

Neural progenitors derived from iPSCs of patients with multiple system atrophy show changes in the mitochondrial morphology, relocation of α‐synuclein to the nucleus, and increased susceptibility to exogenous oxidative stress. The findings suggest early cellular dysfunction preceding α‐synuclein aggregate formation in this disorder.

Keywords: multiple system atrophy, induced pluripotent stem cells, mitochondria, high resolution respirometry, α‐synuclein, oxidative stress
Abstract
Aims
Multiple system atrophy (MSA) is a fatal neurodegenerative disease that belongs to the family of α‐synucleinopathies. At post mortem examination, intracellular inclusions of misfolded α‐synuclein are found in neurons and oligodendrocytes and are considered to play a significant role in the pathogenesis. However, the early steps of the disease process are unknown and difficult to study in tissue derived from end‐stage disease.
Methods
Induced pluripotent stem cells (iPSCs) were generated from patients’ and control skin fibroblasts and differentiated into NCAM‐positive neural progenitor cells (NPCs). The mitochondrial morphology and function were assessed by immunocytochemistry and high resolution respirometry. The ability to cope with exogenous oxidative stress was tested by exposure to different doses of luperox. The expression of α‐synuclein was studied by immunocytochemistry.
Results
We identified increased tubulation of mitochondria with preserved respiration profile in MSA‐derived NPCs. Exposure of these cells to exogenous oxidative stress even at low doses, triggered an excessive generation of reactive oxygen species (ROS) and cleavage of caspase‐3. MSA‐derived NPCs did not present changed levels of SNCA gene expression nor intracellular aggregates of α‐synuclein. However, we identified disease‐related translocation of α‐synuclein to the nucleus.
Conclusions
Our results show early cellular dysfunction in MSA‐derived NPCs. We identified changes in the redox homeostasis which are functionally compensated at baseline but cause increased susceptibility to exogenous oxidative stress. In addition, nuclear translocation of α‐synuclein in MSA‐derived NPCs supports an early cellular stress response which may precede the neurodegenerative process in this disorder.
Abbreviations
- CCCP
carbonyl cyanide m‐chloro phenyl hydrazone
- ETS
electron transfer system
- FCF
Flux control factors
- FCR
Flux control ratios
- GCIs
glial cytoplasmic inclusions
- GNIs
glial nuclear inclusions
- GWAS
Genome‐wide association study
- hESCs
human embryonic stem cells
- iPSCs
induced pluripotent stem cells
- MSA
multiple system atrophy
- NCAM
Neural cell adhesion molecule
- NCIs
neuronal cytoplasmic inclusions
- NNIs
neuronal nuclear inclusions
- NPCs
neural progenitor cells
- PSA‐NCAM
Polysialylated‐neural cell adhesion molecule
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- RCR
respiratory acceptor control ratio
- ROS
reactive oxygen species
- ROX
residual oxygen consumption
- SeV
Sendai virus
- SUIT
substrate‐uncoupler‐inhibitor titrations
Introduction
Multiple system atrophy (MSA) is a fatal and rapidly progressive neurodegenerative disorder. It is a rare disease and the average prevalence is 7.8 per 100,000 among persons older than 40 years of age [1]. MSA is commonly diagnosed in the sixth decade of life with variable combination of autonomic dysfunction and motor symptoms, including Parkinsonism and cerebellar ataxia [2, 3]. The pathological hallmark of the disease is α‐synuclein positive cytoplasmic inclusions in oligodendroglia (glial cytoplasmic inclusions (GCIs)), however neuronal cytoplasmic and neuronal/glial nuclear inclusions (NCIs, NNIs, GNIs) are also common, while Lewy bodies are identified in only 10% of patients [4, 5]. In comparison to Parkinson’s disease, the most common α‐synucleinopathy, MSA has a more rapid progression and death generally occurs 9 years after the diagnosis [3, 6, 7, 8].
The aetiology of MSA remains largely elusive. MSA seems mostly sporadic as proposed by a genome‐wide association screening [9]. Population‐specific point mutations in the COQ2 loci in few cases [10, 11, 12, 13, 14] have suggested a leading pathogenic role of dysfunctional mitochondria in MSA. Other studies have supported the role of mitochondrial deficits in the pathogenesis of MSA regardless of the genotype [15]. A recent epigenome‐wide association study in MSA has further implicated pathways relevant to neurodegeneration, including those related to neuroinflammation and mitophagy, in the pathogenesis of the disease [16].
Disease mechanisms have been further addressed in studies applying post mortem tissue, animal and cellular models [17, 18, 19, 20, 21]. Unfortunately, post mortem analyses provide only a limited time‐window into the end‐stage of the disease. Alternatively, the existing animal and cellular models (cell lines and primary cultures) which are based on the overexpression of α‐synuclein [18, 19, 22, 23] or α‐synuclein propagation [24, 25, 26] are not in the position to identify the initial cellular changes that may contribute to the specific neuronal and oligodendroglial pathology in MSA. The expression of SNCA, the gene encoding α‐synuclein, is generally not changed in MSA brains [27]. The putative prion nature of MSA as proposed in experimental studies is challenged by the fact that endogenous wild‐type α‐synuclein is insufficient to propagate MSA‐derived α‐synuclein pathology and mutated α‐synuclein is needed as a template [24]. Strain differences of α‐synuclein in the different synucleinopathies [28, 29] have been recently proposed to contribute to the more aggressive progression of MSA [30], however their origin and role in triggering disease remain unclear. Pathological and experimental observations have further suggested that early changes in oligodendrocytes associated with relocation of the myelin‐related protein p25α may precede α‐synuclein pathological accumulation in these cells [31, 32, 33]. These data pose the hypothesis that cellular dysfunction preceding α‐synuclein pathology may be present in MSA, however examining the earliest stages of the disease remains challenging.
Induced pluripotent stem cell (iPSC) technologies allow dynamic cellular studies in patient‐specific lines, which may provide insights into the early cellular dysfunction featuring the MSA background. Here, we describe for the first time the use of iPSC‐derived neural progenitor cells (NPCs) to study disease‐related cellular dyshomeostasis in MSA. We demonstrate that MSA‐derived NPCs show changes of mitochondrial morphology and increased sensitivity to oxidative stress. MSA‐derived NPCs show no changes in SNCA expression and no spontaneous α‐synuclein inclusion formation but feature nuclear dislocation of the protein.
Materials and methods
iPSC generation and characterization
Fibroblasts were obtained from skin biopsies of two MSA patients and an age‐matched healthy control (Table S1) at the Department of Neurology, Medical University of Innsbruck with the appropriate consent and ethics approval. Cells were transduced with CytoTune™ 2.0 Sendai reprogramming vectors (A16517, Thermo Fisher Scientific) encoding for the classical Yamanaka’s factors (Klf4, Oct4, Sox2 and c‐Myc). The clearance of the Sendai virus (SeV) vector used for the somatic reprogramming was proven in the iPSCs cultures by quantitative RT‐PCR (qRT‐PCR) and immunocytochemistry and according to manufacturer’s instructions. Karyotyping was performed for each iPSC line by classical G‐banding at the Division of Human Genetics, Medical University of Innsbruck. Cells in metaphase arrest, induced by cocamide solution (0.1 µg/ml) were fixed, and Giemsa staining was performed. Chromosomes were analysed in at least 20 metaphases per cell line.
To confirm the pluripotency of the generated iPSCs, differentiation into the three germ layers (ectoderm, endoderm and mesoderm) was performed using StemMACS™ Trilineage Differentiation Kit (Miltenyi) according to the manufacturer’s protocols. Cells were analysed by immunocytochemistry at day 7 after initiation of the differentiation.
NPCs generation and isolation
For neural induction, 24 h after seeding, iPSCs were cultured in neural induction medium ( 1x NEAA (Thermo Fisher Scientific), 1x Pen/Strep (Thermo Fisher Scientific), 1x B27 (‐Vit A) (Thermo Fisher Scientific), 10 µM SB431542 (StemGent), 250 nM LDN193189 (StemGent), 100 nM RA (Sigma), 25 µg/ml insulin (Sigma), and 1x β‐Mercaptoethanol (Thermo Fisher Scientific) in DMEM/F‐12 Glutamax (Thermo Fisher Scientific). Seven days after neural induction, PSA‐NCAM expressing NPCs were sorted by fluorescence‐activated cell sorting (FACS). Sorted NPCs were further cultured on Geltrex (Thermo Fisher Scientific) coated plates in DMEM/F‐12 Glutamax medium with 1x NEAA, 1x P/S, 1x N2 (Thermo Fisher Scientific), 10 ng/ml FGF (Sigma), 10 ng/ml EGF (Sigma) and 10 µg/ml Heparin (Sigma). The sorted and expanded NPCs were characterized by immunocytochemistry (ICC) for the expression of progenitor, neuronal, and glial markers.
mRNA extraction and quantitative real‐time PCR (qRT‐PCR)
Messenger RNA (mRNA) was isolated from the cells using Dynabeads Oligo (dT)25 (Thermo Fisher Scientific) following the manufacturer’s protocol. Reverse transcription was performed using the High‐capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Control cDNA from hESC line 207 [34] was provided by Dr Nat.
The levels of gene expression were assessed by qRT‐PCR using the iTaq™ Universal Probes Supermix (BioRad) and Gene Expression Assays (Thermo Fisher Scientific, Table S2). RT‐PCR quantification was performed using Bio Rad CFX96 (BioRad). Each experiment was run in two technical replicates. The results were normalized to the housekeeping gene glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) and expressed as ΔCt for each cell line (mean ± SD).
Immunocytochemistry
Cells were washed with Dulbecco’s phosphate buffered saline (DPBS) and fixed with ice cold 4% paraformaldehyde (PFA) for 10 min. Cells were permeabilized with 0.2% Triton X‐100 in PBS (PBS‐T 0.2) at room temperature (RT). After blocking with the blocking solution (10% normal serum, 1% bovine serum albumin (BSA) in PBS‐T 0.1%), the cells were incubated overnight at 4°C with the primary antibody (Table S3) diluted in blocking solution. Next, cells were incubated with the secondary antibody (Table S3) diluted in 2% BSA in PBS‐T 0.1% for 90 min at room temperature. Counterstaining was done with 4',6‐Diamidino‐2‐Phenylindole (DAPI, Thermo Fisher Scientific) and final mounting with Fluoromount‐G® (Southern Biotech).
Fluorescence Activated Cell Sorting (FACS)
Cells were harvested with accutase for 5 min at 37°C. After centrifugation, the pellet was re‐suspended in medium containing 10 µM of ROCK inhibitor (Stemcell Technologies). The cells were incubated with the antibody of interest (Table S4) at 4°C for 10 min, followed by one washing step with Buffer A (0.5% BSA, 2 mM ethylenediaminetetraacetic acid in DPBS) and centrifugation for 5 min at 300 g. Finally, the cells were resuspended in Buffer A and transferred to FACS tubes (Falcon). Settings for the appropriate sorting of PSA‐NCAM‐positive cells were established using the respective isotype controls. The cell sorting was performed at the FACS Facility of the Medical University of Innsbruck with the support of Dr Sieghart Sopper.
Exposure to exogenous oxidative stress
NPCs at P3 were seeded in 24 well plates at a density of 105 cells per well. After 24 h, to induce oxidative stress, the cells were exposed to different concentrations (0 mM, 0.5 mM, 1 mM, 5 mM) of Luperox® (Merk) for 2 h. Three wells per condition were analysed in at least four independent experiments.
Detection of ROS
Measurement of the intracellular superoxide radical generation by the formation of a dark blue formazan deposit resulting from superoxide‐mediated reduction of NBT (nitroblue tetrazolium chloride, Roche Applied Sciences) was performed as previously shown [35, 36]. After exposure to Luperox, the cells were incubated for 30 min at 37°C with NBT (1:100) in DMEM‐F‐12/GLUTAMAX. The staining step was followed by two washes in DPBS and fixation for 10 min with cold 4% PFA at room temperature. After fixation of the cells, they were washed twice with PBS and then counterstained with DAPI (1 mg/ml) diluted in PBS (1:1000).
Image analysis
Random optical fields in three independent replicates per treatment and cell line, and in at least three separate experiments were microphotographed with a 40x objective of a DMI 4000B Leica inverse microscope, provided with digital camera DFC300 FX and LAS V3.8 software (Leica). The images were blinded and further analysed by counting the total number of cells (DAPI staining) and the number of ROS‐positive and cleaved caspase‐3‐positive cells. The results for each cell line and treatment were presented as mean percentage of positive cells for every single experiment. The latter was calculated as the average of the three replicates in the experiment for cell line and treatment.
High‐resolution respirometry
High resolution respirometry was performed at OROBOROS Instruments (Austria) according to a standard substrate‐uncoupler‐inhibitor titrations (SUIT)‐008 D025 protocol (https://bioblast.at/index.php/SUIT‐008_O2_ce‐pce_D025). Cell suspensions were transferred into calibrated Oxygraph‐2k 2 ml‐chambers. Oxygen polarography was performed at 37 ± 0.001 °C (electronic Peltier regulation) in O2k‐chambers. Oxygen concentration (μM), as well as oxygen flux per cell (pmol O2.s−1·cell−1) was recorded in real time using DatLab software.
Shortly, routine respiration was measured after the stabilization of the system. After permeabilization of the cells with digitonin (Dig), nonphosphorylating LEAK‐respiration (CIL) was induced by adding the CI‐linked substrates pyruvate (5 mM) and malate (0.5 mM). Subsequently, OXPHOS capacity of CI‐linked activity (CIP) was measured after addition of a saturating concentration of ADP (2.5 mM). Cytochrome C was added to assess the integrity of the mitochondrial outer membrane. To further evaluate the OXPHOS capacity, GDP and succinate were added (CI&IIP). Stepwise titration of the protonophore carbonyl cyanide m‐chloro phenyl hydrazone (CCCP, 0.5 μM steps) led to proton leakage through the inner mitochondrial membrane, and was used for the measurement of the capacity of the electron transfer system (ETS, CI&IIE), representing the noncoupled state at optimum uncoupler concentration for the maximum oxygen flux. Subsequent inhibition of CI by rotenone (0.5 μM) provided measurement of CII‐linked ETS capacity (CIIE). To control for other oxygen‐consuming processes, CIII was inhibited by Antimycin A. The resulting residual oxygen consumption (ROX) reflected oxygen consumption from undefined sources and was subtracted from mitochondrial respiratory states [37]. ROX was very low, demonstrating the mitochondrial origin of oxygen consumption. Oxygen concentration in the chambers was kept high enough to avoid oxygen limitation of respiration (more than 150 μM O2 until the end of CI&IIP). All reagents used for high‐resolution respirometry were purchased from Sigma‐Aldrich (St. Louis, Missouri, US). Cell‐specific oxygen fluxes were compared in different substrate and coupling states after correction for ROX. Flux control ratios (FCR) were calculated by dividing fluxes in all respiratory states of the SUIT protocol by CI&II‐linked ETS capacity taken as a common reference state [38]. Flux control factors (FCF) express the change of flux in a single step of the SUIT protocol normalized to a reference state with higher flux [37]. Stimulation of ADP‐saturated OXPHOS capacity, P, by optimum uncoupler concentration yields ETS capacity, E. The corresponding FCF is the apparent excess ETS capacity calculated as (E‐P)/E = 1‐P/E. The respiratory acceptor control ratio (RCR = P/L) was obtained in the CI‐linked substrate state. For statistical analysis RCR was transformed to its respective FCF, which is the OXPHOS coupling efficiency calculated as (P‐L)/P = 1‐L/P. The FCF for CI‐linked substrates stimulating CII‐linked respiration was measured as 1–CII/CI&II, in the ETS state in the SUIT protocol. The corresponding FCF for the CII‐linked substrate was calculated as 1–CI/CI&II, determined in the OXPHOS state in the SUIT protocol. Two replicates per cell line were analysed and the mean value per condition was used for the statistical analysis to compare the groups.
Statistical analysis
For statistical analyses we used GraphPad Prism 8 Software. Comparisons between groups were done by t‐test, one– or two‐way ANOVA followed by post hoc analysis, depending on the dataset as indicated. A p‐value lower than 0.05 was considered significant.
Results
Cell model characterization
Two iPSC lines from two MSA‐P patients (MSA1, MSA2) and two iPSC lines from an age‐matched healthy control (C1, C2) were generated, characterized and used for the differentiation and further analysis of MSA and control NCAM‐positive NPCs (Figure 1). Skin fibroblasts were reprogrammed with nonintegrative SeV reprogramming vectors encoding Klf4, Oct4, Sox2 and c‐Myc. The generated iPSCs showed normal karyotype (Figure 1B) and expressed genes of pluripotency at similar levels to hESCs (Figure 1C‐E). Furthermore, the pluripotency of the generated lines was demonstrated by differentiation towards ectodermal, mesodermal and endodermal fate (Figure 1F‐H). All cell lines were SeV free after 20 passages and used for the differentiation and sorting of NCAM‐positive NPCs ((Figure 1I‐K). No differences in the differentiation and yield of NCAM‐positive NPCs were identified between the control (40 ± 16%, n = 7 independent differentiation and sorting experiments) and MSA (51 ± 28%, n = 6 independent differentiation and sorting experiments) lines. The sorted NCAM‐positive NPCs were kept in culture and characterized by immunocytochemistry. In all cases, the NPCs expressed NCAM and β‐III‐tubulin (Figure 1L). A few cells were positive for Olig‐2 (Figure 1L), but no immunoreactivity for GFAP was detected (not shown).
Figure 1.
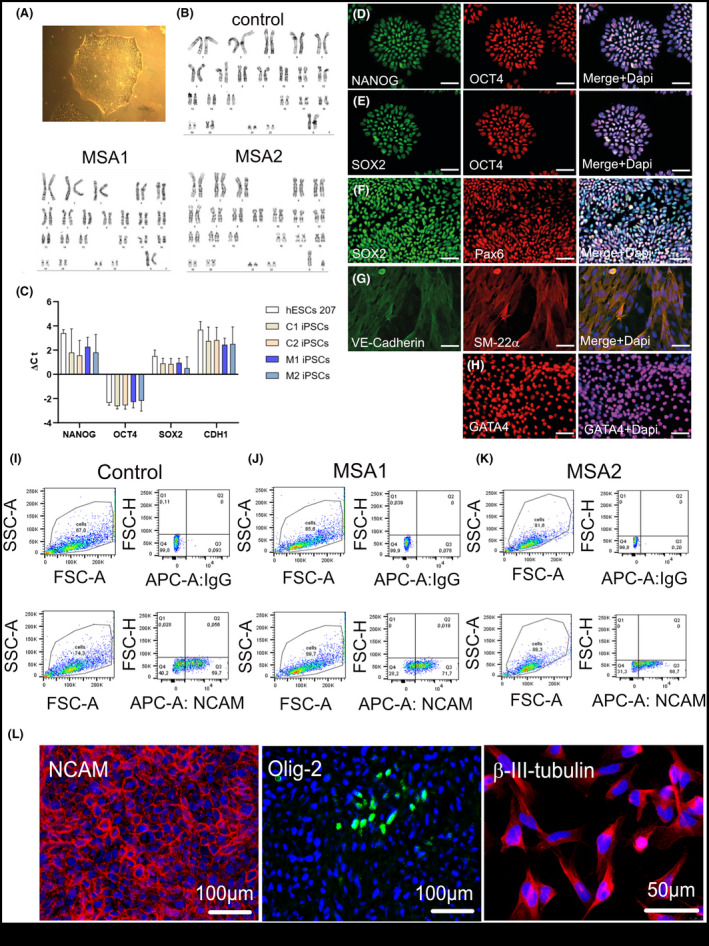
Generation and characterization of iPSCs and NPCs. (A) iPSCs colony of a validated MSA1 line (B) Representative karyotypes of iPSC lines. (C) Histogram depicting the ΔCt of pluripotent genes iPSC lines (C = control; M = MSA; hESC, human embryonic stem cells used as a reference)). Data are presented as Mean ± SD. (D, E) Expression of pluripotency markers NANOG, OCT4 and SOX2 in the generated iPSCs. (F) Ectoderm differentiation of iPSCs and PAX6 expression. (G) Mesoderm differentiation of iPSCs and VE‐Cadherin and SM22α expression. (H) Endoderm differentiation of iPSCs and GATA4 expression. Scale bars 100 µm. (I‐K) FACS plots demonstrating the sorted NCAM‐positive NPCs of control and MSA lines with the respective IgG control. These NPCs were used for the following experiments. (L) Expression of NCAM, Olig‐2, and β‐III‐tubulin in sorted NCAM‐positive NPCs. Counterstaining with DAPI (blue)
Mitochondrial elongation in MSA‐derived NPCs is not linked to changes in respiration
Mitochondrial deficits have been suggested to play a role in MSA pathogenesis [10, 15, 39, 40, 41], therefore we first focussed on the mitochondrial morphology and function in NPCs. We imaged the general morphology and distribution of mitochondria by Tom20 immunocytochemistry (Figure 2A). Mitochondrial morphology was scored as previously suggested by Rambold and co‐workers [42]. We identified a redistribution from predominant intermediate phenotype in control cells towards more tubulated mitochondria in MSA cells (Figure 2B). We then estimated the relative area of mitochondria per cell in the different cell lines but detected no significant differences (Figure 2C). To explore further whether the observed changes in mitochondrial morphology are reflected by their respiratory function, we used high resolution respirometry. This method provides high sensitivity to measure oxygen flux even at 1pmol/s per cm3 in small samples and is offered for diagnostic tests of mitochondrial function in cell culture [43]. The extensive high resolution respirometry analysis identified no significant differences in the respiration of MSA vs control NPCs at any step of the coupling and substrate modulation to measure OXPHOS and ETS capacities (Figure 3).
Figure 2.
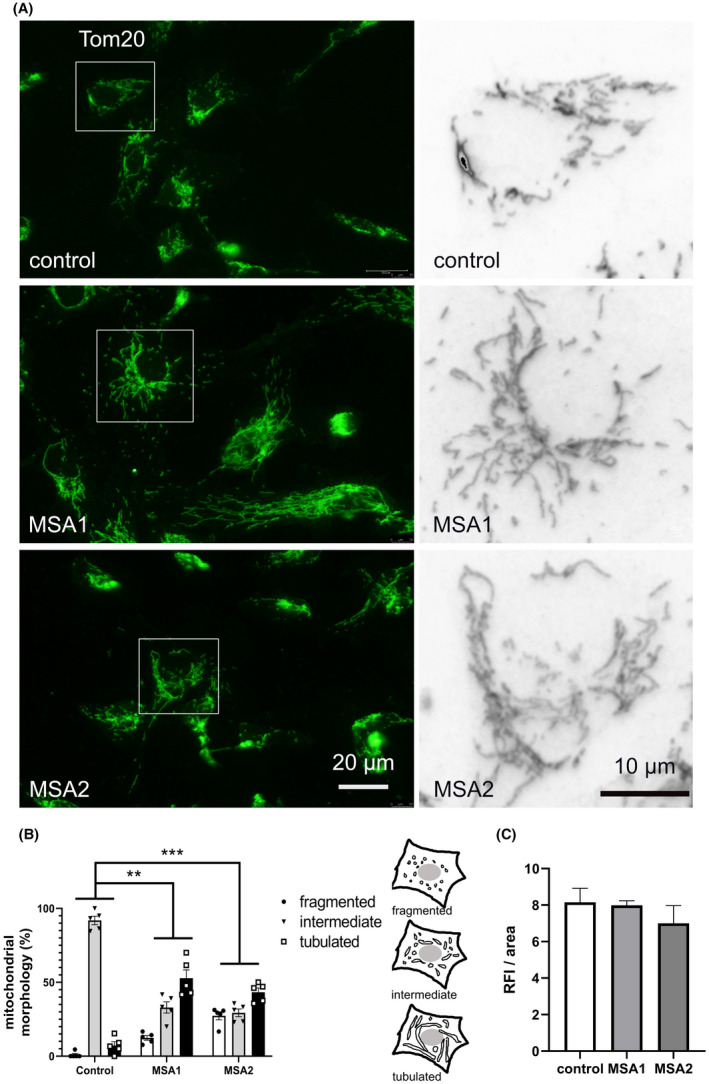
Shift of the mitochondrial morphology in MSA NPCs. (A) Mitochondria were visualized with Tom20 immunofluorescence in control and MSA‐derived NPCs. Insets are shown at higher magnification on the right. (B) The mitochondrial morphology was scored as previously described [42] in five samples per line: fragmented – mainly short and round; intermediate – round and short tubulated; tubular – long with higher connectivity. Data are presented as mean ± SEM. Two‐way ANOVA with Tukey’s posthoc correction for multiple comparison. **P < 0.01; ***P < 0.001. (C) The amount of mitochondria per cell was estimated by measuring the relative fluorescence intensity (RFI) per cell area using ImageJ. Data are presented as mean ± SEM. One‐way ANOVA comparison between the groups showed no significant differences (P = 0.578)
Figure 3.
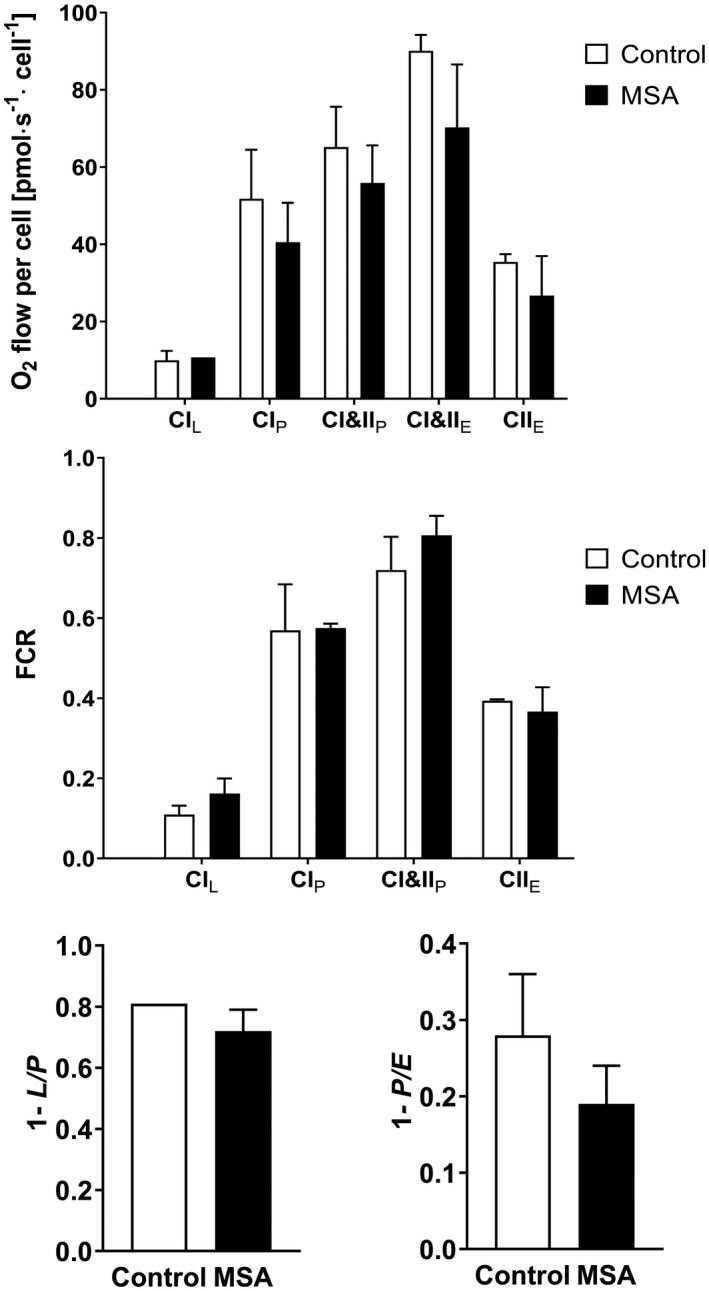
High resolution respirometry of MSA and control NPCs. (A) Absolute oxygen consumption per cell of control and MSA NPCs including CI‐linked (CIL) LEAK state after addition of CI substrate without ADP; CI‐linked (CIP) and CI&II‐linked (CI&IIP) OXPHOS capacity as well as CI&II‐linked (CI&IIE) electron transfer system (ETS) capacity and CII‐linked ETS capacity after addition of an uncoupler (CIIE). (B) Flux control ratios (FCR) for all respiratory states referenced to CI&IIE ETS capacity. (C) Flux control factor (FCF) representing the OXPHOS coupling efficiency (1‐L/P); (D) FCF representing the excess of ETS capacity (1‐P/E). Data are presented as mean ± range. No group differences were observed between the single parameters by two‐way ANOVA or t‐test
Oxidative stress conditions reveal higher susceptibility of MSA‐derived NPCs
Next, we sought to understand whether the morphological changes of mitochondria in MSA NPCs might indicate an adaptation mechanism of diseased cells to cope with energy stress and whether additional mild oxidative stress could be equally well compensated in MSA and healthy control cells. We used different doses of luperox, an organic peroxide, in order to set a threshold of oxidative stress in control and MSA NPCs. MSA NPCs showed an increased generation of ROS as compared to healthy control NPCs (Figure 4A). This increase was significant even after treatment with the lowest dose of luperox used here, while control cells showed significant levels of oxidative stress only after exposure to the highest dose (Figure 4B).
Figure 4.
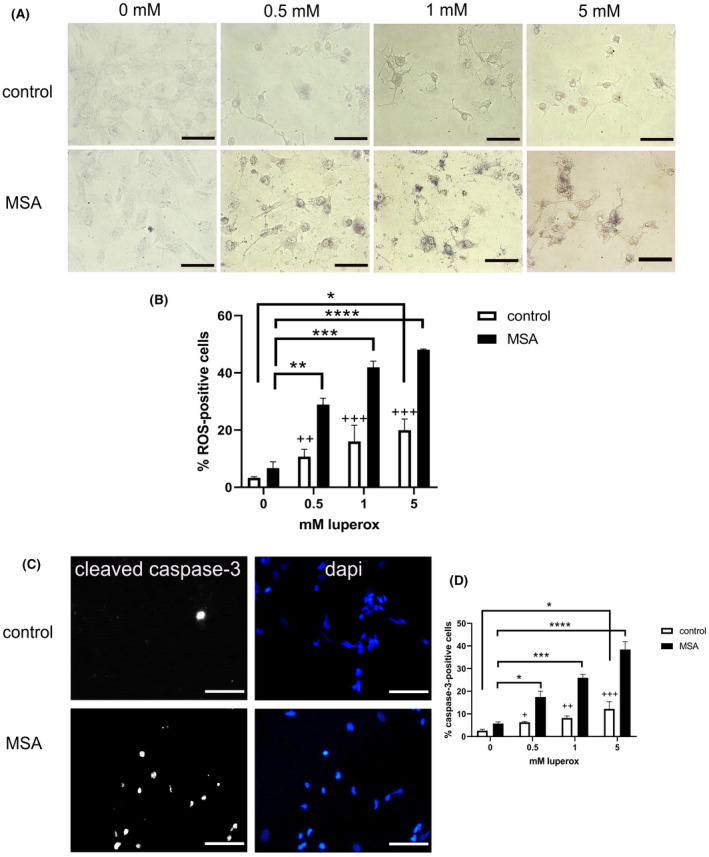
Exogenous oxidative stress in NPCs. (A) Intracellular ROS production (dark blue staining) after exposure to increasing concentrations (0‐5 mM) of luperox in control and MSA NPCs. Scale bars, 50 µm. (B) Percentage of cells with increased dark blue staining (ROS) after exposure to different luperox concentrations. Data are presented as mean ± range. Data were analysed with two‐way ANOVA followed by Sidak’s posthoc multiple comparison test. Crosses indicate the comparison of control and MSA NPCs exposed to the same concentration of luperox: ++P < 0.01; +++P < 0.001. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (C) Representative images of cleaved caspase‐3 activation in control and MSA NPCs exposed to 5 mM luperox. Scale bars, 100 µm. (D) Percentage of cells with positive cleaved caspase‐3 staining in the nucleus. Data are presented as mean ± range. Analysis was performed with two‐way ANOVA followed by Sidak’s posthoc multiple comparison test. Crosses indicate the comparison of control and MSA NPCs exposed to the same concentration of luperox: +P < 0.05; ++P < 0.01; +++P < 0.001. *P < 0.05; ***P < 0.001; ****P < 0.0001
The increased production of ROS was paralleled by increased cleavage of caspase‐3, an effector caspase responsible for the last steps of programmed cell death. The percentage of cells positive for cleaved caspase‐3 was significantly increased in MSA NPCs after exposure to the lowest concentration of luperox, while control cells showed significant increase of caspase‐3 cleavage only after exposure to the highest used dose of luperox (Figure 4C,D).
MSA‐derived NPCs show nuclear translocation of α‐synuclein without changes in the level of SNCA expression
Finally, we were interested to define, if α‐synuclein is expressed in NCAM‐positive NPCs and whether any disease‐related changes may be identified in this very early neural differentiation stage. qRT‐PCR analysis showed no difference in the level of SNCA expression in NCAM‐positive NPCs derived from MSA (ΔCt 7.6 ± 0.5, n = 3) and control (ΔCt 7.8 ± 0.5, n = 3). Interestingly, immunocytochemistry of control NPCs identified cells with a grainy pattern of positive staining throughout the cytoplasm and the nucleus (Figure 5A) as previously observed in mouse neuronal stem cells and different neural cell lines [44]. In the MSA NPCs, we observed translocation of α‐synuclein to the nucleus showing intense nuclear staining (Figure 5A). In both MSA cases, the α‐synuclein nuclear translocation at baseline was significantly higher as compared to control NPCs (Figure 5B). However, no α‐synuclein aggregates or signs of changed α‐synuclein phosphorylation were identified (not shown).
Figure 5.
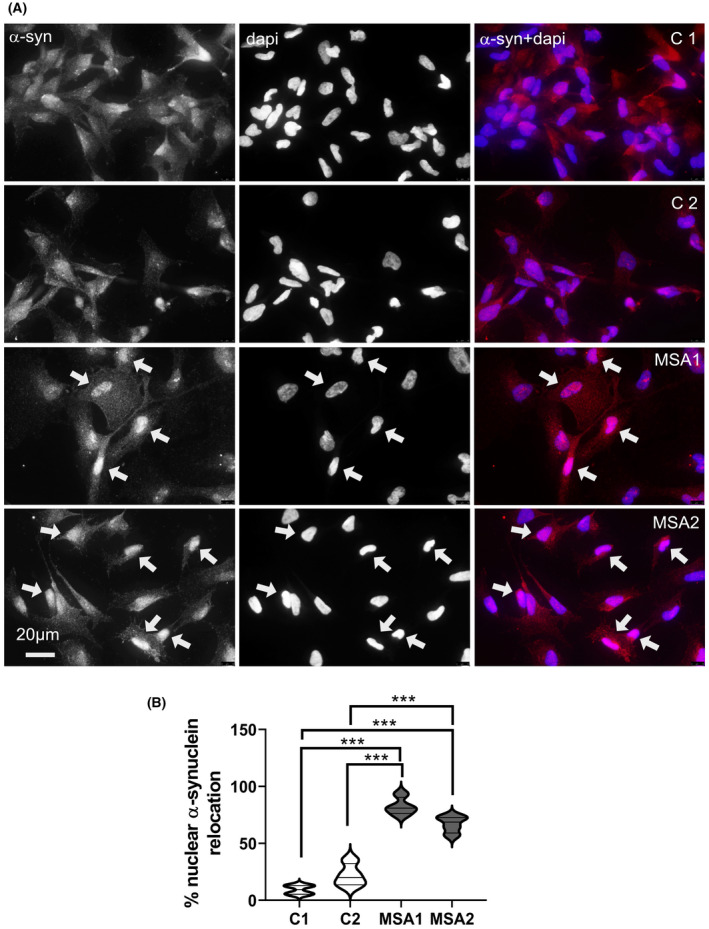
α‐synuclein expression in MSA and control NPCs. (A) Representative immunocytochemistry images show distribution of α‐synuclein in control NPCs C1 and C2 (cytoplasm and nucleus), and two MSA NPC lines (strong nuclear translocation (arrows)). (B) The percentage of NPCs with nuclear translocation of α‐synuclein in control and MSA lines in four samples per line. One‐way ANOVA with Tukey’s multiple comparison posthoc test. ***P < 0.001
Discussion
In this work, we generated NCAM‐positive NPCs from MSA patients and an age‐matched healthy control using the SeV reprogramming system for the generation of iPSCs and a dual SMAD inhibition protocol to generate NPCs. Due to its nonintegrating nature, the virus was diluted by passaging and did not interfere with our further experiments to assess disease‐related fingerprints in MSA‐specific iPSC‐derived NPCs. We show here morphological changes of the mitochondria and nuclear translocation of α‐synuclein in MSA‐derived NPCs without evident changes in respiration, cell viability or differentiation capacity at baseline. However, we identify for the first time a higher susceptibility of MSA‐specific NPCs to oxidative stress as compared to control NPCs, in a dose‐dependent manner.
As previously shown and corroborated in our experiments, PSA‐NCAM‐targeted sorting elicits highly homogeneous and expandable population of NPCs while retaining a typical morphology and molecular characteristics including expression of mostly neuronal (NCAM, β‐III‐tubulin) but also glial (Olig‐2) progenitor markers [45]. The in vivo behaviour of sorted human NCAM‐positive NPCs supports the broad differentiation potential of these cells toward various types of neurons, oligodendrocytes and astrocytes [45]. Therefore, this specific subpopulation of NPCs can inform about disease‐related deficits which may precede/trigger further dysfunction and pathology of neuronal and glial cells as is the case in MSA [21].
Our data indicate the existence of a pathological background in MSA‐derived NPCs, which is compensated functionally at baseline. Despite the changes in the mitochondrial phenotype in MSA‐derived NPCs there were no significant abnormalities in the respiration as measured by high resolution respirometry. Importantly, this method provides dynamic and highly sensitive measurement of metabolic flux in contrast to static determination of molecular components. Furthermore, it yields an integrative measure of the dynamics of complex coupled metabolic pathways and not simple monitoring of isolated enzyme activities [43]. This approach provides a more relevant estimate of the general functional state of the respiratory metabolic system, while large defects in single enzymes [40, 46] may be misleading and result in minor flux changes due to threshold effects. Mitochondrial elongation, which we describe here in MSA NPCs, has been previously reported to represent a putative protective adaptation mechanism against cellular damage [42, 47, 48]. Despite the changes in the morphology, we did not identify differences in the amount of mitochondria per cell, supporting the similar functional profile in high resolution respirometry of control and MSA lines. Interestingly, a previous study suggested that mitochondrial amount and respiratory capacity may be changed in MSA iPSC‐derived dopaminergic [40] suggesting a possible ‘sign of progression’ of the disease in a dish.
In line with this, we report here that the apparent balance of the redox homeostasis in MSA NPCs is easily disrupted by even low levels of exogenous oxidative stress. We selected oxidative stress for its relevance to the pathogenesis of MSA [49, 50, 51, 52] and for the possibility to precisely titrate the cellular exposure in vitro. Our data support the notion of higher susceptibility of MSA NPCs to exogenous stress. Despite the fact that the disease is mostly sporadic and no specific genetic aberrations are detected in GWAS [9], our data suggest that other stress response mechanisms may trigger cellular disbalance and the following pathology in MSA. We examined α‐synuclein expression, since this protein is considered a major player in the pathogenesis of MSA and other synucleinopathies. In these early developmental stages, we did not identify different levels of SNCA gene expression between control and MSA‐derived NPCs, supporting previous studies which concluded no causal relationship between the SNCA gene and MSA [27, 53]. However, we describe here a translocation of α‐synuclein to the nucleus of MSA NPCs. In control NPCs, we observed α‐synuclein expression through the soma and nucleus as described in earlier analyses [44, 54]. Intriguingly, we identified for the first time a nuclear translocation of α‐synuclein in MSA NPCs. Earlier studies by Outeiro and colleagues [44] showed that translocation of α‐synuclein to the nucleus may occur in cell lines under stress conditions, a finding which we confirmed in our control NPCs (Figure S1). For this reason, we believe that the nuclear translocation of α‐synuclein, like the mitochondrial morphological changes demonstrated in MSA NPCs, is yet another early sign of cellular stress. Previous experiments suggested that in cells overexpressing wild‐type α‐synuclein the nuclear location was associated with increased S129 phosphorylation and amyloid formation of α‐synuclein [44]. We were unable to identify similar changes in NPCs derived from MSA iPSCs, however the observed nuclear translocation in these cells may indicate an early event preceding α‐synuclein hyperphosphorylation and inclusion formation typical for MSA pathology [4]. The nuclear location of α‐synuclein has been associated with epigenetic modulation and DNA interaction resulting in transcriptional deregulation [44, 55, 56, 57], especially downregulation of cell cycle‐related genes, therefore supporting the notion that early changes in α‐synuclein cellular location in MSA NPCs may contribute to the observed increased susceptibility to exogenous stress.
All these facts let us hypothesize that in MSA, the initial α‐synuclein pathology is not related to misfolding, aggregate formation or prion properties of the protein, but rather associates with initial early relocation to the nucleus. Through the interactions of α‐synuclein with DNA and histones, it may trigger a preclinical condition which remains counterbalanced (no changes in respiration, no increased apoptotic cell death or ROS production at baseline). However, MSA NPCs are less prone to compensate exogenous stress which results in elicitation of the disease phenotype.
We need to acknowledge the limitations of this study related to the low number of lines used in our experiments linked to the restricted accessibility of cases suffering from the rare disease MSA and to its challenging diagnosis. At this stage, we are not in the position to increase the number of cases, however we believe that the reported data represent a very intriguing observation which deserves further extended evaluation in larger cohorts of patients (possibly in a multicentre study). A rare sporadic disease will necessitate a much larger effort to confirm or discard the observation made here, but we believe that through this report we can trigger the attention to events in MSA, that may precede α‐synuclein accumulation. Further studies in an extended cohort of MSA patients will be needed to expand the current observations and define the exact role of α‐synuclein nuclear translocation in MSA. However, based on the previous observations of Outeiro and colleagues [44] about the subcellular dynamics of α‐synuclein and associated transcriptional deregulation as well as supported by the presence of nuclear inclusions of α‐synuclein in MSA [4, 5, 58], we believe that our findings shed light on the early cellular dysfunction in MSA laying out a novel perspective of the approaches to study this devastating disease. In addition, further experimental data in differentiated neuronal and glial cultures derived from MSA iPSCs will further expand this line of research towards understanding the early stages of MSA pathogenesis.
Ethical Approval
The research has been given ethics approval AN2016‐0105 362/4.16 by the local Ethics Committee.
Author Contributions
MHV: acquisition and analysis of data; drafting and revising the manuscript. AHG: acquisition and analysis of data; revising the manuscript. FK: acquisition of clinical data and material; revising the manuscript. RD: acquisition and analysis of data; revising the manuscript. SB: conception; acquisition of clinical data and material, revising the manuscript. GKW: conception; acquisition of clinical data, interpretation of data; revising the manuscript. NS: conception and design; analysis, and interpretation of data; drafting and revising the manuscript. All authors read and approved the final manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12661.
Supporting information
Table S1. Demographic data.
Table S2. TaqMan Gene Expression Assays used in the study.
Table S3. Antibodies used in ICC
Table S4. Antibodies used for FACS.
Figure S1. Nuclear translocation of α‐synuclein after exposure of NCAM‐positive NPCs derived from a healthy control to oxidative stress (luperox) supporting previous observations by Pinho et al 2019 [44].
Acknowledgement
This work was supported by grant F4414 of the Austrian Science Fund (FWF).
Herrera‐Vaquero M., Heras‐Garvin A., Krismer F., Deleanu R., Boesch S., Wenning G. K. and Stefanova N. (2021) Neuropathology and Applied Neurobiology 47, 268–282 Signs of early cellular dysfunction in multiple system atrophy
Data availability statement
The data that support the findings of this study are available in the main body and the supplementary material of this article.
References
- 1. Fanciulli A, Wenning GK. Multiple‐system atrophy. N Engl J Med 2015; 372: 249–63 [DOI] [PubMed] [Google Scholar]
- 2. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71: 670–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013; 12: 264–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koga S, Dickson DW. Recent advances in neuropathology, biomarkers and therapeutic approach of multiple system atrophy. J Neurol Neurosurg Psychiatry 2018; 89: 175–84 [DOI] [PubMed] [Google Scholar]
- 5. Wakabayashi K, Takahashi H. Cellular pathology in multiple system atrophy. Neuropathology 2006; 26: 338–45 [DOI] [PubMed] [Google Scholar]
- 6. Valera E, Masliah E. The neuropathology of multiple system atrophy and its therapeutic implications. Autonomic neuroscience: basic & clinical 2018; 211: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stefanova N. Translational therapies for multiple system atrophy: Bottlenecks and future directions. Auton Neurosci 2018; 211: 7–14 [DOI] [PubMed] [Google Scholar]
- 8. Stefanova N, Bucke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009; 8: 1172–8 [DOI] [PubMed] [Google Scholar]
- 9. Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, et al. A genome‐wide association study in multiple system atrophy. Neurology 2016; 87: 1591–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collaboration TM‐SAR. Mutations in COQ2 in Familial and Sporadic Multiple‐System Atrophy. N Engl J Med 2013; 369: 233–44 [DOI] [PubMed] [Google Scholar]
- 11. Ogaki K, Fujioka S, Heckman MG, Rayaprolu S, Soto‐Ortolaza AI, Labbe C, et al. Analysis of COQ2 gene in multiple system atrophy. Mol Neurodegener 2014; 9: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen YP, Zhao B, Cao B, Song W, Guo X, Wei QQ, et al. Mutation scanning of the COQ2 gene in ethnic Chinese patients with multiple‐system atrophy. Neurobiol Aging 2015; 36: 1222–11 [DOI] [PubMed] [Google Scholar]
- 13. Schottlaender LV, Houlden H. Mutant COQ2 in multiple‐system atrophy. N Engl J Med 2014; 371: 81 [DOI] [PubMed] [Google Scholar]
- 14. Sharma M, Wenning G, Kruger R. Mutant COQ2 in multiple‐system atrophy. N Engl J Med 2014; 371: 80–1 [DOI] [PubMed] [Google Scholar]
- 15. Maeda N, Honda H, Suzuki SO, Fujii N, Kira JI, Iwaki T. Mitochondrial dysfunction and altered ribostasis in hippocampal neurons with cytoplasmic inclusions of multiple system atrophy. Neuropathology 2018; 38: 361–71 [DOI] [PubMed] [Google Scholar]
- 16. Bettencourt C, Foti SC, Miki Y, Botia J, Chatterjee A, Warner TT, et al. White matter DNA methylation profiling reveals deregulation of HIP1, LMAN2, MOBP, and other loci in multiple system atrophy. Acta Neuropathol 2020; 139: 135–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stefanova N, Wenning GK. Animal models of multiple system atrophy. Clin Auton Res 2015; 25: 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stefanova N, Reindl M, Poewe W, Wenning GK. In vitro models of multiple system atrophy. Mov Disord 2005; 20(Suppl 12): S53–6 [DOI] [PubMed] [Google Scholar]
- 19. Abati E, Di Fonzo A, Corti S. In vitro models of multiple system atrophy from primary cells to induced pluripotent stem cells. J Cell Mol Med 2018; 22: 2536–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Monzio Compagnoni G, Di Fonzo A. Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives. Acta neuropathologica communications 2019; 7: 113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stefanova N, Wenning GK. Review: Multiple system atrophy: emerging targets for interventional therapies. Neuropathol Appl Neurobiol 2016; 42: 20–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stefanova N, Schanda K, Klimaschewski L, Poewe W, Wenning GK, Reindl M. Tumor necrosis factor‐alpha‐induced cell death in U373 cells overexpressing alpha‐synuclein. J Neurosci Res 2003; 73: 334–40 [DOI] [PubMed] [Google Scholar]
- 23. Stefanova N, Klimaschewski L, Poewe W, Wenning GK, Reindl M. Glial cell death induced by overexpression of alpha‐synuclein. J Neurosci Res 2001; 65: 432–8 [DOI] [PubMed] [Google Scholar]
- 24. Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, et al. Evidence for alpha‐synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci USA 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peelaerts W, Bousset L, Baekelandt V, Melki R. a‐Synuclein strains and seeding in Parkinson's disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences. Cell Tissue Res 2018; 373: 195–212 [DOI] [PubMed] [Google Scholar]
- 26. Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. alpha‐Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015; 522: 340–4 [DOI] [PubMed] [Google Scholar]
- 27. Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, et al. Alpha‐synuclein mRNA expression in oligodendrocytes in MSA. Glia 2014; 62: 964–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shahnawaz M, Mukherjee A, Pritzkow S, Mendez N, Rabadia P, Liu X, et al. Discriminating α‐synuclein strains in Parkinson's disease and multiple system atrophy. Nature 2020; 578: 273–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α‐synuclein filaments from multiple system atrophy. Nature 2020. 10.1038/s41586-020-2317-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van der Perren A, Gelders G, Fenyi A, Bousset L, Brito F, Peelaerts W, et al. The structural differences between patient‐derived α‐synuclein strains dictate characteristics of Parkinson's disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol 2020. 10.1007/s00401-020-02157-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Hojrup P, et al. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol 2007; 171: 1291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wenning GK, Stefanova N, Jellinger KA, Poewe W, Schlossmacher MG. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol 2008; 64: 239–46 [DOI] [PubMed] [Google Scholar]
- 33. Mavroeidi P, Arvanitaki F, Karakitsou AK, Vetsi M, Kloukina I, Zweckstetter M, et al. Endogenous oligodendroglial alpha‐synuclein and TPPP/p25α orchestrate alpha‐synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol 2019; 138: 415–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eigentler A, Boesch S, Schneider R, Dechant G, Nat R. Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev 2013; 22: 3271–82 [DOI] [PubMed] [Google Scholar]
- 35. Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, et al. Toll‐like receptor 4 is required for alpha‐synuclein dependent activation of microglia and astroglia. Glia 2013; 61: 349–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reed‐Geaghan EG, Savage JC, Hise AG, Landreth GE. CD14 and Toll‐Like Receptors 2 and 4 Are Required for Fibrillar Aβ‐Stimulated Microglial Activation. J Neurosci 2009; 29: 11982–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burtscher J, Zangrandi L, Schwarzer C, Gnaiger E. Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high‐resolution respirometry. Mitochondrion 2015; 25: 104–12 [DOI] [PubMed] [Google Scholar]
- 38. Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol 2009; 41: 1837–45 [DOI] [PubMed] [Google Scholar]
- 39. Gu M, Gash MT, Cooper JM, Wenning GK, Daniel SE, Quinn NP, et al. Mitochondrial respiratory chain function in multiple system atrophy. Mov Disord 1997; 12: 418–22 [DOI] [PubMed] [Google Scholar]
- 40. Monzio Compagnoni G, Kleiner G, Samarani M, Aureli M, Faustini G, Bellucci A, et al. Mitochondrial Dysregulation and Impaired Autophagy in iPSC‐Derived Dopaminergic Neurons of Multiple System Atrophy. Stem Cell Reports 2018; 11: 1185–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakamoto FK, Okamoto S, Mitsui J, Sone T, Ishikawa M, Yamamoto Y, et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC‐derived neurons from patients with multiple‐system atrophy. Sci Rep 2018; 8: 14215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rambold AS, Kostelecky B, Elia N, Lippincott‐Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 2011; 108: 10190–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pesta D, Gnaiger E. High‐resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods in molecular biology (Clifton, NJ) 2012; 810: 25–58 [DOI] [PubMed] [Google Scholar]
- 44. Pinho R, Paiva I, Jercic KG, Fonseca‐Ornelas L, Gerhardt E, Fahlbusch C, et al. Nuclear localization and phosphorylation modulate pathological effects of alpha‐synuclein. Hum Mol Genet 2019; 28: 31–50 [DOI] [PubMed] [Google Scholar]
- 45. Kim DS, Lee DR, Kim HS, Yoo JE, Jung SJ, Lim BY, et al. Highly pure and expandable PSA‐NCAM‐positive neural precursors from human ESC and iPSC‐derived neural rosettes. PLoS One 2012; 7: e39715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Monzio Compagnoni G, Kleiner G, Bordoni A, Fortunato F, Ronchi D, Salani S, et al. Mitochondrial dysfunction in fibroblasts of Multiple System Atrophy. Biochim Biophys Acta Mol Basis Dis 2018; 1864: 3588–97 [DOI] [PubMed] [Google Scholar]
- 47. Li J, Huang Q, Long X, Guo X, Sun X, Jin X, et al. Mitochondrial elongation‐mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 2017; 36: 4901–12 [DOI] [PubMed] [Google Scholar]
- 48. Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab 2015; 22: 207–18 [DOI] [PubMed] [Google Scholar]
- 49. Kaindlstorfer C, Jellinger KA, Eschlbock S, Stefanova N, Weiss G, Wenning GK. The Relevance of Iron in the Pathogenesis of Multiple System Atrophy: A Viewpoint. J Alzheimers Dis 2018; 61: 1253–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stefanova N, Georgievska B, Eriksson H, Poewe W, Wenning GK. Myeloperoxidase inhibition ameliorates multiple system atrophy‐like degeneration in a transgenic mouse model. Neurotox Res 2012; 21: 393–404 [DOI] [PubMed] [Google Scholar]
- 51. Kikuchi A, Takeda A, Onodera H, Kimpara T, Hisanaga K, Sato N, et al. Systemic increase of oxidative nucleic acid damage in Parkinson's disease and multiple system atrophy. Neurobiol Dis 2002; 9: 244–8 [DOI] [PubMed] [Google Scholar]
- 52. Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective alpha‐synuclein nitration in synucleinopathy lesions. Science 2000; 290: 985–9 [DOI] [PubMed] [Google Scholar]
- 53. Katzeff JS, Phan K, Purushothuman S, Halliday GM, Kim WS. Cross‐examining candidate genes implicated in multiple system atrophy. Acta neuropathologica communications. 2019; 7: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron‐specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 1988; 8: 2804–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kontopoulos E, Parvin JD, Feany MB. Alpha‐synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 2006; 15: 3012–23 [DOI] [PubMed] [Google Scholar]
- 56. Paiva I, Pinho R, Pavlou MA, Hennion M, Wales P, Schütz AL, et al. Sodium butyrate rescues dopaminergic cells from alpha‐synuclein‐induced transcriptional deregulation and DNA damage. Hum Mol Genet 2017; 26: 2231–46 [DOI] [PubMed] [Google Scholar]
- 57. Sturm E, Fellner L, Krismer F, Poewe W, Wenning GK, Stefanova N. Neuroprotection by Epigenetic Modulation in a Transgenic Model of Multiple System Atrophy. Neurotherapeutics 2016; 13: 871–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Burn DJ, Jaros E. Multiple system atrophy: cellular and molecular pathology. Mol Pathol 2001; 54: 419–26 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographic data.
Table S2. TaqMan Gene Expression Assays used in the study.
Table S3. Antibodies used in ICC
Table S4. Antibodies used for FACS.
Figure S1. Nuclear translocation of α‐synuclein after exposure of NCAM‐positive NPCs derived from a healthy control to oxidative stress (luperox) supporting previous observations by Pinho et al 2019 [44].
Data Availability Statement
The data that support the findings of this study are available in the main body and the supplementary material of this article.