

Abstract
Carotid artery atherosclerotic disease (CAAD) is a risk factor for stroke. We used a genome‐wide association (GWAS) approach to discover genetic variants associated with CAAD in participants in the electronic Medical Records and Genomics (eMERGE) Network. We identified adult CAAD cases with unilateral or bilateral carotid artery stenosis and controls without evidence of stenosis from electronic health records at eight eMERGE sites. We performed GWAS with a model adjusting for age, sex, study site, and genetic principal components of ancestry. In eMERGE we found 1793 CAAD cases and 17,958 controls. Two loci reached genome‐wide significance, on chr6 in LPA (rs10455872, odds ratio [OR] (95% confidence interval [CI]) = 1.50 (1.30–1.73), p = 2.1 × 10−8) and on chr7, an intergenic single nucleotide variant (SNV; rs6952610, OR (95% CI) = 1.25 (1.16–1.36), p = 4.3 × 10−8). The chr7 association remained significant in the presence of the LPA SNV as a covariate. The LPA SNV was also associated with coronary heart disease (CHD; 4199 cases and 11,679 controls) in this study (OR (95% CI) = 1.27 (1.13–1.43), p = 5 × 10−5) but the chr7 SNV was not (OR (95% CI) = 1.03 (0.97–1.09), p = .37). Both variants replicated in UK Biobank. Elevated lipoprotein(a) concentrations ([Lp(a)]) and LPA variants associated with elevated [Lp(a)] have previously been associated with CAAD and CHD, including rs10455872. With electronic health record phenotypes in eMERGE and UKB, we replicated a previously known association and identified a novel locus associated with CAAD.
Keywords: carotid artery atherosclerosis, electronic health records, genome‐wide association study
Abbreviations
- CAAD
carotid artery atherosclerosis disease
- CHD
coronary heart disease
- CIMT
carotid initima‐media thickness
- CPT
current procedural terminology
- EHR
electronic health records
- eMERGE
electronic Medical Records and Genomics
- ICD
international classification of disease
- Lp(a)
lipoprotein (a) protein
- MAF
minor allele frequency
- NLP
natural language processing
- PCA
principal components analysis
- PPV
positive predictive value
- SNV
single nucleotide variant
- UKB
United Kingdom biobank
1. INTRODUCTION
Carotid artery atherosclerotic disease (CAAD) is a major cause of ischemic stroke (Goldstein et al., 2006). Risk factors for CAAD include smoking, dyslipidemia, lipoprotein (a) concentration ([Lp(a)]; Clarke et al., 2009), LPA variants (which are associated with [Lp(a)] (Dumitrescu et al., 2011; Ober et al., 2009)), rs1454626 in VLDLR (Crawford et al., 2008), and plasma phospholipid transfer protein activity (Clarke et al., 2009). Relatively little work has been done to identify genetic variants associated with CAAD. Many studies have focused on a correlated trait, carotid intima‐media thickness (CIMT; Forgo et al., 2018). The genetics of another correlated condition, coronary heart disease (CHD), have been well characterized in family and association studies (for a review, see, Khera & Kathiresan, 2017).
CHD and carotid atherosclerosis have related environmental and clinical risk factors, but genetic studies suggest different pathophysiologic mechanisms between them. In one study the consequence of carotid atherosclerosis, ischemic stroke, shared only one genetic risk locus out of 25 with CHD (Lövkvist et al., 2013). In another, ischemic stroke and large artery stroke share three and five loci, respectively, with CHD (Dichgans et al., 2014). This may also be the case within carotid artery‐related phenotypes. For example, different loci are associated with two carotid artery phenotypes associated with atherosclerosis, CIMT and carotid plaque (Bis et al., 2011).
The electronic Medical Records and Genomics (eMERGE) Network is a research network that integrates electronic health records (EHR) and genomic data to drive genomic discovery and advance personalized medicine (McCarty et al., 2011). It allows the identification of phenotypes from many participants using existing health data, providing enough cases to power genome‐wide association studies (GWAS). The purpose of the current study was to identify loci associated with EHR‐defined CAAD using the eMERGE GWAS data set. Data from the UK Biobank (UKB) were used to test the robustness of the associations identified in eMERGE.
2. MATERIALS AND METHODS
2.1. The eMERGE Network
The eMERGE Network is a consortium funded by National Human Genome Research Institute (NHGRI), National Institutes of Health (NIH) since 2007. It includes sites across the United States, with the purpose of linking EHR and genomic data for genomic medicine research. Phenotypes are extracted from EHR by algorithms developed by investigators and applied at each site. It is currently in Phase 3 and includes over 80,000 genotyped participants from nine adults and three pediatric institutions (Chisholm, 2013; Crawford et al., 2014; Gottesman et al., 2013). Enrollment was approved by review boards at each study site and consent obtained, abiding by the Declaration of Helsinki principles.
2.2. Phenotype algorithm
We developed the CAAD phenotype algorithm, including a portable natural language processing (NLP) system at Kaiser Permanente Washington/University of Washington (KPWA/UW) using patients from KPWA. Details of the development of the algorithm and its implementation, including a flow diagram are given in the Supplemental Methods. The algorithm extracts the extent of carotid stenosis from imaging reports. We developed it using the approach previously described for creating eMERGE phenotypes (Kho et al., 2012; Kullo et al., 2010). Inclusion criteria included a minimum number of contacts with a patient (in preceding 5 years to most recent record, two records ≥ 1 year apart, or three in different quarters, or four in different months), and minimum age of 18 years. Cases were defined by one or more of the following criteria: more than or equal to 16% carotid artery stenosis (unilaterally or bilaterally) on carotid imaging, which is a standard ultrasound cutoff (Taylor & Strandness, 1987), or at least one International Classification of Disease (ICD)‐9 or Current Procedural Terminology (CPT) code indicating an endarterectomy (ICD‐9 00.63, 38.12, CPT 35301, 35390), or at least two records indicating CAAD (ICD‐9 433.1) more than or equal to 30 days apart. Cases were excluded if they had lab evidence of maximum total cholesterol greater than 400 mg/dl, which would suggest monogenic familial hyperlipidemia, or a pure hypercholesterolemia diagnosis (ICD‐9 272.0). We used this exclusion because for individuals with carotid stenosis and such high cholesterol, their genetic risk is likely driven by hypercholesterolemia‐associated variants. Controls were defined as having an imaging study showing the lowest category of stenosis bilaterally (0%–15% stenosis) or no evidence of carotid artery imaging, no CAAD diagnoses, and no carotid repair procedures. We also collected index age (age of first CAAD diagnosis for cases and age at latest record for controls), sex, self‐reported race and ethnicity, smoking status, CHD defined by at least two ICD‐9 codes for CHD on encounters at least 60 days apart, and body mass index (BMI).
Algorithm evaluation found that 24 out of 25 cases (96% case positive predictive value [PPV]) and all 25 controls (100% control PPV) were validated with chart review at KPWA, and 100% case and control PPV in 20 cases and 20 controls at Marshfield Clinic (Supplemental Methods). We then applied the developed algorithm to adult participants from the Geisinger, Harvard University, KPWA/University of Washington, Marshfield, Mayo, Mt. Sinai Health System, Northwestern, and Vanderbilt University eMERGE sites. Demographics are presented by case status in Table 1, and by site in Table S1.
Table 1.
Demographics of eMERGE participants included in the CAAD GWAS, separated by CAAD case‐control status
| Case (N = 1793) | Control (N = 17,958) | Total (N = 19,751) | |
|---|---|---|---|
| Age (years) | |||
| Mean (SD) | 71.37 (9.70) | 63.45 (16.08) | 64.16 (15.77) |
| Range | 29.00–98.00 | 19.00–106.00 | 19.00–106.00 |
| Sex | |||
| Female | 667 (37.2%) | 10390 (57.9%) | 11057 (56.0%) |
| Male | 1126 (62.8%) | 7568 (42.1%) | 8694 (44.0%) |
| Racea | |||
| N‐Missb | 5 | 6 | 11 |
| African American | 61 (3.4%) | 4056 (22.6%) | 4117 (20.9%) |
| American Indian/Alaskan Native | 4 (0.2%) | 48 (0.3%) | 52 (0.3%) |
| Asian | 11 (0.6%) | 165 (0.9%) | 176 (0.9%) |
| Caucasian | 1664 (93.1%) | 12,214 (68.0%) | 13,878 (70.3%) |
| Native Hawaiian/Pacific Islander | 0 (0.0%) | 5 (0.0%) | 5 (0.0%) |
| Not reported | 4 (0.2%) | 83 (0.5%) | 87 (0.4%) |
| Unknown/Other | 44 (2.5%) | 1381 (7.7%) | 1425 (7.2%) |
| Ethnicitya | |||
| Hispanic/Latino | 35 (2.0%) | 1192 (6.6%) | 1227 (6.2%) |
| Non Hispanic/Latino | 1578 (88.0%) | 16,304 (90.8%) | 17,882 (90.5%) |
| Unknown | 180 (10.0%) | 462 (2.6%) | 642 (3.3%) |
| Ever smoker | |||
| N‐Miss | 350 | 7938 | 8288 |
| Ever | 1134 (78.6%) | 5024 (50.1%) | 6158 (53.7%) |
| Never | 309 (21.4%) | 4996 (49.9%) | 5305 (46.3%) |
| Median BMI (kg/m2) | |||
| N‐Miss | 279 | 6394 | 6673 |
| Mean (SD) | 28.59 (5.41) | 30.08 (7.47) | 29.90 (7.28) |
| Range | 17.86–56.98 | 14.10–80.86 | 14.10–80.86 |
| Coronary heart disease | |||
| N‐Miss | 138 | 3732 | 3870 |
| No | 631 (38.1%) | 11,051 (77.7%) | 11,682 (73.6%) |
| Yes | 1024 (61.9%) | 3175 (22.3%) | 4199 (26.4%) |
| Site | |||
| Geisinger | 591 (33.0%) | 1601 (8.9%) | 2192 (11.1%) |
| Harvard | 138 (7.7%) | 3732 (20.8%) | 3870 (19.6%) |
| Kaiser/University of Washington | 227 (12.7%) | 2260 (12.6%) | 2487 (12.6%) |
| Marshfield | 165 (9.2%) | 1920 (10.7%) | 2085 (10.6%) |
| Mayo | 407 (22.7%) | 235 (1.3%) | 642 (3.3%) |
| Mt. Sinai | 82 (4.6%) | 4279 (23.8%) | 4361 (22.1%) |
| Northwestern | 56 (3.1%) | 506 (2.8%) | 562 (2.8%) |
| Vanderbilt | 127 (7.1%) | 3425 (19.1%) | 3552 (18.0%) |
Abbreviations: BMI, body mass index; CAAD, Carotid artery atherosclerotic disease; eMERGE, electronic Medical Records and Genomics; GWAS, genome‐wide association studies.
Self‐reported race and ethnicity.
Number of missing values.
2.3. Genotype data
We imputed 83,717 eMERGE participants’ genotype data to Haplotype Reference Consortium version r1.1 (McCarthy et al., 2016) with the Michigan Imputation Server (MIS; Das et al., 2016), as described previously (Stanaway et al., 2019). Single nucleotide variants (SNVs) were filtered for call rate more than 2% and individuals filtered for genotype missingness less than 2%. Related individuals were identified by calculating identity by descent with the plink2‐genome (Chang et al., 2015) function. Only one person per family was retained, prioritizing cases over controls. We typed participants in the CAAD analysis on one of 24 different chips. For each SNV, we calculated mean imputation quality R 2 across all chips, using the R 2 values calculated in the MIS imputation. We also calculated minor allele frequency (MAF) among all CAAD analysis participants and within European and African ancestry subgroups. Principal components analysis (PCA) was performed with plink2 PCA approx method (Chang et al., 2015) on the entire eMERGE cohort, and within European and African ancestry subsets (as described in Stanaway et al, 2019).
2.4. Statistical analysis
For the GWAS, we used an additive genotype model and logistic regression in PLINK 1.9 (Chang et al., 2015). We performed analyses in all participants, and in subsets with genetically determined European or African ancestry, identified through the intersection of the k‐means clustering of PC1 and PC2 and the self‐reported or observed ancestry of the participant (Figure S1; Stanaway et al., 2019). Self‐reported Latino/Hispanics were removed from the ancestry stratified analysis. The main analysis included the covariates age, sex, study site, and PC1–10. For the top associated variants, we included BMI, CHD, and smoking status in the model where possible; these were not included in the main analysis because they were not available from all sites. We filtered results shown including only SNVs with MAF more than 0.05 and imputation quality R 2 > .3. We considered GWAS results statistically significant with a p‐value below 5 × 10−8, as a correction for multiple testing, and carried those passing that threshold forward for replication in the UK Biobank. Other statistical analyses and plots were done with R (R Core Team 2018).
We performed phenome wide association study (PheWAS; Denny et al., 2010) on the significantly associated SNV that had no previously known phenotypic associations, rs6952610 in the larger eMERGE cohort (n = 83,717) to determine if other phenotypes may be associated with this SNP.
2.5. UK Biobank
The UK Biobank (UKB) is a population sample of 502,629 participants in the United Kingdom aged 40–69 years at the time of study recruitment (Sudlow et al., 2015). Participants were recruited between 2006 and 2010 from 22 centers across the United Kingdom, in both urban and rural areas, with a broad mixture of socioeconomic backgrounds. All participants provided written, informed consent under each site's local Institutional Review Board. UKB protocols were approved by the National Research Ethics Service Committee, and all ethical regulations were followed. Analyses carried out in UKB specifically for this study were done under approved application 18,120 (Speliotes).
For the UKB, we determined CAAD status via ICD‐10 codes. We specifically assessed for occlusion and stenosis of the carotid artery (ICD‐10 code I65), which includes the subclasses of occlusion and stenosis of the right (I65.21), left (I65.22), bilateral (I65.23), or unspecified (I65.29) carotid arteries. All participants without those codes were included as controls. Due to data limitations, individuals with hypercholesterolemia (max total cholesterol > 400 mg/dl or a coded diagnosis of hypercholesterolemia) were not excluded from the UKB analysis. A total of 934 cases and 408,027 controls had complete genotype, phenotype, and covariate data.
2.6. UKB Genotyping and imputation
Participant genotyping, data collection, and quality control has previously been described (Sudlow et al., 2015). In brief, participants were genotyped on either the Affymetrix UK BiLEVE Axiom Array (N = 50,520) or the Affymetrix UK Biobank Axiom Array (N = 438,692) with 95% overlap between markers on each array. Haplotype phasing of the data was performed with SHAPEIT2 (Delaneau, Howie, Cox, Zagury, & Marchini, 2013; Delaneau, Zagury, & Marchini, 2013). Reference data from the Haplotype Reference Consortium (Loh et al., 2016) was used for imputation (Das et al., 2016).
The largest ancestry group in UKB are self‐identified “White British” that also group closely by genotype PCA (Price, Zaitlen, Reich, & Patterson, 2010). The first 10 principal components in such a large sample will not capture the substructure in all possible ancestries. To increase sensitivity, we included only that group. We filtered genetic variants for a minimum imputation information score of 0.85 and a minimum allele count of 20. Following quality control, 17,981,292 genetic variants in 408,961 participants remained for analysis.
2.7. UKB Genome‐wide association analyses
For the GWAS of CAAD in UKB, we used a linear mixed model with saddle point‐correction implemented in the package SAIGE. This approach decreases p‐value inflation commonly observed in rare outcome and rare genetic variant associations (Zhou et al., 2018), and is appropriate for the imbalanced case–control ratio in the UKB data. Analyses were adjusted for age, sex, and the first 10 principal components (Price et al., 2006), and the results reported were filtered for MAF more than 0.05.
2.8. Annotation of results
We annotated SNVs passing the genome‐wide significance threshold with Genotype‐Tissue Expression project data (GTEx; https://gtexportal.org/home/), European Bioinformatics Institute GWAS catalog (https://ebi.ac.uk/gwas/), and Haploreg (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php).
3. RESULTS
3.1. Phenotype and demographics
We identified 1793 cases and 17,958 controls in the eight participating eMERGE sites. Demographics and site of origin for cases versus controls are shown in Table 1. Of the cases, 950 (53.0%) had more than 50% stenosis and/or at least one procedure code for endarterectomy. Of the controls, 411 (2.3%) had an imaging study showing less than or equal to 15% carotid artery stenosis. Participants were 56.0% female, 70.3% self‐reported Caucasian, and 53.7% were ever a smoker.
3.2. GWAS results
3.2.1. Pooled Association Results
Two loci were associated with CAAD in the full data set, adjusting for age, sex, eMERGE site, and the first 10 ancestry principal components, and with a minimum p‐value of 5 × 10−8 (Table 2). These were a locus in the 25th intron of LPA (lead SNV rs10455872), and an intergenic region on chromosome 7 between TYW1 and AUTS2 (lead SNV rs6952610; Table 2, Figure 1). LPA variant rs10455872 is a GTEx eQTL for increased expression of SLC22A3 in “Skin – Sun Exposed (Lower leg)” (p = 2 × 10−6), and alters four predicted transcription factor binding motifs: Foxd1_1, Foxf1, Foxj1_2, and Foxo_1. It has entries for association with 16 traits in the GWAS catalog, mostly cardiovascular and lipid phenotypes. Intergenic variant rs6952610 is not a GTEx eQTL, and has no associations in the GWAS catalog. It changes two predicted transcription factor binding motifs: GR_disc1 and GR_known3 (from Haploreg; Ward & Kellis, 2012), data from ENCODE (ENCODE Project Consortium, 2012).
Table 2.
SNVs with p < 5 × 10−8 in the eMERGE CAAD GWAS with covariates age, sex, study site, and first 10 principal components
| rsID | Chr | Position (hg19) | A1 a | A2 a | OR (95% CI) | p | MAF | MAF EUR b | MAF AFR b | Mean R2 c | N chips genotyped d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs118039278 | 6 | 160985526 | A | G | 1.49 (1.3–1.72) | 2.32e−08 | 0.0535 | 0.067 | 0.0115 | 0.871 | 0 |
| rs74617384 | 6 | 160997118 | T | A | 1.49 (1.29–1.72) | 2.91e−08 | 0.0535 | 0.067 | 0.0115 | 0.864 | 0 |
| rs55730499 | 6 | 161005610 | T | C | 1.48 (1.29–1.7) | 3.9e−08 | 0.0546 | 0.069 | 0.0117 | 0.855 | 0 |
| rs10455872 | 6 | 161010118 | G | A | 1.5 (1.3–1.73) | 2.12e−08 | 0.0533 | 0.067 | 0.0117 | 0.858 | 3 |
| rs7792316 | 7 | 68296349 | A | G | 1.25 (1.15–1.36) | 4.91e−08 | 0.433 | 0.36 | 0.683 | 0.966 | 1 |
| rs6952610 | 7 | 68299862 | T | C | 1.25 (1.16–1.36) | 4.27e−08 | 0.429 | 0.36 | 0.669 | 0.964 | 0 |
Abbreviations: CAAD, Carotid artery atherosclerotic disease; CI, confidence interval; eMERGE, electronic Medical Records and Genomics; GWAS, genome‐wide association studies; SNVs, single nucleotide variants; OR, odds ratio.
A1 is the effect and minor allele, A2 the reference allele.
Minor allele frequency (MAF) for EUR = European ancestry, AFR = African ancestry subsets, as defined by k‐means clustering of genotypes.
Mean imputation quality R 2 for that variant.
Number of chips of 24 total where the SNV was typed rather than imputed.
Figure 1.
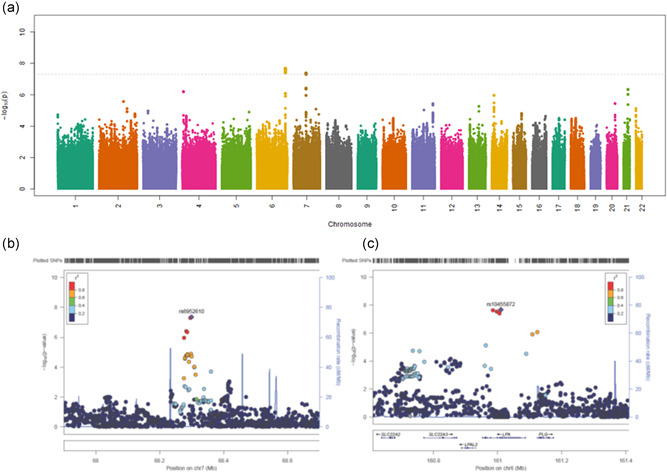
Plots of eMERGE CAAD GWAS results, SNVs arranged by chromosome position and 25 filtered for MAF more than 0.05, imputation quality R 2 more than .3. (a) Manhattan plot of all result. (b) 26 LocusZoom plot of chromosome 6 locus. (c) LocusZoom plot of choromosome 7 locus. CAAD, Carotid artery atherosclerotic disease; eMERGE, electronic Medical Records and Genomics; GWAS, genome‐wide association studies; SNVs, single nucleotide variants
3.2.2. Associations through ancestry stratification
We defined ancestry groups via k‐means clustering of genetic markers only, as many participants were missing self‐reported ancestry. The LPA association remained statistically significant in the European ancestry subset (1705 cases, 13,198 controls; p = 1.52 × 10−8) but the TYW1–AUTS2 association did not reach genome‐wide significance (p = 7.45 × 10−7). Only 89 (5.0%) cases were of non‐European ancestry, 70 of those were of genetically determined African ancestry. As expected, given the limited power, we found no significant associations in the African ancestry subset (results not shown).
3.2.3. Other covariates
The addition of covariates other than age, sex, site, and PC1–10 did not substantially change the results. We individually added BMI, CHD, and smoking to the logistic model, as well as combining all covariates in a single model (Table 3). Sample sizes differed in these analyses due to missing data. When we added an LPA SNV (rs118039278) as a covariate, the TYW1–AUTS2 SNV rs6952610 remained associated (p = 4.3 × 10−8).
Table 3.
Secondary analyses of two lead SNVs from eMERGE CAAD GWAS, rs10455872 and rs6952610, with additional covariates added to the main, and a combined model including all
| Covariate | OR (95% CI) | p | N cases | N controls |
|---|---|---|---|---|
| rs10455872LPA | ||||
| Age, sex, site, PC1–10 (main) | 1.5 (1.3–1.73) | 2.12e−8 | 1793 | 17,958 |
| Main + BMI | 1.5 (1.3–1.73) | 4.01e−08 | 1746 | 17,072 |
| Main + CHD | 1.39 (1.19–1.63) | 3.27e−05 | 1655 | 14,223 |
| Main + ever smoker | 1.44 (1.23–1.7) | 1.19e−05 | 1443 | 10,018 |
| Main + BMI, CHD, ever smoker | 1.4 (1.17–1.67) | .000169 | 1417 | 9703 |
| rs6952610 TYW1–AUTS2 | ||||
| Age, sex, site, PC1–10 (main) | 1.25 (1.16–1.36) | 4.27e−08 | 1793 | 17,958 |
| Main + BMI | 1.27 (1.17–1.38) | 1.37e−08 | 1746 | 17,072 |
| Main + CHD | 1.27 (1.17–1.39) | 5.08e−08 | 1655 | 14,223 |
| Main + ever smoker | 1.26 (1.15–1.38) | 8.42e−07 | 1443 | 10,018 |
| Main + BMI, CHD, ever smoker | 1.29 (1.17–1.42) | 2.19e−07 | 1417 | 9703 |
Abbreviations: BMI, body mass index; CAAD, Carotid artery atherosclerotic disease; CHD, coronary heart disease; CI, confidence interval; eMERGE, electronic Medical Records and Genomics; GWAS, genome‐wide association studies; OR, odds ratio; SNVs, single nucleotide variants.
3.2.4. Coronary heart disease
We also evaluated the association of CHD with the putative CAAD loci in the same data set. There were 4199 cases and 11,684 controls, defined by the presence of a CHD diagnosis. The LPA variant was significantly associated with CHD, (OR (95% CI) = 1.27 (1.13–1.43), p = 5.5 × 10−5), whereas the intergenic chr7 SNV was not (OR (95% CI) = 1.03 (0.97–1.09), p = .30).
3.2.5. Replication in the UKB
We tested the six SNVs with p < 5 × 10−8 in our main analysis in the UKB (Table 4). The LPA association replicated (rs10455872 p = 3.4 × 10−6), as did the chr7 SNV (rs6952610 p = .0096). Both were statistically significant at the 5% level, after adjustment for the two contrasts.
Table 4.
Association of significant eMERGE SNVs with CAAD in the UK Biobank
| rsID | Chr | Position (hg19) | eMERGE p | UKB p | UKB β | UKB SE | UKB MAF | eMERGE MAF |
|---|---|---|---|---|---|---|---|---|
| rs118039278 | 6 | 160985526 | 2.32e−08 | 2.61e−06 | .41 | 0.088 | 0.081 | 0.054 |
| rs74617384 | 6 | 160997118 | 2.91e−08 | 3.04e−06 | .41 | 0.088 | 0.081 | 0.054 |
| rs55730499 | 6 | 161005610 | 3.9e−08 | 3.87e−06 | .40 | 0.087 | 0.081 | 0.055 |
| rs10455872 | 6 | 161010118 | 2.12e−08 | 3.43e−06 | .41 | 0.087 | 0.081 | 0.053 |
| rs7792316 | 7 | 68296349 | 4.91e−08 | .00792 | .12 | 0.048 | 0.37 | 0.43 |
| rs6952610 | 7 | 68299862 | 4.27e−08 | .00962 | .13 | 0.048 | 0.37 | 0.43 |
Abbreviations: CAAD, Carotid artery atherosclerotic disease; eMERGE, electronic Medical Records and Genomics; MAF, minor allele frequency; SNVs, single nucleotide variants.
3.2.6. GWAS in the UKB
As a secondary analysis, we performed a GWAS within the UKB with the ICD‐10 defined phenotype. Two loci reach genome‐wide significance, on chromosomes 1 and 7 (see Figure S2, Manhattan plot and Table S2 of the most significant results). The lead SNV on chr7 was rs2526620, which is intergenic between HDAC9 and TWIST1, and is not near the chr7 SNV in the eMERGE GWAS. The lead SNV on chromosome 1, rs682112, is in an intron of neuron navigator 1 (NAV1). NAV1 is expressed more in the aorta than other GTEx tissues (gtexportal. org). rs682112 is in the GTEx catalog as an eQTL for NAV1 in Heart—Atrial Appendage (normalized effect size = 0.22, p = 5.6 × 10−8) and Esophagus—Mucosa (normalized effect size = −0.18, p = 2.9 × 10−5). These did not replicate in the eMERGE data.
4. DISCUSSION
4.1. GWAS results
We found two loci, tagged by rs10455872 and rs6952610, associated with CAAD in the eMERGE Network participants. Both replicated in UKB with a simple ICD‐only phenotype, despite differences in the patient demographics and phenotype definition. UKB is a younger, population‐based cohort with less depth of longitudinal data than most eMERGE sites (Sudlow et al., 2015). Longitudinal data can result in deeper phenotyping for participants, allowing improved assignment of case–control status. In addition, no NLP or image analysis was possible in UKB. Due to these differences, proportionally more cases were found in eMERGE: 9.1% in eMERGE versus 0.23% in UKB. Even when comparing the more severe cases, eMERGE cases with more than or equal to 50% stenosis and/or at least one endarterectomy, there were more than UKB: 950, or 4.8% of participants. In KPWA, where validation was performed, 76 of 300 cases were included due to the ICD‐9 code 433.1; the rest had imaging studies or endarterectomy.
Both SNVs were associated in the eMERGE all‐participant analysis and the European ancestry subset, although the chr7 SNV did not pass genome‐wide significance in European ancestry alone. Other ancestry subsets were too small for subgroup analyses. The [Lp(a)]‐associated risk for CAAD varies by race/ethnicity; the burden of risk is higher in individuals with European than African ancestry, but still unclear in Hispanic and Asian populations (Steffen et al., 2019). More work on the risk and genetic predictors of CAAD in non‐European ancestry groups is warranted.
The addition of BMI as a covariate had little impact on the association of either locus. Adding smoking and CHD to the model altered effect sizes and p‐values; however, these results are difficult to compare, primarily due to the large drop in sample size due to missing data, as well as due to possible pleiotropic effects of LPA variation on both CHD and CAAD. The lead LPA SNV was also associated with CHD, but the chr7 SNV rs6952610 was not. Based on the current study and others (Bis et al., 2011; Dichgans et al., 2014; Lövkvist et al., 2013), these phenotypes appear to have overlapping, but also distinct genetic risk factors.
4.2. LPA variant
LPA variant rs10455872 has been shown to explain 17% of the variance in [Lp(a)] (Ronald et al., 2011). Lp(a) is a small low‐density lipoprotein (LDL)‐like particle formed by apolipoprotein B (apoB) covalently bound to apolipoprotein(a). Rs10455872 is associated with CHD (Nelson et al., 2017), large artery stroke and ischemic stroke (Dichgans et al., 2014), and myocardial infarction (Nikpay et al., 2015). In addition, rs10455872‐G is associated with CHD events independent of statin‐induced lowering of LDL‐C (Khera et al., 2014; Wei et al., 2018), and with smaller LDL‐C reduction with rosuvastatin (Chasman et al., 2012). Other LPA variants that predict elevated [Lp(a)] have previously been shown to be associated with CAAD (Ronald et al., 2011). The minor alleles of rs10455872 mark haplotypes carrying short KIV2 kringle repeat alleles that are associated with elevated plasma [Lp(a)] (Ronald et al., 2011), due to increased liver secretion. The KIV4 repeat has been shown to account for 69% of [Lp(a)] plasma variation (Emdin et al., 2016); thus, we expect that rs10455872 predicts both [Lp(a)] and CAAD due to its association with KIV4 repeat length.
Measured and genetically predicted [Lp(a)] is associated with CHD risk (Emdin et al., 2016), but [Lp(a)] is not often measured clinically (Burgess et al., 2018), in part because it is not very responsive to current treatments. People with high [Lp(a)] can have their risk managed with other approaches, and, may benefit from new drugs being developed (Nordestgaard, Nicholls, Langsted, Ray, & Tybjærg‐Hansen, 2018). Burgess et al. (2018) analyzed CHD risk based on genetically predicted Lp(a) and concluded that pharmacological lowering of [Lp(a)] by 100 mg/dl would reduce risk of CHD by 22%–25% in a 3–5 year trial (Burgess et al., 2018). Our results suggest an etiological relationship between elevated [Lp(a)] and CAAD; thus, lowering [Lp(a)] also has the potential to reduce CAAD risk. These data could be used for prevention efforts, including constructing polygenic risk scores.
4.3. TYW1–AUTS2
Unlike LPA, the chr7 intergenic locus does not have an obvious nearby candidate gene or regulatory epigenetic role, other than altering predicted transcription factor binding motifs of unknown relevance. This SNV is intergenic between TYW1–AUTS2. TRNA‐YW synthesizing protein 1 homolog (TYW1) codes for a protein that stabilizes codon–anticodon interactions in the ribosome. AUTS2, autism susceptibility candidate 2, is an activator of transcription, has been shown to regulate neuronal migration and have a role in brain development (Hori & Hoshino, 2017; Sultana et al., 2002). Variation in this gene has been associated with intellectual disability, microcephaly, autism, and other behavioral phenotypes (Beunders et al., 2016). A PheWAS of rs6952610 in a larger segment of eMERGE (N = 83,717) yielded no phenome‐wide significant results. However, the top four phecodes were “Musculoskeletal symptoms referable to limbs,” “Other disorders of middle ear and mastoid,” “Nephritis and nephropathy with pathological lesion,” and “Cerebral ischemia,” which is a condition caused by insufficient blood flow to the brain. The later result may suggest cerebral ischemia due to CAAD; however, this would require further evidence as it is not statistically significant when the multiple tests are considered. See Figure S3, the Phewas Manhattan plot and Table S3 of the top Phewas results.
4.4. Use of natural language processing
The portable NLP system's built‐in support for local evaluation of NLP performance, combined with centralized modification and re‐distribution of site‐tailored versions of the NLP system, allowed for accurate extraction of quantitative stenosis severity measures from heterogeneously sourced free‐text imaging reports, a known challenge in multisite application of clinical NLP (Carrell et al., 2017).
4.5. Limitations
Electronic health records provide an efficient way to identify cases and controls. However, people with no evidence of CAAD in their medical record may have subclinical carotid atherosclerosis, but would be included as controls. This would reduce the power and thus the effect size we see from true associations. As [Lp(a)] is not often clinically measured, we do not have it for these participants. Therefore, we were unable to show that inclusion of this phenotype would eliminate the effect of the genotype, as has been previously reported (Ronald et al., 2011). As discussed above, other limitations were the different phenotype definitions in eMERGE and UKB and the lack of ancestral diversity in these cohorts.
4.6. Conclusions
In summary, we identified and replicated two loci associated with CAAD, a risk factor for stroke. The LPA association validates prior work that LPA variation which affects [Lp(a)] is associated with carotid artery disease and suggests [Lp(a)] is an important therapeutic target for CAAD. The second association, on chr7, is novel and its mechanism of action is unknown.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
The following authors made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; were involved in drafting the manuscript or revising it critically for important intellectual content: David R. Crosslin, Elisabeth A. Rosenthal, David S. Carrell, David J. Cronkite, Adam Gordon, Xiaomeng Du, Yatong K. Li, Marc S. Williams, Chunhua Weng, Qiping Feng, Rongling Li, Sarah A. Pendergrass, Hakon Hakonarson, David Fasel, Sunghwan Sohn, Patrick Sleiman, Samuel K. Handelman, Elizabeth Speliotes, Iftikhar J. Kullo, and Eric B. Larson. In addition to the above, the following authors also gave final approval of the version to be published, Melody R. Palmer, Daniel Seung Kim, Ian B. Stanaway, and Gail P. Jarvik.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
In eMERGE network (Phase 3 ascertainment), this phase of the eMERGE Network was initiated and funded by the NHGRI through the following grants: U01HG8657 (Kaiser Washington/University of Washington); U01HG8685 (Brigham and Women's Hospital); U01HG8672 (Vanderbilt University Medical Center); U01HG8666 (Cincinnati Children's Hospital Medical Center); U01HG6379 (Mayo Clinic); U01HG8679 (Geisinger Clinic); U01HG8680 (Columbia University Health Sciences); U01HG8684 (Children's Hospital of Philadelphia); U01HG8673 (Northwestern University); U01HG8701 (Vanderbilt University Medical Center serving as the Coordinating Center); U01HG8676 (Partners Healthcare/Broad Institute); and U01HG8664 (Baylor College of Medicine). In eMERGE network (Phase 1 and 2 ascertainment), the eMERGE Network was initiated and funded by NHGRI through the following grants: U01HG006828 (Cincinnati Children's Hospital Medical Center/Boston Children's Hospital); U01HG006830 (Children's Hospital of Philadelphia); U01HG006389 (Essentia Institute of Rural Health, Marshfield Clinic Research Foundation and Pennsylvania State University); U01HG006382 (Geisinger Clinic); U01HG006375 (Group Health Cooperative/University of Washington); U01HG006379 (Mayo Clinic); U01HG006380 (Icahn School of Medicine at Mount Sinai); U01HG006388 (Northwestern University); U01HG006378 (Vanderbilt University Medical Center); and U01HG006385 (Vanderbilt University Medical Center serving as the Coordinating Center) with U01HG004438 (CIDR) and U01HG004424 (the Broad Institute) serving as Genotyping Centers. Help from James Linneman and NIH support from NIH NHGRI 5U01HG008701 and NIH NCATS UL1TR002373. The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 02/26/19. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Palmer MR, Kim DS, Crosslin DR, et al. Loci identified by a genome‐wide association study of carotid artery stenosis in the eMERGE network. Genetic Epidemiology. 2021;45:4–15. 10.1002/gepi.22360
DATA AVAILABILITY STATEMENT
These data have been posted to dbGaP, study ID phs000888.v1.p1. This study ID contains the eMERGE genotyped and imputed SNV data. The CAAD phenotype can be found with variable accession number phv00225976.v1.p1.
REFERENCES
- Beunders, G. , van de Kamp, J. , Vasudevan, P. , Morton, J. , Smets, K. , Kleefstra, T. , … Sistermans, E. A. (2016). A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. American Journal of Medical Genetics, 53(8), 523–532. 10.1136/jmedgenet-2015-103601 [DOI] [PubMed] [Google Scholar]
- Bis, J. C. , Kavousi, M. , Franceschini, N. , Isaacs, A. , Abecasis, G. R. , Schminke, U. , … O'Donnell, C. J. , CARDIoGRAM Consortium . (2011). Meta‐analysis of genome‐wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nature Genetics, 43(10), 940–947. 10.1038/ng.920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, S. , Ference, B. A. , Staley, J. R. , Freitag, D. F. , Mason, A. M. , Nielsen, S. F. , … Danesh, J. , European Prospective Investigation Into Cancer and Nutrition–Cardiovascular Disease (EPIC‐CVD) Consortium . (2018). Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)‐Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiology, 3(7), 619–627. 10.1001/jamacardio.2018.1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell, D. S. , Schoen, R. E. , Leffler, D. A. , Morris, M. , Rose, S. , Baer, A. , … Mehrotra, A. (2017). Challenges in adapting existing clinical natural language processing systems to multiple, diverse health care settings. Journal of the American Medical Informatics Association, 24(5), 986–991. 10.1093/jamia/ocx039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C. C. , Chow, C. C. , Tellier, L. C. , Vattikuti, S. , Purcell, S. M. , & Lee, J. J. (2015). Second‐generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience, 4, 4 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chasman, D. I. , Giulianni, F. , MacFayden, J. , Barratt, B. J. , Nyberg, F. , & Ridker, P. M. (2012). Genetic determinants of statin‐induced low‐density lipoprotein cholesterol reduction: The Justification for the use of statins in prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circulation: Cardiovascular Genetics, 5(2), 257–264. 10.1161/CIRCGENETICS.111.961144 [DOI] [PubMed] [Google Scholar]
- Chisholm, R. L. (2013). At the interface between medical informatics and personalized medicine: The eMERGE network experience. Healthcare Informatics Research, 19(2), 67–68. 10.4258/hir.2013.19.2.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, R. , Peden, J. F. , Hopewell, J. C. , Kyriakou, T. , Goel, A. , Heath, S. C. , … Farrall, M. , PROCARDIS Consortium . (2009). Genetic variants associated with Lp(a) lipoprotein level and coronary disease. New England Journal of Medicine, 361(26), 2518–2528. 10.1056/NEJMoa0902604 [DOI] [PubMed] [Google Scholar]
- Crawford, D. C. , Crosslin, D. R. , Tromp, G. , Kullo, I. J. , Kuivaniemi, H. , Hayes, M. G. , … Ritchie, M. D. (2014). eMERGEing progress in genomics—the first seven years. Frontiers in Genetics, 5, 184 10.3389/fgene.2014.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, D. C. , Nord, A. S. , Badzioch, M. D. , Ranchalis, J. , McKinstry, L. A. , Ahearn, M. , … Jarvik, G. P. (2008). A common VLDLR polymorphism interacts with APOE genotype in the prediction of carotid artery disease risk. Journal of Lipid Research, 49(3), 588–596. 10.1194/jlr.M700409-JLR200 [DOI] [PubMed] [Google Scholar]
- Das, S. , Forer, L. , Schoenherr, S. , Sidore, C. , Locke, A. E. , Kwong, A. , … Fuchsberger, C. (2016). Next‐generation genotype imputation service and methods. Nature Genetics, 48(10), 1284–1287. 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau, O. , Howie, B. , Cox, A. J. , Zagury, J. F. , & Marchini, J. (2013). Haplotype estimation using sequencing reads. American Journal of Human Genetics, 93(4), 687–696. 10.1016/j.ajhg.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau, O. , Zagury, J. F. , & Marchini, J. (2013). Improved whole‐chromosome phasing for disease and population genetic studies. Nature Methods, 10(1), 5–6. 10.1038/nmeth.2307 [DOI] [PubMed] [Google Scholar]
- Denny, J. C. , Ritchie, M. D. , Basford, M. A. , Pulley, J. M. , Bastarache, L. A. , Brown‐Gentry, K. , … Crawford, D. C. (2010). PheWAS: Demonstrating the feasibility of a phenome‐wide scan to discover gene‐disease associations. Bioinformatics, 26(9), 1205–1210. 10.1093/bioinformatics/btq126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichgans, M. , Malik, R. , König, I. R. , Rosand, J. , Clarke, R. , Gretarsdottir, S. , … Schunkert, H. , METASTROKE Consortium; CARDIoGRAM Consortium; C4D Consortium; C4D Consortium; International Stroke Genetics Consortium . (2014). Shared genetic susceptibility to ischemic stroke and coronary artery disease: A genome‐wide analysis of common variants. Stroke, 45(1), 24–36. 10.1161/STROKEAHA.113.002707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu, L. , Glenn, K. , Brown‐Gentry, K. , Shephard, C. , Wong, M. , Rieder, M. J. , … Crawford, D. C. (2011). Variation in LPA is associated with Lp(a) levels in three populations from the Third National Health and Nutrition Examination Survey. PLoS One, 6(1), e16604 10.1371/journal.pone.0016604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdin, C. A. , Kherva, A. V. , Natarajan, P. , Klarin, D. , Won, H. H. , Peloso, G. M. , … Kathiresan, S. , CHARGE–Heart Failure Consortium and CARDIoGRAM Exome Consortium . (2016). Phenotypic characterization of genetically lowered human lipoprotein(a) Levels. Journal of the American College of Cardiology, 68(25), 2761–2772. 10.1016/j.jacc.2016.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium . (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgo, B. , Medda, E. , Hernyes, A. , Szalontai, L. , Tarnoki, D. L. , & Tarnoki, A. D. (2018). Carotid artery atherosclerosis: A review on heritability and genetics. Twin Research and Human Genetics, 21(5), 333–346. 10.1017/thg.2018.45 [DOI] [PubMed] [Google Scholar]
- Goldstein, L. B. , Adams, R. , Alberts, M. J. , Appel, L. J. , Brass, L. M. , Bushnell, C. D. , … Sacco, R. L. , American Academy of Neurology . (2006). Primary prevention of ischemic stroke: A guideline from the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline. Stroke, 37(6), 1583–1633. 10.1161/01.STR.0000223048.70103.F1 [DOI] [PubMed] [Google Scholar]
- Gottesman, O. , Kuivaniemi, H. , Tromp, G. , Faucett, W. A. , Li, R. , Manolio, T. A. , … Williams, M. S. , eMERGE Network . (2013). The electronic Medical Records and Genomics (eMERGE) Network: Past, present, and future. Genetics in Medicine, 15(10), 761–771. 10.1038/gim.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori, K. , & Hoshino, M. (2017). Neuronal migration and AUTS2 syndrome. Brain Sciences, 7(5), 54 10.3390/brainsci7050054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera, A. V. , Everett, B. M. , Caulfield, M. P. , Hantash, F. M. , Wohlgemuth, J. , Ridker, P. M. , & Mora, S. (2014). Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation, 129(6), 635–642. 10.1161/CIRCULATIONAHA.113.004406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera, A. V. , & Kathiresan, S. (2017). Genetics of coronary artery disease: Discovery, biology and clinical translation. Nature Reviews Genetics, 18(6), 331–334. 10.1038/nrg.2016.160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho, A. N. , Hayes, M. G. , Rasmussen‐Torvik, L. , Pacheco, J. A. , Thompson, W. K. , Armstrong, L. L. , … Lowe, W. L. (2012). Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome‐wide association study. Journal of the American Medical Informatics Association, 19(2), 212–218. 10.1136/amiajnl-2011-000439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullo, I. J. , Fan, J. , Pathak, J. , Savova, G. K. , Ali, Z. , & Chute, C. G. (2010). Leveraging informatics for genetic studies: Use of the electronic medical record to enable a genome‐wide association study of peripheral arterial disease. Journal of the American Medical Informatics Association, 17(5), 568–574. 10.1136/jamia.2010.004366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh, P. R. , Danecek, P. , Palamara, P. F. , Fuchsberger, C. , Reshef, Y. A. , Finucane, H. K. , … Price, A. L. (2016). Reference‐based phasing using the Haplotype Reference Consortium panel. Nature Genetics, 48(11), 1443–1448. 10.1038/ng.3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lövkvist, H. , Sjögren, M. , Höglund, P. , Engström, G. , Jern, C. , Olsson, S. , … Lindgren, A. (2013). Are 25 SNPs from the CARDIoGRAM study associated with ischaemic stroke? European Journal of Neurology, 20(9), 1284–1291. 10.1111/ene.12183 [DOI] [PubMed] [Google Scholar]
- McCarthy, S. , Das, S. , Kretzschmar, W. , Delaneau, O. , Wood, A. R. , Teumer, A. , … Marchini, J. (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nature Genetics, 48(10), 1279–1283. 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, C. A. , Chisholm, R. L. , Chute, C. G. , Kullo, I. J. , Jarvik, G. P. , Larson, E. B. , … Wolf, W. A. , eMERGE Team . (2011). The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Medical Genomics, 4, 13 10.1186/1755-8794-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C. P. , Goel, A. , Butterworth, A. S. , Kanoni, S. , Webb, T. R. , Marouli, E. , … Deloukas, P. (2017). Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nature Genetics, 49(9), 1385–1391. 10.1038/ng.3913 [DOI] [PubMed] [Google Scholar]
- Nikpay, M. , Goel, A. , Won, H. H. , Hall, L. M. , Willenborg, C. , Kanoni, S. , … Farrall, M. (2015). A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nature Genetics, 47(10), 1121–1130. 10.1038/ng.3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard, B. G. , Nicholls, S. J. , Langsted, A. , Ray, K. K. , & Tybjærg‐Hansen, A. (2018). Advances in lipid‐lowering therapy through gene‐silencing technologies. Nature Reviews Cardiology, 15(5), 261–272. 10.1038/nrcardio.2018.3 [DOI] [PubMed] [Google Scholar]
- Ober, C. , Nord, A. S. , Thompson, E. E. , Pan, L. , Tan, Z. , Cuzanovich, D. , … Nicolae, D. L. (2009). Genome‐wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. Journal of Lipid Research, 50(5), 798–806. 10.1194/jlr.M800515-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A. L. , Patterson, N. , Plenge, R. M. , Weinblatt, M. E. , Shadick, N. A. , & Reich, D. (2006). Principal components analysis corrects for stratification in genome‐wide association studies. Nature Genetics, 38(8), 904–909. 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- Price, A. L. , Zaitlen, N. A. , Reich, D. , & Patterson, N. (2010). New approaches to population stratification in genome‐wide association studies. Nature Reviews Genetics, 11(7), 459–463. 10.1038/nrg2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronald, J. , Rajagopalan, R. , Cerrato, F. , Nord, A. S. , Hatsukami, T. S. , Kohler, T. , … Jarvik, G. P. (2011). Genetic variation in LPAL2, LPA, and PLG predicts plasma lipoprotein(a) level and carotid artery disease risk. Stroke, 42(1), 2–9. 10.1161/STROKEAHA.110.591230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanaway, I. B. , Hall, T. O. , Rosenthal, E. A. , Palmer, M. , Naranbhai, V. , Knevel, R. , … Crosslin, D. R. (2019). The eMERGE genotype set of 83,717 subjects imputed to ~40 million variants genome wide and association with the herpes zoster medical record phenotype. Genetic Epidemiology, 43(1), 63–81. 10.1002/gepi.22167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen, B. T. , Thanassoulis, G. , Duprez, D. , Stein, J. H. , Karger, A. B. , Tattersall, M. C. , … Tsai, M. Y. (2019). Race‐based differences in lipoprotein(a)‐associated risk of carotid atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(3), 523–529. 10.1161/ATVBAHA.118.312267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow, C. , Gallacher, J. , Allen, N. , Beral, V. , Burton, P. , Danesh, J. , … Collins, R. (2015). UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Medicine, 12(3), e1001779 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana, R. , Yu, C. E. , Munson, J. , Chen, D. , Hua, W. , Estes, A. , … Villacres, E. C. (2002). Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics, 80(2), 129–134. 10.1006/geno.2002.6810 [DOI] [PubMed] [Google Scholar]
- Taylor, D. C. , & Strandness, D. E. J. (1987). Caritod artery duplex scanning. Journal of Clinical Ultrasound, 15(9), 635–644. 10.1002/jcu.1870150906 [DOI] [PubMed] [Google Scholar]
- Ward, L. D. , & Kellis, M. (2012). HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Research, 40, D930–D934. 10.1093/nar/gkr917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, W. Q. , Li, X. , Feng, Q. , Kubo, M. , Kullo, I. J. , Peissig, P. L. , … Denny, J. C. (2018). LPA variants are associated with residual cardiovascular risk in patients receiving statins. Circulation, 138(17), 1839–1849. 10.1161/CIRCULATIONAHA.117.031356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, W. , Nielsen, J. B. , Fritsche, L. G. , Dey, R. , Gabrielsen, M. E. , Wolford, B. N. , … Lee, S. (2018). Efficiently controlling for case‐control imbalance and sample relatedness in large‐scale genetic association studies. Nature Genetics, 50(9), 1335–1341. 10.1038/s41588-018-0184-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
These data have been posted to dbGaP, study ID phs000888.v1.p1. This study ID contains the eMERGE genotyped and imputed SNV data. The CAAD phenotype can be found with variable accession number phv00225976.v1.p1.