

Abstract
Background and Aims
Polycystic liver diseases (PLDs) are genetic disorders characterized by progressive development of symptomatic biliary cysts. Current surgical and pharmacological approaches are ineffective, and liver transplantation represents the only curative option. Ursodeoxycholic acid (UDCA) and histone deacetylase 6 inhibitors (HDAC6is) have arisen as promising therapeutic strategies, but with partial benefits.
Approach and Results
Here, we tested an approach based on the design, synthesis, and validation of a family of UDCA synthetic conjugates with selective HDAC6i capacity (UDCA‐HDAC6i). Four UDCA‐HDAC6i conjugates presented selective HDAC6i activity, UDCA‐HDAC6i #1 being the most promising candidate. UDCA orientation within the UDCA‐HDAC6i structure was determinant for HDAC6i activity and selectivity. Treatment of polycystic rats with UDCA‐HDAC6i #1 reduced their hepatomegaly and cystogenesis, increased UDCA concentration, and inhibited HDAC6 activity in liver. In cystic cholangiocytes UDCA‐HDAC6i #1 restored primary cilium length and exhibited potent antiproliferative activity. UDCA‐HDAC6i #1 was actively transported into cells through BA and organic cation transporters.
Conclusions
These UDCA‐HDAC6i conjugates open a therapeutic avenue for PLDs.
Abbreviations
- 2D
two‐dimensional
- 3D
three‐dimensional
- ADPKD
autosomal‐dominant polycystic kidney disease
- BA
bile acid
- ERK
extracellular signal‐regulated kinase
- H3K9
lysine 9 of histone 3
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- HDAC1
histone deacetylase 1
- HDAC6
histone deacetylase 6
- HDAC6is
histone deacetylase 6 inhibitors
- IC50
half maximal inhibitory concentration
- OATP
organic anionic transporting polypeptide
- OCT
organic cation transporter
- pERK1/2
phosphorylated extracellular signal‐regulated kinase 1 and 2
- PKD
polycystic kidney disease
- PKHD1
polycystic kidney and hepatic disease 1
- PLDs
polycystic liver diseases
- UDCA
ursodeoxycholic acid
- UDCA‐HDAC6i
synthetic conjugate of ursodeoxycholic acid
- WT
wild type
Polycystic liver diseases (PLDs) constitute a heterogeneous group of genetic disorders characterized by progressive development of multiple fluid‐filled biliary cysts (>10), which are the main cause of morbidity.( 1 , 2 ) PLDs are inherited as dominant (i.e., autosomal‐dominant polycystic liver disease [~1:100,000 prevalence] and autosomal‐dominant polycystic kidney disease [ADPKD; ~1:1,000 prevalence]) or recessive (i.e., autosomal‐recessive polycystic kidney disease [ARPKD; ~1:20,000 prevalence]) forms. Depending on the gene affected and the type of mutation, cysts arise exclusively in liver (i.e., protein kinase C substrate 80K‐H, translocation protein SEC63 homolog, low‐density lipoprotein receptor‐related protein 5, neutral alpha‐glucosidase AB [GANAB], protein transport protein Sec61 subunit beta, and asparagine‐linked glycosylation 8 genes) or also in kidneys (polycystic kidney disease [PKD]; PKD1, PKD2, GANAB, and polycystic kidney and hepatic disease 1 [PKHD1] genes).( 1 , 2 ) Available treatments are mainly surgical resection of symptomatic cysts and systemic treatment with somatostatin analogues, which show short‐term and modest benefits. Liver transplantation represents the only curative option.( 1 , 2 ) Therefore, therapeutic approaches based on a deeper understanding of the molecular mechanisms of pathogenesis are required. Cystogenesis in PLDs is characterized by diverse functional alterations in the bile duct cells (i.e., cholangiocytes),( 1 , 2 ) which include hyperproliferation and ‐secretion, increased matrix metalloproteolytic activity, changes in microRNA expression patterns, and morphological and functional alterations of the primary cilium, a sensory organelle. These pathological events are intracellularly mediated by increased levels of cAMP and decreased Ca2+, their regulation being postulated as a potentially therapeutic target.( 1 , 2 , 3 )
The use of somatostatin analogues aimed to down‐regulate the characteristic increased intracellular cAMP levels in cystic cholangiocytes, and their resultant hyperproliferation and ‐secretion, is the only current pharmacological option with limited therapeutic benefits and several side effects. Ursodeoxycholic acid (UDCA), an endogenous bile acid (BA) with choleretic and hepatoprotective properties, is able to halt hepatic cystogenesis in experimental models (in vitro and in vivo in PCK rats) and patients with advanced ADPKD, by normalizing intracellular calcium levels in cystic cholangiocytes and by clearing the liver from accumulation of cytotoxic and promitotic BAs.( 4 , 5 ) Of note, UDCA treatment improved symptomatology in both PCK rats and patients with ADPKD.( 4 , 5 ) On the other hand, the therapeutic potential of histone deacetylase (HDAC) 6 (HDAC6) inhibitors (HDAC6is), such as tubastatin‐A and ACY1215, for treatment of both PLDs and PKDs has been described in vitro ( 6 , 7 , 8 ) and in vivo,( 9 ) given that cystogenesis is characterized by cytoplasmic overexpression of HDAC6, which promotes the deacetylation of α‐tubulin, one of the main structural proteins of the primary cilium.( 9 ) Consequently, decreased acetylation of α‐tubulin results in morphological and functional abnormalities of the primary cilium and promotes cell proliferation.( 9 )
The hypothesis of the present study was that UDCA, owing to its particular structural features and ability to conjugate taurine or glycine, could be an excellent external van der Waals cap molecule for the design of HDAC6is (i.e., UDCA‐HDAC6i [synthetic conjugate of ursodeoxycholic acid]). It is known that preorganized macrocyclic systems can exhibit high noncovalent binding constants with the area close to the rim that connects the active sites of HDACs with the surface of these enzymes.( 10 , 11 ) We reasoned that a polycyclic molecule such as UDCA could be a suitable conformationally restricted analogue to these macrocyclic systems. This polycyclic moiety should be covalently bound to a spacer able to fill the tube that connects the active site of the enzyme with the rim.( 12 ) Finally, this kind of HDAC inhibitor (HDACi) should include a chelating group able to bind the Zn2+ present in the active site.( 13 ) The presence of UDCA in the UDCA‐HDAC6i structure could also improve the hepatotropism of these molecules and their targeting toward cystic cholangiocytes.( 14 , 15 , 16 ) Moreover, UDCA‐HDAC6i could retain some of the previously observed therapeutic benefits of UDCA on PLDs, and the abundance of UDCA in the BA pool could be increased because of intestinal reuptake after UDCA‐HDAC6i hydrolysis by the gut microbiota.( 17 ) In contrast to conventional HDAC6i, UDCA‐HDAC6i biotransformation is not expected to result in toxic intermediate molecules. Therefore, this strategy would provide a single‐molecule–based combination therapy.
Materials and Methods
Docking and Homology Modeling
The X‐ray structure of the human HDAC6 catalytic domain 2 was obtained from the RCSB Protein Data Bank (PDB entry: 5EDU). The protein structure was optimized and minimized using the protein preparation wizard software included in the Maestro 10.4 package (Schrödinger, LLC, New York, NY). Ligands were represented with a Maestro graphic interface and optimized with the LigPrep tool included in the Maestro 10.4 package. Flexible docking experiments were carried out using Glide software included in the Maestro 10.4 package, using extra precision mode to maximize accuracy of the obtained results. Obtained structures and binding‐predicted energies were analyzed using Maestro and Pymol graphic interfaces. The homology model of rat HDAC6 catalytic domain 2 (Uniprot entry: A0A0G2QC41) was generated based on human HDAC6 catalytic domain 2 (PDB entry: 5EDU) with Prime software, also included in the Maestro 10.4 package (Schrödinger, LLC). For docking studies, the same procedure as for the human HDAC6 catalytic domain 2 was followed.
HDAC Activity Assays
HDAC activity assays were performed using acetylated peptide substrates labeled with 7‐amino‐4‐methylcoumarin at Reaction Biology Corporation (Malvern, PA). All the enzymes and substrates used are summarized in Supporting Table S9, and the details of the assays and protocols are described in the Supporting Information.
Treatment of PCK Rats With HDAC6i‐UDCA #1
The PCK rat (Charles River Laboratories Inc, Wilmington, MA) is an animal model of ARPKD, presenting a mutation in the human orthologue Pkhd1 gene and spontaneously developing both hepatic and renal cysts. Both wild‐type (WT; n = 12) and PCK rats (n = 20) were 8 weeks old at the beginning of the experiment. PCK rats were randomized into HDAC6i‐UDCA #1–treated (n = 8; 15 mg/kg/d by oral gavage for 5 months) and nontreated (n = 12) groups. After treatment, animals were euthanized and blood was collected, and serum was further isolated to determine biochemical markers of liver and kidney disease. Liver and kidneys were collected, weighted, and either fixed in 4% paraformaldehyde or stored at −80°C for further analysis. All in vivo experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee of Biodonostia Health Research Institute.
BA Measurement
Concentrations of BA species in liver, bile, peripheral, and portal blood were measured after methanol precipitation/extraction by high‐performance liquid chromatography with tandem mass spectrometry using a modification( 18 ) of a described method.( 19 )
Cells and Culture Conditions
Primary cultures of normal and cystic human cholangiocytes, previously isolated and characterized by our group,( 20 ) were used for in vitro assays. Culture conditions, and the three‐dimensional (3D) culturing of cystic cholangiocytes, are described in the Supporting Information.
Experimental Overexpression of Human Transporters in Cells and Transport Assays
Experimental overexpression of the human transporters in cells and transport assays were performed as described in the Supporting Information.
qPCR
Total RNA extraction and retrotranscription and further qPCRs were performed as described in the Supporting Information. All the primer sequences are summarized in Supporting Table S11.
Immunoblotting
Analysis of protein expression was performed as described in the Supporting Information and using the antibodies listed in Supporting Table S12.
Primary Cilia Analysis
Primary cilia length and number of ciliated cholangiocytes were analyzed by immunofluorescence as described in the Supporting Information.
Cell Proliferation
Evaluation of proliferation in cystic cholangiocytes was carried out as described in the Supporting Data.
Statistical Analysis
GraphPad Prism software was applied for statistical analysis (version 6; GraphPad Software Inc., La Jolla, CA). Data are expressed as mean ± SEM. Unless otherwise indicated, for comparisons between two groups, a two‐tailed parametric Student t test or nonparametric Mann‐Whitney U test were used. In all cases, measurements were taken from distinct samples (i.e., cell cultures, tissue samples, and individual cystic cholangioids). Statistically significant data, represented as asterisks (*, **, and ***), denote a P value <0.05, <0.01, and <0.001, respectively.
Results
Synthetic UDCA Conjugates Selectively Inhibit HDAC6 Activity
We applied our previously described HDAC inhibition model( 21 ) to the X‐ray structure of the catalytic domain 2 of human HDAC6 (PDB code: 5EDU)( 22 ) (Fig. 1A). HDAC6 active sites can be schematically described as an ensemble of external shallow cavities connected through a hydrophobic channel with a Zn2+‐containing catalytic site. Following this general model, we designed a family of synthetic UDCA conjugates, in which the steroid skeleton of UDCA acted as the external van der Waals cap of the inhibitor, followed by a number of different hydrophobic spacers ended on a Zn2+ chelating group (Fig. 1A).
FIG. 1.
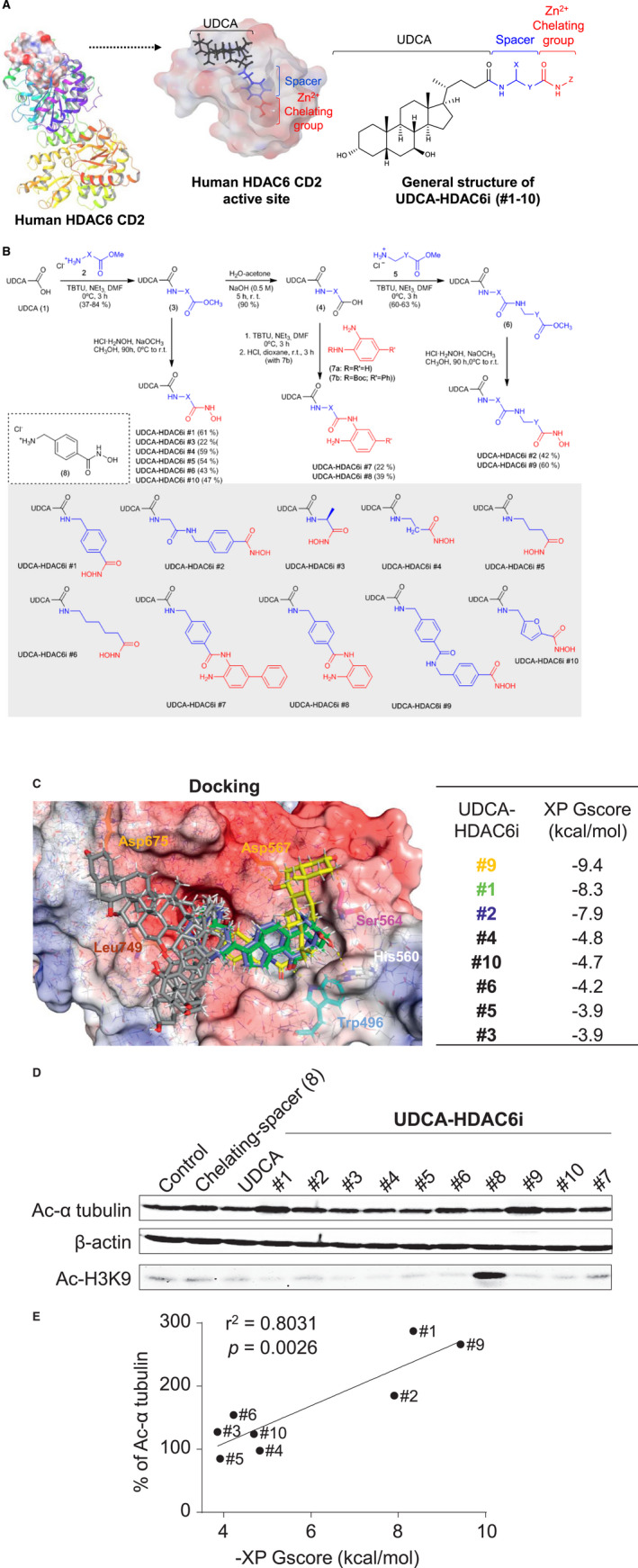
Design, synthesis, in silico prediction, and in vitro characterization of the HDACi activity of UDCA synthetic conjugates. (A) Scheme of the designing of UDCA‐HDAC6i molecules based on HDAC6 structure. (B) Chemical synthesis of the 10 different UDCA‐HDAC6i molecules. (C) Docking studies. Graphical representation of the biding modes of UDCA‐HDAC6 is (yellow #9, green #1, blue #2, and grey #4, #10, #6, #5, and #3) to the active site of human HDAC6 CD2. Dashed yellow lines represent hydrogen bonds between hydroxyl groups of UDCA and amino acids of the protein surface. Table displays the predicted binding energies for UDCA‐HDAC6is. (D) Representative immunoblotting of acetylation (Ac) levels of α‐tubulin and H3K9 after 24 hours of treatment with 2 μM of each compound. (E) Experimental validation of the in silico model. Correlation between the predicted biding energies (HDAC6i) and levels (%) of acetylated α‐tubulin in PLD cells treated with each UDCA‐HDAC6i. Abbreviations: Asp567, aspartate; DMF, N,N‐dimethylformamide; His, histidine; Leu, leucine; Ser, serine; TBTU, N,N,N′,N′‐tetramethyl‐O‐(benzotriazol‐1‐yl)uronium tetrafluoroborate; Trp, tryptophan; XP Gscore, XP GlideScore.
Chemical synthesis of the UDCA‐HDAC6i molecules (Fig. 1B) started with the coupling reaction of UDCA (1) with amines (2) to yield amide esters (3). Cleavage of the methoxy groups of adducts (3) and in situ reaction with hydroxylamine permitted the isolation and characterization of UDCA‐hydroxamic acids UDCA‐HDAC6i #1, #3‐6, and #10, which possess different lengths and stereoelectronic features. Alternatively, hydrolysis of esters (3) permitted the preparation of acid (4). Subsequent coupling with orthophenylenediamines (7) yielded amide candidates UDCA‐HDAC6i #7 and #8. The spacer between the chelating group and the UDCA component was elongated by means of an additional hydrolysis‐coupling sequence (5), thus yielding esters (6) whose cleavage and reaction with hydroxylamine permitted the isolation and characterization of candidates UDCA‐HDAC6i #2 and #9.
The potential HDAC6 inhibitory capacity of this family of UDCA synthetic conjugates was first evaluated in silico with a docking model (Fig. 1C). Significant binding affinities to HDAC6 were predicted for eight of these compounds, with UDCA‐HDAC6i #1, #2, and #9 being the ones that displayed the highest energy scores (−8.3, −7.9, and −9.4 kcal/mol, respectively; Fig. 1C). Interestingly, these three compounds oriented the hydrocarbon skeleton of UDCA toward the same region of the protein surface (Fig. 1C). We decomposed the total binding energy of each synthetic derivative component into the contribution of the different descriptors (i.e., hydrogen bonds, van der Waals interactions) to the overall score (Supporting Tables S1 and S2A,B). Importantly, the contribution of UDCA to the final binding energy of UDCA‐HDAC6i #1, #2, and #9 was approximately one‐third of the total value, and was mainly integrated by van der Waals interactions, and hydrogen bonds established by the two hydroxyl groups of the steroid skeleton of the BA (Fig. 1C; Supporting Tables S1 and S2A,B).
Given that cytoplasmic levels of acetylated α‐tubulin in cholangiocytes are directly linked to the degree of HDAC6 expression and activity,( 9 , 23 ) cystic human cholangiocytes in culture were incubated with UDCA‐HDAC6i and protein expression of acetylated α‐tubulin was measured by immunoblotting. In addition, acetylation levels of lysine 9 of nuclear histone 3 (H3K9) were assessed as a selectivity test (negative control). The results (Fig. 1D) revealed that, at low concentration (2 μM), UDCA‐HDAC6i #1, #2, #6, and #9 increased acetylated α‐tubulin levels in cystic cholangiocytes, without affecting H3K9 acetylation, thus indicating a selective inhibitory activity on HDAC6. In contrast, UDCA‐HDAC6i #8 increased H3K9 acetylation, without affecting α‐tubulin acetylation. Of note, the predicted binding affinity (i.e., energy values by docking) of the aforementioned UDCA‐HDAC6i for HDAC6 and their effect on HDAC6‐dependent acetylated α‐tubulin levels in cystic cholangiocytes showed significant correlation (r 2 = 0.8031), supporting the accuracy of the computational approach used here (Fig. 1E).
Next, inhibitory capacity of UDCA‐HDAC6i #1, #2, #6, #8, and #9 on purified histone deacetylase 1 (HDAC1) and HDAC6 enzymes was measured by monitoring the dose‐dependent deacetylation of a fluorogenic acetylated peptide (Fig. 2A). UDCA‐HDAC6i #1, #2, #6, and #9 displayed low half maximal inhibitory concentration (IC50) values (58, 56.1, 82.1, and 4.26 nM, respectively) for HDAC6 inhibition, concomitantly with a high degree of selectivity, particularly in the case of compounds #1, #2, and #9 (Fig. 2A). As expected, UDCA‐HDAC6i #8 showed both high inhibitory activity and selectivity for HDAC1 (IC50 = 140 nM; Fig. 2A). Importantly, UDCA itself did not exhibit any direct HDAC inhibitory activity; however, its formal coupling with the separate spacer‐chelating component, 4‐(aminomethyl)‐N‐hydroxybenzamide hydrochloride [Fig. 2B, chelating‐spacer (8)], to form UDCA‐HDAC6i #1 significantly increased (~12‐fold) the HDAC6 inhibitory capacity of the chemical entity (IC50 = 689 vs. 58 nM; Fig. 2B), highlighting the active contribution of UDCA to the inhibitory activity of the UDCA‐HDAC6is (Fig. 2B and Supporting Fig. S1). In addition, the effect of UDCA‐HDAC6i #1 and #2 was also evaluated on purified HDAC2‐5 and HDAC7‐11 enzymes, showing quite similar inhibitory capacity to HDAC1 and significantly less compared to HDAC6 (Supporting Table S3). Of note, the 3D structural alignment of the catalytic domains of human HDAC1, 2, 3, 4, 7, and 8 and zebrafish HDAC10 (PDB entries: 5ICN, 6G3O, 4A69, 4CBT, 3ZNR, 5VI6, and 5TD7) with our docking model of human HDAC6 CD2 revealed that although all the analyzed HDAC active sites fitted well to the general model previously mentioned (Fig. 1A), they possessed important differences in the architectures of their outer rims, hydrophobic channels, and catalytic sites (Fig. 2C). In this regard, we found particularly relevant the high degree of sequence disparity between HDAC6 and the rest of the isoforms in the surface area occupied by UDCA in the three most active/selective inhibitors (Supporting Fig. S2), resulting in the absence of this cavity in the other HDAC isoforms, whereas the other two cavities occupied by less active/selective UDCA‐HDAC6is (e.g., UDCA‐HDAC6i #6) remained almost intact (Fig. 2D and Supporting Fig. S2).
FIG. 2.
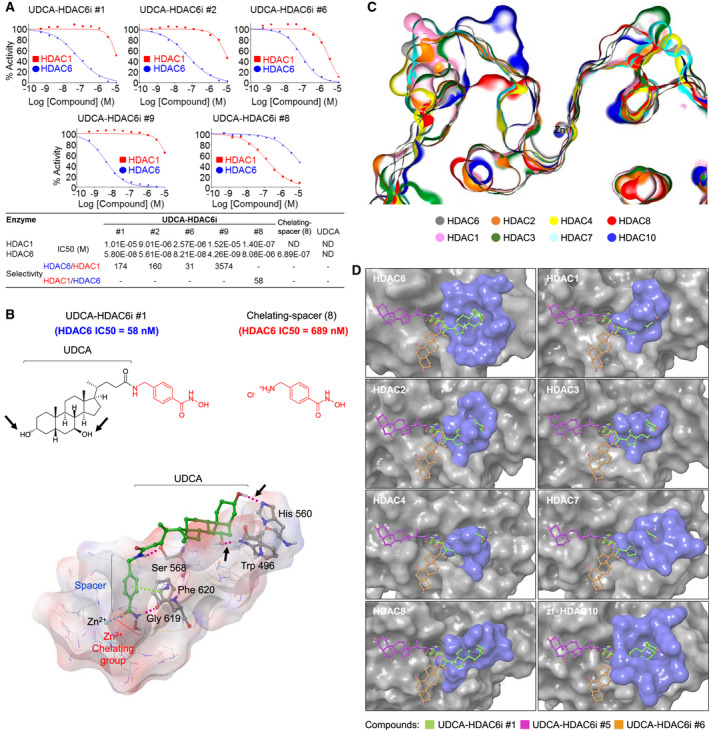
In vitro and in silico characterization of the critical role of UDCA for the HDAC6 inhibitory activity and selectivity of UDCA‐HDAC6is. (A) Dose‐response curves of the inhibitory activity of UDCA‐HDAC6i #1 (n = 9), #2 (n = 9), #6 (n = 3), #8 (n = 3), and #9 (n = 3) on HDAC6 and HDAC1. For curve fitting and IC50 calculation, log (inhibitor) versus normalized response with variable slope equation was used in GraphPad software (version 6; GraphPad Software Inc., La Jolla, CA). Extended information about HDAC IC50 values is provided in Supporting Table S3. (B) Comparison of the IC50 values of UDCA‐HDAC6i #1 and 4‐(aminomethyl)‐N‐hydroxybenzamide hydrochloride (8) for HDAC6 and graphical representation of the main interactions taking place in the binding of UDCA‐HDAC6i #1 to the catalytic domain 2 of HDAC6 (hydrogen bonds in purple and π‐π stacking interactions in green). Black arrows point to the two hydroxyl groups of UDCA within UDCA‐HDAC6i #1 and the hydrogen bonds established between these and tryptophan 496 and histidine 560 of the catalytic domain 2 of human HDAC6. (C) Cross‐sectional view of 3D‐aligned active sites of different HDAC isoforms (HDAC1, 2, 3, 4, 6, 7, and 8 and zebrafish HDAC10 [PDB entries: 5ICN, 6G3O, 4A69, 4CBT, 5EDU, 3ZNR, 5VI6, and 5TD7]). (D) 3D structural alignments of the docking model for the catalytic domain 2 of human HDAC6 with the catalytic domains of different HDAC isoforms. In blue, surface region of each isoform aligned to the cavity in which the three most active/selective UDCA‐HDAC6is accommodate UDCA in HDAC6. Abbreviations: Gly, glycine; His, histidine; ND, not determined; Phe, phenylalanine; Ser, serine; Trp, tryptophan.
Once the affinity and selectivity for HDAC6 inhibition was confirmed in four of the synthesized UDCA‐HDAC6i molecules, we generated a homology model of the catalytic domain 2 of rat HDAC6 in order to predict the inhibitory capacity of these compounds between species (Supporting Fig. S3). Sequence alignment of the catalytic domains of both species revealed an identity of 88.4%. Importantly, as for the human HDAC6, UDCA‐HDAC6i #1, #2, and #9 were the compounds with the highest binding energies in the rat homology model (Supporting Fig. S3), suggesting that their inhibitory potential would be maintained in the rat. This points to a direct correspondence between our results and those expected in humans.
Next, UDCA‐HDAC6i #1 was selected for further in vivo analysis (Supporting Table S4). In particular, we hypothesized that the UDCA‐HDAC6i molecules could benefit from the intrinsic hepatotropic features of UDCA, concentrating them preferentially into the liver and in cystic cholangiocytes after oral administration and gastrointestinal absorption. In addition, these molecules could also be reabsorbed through the enterohepatic/bile circulation. In this regard, we applied the QikProp software included in the Maestro package 10.4 (Schrödinger, LLC) to in silico predict several pharmacokinetic parameters for all UDCA‐HDAC6i molecules, obtaining considerably better values for UDCA‐HDAC6i #1 than for the other three active candidates in terms of oral absorption. UDCA‐HDAC6i #1 displayed values within the recommended range for all the parameters analyzed, whereas UDCA‐HDAC6i #2 and #9 presented several potential limitations.
UDCA‐HDAC6i #1 Halts Hepato‐ and Nephromegaly in PCK Rats
The therapeutic value of UDCA‐HDAC6i #1 was evaluated in PCK rats (Pkhd1 mutant), an animal model for hepatorenal cystogenesis (Fig. 3).( 24 , 25 ) Eight‐week‐old PCK rats were treated with UDCA‐HDAC6i #1 (15 mg/kg/d by oral gavage) for 5 months. Remarkably, the characteristic hepato‐ and nephromegaly that PCK rats develop over time was significantly reduced after treatment with UDCA‐HDAC6i #1. Liver‐ and kidney‐to‐body weight ratios were also reduced compared to untreated PCK rats (Fig. 3A,B, respectively). These effects were associated with an improvement of both liver and kidney functions, as reflected by increased serum albumin and reduced urea levels, respectively (Fig. 3A,B). At the end of treatment, UDCA‐HDAC6i #1 was detected in liver, bile, and portal and peripheral blood, but was particularly abundant in bile (Fig. 3C). Moreover, an increase in acetylated α‐tubulin levels was found in both liver and kidney of animals receiving UDCA‐HDAC6i #1 compared to untreated PCK rats (Fig. 3D).
FIG. 3.
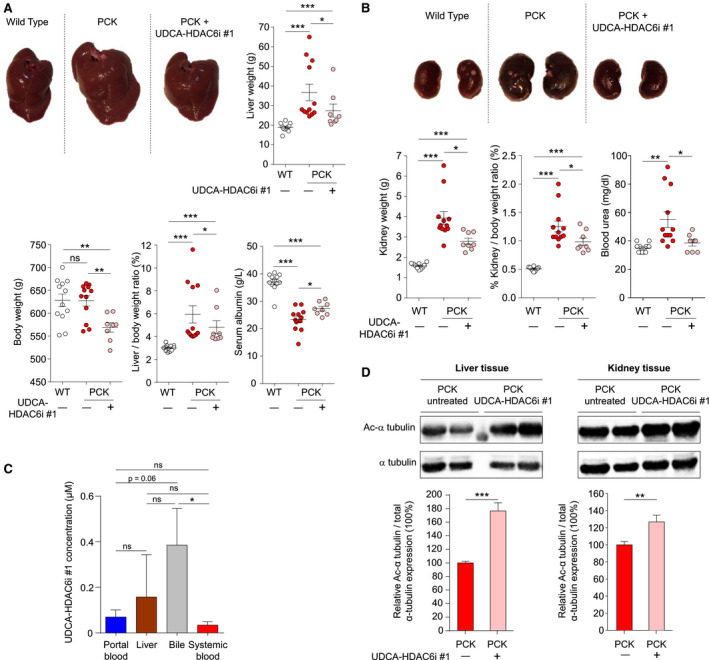
Effect of UDCA‐HDAC6i #1 on hepatorenal cystogenesis in vivo. (A) Representative liver images from a normal rat, as well as nontreated and UDCA‐HDAC6i #1–treated PCK rats. Graphical representation of relative comparisons of liver weight, total body weight, liver/body weight ratio, and serum albumin concentration in the three experimental groups. A statistical unpaired two‐tailed t‐test analysis was applied to determine significance in all cases, with the exception of PCK treated versus nontreated liver/body weight in which an unpaired one‐tailed t‐test analysis was applied. (B) Representative kidney images from a normal rat, as well as nontreated and UDCA‐HDAC6i #1–treated PCK rats. Graphical representation of relative comparisons of kidney weight, kidney/body weight ratio, and serum urea concentration in the three experimental groups. A statistical unpaired two‐tailed t test was applied to determine significance. (C) Analysis of UDCA‐HDAC6i #1 pharmacokinetics. Graph represents the bioavailability profile of UDCA‐HDAC6i #1 in blood (n = 8), liver (n = 8), bile (n = 7), and peripheral blood (n = 8) during chronic oral administration. An unpaired two‐tailed t test was applied to assess for significance. (D) Representative immunoblotting of the acetylated (Ac) α‐tubulin levels in liver and kidney of treated (n = 8) and nontreated (n = 12) PCK rats. Graph represents the relative amount of acetylated α‐tubulin in liver and kidney using total tubulin as normalizing control. For statistical significance assessment, an unpaired two‐tailed t test was used. Abbreviation: ns, not significant.
UDCA‐HDAC6i #1 Modulates the BA Pool and Increases Levels of Unconjugated UDCA in PCK Rats
BA concentrations and pool composition were determined in liver, bile, and both portal and peripheral blood. Consistent with previous reports,( 4 , 5 ) total BA concentrations in both liver and peripheral blood were higher in PCK rats than in normal animals. These values were not modified in PCK rats treated with UDCA‐HDAC6i #1 (Fig. 4). Moreover, total BA concentrations in portal blood and bile were similar in treated and untreated groups (Fig. 4). However, some differences in composition of the BA pool were found. Thus, the described( 4 , 5 ) decreased UDCA levels in liver of PCK rats were restored upon UDCA‐HDAC6i #1 treatment (Fig. 4). Similarly, increased levels of UDCA were found in portal and peripheral blood, as well as in bile, of PCK rats treated with UDCA‐HDAC6i #1 compared to both normal and PCK untreated animals (Fig. 4). Likewise, a normalization of major unconjugated bile acids (i.e., cholic acid [CA], α/β murocholic acid, and sulpholithocholic in liver; CA and hyodeoxycholic acid in bile) in treated PCK animals was found (Fig. 4; Supporting Tables S6 and S7).
FIG. 4.
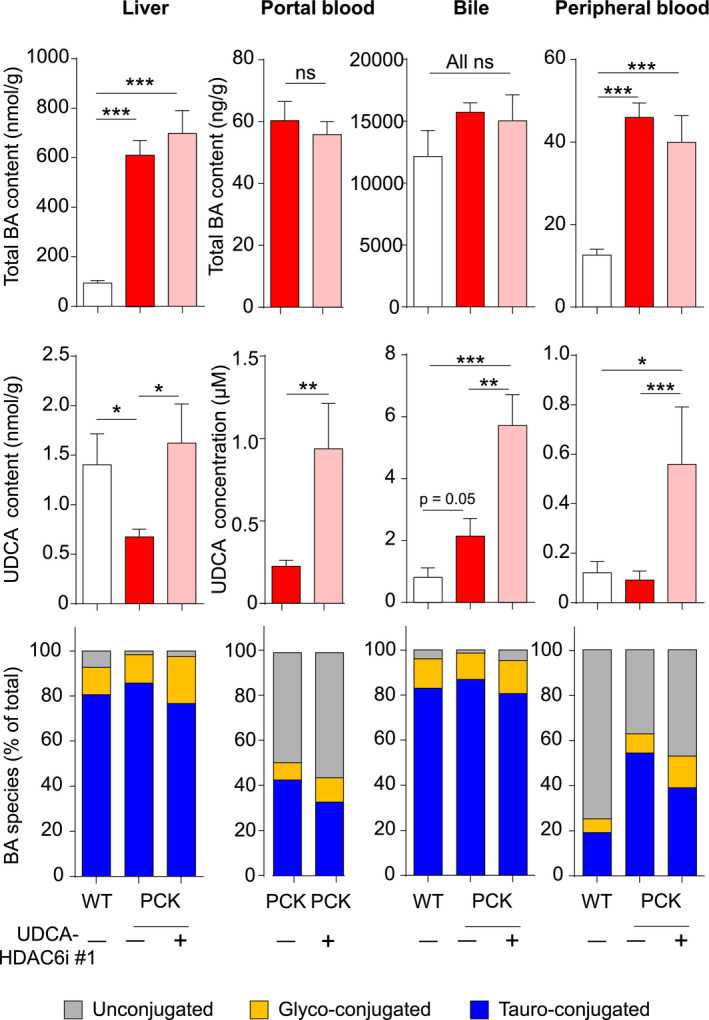
BA pool in portal blood, liver, bile, and peripheral blood. Bar graphs represent total BA concentration, unconjugated UDCA concentration, and relative abundance of conjugated BAs in each biological sample. Quantitative and statistical information about the relative abundancy of single BAs is depicted in Supporting Tables S5‐S8. A statistical unpaired two‐tailed t test was applied to determine significance. Abbreviation: ns, not significant.
UDCA‐HDAC6i #1 Halts Liver Cystogenesis In Vivo, Restores Primary Cilium Length, and Inhibits Cystic Cholangiocyte Proliferation
Histological analysis of the cholangiocyte marker, cytokeratin 19 (CK19), revealed a reduction in magnitude of cystic areas in liver of UDCA‐HDAC6i #1–treated animals compared to PCK untreated rats (Fig. 5A). HDAC6 inhibitory activity of UDCA‐HDAC6i #1 increased (64%) the primary cilium length of cystic cholangiocytes and the number of ciliated cholangiocytes (182%) in liver of the treated animals (Fig. 5B); similarly, in vitro, UDCA‐HDAC6i #1 increased levels of acetylated α‐tubulin (Fig. 5C) and restored the primary cilium length of cystic cholangiocytes (~40% increase; Fig. 5D), but induced no changes in normal cholangiocytes (Fig. 5D).
FIG. 5.
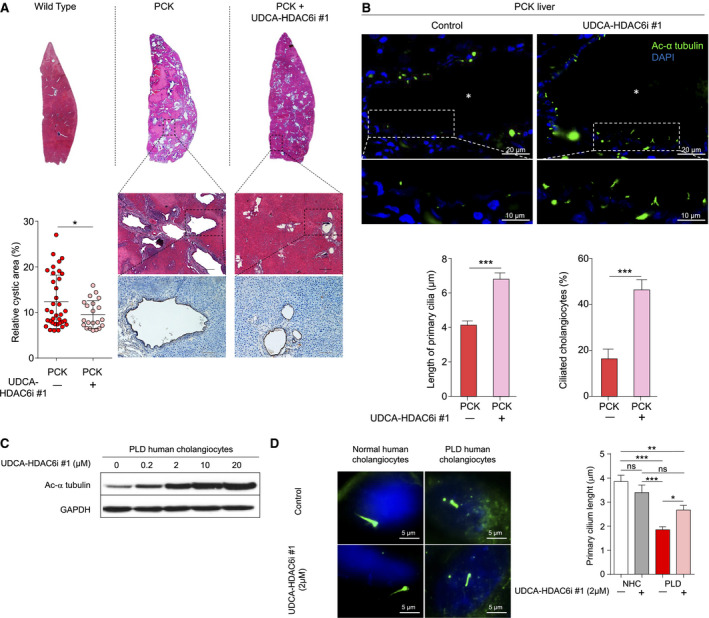
Effect of UDCA‐HDAC6i #1 on liver cystogenesis and primary cilium length. (A) Representative images of hematoxylin/eosin staining (scale bar = 250 µm) and CK19 immunohistochemistry (scale bar = 100 µm). Graph represents the cystic area relative to total tissue area of individual hepatic lobes (three for each animal). A statistical unpaired two‐tailed t test was applied to determine significance. (B) Representative images of liver sections from untreated and UDCA‐HDAC6i #1–treated PCK rats. Cilia are labeled with antibodies against acetylated (Ac) α‐tubulin (green) and nuclei are stained (blue) with DAPI. Bar graphs represent length of primary cilia and percentage of ciliated cholangiocytes. White asterisks indicate cyst lumen. Cysts were randomly selected from untreated (n = 6) and treated rats (n = 9). A statistical unpaired two‐tailed t test was applied to determine significance. Data are expressed as mean ± SEM. (C) Representative immunoblotting of the effect of increasing doses of UDCA‐HDAC6i #1 in levels of acetylated α‐tubulin in cultured PLD cholangiocytes. GAPDH was used as normalizing control. (D) Representative immunofluorescence images of acetylated α‐tubulin in cholangiocytes (primary cilium in green and nucleus in blue stained with DAPI). Bar graph represents the mean cilia length of normal (nontreated [n = 23] and 2 µM of UDCA‐HDAC6i #1 treated [n = 21]) and PLD (nontreated [n = 30] and 2 µM of UDCA‐HDAC6i #1 treated [n = 27]) human cholangiocytes. A statistical one‐way ANOVA was used. Abbreviations: ANOVA, analysis of variance; DAPI, 4′,6‐diamidino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; NHC, normal human cholangiocytes; ns, not significant.
Given that the primary cilium promotes epithelial cell polarity and prevents cell‐cycle initiation,( 26 , 27 ) we analyzed the effect of UDCA‐HDAC6i #1 on proliferation/expansion of cystic cholangiocytes. UDCA‐HDAC6i #1 inhibited the 3D growth of cystic cholangioids compared to control conditions (Fig. 6A). Similarly, UDCA‐HDAC6i #1 inhibited two‐dimensional (2D) cystic cholangiocyte proliferation in vitro in a dose‐dependent manner (Fig. 6B), which was associated with phosphorylated extracellular signal‐regulated kinase (ERK) 1/2 (pERK1/2) regulation (Fig. 6C). Importantly, this antiproliferative effect was significantly higher than that observed upon combined treatment with UDCA plus the separate spacer‐chelating component (8) (Fig. 6B), demonstrating that UDCA‐HDAC6i #1 has intrinsic properties.
FIG. 6.
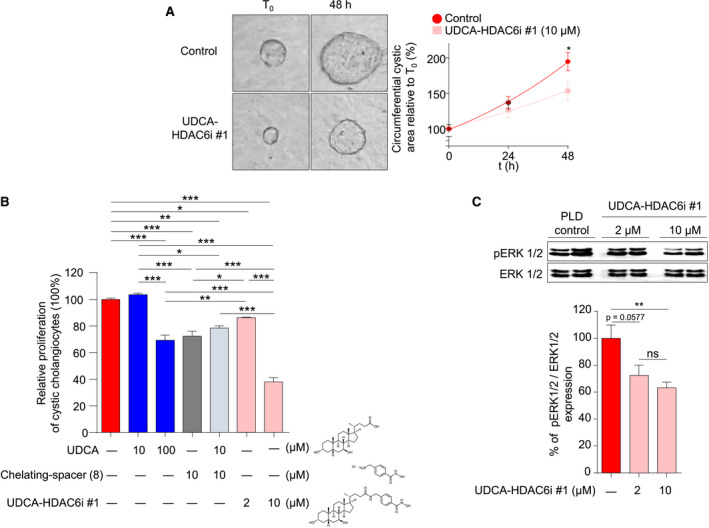
Effect of UDCA‐HDAC6i #1 on proliferation of 2D‐ and 3D‐cultured PLD cholangiocytes. (A) 3D culture in collagen type I of bile ducts/cysts isolated from PCK rats. Representative images and graph representing the relative circumferential area of nontreated (n = 20) and UDCA‐HDAC6i #1–treated (n = 20; 10 µM) cystic cholangioids along time. (B) Proliferation of PLD cholangiocytes by flow cytometry. Bar graph represents the percentage of cell proliferation after 48 hours of treatment with 10 µM of UDCA (n = 7), 100 µM of UDCA (n = 22), 2 µM of UDCA‐HDAC6i #1 (n = 16), 10 µM of UDCA‐HDAC6i #1 (n = 23), 10 µM of spacer chelating component (8) (n = 23), or combined treatment with 10 µM of UDCA + 10 µM of spacer chelating component (8) (n = 7) relative to control PLD cholangiocytes (n = 23) without treatment. (C) Representative immunoblotting of phosphorylated and total ERK1/2 protein levels from cell lysates of nontreated and UDCA‐HDAC6i #1–treated PLD cholangiocytes. Graph represents the relative abundance of phosphorylated ERK1/2 in baseline (n = 5) and 2 µM (n = 5) or 10 µM (n = 5) of UDCA‐HDAC6i #1–treated PLD cholangiocytes using total ERK as normalizing control. In all cases, a statistical unpaired two‐tailed t test was applied to determine significance. Abbreviation: ns, not significant.
Hepatotropic Properties of UDCA‐HDAC6i #1
To investigate the mechanism of UDCA‐HDAC6i #1 uptake by cystic cholangiocytes (i.e., passive diffusion or membrane protein transport), time‐dependent compound internalization was measured in cystic cholangiocytes exposed to UDCA‐HDAC6i #1 at 4°C or 37°C, respectively. Importantly, significant differences in uptake ratios at both temperatures were observed, suggesting that an important part of UDCA‐HDAC6i #1 internalization occurs through plasma membrane proteins (37°C), rather than by passive diffusion across the lipid bilayer (4°C; Fig. 7A). Therefore, in order to assess the specific transporters involved in the UDCA‐HDAC6i #1 internalization process, we next used cell models experimentally overexpressing plasma membrane transporters in substrate competition assays (Supporting Fig. S4). The results indicated that UDCA‐HDAC6i #1 was transported by the organic cation transporters (OCTs), OCT1 and OCT3, (Fig. 7B) as well as by the sodium‐dependent BA transporter (Na+‐taurocholate cotransporting polypeptide), commonly expressed in hepatocytes (Supporting Fig. S5). In contrast, neither the apical sodium‐dependent bile salt transporter expressed in cholangiocytes nor any of the assayed organic anion transporters (i.e., organic anionic transporting polypeptide [OATP] 1A2, OATP1B1, OATP1B3, and OATP2B1) mediated UDCA‐HDAC6i #1 uptake (Supporting Fig. S5). Interestingly, the analysis of expression (RT‐qPCR) of these transporters marked up‐regulation of OCT1 and OCT3 in cystic human cholangiocytes compared to normal controls (Fig. 7C). Furthermore, we confirmed by immunohistochemistry the expression (plasma membrane and cytoplasm) of OCT1 and OCT3 transporters in cyst‐lining cholangiocytes (and hepatocytes; Fig. 7D) of PCK rat liver tissue.
FIG. 7.
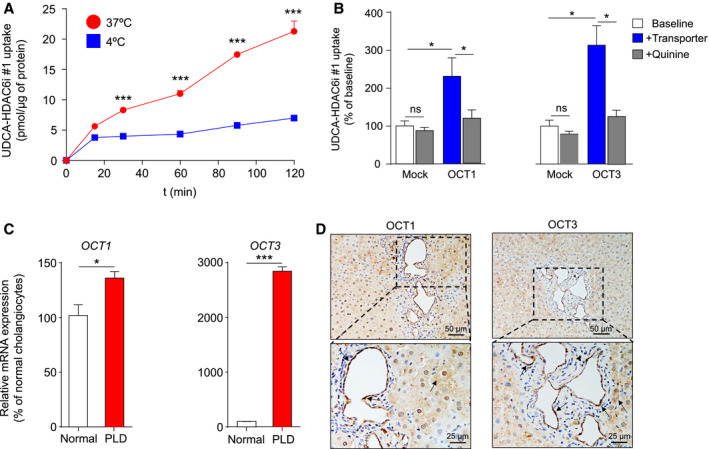
Mechanisms of UDCA‐HDAC6i #1 uptake into cells. (A) Time‐course analysis of UDCA‐HDAC6i #1 uptake in cystic cholangiocytes incubated with 100 µM of UDCA‐HDAC6i #1 at 37°C or 4°C. A statistical unpaired two‐tailed t test between both conditions at each time point was applied to determine significance. Data are expressed as mean ± SEM. (B) Bar graphs representing UDCA‐HDAC6i #1 uptake in HepG2 or CHO Mock cells or stably expressing OCT1 (n = 4) or OCT3 (n = 5), respectively, in the presence or absence of quinine as a competing substrate. The paired two‐tailed t test was used to assess statistical significance. (C) RT‐qPCR analysis of mRNA expression of OCT1 and OCT3 transporters in normal (n = 6) and PLD (n = 6) human cholangiocytes. The RPL22 gene was used as the normalizing control. An unpaired two‐tailed t test was applied to determine statistical significance. (D) Immunohistochemical analysis of OCT1 and OCT3 in liver sections of PCK rats. Abbreviations: ns, not significant; RPL22, 60S ribosomal protein L22.
Discussion
Oral administration of UDCA is well tolerated, safe, and is currently the first‐line therapy internationally approved for the treatment of primary biliary cholangitis.( 28 ) Chronic treatment with UDCA inhibits hepatic cyst growth in experimental models and also in a subset of patients with advanced PLD (CURSOR [Ursodeoxycholic Acid as Treatment for Polycystic Liver Disease] phase II clinical trial), improving their symptomatology.( 4 , 5 ) On the other hand, inhibition of HDAC6 activity has been proposed as a therapeutic strategy for PLD.
The currently available HDAC6is do not show preferential targeting to the liver and, in particular, to biliary cells. Most HDAC6is developed to date comprise on their structure a hydrophobic linker ended on a Zn2+ chelating group that enters into the catalytic sites of HDAC6 and an external cap that accommodates on the surface cavities.( 21 ) HDAC6i molecules usually contain aromatic peptide derivatives and hydrocarbons acting as the closing cap of the inhibitor,( 29 , 30 , 31 , 32 ) which are prone to be released into systemic circulation after their hepatic metabolism. These residues have been largely demonstrated to be potentially toxic in humans. Most frequently observed manifestations of toxicity in clinical trials are fatigue, nausea, diarrhea, and cardiotoxicity, including ventricular arrhythmia, being the most worrisome adverse effects.( 33 , 34 ) Considering these limitations for combination therapies,( 35 ) and taking advantage of our experience in the designing of HDACis through innovative in silico docking analyses,( 21 ) we hypothesized that UDCA could be an appropriate external cap to reinforce the HDAC6 inhibitory capacity of the chelating groups, improve their hepatotropism, decrease their toxicity, and provide some of the aforementioned UDCA benefits, because: (1) Our in silico docking experiments indicate that UDCA may actively contribute to reinforce the inhibitory potential of the hydrophobic linker + Zn2+ chelating group through the establishment of van der Waals interactions and hydrogen bonds between the two hydroxyl groups present in the UDCA structure and the side chains of several amino acid residues located on the external cavities of HDAC6 close to the catalytic domains; (2) UDCA would provide hepatotropic features to these new UDCA‐HDAC6i entities by promoting its enterohepatic transport through BA and/or OCTs; (3) UDCA is usually amidated with taurine (tauroursodeoxycholic acid) or glycine (glycoursodeoxycholic acid) by hepatocytes,( 36 , 37 ) so this residue could be used for the formal conjugation to the spacer‐chelating group; and (4) the gut microbiome deconjugates the taurine or glycine residues of UDCA,( 38 ) and, consequently, UDCA‐HDAC6i present in bile and entering the gut could be deconjugated by this process, resulting in a secondary increase of the enterohepatic levels of UDCA that would additionally inhibit PLD progression.
Data of the present study showed that UDCA has no HDAC inhibitory activity. However, its formal coupling to the spacer‐chelating component [Fig. 2B, chelating‐spacer (8)] (i.e., UDCA‐HDAC6i #1) reduced 1 order of magnitude the IC50 value of [Fig. 2B, chelating‐spacer (8)] for HDAC6 inhibition. Of note, the three UDCA‐HDAC6i molecules displaying the highest binding affinities to HDAC6 (i.e., UDCA‐HDAC6i #1, #2, and #9) were those containing one or two 1,4‐phenylene spacers and hydroxamates as chelating groups. These three molecules oriented the hydrocarbon skeleton of UDCA toward the same region of the protein surface; indeed, the difference in binding energies with the other compounds, which accommodated UDCA in other surface cavities, was >3 kcal/mol. Importantly, predicted inhibitory activity of the UDCA‐HDAC6i molecules on HDAC6 was further validated in both cystic cholangiocytes (i.e., acetylated α‐tubulin expression) and enzymatic assays (specific HDAC6 vs. HDAC1 inhibitory activity) in vitro. Of note, replacement of the hydroxamic acid moiety of UDCA‐HDAC6i #1 by the ortho‐phenylenediamine group present in UDCA‐HDAC6i #8 resulted in a complete switch in both the inhibitory potential and selectivity toward nuclear HDACs instead of HDAC6. These data highlight the accuracy of our in silico predictive model for molecules to inhibit HDAC6. In this regard, the importance of certain regions of the HDAC6 CD2 surface to determine inhibitor selectivity has been recently reported.( 39 ) In accordance, structure alignment of different HDAC isoforms with our docking model revealed that the surface cavity occupied by UDCA in the three most HDAC6 active/selective compounds is not present in the other HDAC isoforms, whereas the cavity occupied by a still active, but much less selective, compound (i.e., UDCA‐HDAC6i #6) remained fully accessible in all isoforms. All these data highlight the critical role that UDCA plays in dictating both activity and selectivity of UDCA‐HDAC6is.
Once the inhibitory potential of the three best candidates was confirmed in human and rat HDAC6 by using a homology model of the catalytic domain 2, UDCA‐HDAC6 #1 was selected for further in vivo and in vitro evaluation based on comparative analysis of its selective HDAC6 inhibitory capacity (IC50), potential pharmacokinetics, and synthetic cost. Oral administration of UDCA‐HDAC6i #1 to PCK rats considerably reduced the hepato‐ and nephromegaly characteristic on these animals, as well as hepatic cystogenesis. Of note, this new molecule partially normalized the decreased serum levels of albumin, suggesting an improvement of liver function of these animals. In addition, levels of urea in serum were reduced upon treatment, indicating an improvement in renal function. On the other hand, no signs of toxicity were observed at biochemical and histological levels. The therapeutic effects of UDCA‐HDAC6i #1 in the liver seem to be comparable (i.e., hepatic cystogenesis) or even superior (i.e., hepatomegaly) to the ones previously reported for UDCA( 4 , 5 ) in the same animal model, even at substantially lower doses (i.e., 15 vs. 25 mg/kg/d for 5 months, respectively; Supporting Fig. S6). Moreover, in contrast to UDCA, UDCA‐HDAC6i #1 also provides therapeutic effects on nephromegaly and renal function of PCK rats (Supporting Fig. S6).
At the histological level, increased acetylated α‐tubulin was obtained in both liver and kidney of treated PCK rats compared to untreated animals, supporting that UDCA‐HDAC6i #1 is properly absorbed by the gut and is able to reach both the liver and kidneys, leading to HDAC6 inhibition. Importantly, this effect was mirrored by a significant increase in the number of ciliated cholangiocytes and primary cilium length in treated animals, providing important insights into the biological mechanisms behind the therapeutic effects. In line with this result, UDCA‐HDAC6i #1 restored the primary cilium length of cystic cholangiocytes in an acetylated α‐tubulin–dependent manner, whereas it had no effect on primary cilium length of normal cholangiocytes in vitro. Moreover, UDCA‐HDAC6i #1 inhibited the proliferation of cystic cholangiocytes cultured in 2D and 3D conditions, and this was more significant than the combined treatment with its forming components (i.e., UDCA + (8)) at the same concentration, indicating that UDCA reinforces the HDAC6 inhibitory activity of the spacer‐chelating component. This observation was also accompanied by a significant reduction of ERK1/2 phosphorylation, which mediates cystic cholangiocyte hyperproliferation.( 3 ) All these data are consistent with the fact that the primary cilium acts as a check‐point inhibitor of the cell cycle, its restoration in cystic cholangiocytes being a therapeutic strategy.( 26 , 27 , 40 )
Regarding BA metabolism, and similarly to our previous data with UDCA,( 4 ) no significant changes in total BA concentration were observed between UDCA‐HDAC6i #1–treated and nontreated animals. However, changes in certain BA species in bile, and to a lesser extent in liver, were found. Of note, increased levels of free UDCA in liver, bile, and serum were found in PCK rats treated with UDCA‐HDAC6i #1, reinforcing the idea that biliary UDCA‐HDAC6i #1 could be further metabolized in the gut, leading to excision of the amide bond and thus resulting in the release of free UDCA that may enter the enterohepatic circulation in a second round and induce additional therapeutic benefit on hepatic cystogenesis.
As indicated above, two important limitations of currently available HDAC6is are their lack of selective affinity toward hepatic tissue as well as the presence of aromatic hydrocarbons or heterocycles in their structures,( 29 , 30 , 31 , 32 ) which can be released as potentially toxic metabolites. Our data reveal that, after oral administration, UDCA‐HDAC6i #1 accumulates preferentially in liver and bile, rather than in peripheral blood, indicating a superior targeting to the liver and posterior secretion into bile, thus potentially diminishing the off‐target effects in other tissues. This preferential biodistribution to the liver could be favored by specific transport activities driven by transport proteins located at the plasma membrane of cystic cholangiocytes.
In summary, we have designed, synthesized, and functionally analyzed a family of UDCA synthetic conjugates with selective HDAC6 inhibitory activity. UDCA‐HDAC6i #1 inhibited hepatorenal cystogenesis in vivo, improved ciliogenesis in cystic cholangiocytes, and inhibited their hyperproliferation. These therapeutic effects are superior to the simple sum of the individual or combined effects of its constituting pharmacologically active elements. These UDCA synthetic conjugates have preferential targeting to the liver through specific BA and organic cation transport properties and are highly concentrated into the enterohepatic circulation. These particular features make UDCA‐HDAC6i molecules especially promising for the treatment of hepatic and gastrointestinal disorders where HDAC6 inhibition is considered a therapeutic target, such as acute liver failure, hepatocellular carcinoma, cholangiocarcinoma, pancreatic cancer, or colon cancer, among others,( 23 , 41 , 42 , 43 , 44 , 45 ) highlighting the high degree of translational potential of this family of chemical entities.
Author Contributions
F.J.C.‐C., I.R., E.H., O.B., A.S.‐L., L.I.‐S., P.Y.L.‐L., P.M.R., P.M.‐G., S.J., E.P., S.R., S.G., M.J.P., M.E., L.B., J.J.G.M., J.M.B., F.P.C.: study concept and design, acquisition of data, statistical analysis, analysis and interpretation of data, drafting of the manuscript. L.B., J.J.G.M., J.M.B., F.P.C.: obtained funding.
Supporting information
Supplementary Material
Video S1
Supported by the Spanish Carlos III Health Institute (ISCIII; J.M. Banales: FIS PI15/01132, PI18/01075 and Miguel Servet Program CON14/00129; M.J. Perugorria: PI14/00399, PI17/00022; J.J.G. Marin: FIS PI16/00598) cofinanced by “Fondo Europeo de Desarrollo Regional” (FEDER); CIBERehd (ISCIII): J.M. Banales, M.J. Perugorria, L. Bujanda, and J.J.G. Marin; Spanish Ministry of Economy and Competitiveness (M.J. Perugorria: Ramon y Cajal Program RYC‐2015‐17755); IKERBASQUE, Basque foundation for Science (M.J. Perugorria and J.M. Banales), Spain; “Junta de Castilla y Leon” (J.J.G. Marin: SA06P17); “Diputación Foral Gipuzkoa” (J.M. Banales: DFG15/010, DFG16/004; M.J. Perugorria: DFG18/114, DFG19/081), BIOEF (Basque Foundation for Innovation and Health Research: EiTB Maratoia BIO15/CA/016/BD to J.M. Banales), Department of Health of the Basque Country (J.M. Banales: 2017111010; M.J. Perugorria: 2019111024), and Euskadi RIS3 (J.M. Banales: 2016222001, 2017222014, and 2018222029; 2019222054); La Caixa Scientific Foundation (J.M. Banales: HR17‐00601); “Fundación Científica de la Asociación Española Contra el Cáncer” (AECC Scientific Foundation, to J.M. Banales and J.J.G. Marin); and “Centro Internacional sobre el Envejecimiento”, Spain (J.J.G. Marin: OLD‐HEPAMARKER, 0348‐CIE‐6‐E). F.J. Caballero‐Camino was funded by the Spanish Ministry of Science and Innovation (BES‐2014‐069148), A. Santos‐Laso by the Basque Government (PRE_2018_2_0195), and Pui Y. Lee‐Law by the European Association for the Study of the Liver (EASL; Sheila Sherlock Award). The Spanish Ministry of Science and Innovation supported F.P. Cossío: (CTQ2016‐80375‐P and CTQ2014‐51912‐REDC) as well as the Basque Government (F.P. Cossío: IT‐324‐07). I. Rivilla had a postdoctoral contract from the Donostia International Physics Center.
Potential conflict of interest: Nothing to report.
Contributor Information
Jesus M. Banales, Email: jesus.banales@biodonostia.org.
Fernando P. Cossío, Email: fp.cossio@ehu.es.
References
Author names in bold designate shared co‐first authorship.
- 1. Gevers TJ, Drenth JP. Diagnosis and management of polycystic liver disease. Nat Rev Gastroenterol Hepatol 2013;10:101‐108. [DOI] [PubMed] [Google Scholar]
- 2. Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, LaRusso NF, et al. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol 2014;11:750‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology 2009;49:160‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Munoz‐Garrido P, Marin JJ, Perugorria MJ, Urribarri AD, Erice O, Saez E, et al. Ursodeoxycholic acid inhibits hepatic cystogenesis in experimental models of polycystic liver disease. J Hepatol 2015;63:952‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D’Agnolo HM, Kievit W, Takkenberg B, Riaño I, Bujanda L, Neijenhuis MK, et al. Ursodeoxycholic acid in advanced polycystic liver disease: an international multicenter randomized controlled phase 2 trial. J Hepatol 2016;65:601‐607. [DOI] [PubMed] [Google Scholar]
- 6. Wu M, Mei C. Histone deacetylases 6 increases the cyclic adenosine monophosphate level and promotes renal cyst growth. Kidney Int 2016;90:20‐22. [DOI] [PubMed] [Google Scholar]
- 7. Yanda MK, Liu Q, Cebotaru L. An inhibitor of histone deacetylase 6 activity, ACY‐1215, reduces cAMP and cyst growth in polycystic kidney disease. Am J Physiol Renal Physiol 2017;313:F997‐F1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lorenzo Pisarello M, Masyuk TV, Gradilone SA, Masyuk AI, Ding JF, Lee PY, et al. Combination of a histone deacetylase 6 inhibitor and a somatostatin receptor agonist synergistically reduces hepatorenal cystogenesis in an animal model of polycystic liver disease. Am J Pathol 2018;188:981‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gradilone SA, Habringer S, Masyuk TV, Howard BN, Masyuk AI, Larusso NF. HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis. Am J Pathol 2014;184:600‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng K, Li S, Liao C. Progress in the discovery of macrocyclic histone deacetylase inhibitors for the treatment of cancer. Curr Med Chem 2017;24:4166‐4179. [DOI] [PubMed] [Google Scholar]
- 11. Mwakwari SC, Patil V, Guerrant W, Oyelere AK. Macrocyclic histone deacetylase inhibitors. Curr Top Med Chem 2010;10:1423‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem 2003;46:5097‐5116. [DOI] [PubMed] [Google Scholar]
- 13. Hodawadekar SC, Marmorstein R. Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene 2007;26:5528‐5540. [DOI] [PubMed] [Google Scholar]
- 14. Monte MJ, Dominguez S, Palomero MF, Macias RI, Marin JJ. Further evidence of the usefulness of bile acids as molecules for shuttling cytostatic drugs toward liver tumors. J Hepatol 1999;31:521‐528. [DOI] [PubMed] [Google Scholar]
- 15. Lozano E, Monte MJ, Briz O, Hernandez‐Hernandez A, Banales JM, Marin JJ, et al. Enhanced antitumour drug delivery to cholangiocarcinoma through the apical sodium‐dependent bile acid transporter (ASBT). J Control Release 2015;216:93‐102. [DOI] [PubMed] [Google Scholar]
- 16. Dominguez MF, Macias RI, Izco‐Basurko I, de La Fuente A, Pascual MJ, Criado JM, et al. Low in vivo toxicity of a novel cisplatin‐ursodeoxycholic derivative (Bamet‐UD2) with enhanced cytostatic activity versus liver tumors. J Pharmacol Exp Ther 2001;297:1106‐1112. [PubMed] [Google Scholar]
- 17. Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 2016;24:41‐50. [DOI] [PubMed] [Google Scholar]
- 18. Nytofte NS, Serrano MA, Monte MJ, Gonzalez‐Sanchez E, Tumer Z, Ladefoged K, et al. A homozygous nonsense mutation (c.214C‐>A) in the biliverdin reductase alpha gene (BLVRA) results in accumulation of biliverdin during episodes of cholestasis. J Med Genet 2011;48:219‐225. [DOI] [PubMed] [Google Scholar]
- 19. Steiner C, von Eckardstein A, Rentsch KM. Quantification of the 15 major human bile acids and their precursor 7alpha‐hydroxy‐4‐cholesten‐3‐one in serum by liquid chromatography‐tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2010;878:2870‐2880. [DOI] [PubMed] [Google Scholar]
- 20. Urribarri AD, Munoz‐Garrido P, Perugorria MJ, Erice O, Merino‐Azpitarte M, Arbelaiz A, et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut 2014;63:1658‐1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zubia A, Ropero S, Otaegui D, Ballestar E, Fraga MF, Boix‐Chornet M, et al. Identification of (1H)‐pyrroles as histone deacetylase inhibitors with antitumoral activity. Oncogene 2009;28:1477‐1484. [DOI] [PubMed] [Google Scholar]
- 22. Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol 2016;12:741‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res 2013;73:2259‐2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mason SB, Liang Y, Sinders RM, Miller CA, Eggleston‐Gulyas T, Crisler‐Roberts R, et al. Disease stage characterization of hepatorenal fibrocystic pathology in the PCK rat model of ARPKD. Anat Rec (Hoboken) 2010;293:1279‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, et al. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol 2004;165:1719‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mansini AP, Peixoto E, Thelen KM, Gaspari C, Jin S, Gradilone SA. The cholangiocyte primary cilium in health and disease. Biochim Biophys Acta 2018;1864:1245‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pan J, Snell W. The primary cilium: keeper of the key to cell division. Cell 2007;129:1255‐1257. [DOI] [PubMed] [Google Scholar]
- 28. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol 2015;62(1 Suppl.):S25‐S37. [DOI] [PubMed] [Google Scholar]
- 29. Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc 2010;132:10842‐10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain‐selective small‐molecule inhibitor of histone deacetylase 6 (HDAC6)‐mediated tubulin deacetylation. Proc Natl Acad Sci U S A 2003;100:4389‐4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY‐1215, in combination with bortezomib in multiple myeloma. Blood 2012;119:2579‐2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dallavalle S, Pisano C, Zunino F. Development and therapeutic impact of HDAC6‐selective inhibitors. Biochem Pharmacol 2012;84:756‐765. [DOI] [PubMed] [Google Scholar]
- 33. Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, et al. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res 2006;12:3997‐4003. [DOI] [PubMed] [Google Scholar]
- 34. Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009;27:5459‐5468. [DOI] [PubMed] [Google Scholar]
- 35. Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol 2018;8:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hofmann AF. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand J Gastroenterol Suppl 1994;204:1‐15. [DOI] [PubMed] [Google Scholar]
- 37. Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci 2008;65:2461‐2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res 2017;1:3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, et al. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol 2016;12:748‐754. [DOI] [PubMed] [Google Scholar]
- 40. Masyuk TV, Masyuk AI, LaRusso NF. Therapeutic targets in polycystic liver disease. Curr Drug Targets 2017;18:950‐957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding G, Liu HD, Huang Q, Liang HX, Ding ZH, Liao ZJ, et al. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett 2013;587:880‐886. [DOI] [PubMed] [Google Scholar]
- 42. Zhang WB, Zhang HY, Jiao FZ, Wang LW, Zhang H, Gong ZJ. Histone deacetylase 6 inhibitor ACY‐1215 protects against experimental acute liver failure by regulating the TLR4‐MAPK/NF‐κB pathway. Biomed Pharmacother 2018;97:818‐824. [DOI] [PubMed] [Google Scholar]
- 43. Li D, Sun X, Zhang L, Yan B, Xie S, Liu R, et al. Histone deacetylase 6 and cytoplasmic linker protein 170 function together to regulate the motility of pancreatic cancer cells. Protein Cell 2014;5:214‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arlt A, Muerkoster SS, Schafer H. Targeting apoptosis pathways in pancreatic cancer. Cancer Lett 2013;332:346‐358. [DOI] [PubMed] [Google Scholar]
- 45. Kaliszczak M, Trousil S, Aberg O, Perumal M, Nguyen QD, Aboagye EO. A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth. Br J Cancer 2013;108:342‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Video S1