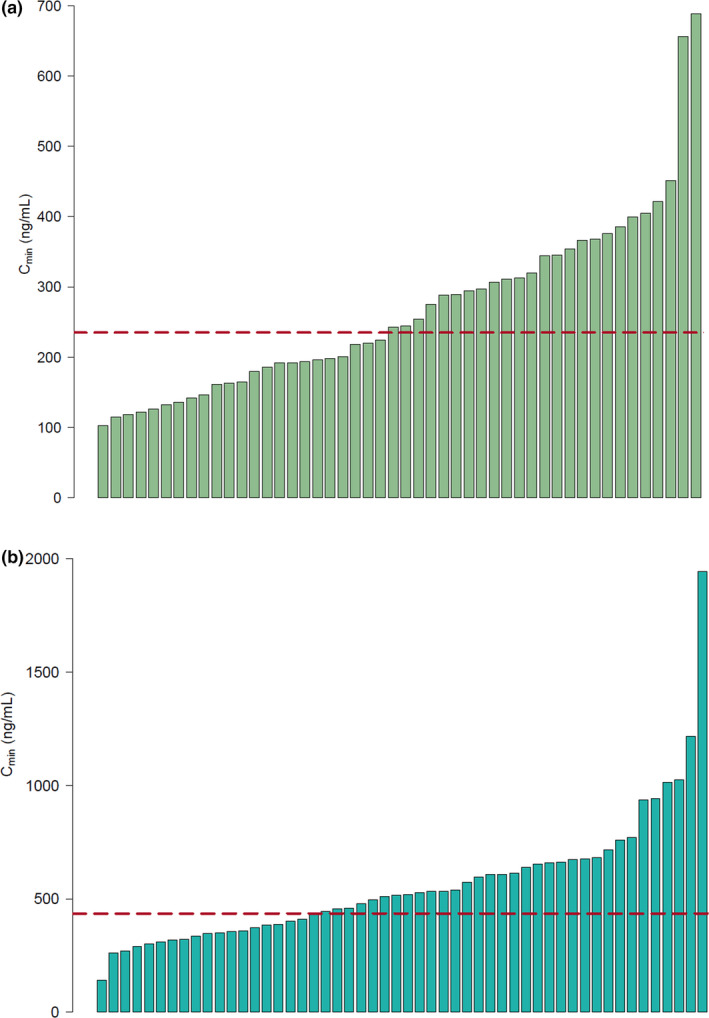
Bar plots of median crizotinib and alectinib Cmin per patient. (a) Median crizotinib Cmin per patient. Each bar represents one patient. The dotted line indicates the threshold of 235 ng/mL. Twenty‐three patients (48%) have a pharmacokinetic exposure below this threshold. Interindividual and intraindividual variability were 45% and 20%, respectively. (b) Median alectinib Cmin per patient. Each bar represents one patient. The dotted line indicates the threshold of 435 ng/mL. Nineteen patients (37%) have a pharmacokinetic exposure below this threshold. Interindividual and intraindividual variability were 57% and 27%, respectively. Cmin, minimum plasma concentration. [Colour figure can be viewed at wileyonlinelibrary.com]
Exposure–efficacy analysis
Of the ALK‐positive patients treated with crizotinib (n = 48), 37 patients (77%) progressed, i.e., 20 patients (87%) in the group with median Cmin < 235 ng/mL and 17 patients (68%) in the group with median Cmin ≥ 235 ng/mL. Intracranial progression occurred in 17 patients, i.e., 8 patients in the group with median Cmin < 235 ng/mL and 9 patients in the group with median Cmin ≥ 235 ng/mL. Median PFS in patients with crizotinib Cmin < 235 ng/mL was 5.7 months (95% confidence interval (CI), 5.0–26.8 months), compared with 17.4 months (95% CI, 16.9–not estimable months) in patients with Cmin ≥ 235 ng/mL (P = 0.08, log‐rank test, Figure 2a ). In multivariable analysis, Cmin < 235 ng/mL resulted in a hazard ratio (HR) of 1.79 (95% CI, 0.90–3.59, P = 0.10) when WHO performance status and the number of prior lines of treatment were taken into account (Table 3 ). A swimmer plot is shown in Figure S2 , which illustrates the treatment duration, dose reductions, and resistance mutations (when available) for each individual patient.
Figure 2.
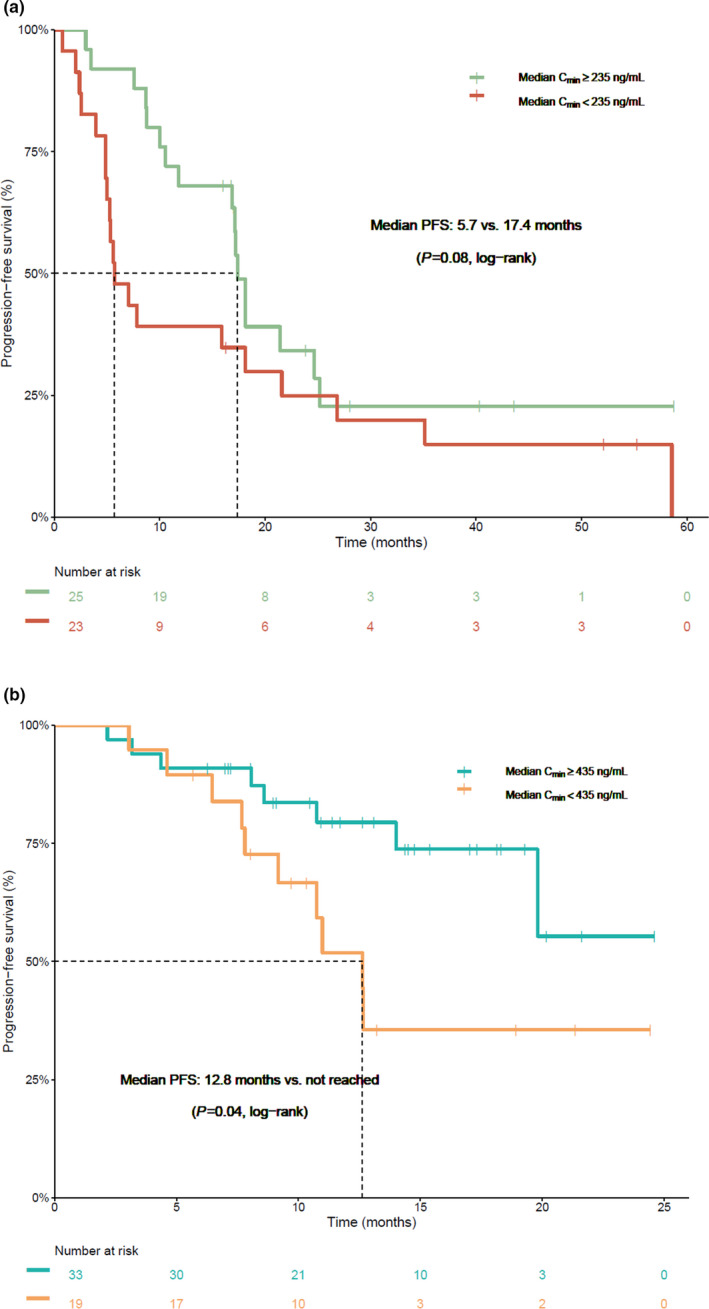
Progression‐free survival in patients treated with crizotinib and alectinib with an exposure above and below the proposed efficacy thresholds. (a) Kaplan‐Meier curve indicating the progression‐free survival in patients treated with crizotinib with a median Cmin above and below the threshold of 235 ng/mL. (b) Kaplan‐Meier curve indicating the progression‐free survival in patients treated with alectinib with a median Cmin above and below the threshold of 435 ng/mL. Cmin, minimum plasma concentration; PFS, progression‐free survival. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 3.
Univariable and multivariable Cox regression analysis for progression‐free survival in patients treated with crizotinib
| Variable | Univariable | Multivariable | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | |
| Cmin < 235 ng/mL | 1.76 | 0.92–3.39 | 0.089 | 1.79 | 0.90–3.59 | 0.100 |
| Age (years) | 0.99 | 0.97–1.01 | 0.295 | |||
| Gender, female | 0.65 | 0.34–1.27 | 0.212 | |||
| WHO performance status | 1.34 | 0.68–2.66 | 0.398 | 1.97 | 0.93–4.15 | 0.076 |
| Number of prior lines of treatment | 1.57 | 1.00–2.48 | 0.052 | 1.61 | 1.01–2.58 | 0.046 |
CI, confidence interval; Cmin, minimum plasma concentration; HR, hazard ratio; WHO, World Health Organization.
In the pooled analysis of all patients treated with crizotinib (i.e., ALK‐positive, ROS1‐positive, and cMET dysregulation, n = 79), median PFS in patients with crizotinib Cmin < 235 ng/mL was 5.3 months (95% CI, 4.9–15.9 months), compared with 11.8 months (95% CI, 8.7–18.1 months) in patients with Cmin> 235 ng/mL (P = 0.04, log‐rank test, Figure S1 ). In multivariable analysis, Cmin> 235 ng/mL resulted in an HR of 2.15 (95% CI, 1.21–3.84, P = 0.009) when mutation status, WHO performance status, and the number of prior lines of treatment were taken into account (Table S2 ).
Of the patients treated with alectinib, 18 patients (35%) progressed, i.e., 10 patients (53%) in the group with median Cmin < 435 ng/mL and 8 patients (24%) in the group with median Cmin ≥ 435 ng/mL. Intracranial progression occurred in four patients, i.e., three patients in the group with median Cmin < 435 ng/mL and one patient in the group with median Cmin ≥ 435 ng/mL. Median PFS in patients with alectinib Cmin < 435 ng/mL was 12.6 months (95% CI, 9.2–NA months), compared with not reached (95% CI, 19.8–NA months) in patients with Cmin ≥ 435 ng/mL (P = 0.04, log‐rank test, Figure 2b ). In multivariable analysis, Cmin ≥ 435 ng/mL resulted in an HR of 4.29 (95% CI, 1.33–13.90, P = 0.015) when WHO performance status and prior treatment with ALK inhibitor(s) were taken into account (Table 4 ). A swimmer plot is shown in Figure S3 , which illustrates the treatment duration, dose reductions, and resistance mutations (when available) for each individual patient.
Table 4.
Univariable and multivariable Cox regression analysis for progression‐free survival in patients treated with alectinib
| Variable | Univariable | Multivariable | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P value | HR | 95% CI | P value | |
| Cmin < 435 ng/mL | 2.58 | 1.01–6.59 | 0.047 | 4.29 | 1.33–13.90 | 0.015 |
| Age (years) | 0.99 | 0.95–1.02 | 0.388 | |||
| Gender, female | 0.35 | 0.13–0.93 | 0.035 | |||
| WHO performance status | 1.28 | 0.69–2.38 | 0.428 | 2.35 | 1.07–5.16 | 0.034 |
| Number of prior lines of treatment | 1.65 | 1.05–2.61 | 0.030 | |||
| Prior treatment with ALK inhibitor(s) | 3.08 | 0.70–13.51 | 0.136 | 2.81 | 0.57–13.94 | 0.205 |
Data were missing for two patients regarding WHO performance status.
ALK, anaplastic lymphoma kinase; CI, confidence interval; Cmin, minimum plasma concentration; HR, hazard ratio; WHO, World Health Organization.
Exposure–toxicity analysis
For crizotinib, 13 patients experienced clinically relevant toxicities (10 dose reductions, 7 dose interruptions, and 1 treatment discontinuation), including liver toxicity (n = 5), gastrointestinal toxicity (n = 2), pneumonitis (n = 1), neuropathy (n = 1), renal insufficiency (n = 1), neutropenia (n = 1), and fatigue (n = 1). In six of these patients, the toxicity event occurred before the first PK sample was collected. Median Cmin before the toxicity event was 338 ng/mL (range: 185–678 ng/mL), compared with 264 ng/mL (range: 118–688 ng/mL) in patients without clinically relevant toxicities (P = 0.281). The two patients with the highest median Cmin did not have clinically relevant toxicities with the currently used definition. However, the first patient (median Cmin 656 ng/mL) discontinued treatment due to cerebral progression, while at the same time she experienced symptoms possibly related to crizotinib (i.e., muscle weakness, ground glass opacities in the lungs, and progression of kidney cysts). The second patient had a median Cmin of 688 ng/mL and died at the intensive care unit with an unknown cause of death, possibly due to cardiac arrhythmia, a recognized crizotinib toxicity.
For alectinib, 16 patients experienced clinically relevant toxicities (15 dose reductions, 7 dose interruptions), including edema (n = 6), fatigue (n = 4), myalgia (n = 3), gastrointestinal toxicity (n = 3), bradycardia (n = 2), liver toxicity (n = 2), skin rash (n = 1), anemia (n = 1), and renal insufficiency (n = 1). In addition, six patients started at a lower dose, due to severe toxicity during prior treatment with crizotinib (n = 4), elevated liver enzymes, (n = 1) and miscommunication between patient and physician (n = 1). Median Cmin in patients with and without clinically relevant toxicities was 539 ng/mL and 431 ng/mL, respectively (P = 0.205).
DISCUSSION
In this observational study we investigated whether pharmacokinetic exposure to ALK inhibitors crizotinib and alectinib is related to treatment efficacy and toxicity. Patients with a median alectinib Cmin ≥ 435 ng/mL had a significantly longer median PFS compared with patients with an exposure below this threshold (12.6 months vs. not reached yet, Figure 2b and Table 4 ). For crizotinib, median PFS was also numerically longer in patients with a median Cmin ≥ 235 ng/mL (5.7 vs. 17.4 months), which is a clinically relevant difference, although this difference was not statistically significant (Figure 2a and Table 3 ). In the pooled analysis, which also included ROS1‐positive and c‐MET dysregulated patients, statistical significance was reached (Figure S1 and Table S2 ). Exposure to both crizotinib and alectinib was not significantly related to clinically relevant toxicities.
Interindividual variability in pharmacokinetic exposure was found to be considerable (i.e., 45–57%), which is in line with previous literature. 6 , 7 As a consequence, 48% of the patients treated with crizotinib and 37% of the patients treated with alectinib had an exposure below the efficacy threshold, and were found at risk of decreased treatment efficacy. This implies that individualized dosing is indicated in this subgroup of patients with a low exposure to improve treatment outcomes, for which we provide practical recommendations in Figure 3 .
Figure 3.

Practical recommendations for precision dosing of crizotinib and alectinib. Patients start treatment at the approved dose of 250 mg b.i.d. for crizotinib and 600 mg b.i.d. for alectinib. PK samples will be collected 4, 8, and 12 weeks after start of treatment and every 12 weeks thereafter. In case of (calculated) Cmin below the TDM target of 235 ng/mL for crizotinib or 435 ng/mL for alectinib and manageable toxicity, the dose will be increased by one dose level (after checking treatment adherence and drug–drug interactions). Maximum dose levels are based on data from phase I dose finding studies. b.i.d., twice daily; Cmin, minimum plasma concentration; PK, pharmacokinetic; q.d., once daily; TDM, therapeutic drug monitoring; W, week. [Colour figure can be viewed at wileyonlinelibrary.com]
Apart from the high interindividual variability, intraindividual variability in alectinib exposure was also found to be large (i.e., 27%). This could be caused by the substantial food effect of alectinib, as its exposure increases more than fourfold when administered with a high‐fat meal compared with fasting conditions. 16 According to the label, alectinib is administered concomitantly with food, but the content and volume of these meals could vary substantially within patients over time.
Many patients needed a dose reduction due to toxicity or started treatment at a lower dose (i.e., 23% for crizotinib and 40% for alectinib, Tables 1 and 2 ). It is notable that, especially for alectinib, many of these patients still had an adequate exposure. Of the patients with a low exposure to crizotinib and alectinib, 30% and 16%, respectively, received a prior dose reduction due to toxicity. This means that 70% and 84% of the low exposed patients, respectively, in the absence of toxicity, would potentially benefit from dose escalation.
Notably, patients with a low exposure to alectinib were more often pretreated with other ALK inhibitors (84% compared with 58% of patients with adequate exposure). Similar underlying factors (i.e., increased clearance or decreased absorption) could have caused a low exposure to both alectinib and the previous ALK inhibitor(s). These patients may, therefore, have failed treatment with the previous ALK inhibitor(s) earlier due to their low exposure and needed subsequent treatment with alectinib sooner. In addition, an inherently lower treatment adherence in these patients could have played a role.
While alectinib is indicated only for the treatment of ALK‐positive NSCLC, crizotinib is also approved for ROS1‐positive NSCLC and used off‐label in the treatment of patients with cMET dysregulation (i.e., amplification or exon 14 skipping). Due to the similarity of ALK and ROS1 kinase domains, crizotinib has similar half maximal inhibitory concentration values of 40–60 nM and 60 nM against ALK and ROS1, respectively, while the half maximal inhibitory concentration against cMET was notably lower (i.e., 8 nM). 17 It could, therefore, be hypothesized that the efficacy threshold of Cmin ≥ 235 ng/mL, that was established in ALK‐positive patients, will also hold true for ROS1‐positive patients, while a lower threshold might be sufficient for patients with cMET dysregulation. Since these subgroups have a different prognosis and the efficacy threshold might be different, exposure–efficacy analyses should preferably be performed separately for each subgroup. However, to further increase our sample size, we did perform a pooled analysis, in which we accounted for mutation status, resulting in a statistically significant exposure–response relationship.
In a previously performed exposure–response analysis in patients treated with alectinib (n = 207), no association has been identified between median Cmin of alectinib and its active metabolite M4 and overall survival. 18 Although overall survival is regarded as the gold standard metric of treatment outcome, an exposure–efficacy relationship can easily be diluted by the effects of successive treatment lines. 4 In this study, a potential relationship with PFS has not been evaluated. Furthermore, no threshold was tested and adjustments to measured concentrations were performed to account for analytical bias (due to cross‐validation issues). 18 More recent analyses across the phase III studies did demonstrate a relationship between exposure and PFS. They identified an optimal pharmacokinetic threshold of 1,040 nM for the sum of alectinib and M4, corresponding to an alectinib Cmin of approximately 370 ng/mL. 19 Since only limited data on this analysis is currently available in the literature, we did not use this cutoff value in our analyses.
An important strength of the current study is that data were collected from real‐life patients, instead of from highly selected patients in a clinical trial. This is the first time that an exposure–response relationship is described outside the context of a clinical trial for ALK inhibitors, which is relevant, since treatment in a real‐life setting may differ considerably from treatment in a clinical trial setting (i.e., no drug accountability, more complex patients who would not be eligible for a trial). In addition, multiple samples over time were available for most patients, providing an adequate reflection of pharmacokinetic exposure during treatment.
On the other hand, limitations of this study include that the applied method to estimate Cmin assumes an equal alectinib clearance in all patients, not taking into account interindividual differences in the elimination half‐life. In addition, part of the samples (up to 59%, Figure S4 ) were collected during the absorption or distribution phase (i.e., before the time to maximum concentration), resulting in an underestimation of Cmin. However, due to the long t 1/2 (i.e., 42 hours for crizotinib and 32 hours for alectinib) in respect to the dosing interval (i.e., 12 hours), differences between Cmin and Cmax are small with a peak‐to‐trough ratio of approximately 1.3. 20 , 21 Therefore, the deviations from the actual trough levels can be considered acceptable. These imprecisions could have been circumvented by drawing actual trough levels, but this is less convenient for patients as the collection of PK samples is usually combined with regular visits to the outpatient clinic. Another approach would be to use population PK models to estimate Cmin, which would allow for taking into account the interpatient variability in PK parameters. However, disadvantages of this approach are its increased complexity and the fact that Bayesian estimation based on limited sampling will result in shrinkage towards the typical value. 22 Furthermore, another limitation is that selection bias could have played a role, as some patients may have discontinued treatment before the first sample was drawn. These patients may have had early progression due to low exposure. Finally, Cmin might not be the most appropriate PK parameter to assess exposure–toxicity relationships. Although trough levels are critical to ensure maximal target engagement during the complete dosing interval at the tumor level, other PK parameters may better reflect the potential relationship between exposure and side effects (i.e., AUC or Cmax).
It is known that the emergence of resistance mutations causes treatment failure. 23 An interesting concept that needs to be further elucidated is whether the prevalence of these resistance mutations is equally high in patients with a low pharmacokinetic exposure compared with patients with an adequate pharmacokinetic exposure. In Figures S2 , and S3 we report the identified resistance mutations in our patient cohorts. However, as resistance mutation analysis was only performed in a small subset of patients (n = 17), we have insufficient data to answer this question.
Future steps will be to evaluate the feasibility, tolerability, and efficacy of individualized dosing of crizotinib and alectinib, which will be studied in an ongoing prospective study on therapeutic drug monitoring (i.e., adjusting the dose based on measured drug levels) of oral anticancer drugs. 24 Although a randomized controlled trial comparing a therapeutic drug monitoring strategy to a flat dosing strategy could be considered the gold standard, this also assumes an equipoise between treatment arms. Therefore, it could be questioned if performing a randomized controlled trial is ethical where a clear exposure–response relationship exists.
In addition, it would be interesting to investigate the role of the active metabolite of alectinib (i.e., M4). This metabolite has a similar potency as alectinib itself, but a much lower abundance (± 30% of the parent). 7 Concentrations of M4 follow the alectinib concentrations, although data are very limited. Concomitant administration of alectinib with cytochrome P450 3A4 isozyme inhibitors or inducers results in inverse changes in alectinib and M4 concentrations, without affecting the total exposure to a clinically relevant degree. 25 This should be kept in mind when therapeutic drug monitoring of alectinib is applied without measuring M4. It should be noted that all patients in the current study were carefully monitored for drug–drug interactions in clinical practice and, therefore, strong or moderate cytochrome P450 3A4 isozyme inhibitors or inducers were not used in this cohort. A combined threshold of the sum concentration of alectinib plus M4 may further improve precision dosing of alectinib. But given the above considerations, we think the addition of M4 will not relevantly change the finding of this study.
In conclusion, here we demonstrated that exposure to crizotinib and alectinib is related to efficacy in a real‐life patient cohort, providing a strong rationale for therapeutic drug monitoring. Individualized dosing based on therapeutic drug monitoring may improve treatment outcomes for the subgroup of patients with a Cmin below the efficacy thresholds of 235 ng/mL for crizotinib and 435 ng/mL for alectinib.
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
J.H.B. is a part‐time employee, stockholder and patent holder of Modra Pharmaceuticals (a spin‐off company developing oral taxane formulations, not related to this study). All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
S.L.G. wrote the manuscript. S.L.G., D.R.G., A.D.R.H., and N.S. designed the research. S.L.G., D.R.G., J.M.J., N.d.V., H.R., J.H.B., J.A.B., E.F.S., A.H., and N.S. performed the research. S.L.G., D.R.G., A.D.R.H., and N.S. analyzed the data.
Supporting information
Acknowledgments
We thank everyone who contributed to the logistics of collecting and measuring the crizotinib and alectinib concentrations, in particular Remy Verheijen, Laura Molenaar‐Kuijsten, and René Boosman. This work was presented in part at the 2019 ESMO Annual Meeting, Barcelona (Annals of Oncology, Volume 30, Supplement 5, October 2019, mdz260.008, https://doi.org/10.1093/annonc/mdz260.008).
References
- 1. Soda, M. et al A prospective PCR‐based screening for the EML4‐ALK oncogene in non‐small cell lung cancer. Clin. Cancer Res. 18, 5682–5689 (2012). [DOI] [PubMed] [Google Scholar]
- 2. Horn, L. & Pao, W. EML4‐ALK: honing in on a new target in non‐small‐cell lung cancer. J. Clin. Oncol. 27, 4232–4235 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao, F. et al Clinicopathological characteristics of patients with non‐small‐cell lung cancer who harbor EML4‐ALK fusion gene: a meta‐analysis. PLoS One 10, e0117333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Solomon, B.J. et al Final overall survival analysis from a study comparing first‐line crizotinib versus chemotherapy in ALK‐mutation‐positive non–small‐cell lung cancer. J. Clin. Oncol. 36, 2251–2258 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Peters, S. et al Alectinib versus crizotinib in untreated ALK‐positive non–small‐cell lung cancer. N. Engl. J. Med. 377, 829–838 (2017). [DOI] [PubMed] [Google Scholar]
- 6. US Food and Drug Administration. Center for Drug Evaluation and Research . Crizotinib clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000ClinPharmR.pdf> (2011).
- 7. Food and Drug Administration. Center for Drug Evaluation and Research . Alectinib clinical pharmacology and biopharmaceutics review <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208434Orig1s000ClinPharmR.pdf> (2016).
- 8. Mitchell, A.P. et al Clinical trial participants with metastatic renal cell carcinoma differ from patients treated in real‐world practice. J. Oncol. Pract. 11, 491–497 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Herbrink, M. et al Development and validation of a liquid chromatography – tandem mass spectrometry analytical method for the therapeutic drug monitoring of eight novel anticancer drugs. Biomed. Chromatogr. 32, e4147 (2018). [DOI] [PubMed] [Google Scholar]
- 10. Janssen, J.M., de Vries, N., Venekamp, N., Rosing, H., Huitema, A.D.R. & Beijnen, J.H . Development and validation of a liquid chromatography‐tandem mass spectrometry assay for nine oral anticancer drugs in human plasma. J. Pharm. Biomed. Anal. 174, 561–566 (2019). [DOI] [PubMed] [Google Scholar]
- 11. Wang, Y. , Chia, Y.L., Nedelman, J., Schran, H., Mahon, F.‐X. & Molimard, M. A therapeutic drug monitoring algorithm for refining the imatinib trough level obtained at different sampling times. Ther. Drug Monit. 31, 579–584 (2009). [DOI] [PubMed] [Google Scholar]
- 12. US Food and Drug Administration. Center for Drug Evaluation and Research . Imatinib clinical pharmacology and biopharmaceutics review <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21‐335_Gleevec_biopharmr_P1.pdf> (2001).
- 13. Verheijen, R.B. , Yu, H., Schellens, J.H.M., Beijnen, J.H., Steeghs, N. & Huitema, A.D.R. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin. Pharmacol. Ther. 102, 765–776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. R Core Development Team . A language and environment for statistical computing. R Found Statistical Computing; <https://www.r‐project.org/> (2016) [Google Scholar]
- 15. Schemper, M. & Smith, T.L. A note on quantifying follow‐up in studies of failure time. Control. Clin. Trials 17, 343–346 (1996). [DOI] [PubMed] [Google Scholar]
- 16. Morcos, P.N. et al Effect of food and esomeprazole on the pharmacokinetics of alectinib, a highly selective ALK inhibitor, in healthy subjects. Clin. Pharmacol. Drug Dev. 6, 388–397 (2017). [DOI] [PubMed] [Google Scholar]
- 17. Lin, J.J. & Shaw, A.T. Recent advances in targeting ROS1 in lung cancer. J. Thorac. Oncol. 12, 1611–1625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morcos, P.N. et al Exposure–response analysis of alectinib in crizotinib‐resistant ALK‐positive non‐small cell lung cancer. Cancer Chemother. Pharmacol. 82, 129–138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smart, K. et al Alectinib exposure‐response (ER) in ALK‐inhibitor naive ALK‐positive NSCLC patients: pooled analysis across phase III studies. J. Clin. Oncol. 37, e20575 (2019). [Google Scholar]
- 20. Hirota, T. , Muraki, S. & Ieiri, I. Clinical pharmacokinetics of anaplastic lymphoma kinase inhibitors in non‐small‐cell lung cancer. Clin. Pharmacokinet. 58, 403–420 (2019). [DOI] [PubMed] [Google Scholar]
- 21. Gadgeel, S.M. et al Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib‐resistant ALK‐rearranged non‐small‐cell lung cancer (AF‐002JG): results from the dose‐finding portion of a phase 1/2 study. Lancet Oncol. 15, 1119–1128 (2014). [DOI] [PubMed] [Google Scholar]
- 22. Janssen, J.M. , Dorlo, T.P.C. , Beijnen, J.H. & Huitema, A.D.R. Evaluation of extrapolation methods to predict trough concentrations to guide therapeutic drug monitoring of oral anticancer drugs. Ther. Drug Monit. 42, 532–539 (2020). [DOI] [PubMed] [Google Scholar]
- 23. Gainor, J.F. et al Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK ‐rearranged lung cancer. Cancer Discov. 6, 1118–1133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Groenland, S.L. et al Therapeutic drug monitoring of oral anticancer drugs: the Dutch Pharmacology Oncology Group–Therapeutic Drug Monitoring protocol for a prospective study. Ther. Drug Monit. 41, 561–567 (2019). [DOI] [PubMed] [Google Scholar]
- 25. Morcos, P.N. et al Clinical drug–drug interactions through cytochrome P450 3A (CYP3A) for the selective ALK inhibitor alectinib. Clin. Pharmacol. Drug Dev. 6, 280–291 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials