

Abstract
The three human RAS proteins are mutated and constitutively activated in ~20% of cancers leading to cell growth and proliferation. For the past 3 decades, many attempts have been made to inhibit these proteins with little success. Recently; however, multiple methods have emerged to inhibit KRAS, the most prevalently mutated isoform. These methods and the underlying biology will be discussed in this review with a special focus on KRAS-plasma membrane interactions.
1. RAS Isoforms, Structure and Posttranslational Modifications
RAS proteins are part of a superfamily of more than 150 low molecular weight GTP binding proteins [1, 2] that include the RAB, RAN, RHO and ARF families [3]. RAS proteins oscillate between an active GTP-bound and inactive GDP-bound state to modulate cell survival, proliferation and differentiation [4]. RAS is activated by guanine nucleotide exchange factors (GEFs) that facilitate GDP unloading; this promotes GTP binding because GTP concentrations in the cell are 10-fold higher than GDP [5]. RAS proteins possess a slow intrinsic GTPase activity [6, 7] which is enhanced 105-fold by GTPase-activating proteins (GAPs). GTP hydrolysis returns active GTP-bound RAS to the inactive GDP-bound ground state.
There are 3 RAS genes ubiquitously expressed in human cells. HRAS and KRAS were first identified as oncogenes in the Harvey and Kirsten Rat Sarcoma retroviruses [8]. NRAS was identified in transformation assays using DNA from a Neuroblastoma [9]. Due to alternative splicing, the KRAS gene generates two protein isoforms, KRAS4A and KRAS4B (hereafter KRAS), the latter of which is generally accepted to be expressed at higher levels [10]; however, some studies have shown that KRAS4A is equally expressed in a variety of cancer cell lines and human colorectal cancers [11]. The RAS isoforms share 90% sequence homology in the G domain, which binds the guanine nucleotide [12, 13]. The G domain comprises an effector lobe (residues 1–86) and an allosteric lobe (residues 87–165). The effector lobe interacts with RAS effectors such as RAF and PI3K and has two regions called Switch I (residues 30–40) and Switch II (60–76) [12] that undergo major conformational reorganization on GTP binding [14, 15]. The GTP-bound form exists in two states: state 1 is an open conformation that promotes nucleotide exchange and discourages effector binding; while state 2 is a closed conformation that encourages GTP hydrolysis and effector binding [16]. GTP binding also reorients the RAS protein with respect to the membrane to allow for effector protein interaction [15, 17, 18]. Despite extensive sequence homology in the G-domain, the RAS isoforms differ substantially in their C-terminal hypervariable region (HVR) (residues 166–189) which regulates subcellular localization, trafficking and plasma membrane (PM) spatiotemporal organization [19, 20].
All RAS proteins have a C-terminal “CAAX” sequence where C is cysteine, A is an aliphatic amino acid and X is Methionine or Serine. Nascent RAS proteins synthesized in the cytosol are processed by farnesyl transferase (FTase) that catalyzes the addition of a 15-carbon (farnesyl) isoprenoid to the C-terminal cysteine [21] (Figure 1). This modification targets RAS to the cytosolic surface of the endoplasmic reticulum (ER) where the endoprotease RAS converting enzyme 1 (RCE1), cleaves the “AAX” tripeptide from the prenylated cysteine [22–26]. Finally, isoprenylcysteine carboxyl-methyltransferase (ICMT) methyl-esterifies the a-carboxyl group of the now C-terminal farnesylated cysteine [27–33]. Following CAAX processing, the RAS isoforms differ in membrane trafficking and localization due to different “second signals” in the HVR upstream of the processed CAAX motif [34]. The HVR of NRAS and HRAS, contain one or two, respectively, cysteine residues that are palmitoylated by the heterodimeric Golgi palmitoyl acyltransferase DHHC9 and GCP16 [35, 36] and are then transported to the inner leaflet of the plasma membrane (PM) via the exocytic pathway [27, 37, 38]. The HVR of KRAS, contains a polybasic domain of 6 contiguous lysine residues that targets KRAS to the largely negatively-charged inner leaflet of the PM via the endosome, bypassing the Golgi [38]. Hence, the HVR contributes to differential signaling between RAS proteins due to different post-translational modifications these regions undergo that dictate RAS membrane trafficking and localization [5, 19]. This can clearly be seen in studies showing that KRAS but, not N- or HRAS, is essential for normal development in mice [24, 39–44] and that each isoform activates a common set of effectors with varying efficiencies in vitro [42].
Figure 1. Schematic of RAS posttranslational processing, plasma membrane targeting and recycling.
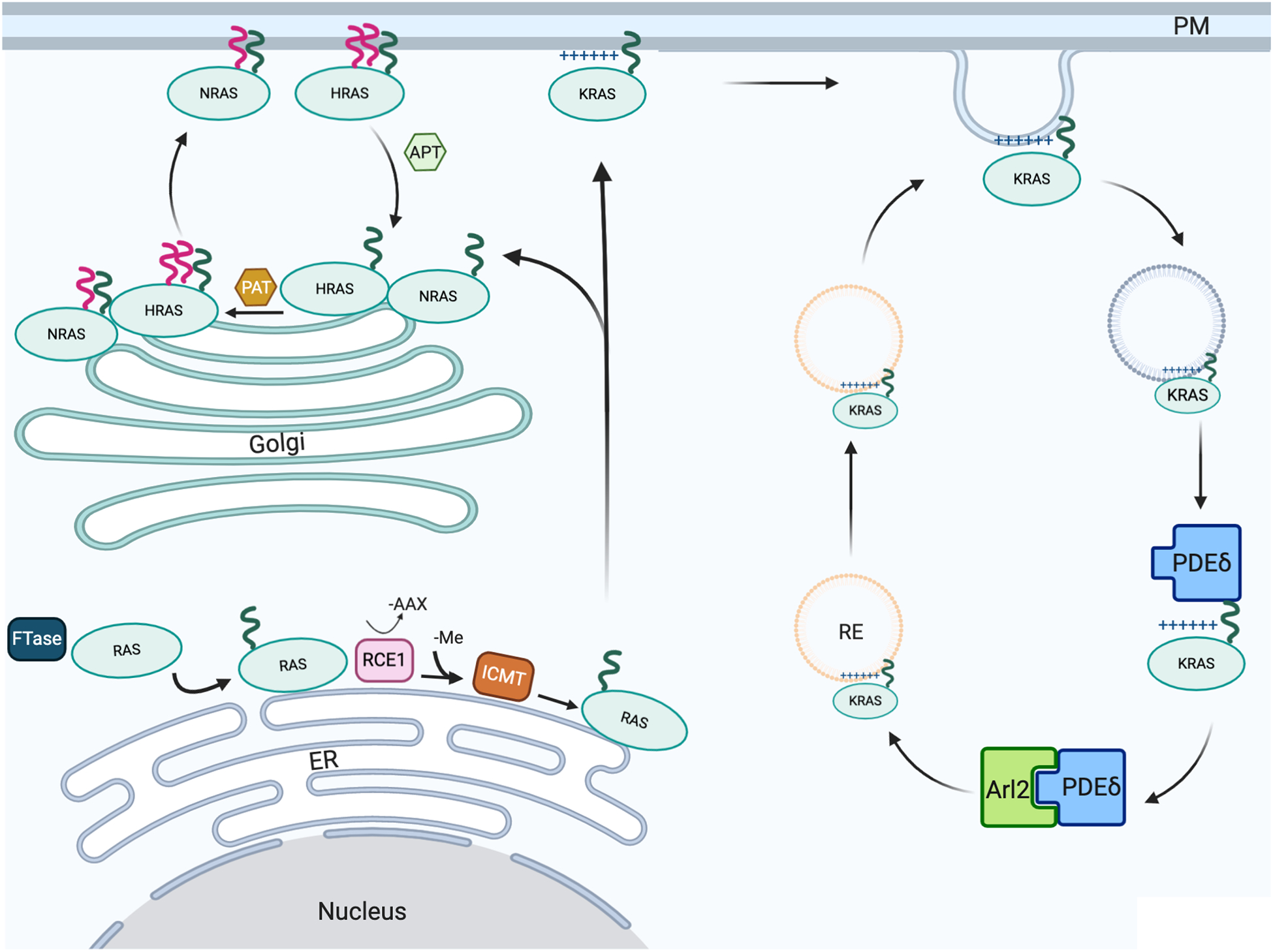
Following mRNA translation in the cytosol, the three RAS isoforms (HRAS, NRAS and KRAS) are trafficked to the PM in a series of steps in specific subcellular localizations. HRAS and NRAS are recycled through palmitoylation-depalmitoylation cycles; KRAS is recycled via the recycling endosome which is enriched with PtdSer. Green and red lines indicate farnesyl and palmitate, respectively. The (+) symbol denotes polybasic residues of the KRAS hypervariable region. FTase, farnesyl transferase; RCE, Ras-converting enzyme 1 protease; ICMT, isoprenylcysteine carboxylmethyltransferase; PAT, palmitoyl acyl transferase; APT, acyl-protein thioesterase; PDEδ, phosphodiesterase delta; Arl2, ADP-ribosylation factor-like protein 2; ER, endoplasmic reticulum; Golgi, Golgi apparatus; RE, recycling endosome; PM, plasma membrane; -Me, methyl group.
RAS proteins undergo constant cycles of solubilization and membrane binding that are required to maintain the fidelity of PM localization. Following endocytosis, HRAS and NRAS undergo de-palmitoylation by acyl-protein thioesterase (APT) releasing the proteins into the cytosol for recycling back to the Golgi where they are repalmitoylated and returned to the PM by vesicular transport via the exocytic pathway (Figure 1). PM-bound KRAS also undergoes endocytosis but has different outcomes depending on the phosphorylation state of the KRAS HVR at Ser181 by PKC and PKG2 [24, 45–49]. The endocytic vesicle is initially rich in the anionic phospholipid phosphatidylserine (PtdSer) but loses this asymmetry, decreasing the negative charge after endocytosis [50–53], releasing KRAS into the cytosol. Soluble KRAS is then captured by the prenyl-binding protein phosphodiesterase delta (PDEδ) which unloads KRAS at the perinuclear region where the Arl2 GTPase is concentrated. Arl2 interacts with PDEδ inducing a conformational change that promotes KRAS release [49]. Non-phosphorylated KRAS is then loaded onto PtdSer-rich recycling endosomes (REs) by electrostatic trapping for forward transport to the PM by vesicular transport (Figure 1). By contrast, electrostatic repulsion likely operates to prevent phosphorylated KRAS from binding to the negatively charged membrane of the RE [54], leading to the accumulation of phosphorylated KRAS in the cytosol and other endomembranes [5, 48, 55]. Therefore, phosphorylation of membrane-bound KRAS does not directly trigger dissociation from the PM but rather decreases KRAS PM content by inhibiting recycling back to the PM [48].
2. RAS in Cancer
2.1. Mutational Profiles
The RAS genes are the most frequently mutated oncogenes in cancer [13, 56] and as such have long been considered a drug target [5, 57, 58]. The prevalence of mutations varies substantially across the isoforms with KRAS, NRAS and HRAS being implicated in 85%, 12% and 3% of cancers, respectively [5, 59]. Additionally, mutational bias between the isoforms also exists with the majority of mutations occurring at codons 12 and 61 for KRAS and NRAS, respectively, while HRAS is mutated at both these sites at a similar frequency. Overall, the 3 oncogenic mutational hotspots in RAS genes are G12, G13 and Q61 [13]. G12 and G13 mutations displace crucial interactions between RAS and GAPs required to form a transition-state complex, without which an increase in GTPase activity cannot occur, effectively maintaining KRAS in its “on” state leading to constitutive downstream signaling [60–62]. The Q61 mutation inhibits GTP hydrolysis of GTP-RAS by interfering with a catalytic water molecule essential for this process [60, 61]. Together, these mutations render RAS primarily GTP-bound and relatively insensitive to GAPs [62], with a 10-fold decrease in the rate of intrinsic GTP hydrolysis [7, 63–65].
KRAS is the isoform predominantly mutated in human cancers and occurs in some of the more deadly cancers in the U.S: pancreatic cancer (~90%), colorectal cancer (~50%) and non-small cell lung cancer (~25%). These cancers originate from the endodermal layer which correlate with comparative studies that showed that KRAS G12V mutations lead to enhanced proliferation and inhibited differentiation of endodermal stem cells, increasing the pool of progenitor cancer cells in these organs [66–68]. Since KRAS represents the critical clinical concern, it will be the focus of this review.
2.2. KRAS Effector Signaling
Multiple RAS effectors that bind GTP-RAS proteins via a RAS-binding domain (RBD) or a RAS association (RA) domain have been described [69]. Among these, the most important in KRAS oncogenesis are components of the RAF-MEK-ERK (MAPK) pathway and the PI3K-AKT-mTOR pathway that promote cell proliferation and survival, respectively [3, 13, 70].
The RAF proteins (CRAF, BRAF and ARAF) are serine/threonine kinases that, on activation, phosphorylate and activate MEK1 and MEK2 which in turn phosphorylate and activate ERK1 and ERK2 [71, 72]. Activated ERK1/2 then phosphorylate >200 substrates including members of the ETS family of transcription factors and c-FOS which activates the AP1 transcription factor. Together, these events lead to the expression of D-type cyclins and other cell cycle regulatory proteins to promote cell cycle progression through the G1 phase [73–77].
Interaction of GTP-KRAS with the catalytic subunits of type I phosphatidylinositol 3-kinase (PI3K), p110α/δ/γ [78, 79], recruits the kinase to the PM where it phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3) [80, 81]. PIP3 then binds to and activates the serine/threonine kinase AKT, which phosphorylates and activates many downstream targets including the NF-κB transcription factor involved in cell survival, and FOXOs involved in DNA damage repair [69, 82–85]. RAS activation of PI3K is important because it mimics a PI3K-dependent survival signal normally induced by extracellular matrix (ECM) attachment, which is lost in cancer cells [85] (reviewed in [86]). Under normal physiological conditions, loss of ECM attachment induces BAX translocation to the mitochondria and cytochrome c release, leading to apoptosis [87]. PI3K signaling inhibits cytochrome c release through AKT-mediated phosphorylation of I-kB kinase (IKK), releasing NF-κB, leading to the production of anti-apoptotic proteins [88]. NF-κB activation through PI3K-RAC signaling can attenuate the toxic effects of reactive oxygen species, modulate oxidative stress [87, 89–91] and lead to the production of IAPs (inhibitor of apoptosis proteins) (reviewed in [92, 93]).
In vitro experiments show that HRAS and KRAS more effectively activate the PI3K and MAPK pathways, respectively [94, 95]. Several lines of evidence also suggest that KRAS oncogenesis predominantly depends on the MAPK pathway: First, KRAS-WT pancreatic ductal adenocarcinomas (PDACs) have BRAF mutations, which phenocopy KRAS rather than PI3K pathway mutations [96–98]. Second, PI3K mutations co-occur with KRAS mutations indicating that KRAS does not potently activate the PI3K/AKT pathway [99]. Finally, only activated RAF and MEK can restore the growth of RASless mouse embryonic fibroblasts (MEFs) [100].
3. KRAS localization and nanoclustering
KRAS signaling occurs predominantly on the PM to where effectors are recruited and activated. A prerequisite for effector recruitment is the formation of transient (t<1s) GTP-KRAS nanoclusters, containing 5–6 KRAS proteins which selectively sort PM lipids [101–104]. Though it has historically been thought that the KRAS polybasic domain (PBD) associates with anionic PM phospholipids purely through electrostatic interactions, recent work has shown that the PBD-farnesyl anchor actually exhibits exquisite binding specificity for PtdSer. Indeed, the precise amino acid sequence of the PBD and the prenyl group define a cryptic code for lipid binding specificity [102, 104]. Differential interactions between different lysine side chains and PM lipids lead to a realized tertiary structure on the PM with a very specific binding preference for PtdSer [105]. Moreover the KRAS anchor binds specifically to asymmetric PtdSer containing one saturated and one desaturated tail, thus the anchor also recognizes the acyl chain structure of PtdSer [106]. In consequence, KRAS PM binding and spatial organization is highly sensitive to PM PtdSer content such that decreasing levels of endogenous PtdSer decreases KRAS PM levels, nanoclustering, and signaling [104, 106–108]. A role for G-domain homodimerization in KRAS spatial organization has also been proposed. Dimerization surfaces have been identified by molecular dynamics (MD) simulations that allow the building of membrane-bound KRAS oligomers up to and including pentamers [109, 110] To some extent these surfaces have been validated by mutating interacting residues, which leads to reduced nanoclustering in intact cells and altered biology in more complex analyses [109, 110]. What is clear is that dimers involve low affinity protein interactions and only exist in the constrained two diffusional space of a membrane surface. However, the lipid composition of that membrane is also critical; reconstitution experiments with simple bilayers have failed to visualize any evidence of RAS dimerization or oligomerization [111], whereas more complex supported bilayers that emulate the lipid composition of plasma membrane faithfully recapitulate KRAS nanoclustering behavior [112]. Similarly, isolated KRAS anchors also display lipid-dependent nanoclustering behavior. In sum, anchor-lipid interactions are likely the driving force for KRAS nanoclustering on the PM with additional regulation, or modulation, provided through G-domain-lipid and G-domain-G-domain interactions. A thorough review of RAS PM targeting, spatial organization and anchor-induced lipid sorting can be found in [105].
KRAS signaling through the MAPK pathway occurs primarily on the PM, but has also been reported to occur from intracellular membranes such as the Golgi [27, 37], ER, endosomes [113, 114] and mitochondria [115, 116]. For example, EGFR activation leads to initial RAS activation on the PM and subsequently on Golgi membranes. RAS activation on the PM is rapid and transient (1–10 minutes) while activation on the Golgi is delayed and sustained (>20 minutes) [117]. The nature of MAPK signaling from these different platforms also varies. GTP-KRAS nanoclusters on the PM operate as digital switches which translate a graded/analog input (RAF kinase activities) into amplified fixed/digital output (pp-ERK) [103, 118, 119]. In contrast, RAS signaling on the Golgi generates a graded pp-ERK output, indicating the requirement of a higher activation threshold for the MAPK module in the Golgi compared to at the PM [120]. Interestingly, high-threshold graded Golgi MAPK signaling has clear biological relevance in the context of thymic deletion of highly autoreactive T-cells during fetal development [121]. The fact that one signaling module can generate different signaling dynamics and produce fundamentally different outputs points towards the importance of the nanoscale heterogeneity present within membrane environments.
4. KRAS Inhibitors
The quest to identify KRAS inhibitors has taken over 30 years, a journey that earned KRAS an “undruggable” status [122]. The first crystal structure of RAS solved in 1989 showed that, other than the nucleotide binding site, KRAS lacks any deep pockets that can be susceptible to small molecule binding [99, 123, 124]. Nevertheless, many combination therapies and new approaches to inhibiting KRAS signaling are emerging which can broadly be categorized into three strategies: (1) Direct inhibition of KRAS by targeting the nucleotide binding pocket, or allosterically modulating KRAS activity by binding newly identified surface pockets, (2) inhibition of downstream effectors of KRAS, and (3) inhibition of KRAS-PM interactions.
4.1. Direct KRAS Targeting:
4.1.1: Non-nucleotide Site Binding
RAS proteins have a picomolar affinity for GTP, which when combined with the cellular concentration of GTP in the millimolar range, essentially eliminates the feasibility of developing of GTP-competitive inhibitors for KRAS [69, 125]. This contrasts with the relative ease of designing ATP-competitive inhibitors against receptor tyrosine kinases (RTKs) that have binding affinities for ATP in the low micromolar to millimolar range [126]. Many attempts have been made to engineer proteins that specifically bind KRAS, including whole antibodies [127], single chain variable fragments [128], affibodies [129], monobodies [130, 131], DARPins [132], anticalins and other biologics that bind to allosteric sites on RAS (reviewed in [133]). Allosteric inhibitors targeting RAS dimerization and protein-protein interactions, rather than nucleotide analogs, seem to offer a feasible approach to inhibit RAS function [130, 131, 134, 135]. Moving forward, some degree of isoform and mutant specificity may be required to limit potential toxicity that may arise from inhibiting all RAS proteins [136]. For example, some pan-RAS inhibitors disrupt RAS-RAF interaction by binding to sites adjacent to the effector binding site and have shown promising results in vitro and in vivo; however, concern with toxicity in normal cells arise when targeting all isoforms [137].
KRAS allosteric sites were discovered by Gorfe et al. and colleagues through ensemble docking, MD simulations, bioinformatics, and in vitro validation of potential inhibitors. They showed that helix 5 and loop 7 are involved in allosteric regulation of the nucleotide-binding switch, and identified three adjacent transient binding pockets, termed p1, p2 and p3 (Figure 2). Virtual screening and ensemble docking were then used to identify novel small molecule binders to these pockets that could not be captured by traditional crystallography [138, 139]. Their efforts led to the identification of a novel pyrazolopyrimidine-based allosteric KRAS inhibitor, compound 11 (Figure 3A), that binds GTP-KRAS at the “p1” pocket with an IC50 of ~1–5 μM, disrupting RAS-RAF binding, inhibiting KRAS signaling and cancer cell proliferation [140].
Figure 2. Druggable allosteric pockets on KRAS.
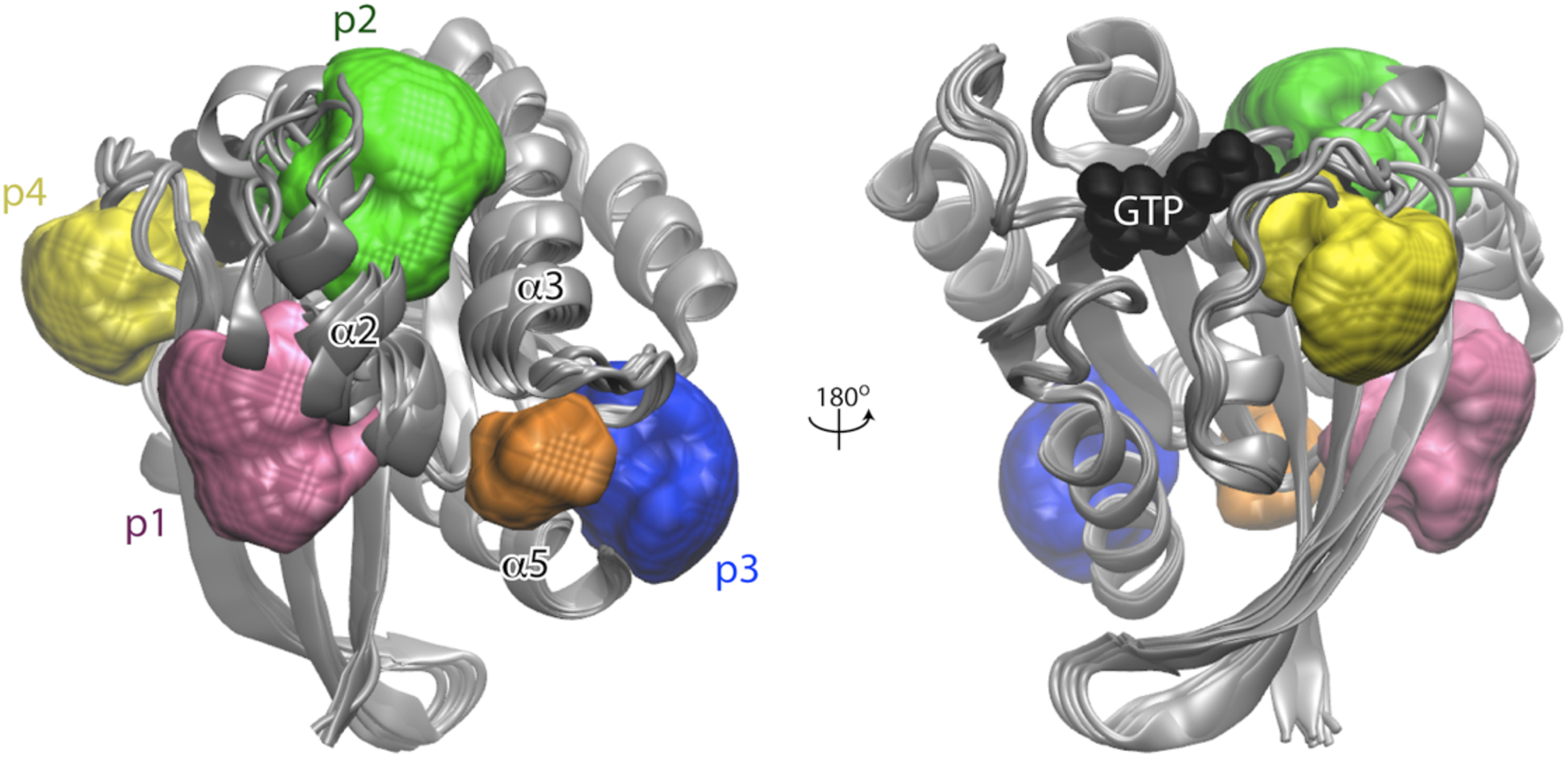
Ensemble fragment mapping analysis identified four allosteric binding pockets, p1, p2, p3 and p4 (pink, green, blue and yellow, respectively), on the catalytic domain of KRAS. P1 is located near the core-beta sheet while p2 is between helix 3 switch 2. P3 and p4 are near the C-terminus and behind switch 1, respectively. Image reproduced from Grant et al. with permission [356].
Figure 3. Chemical structures of several KRAS signaling inhibitors.
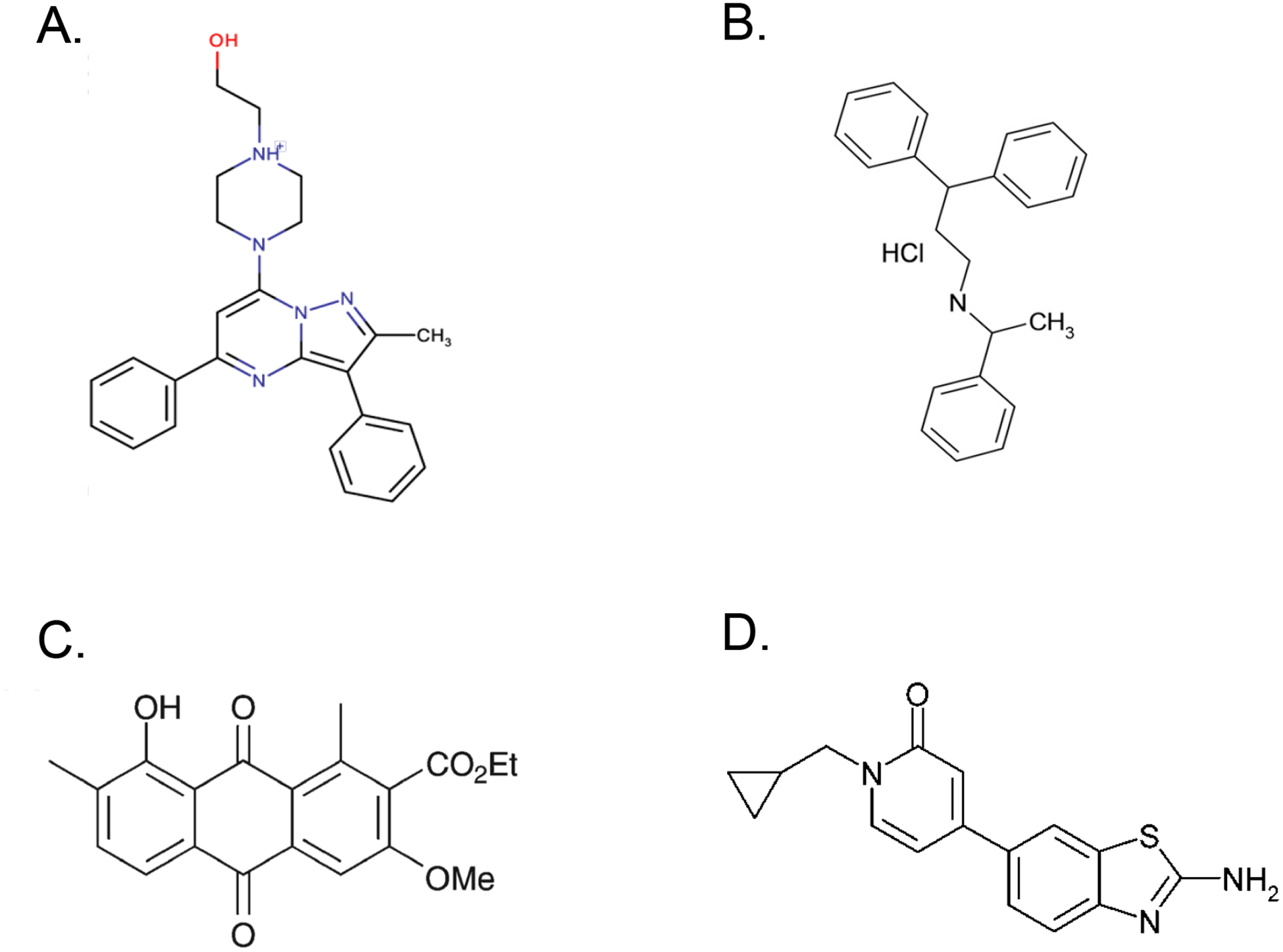
(A) Compound 11 binds to the p1 allosteric pocket on KRAS to inhibit its function; image reused with permission from McCarthy et al. [140] (https://pubs.acs.org/doi/10.1021/acsomega.8b03308). (B) Fendiline inhibits acid sphingomyelinase in the lysosome, resulting in PtdSer and KRAS mislocalization from the PM. (C) G01 inhibits acid sphingomyelinase as well but also perturbs RAS recycling to the PM through the recycling endosome pathway by inhibiting APEH; republished with the permission of ASBMB, from “An oxanthroquinone derivative that disrupts RAS plasma membrane localization inhibits cancer cell growth”, Tan et al., vol. 293, issue 35, 2018; permission conveyed through Copyright Clearance Center, Inc. [271]. (D) C7 selectively inhibits PI4KIIIα leading to depletion of PI4P from the PM and concomitant mislocalizaion of PtdSer and KRAS; image reused from Raubo et al. with permission from Royal Society of Chemistry [340].
Macromolecule inhibitors bind their targets with high specificity and affinity but their large size hinders uptake by cells. While small molecule drugs overcome this issue, their small size means a smaller surface area to bind targets and lower target affinity [141–143]. To circumvent these issues, Quevedo et al. used intracellular antibody capturing technology [144, 145] to develop an antibody fragment that binds to all RAS isoforms in the GTP-bound conformation. They showed that their product bound to RAS with low Kd, high Kon and low Koff, and inhibited tumor growth in vivo [146]. Since it bound to RAS with such desirable properties, they used it as a starting point to screen for other compounds that bound RAS at the same location and discovered a compound, Abd-7, that bound RAS adjacent to the effector binding region. Treatment with Abd-7 reduced signaling through the PI3K and MAPK pathways as measured by decreased levels of p-AKT and pp-ERK in colorectal and non-small cell lung cancer cell lines. Their work showed that using intracellular antibody fragments as starting points for drug development represents a viable option for the generation of small high-affinity compounds [147].
Another intriguing mechanism visualizes targeting RAS-membrane interaction sites to lock KRAS in an orientation with respect to the PM that occludes access to the effector binding site. This was demonstrated by the compound, Cmpd2, discovered using nanodiscs coupled with NMR spectroscopy to generate and characterize atomic-scale structures of KRAS on a 20% PtdSer lipid bilayer [148].
4.1.2: KRAS G21C Inhibitors
Mutant G12C KRAS is found in approximately 13% of non-small cell lung cancer (NSCLC), 3% of colorectal cancer and 2% of other solid tumors [149]. Many compounds have been developed to target KRAS G12C following the pioneering work of Shokat and his group [150] who developed a thiol-reactive compound which binds to a shallow pocket below switch II and forms a disulfide bond with the mutant cysteine to lock KRAS in the GDP-bound state [151–155]. One such KRAS G12C inhibitor, ARS-853, was among the first compounds to show drug-like qualities in potency and selectivity [154]. KRAS G12C has a much faster nucleotide cycling time (t1/2 of nucleotide release = 9.9 minutes, t1/2 hydrolysis = 27 minutes) compared to G12D and G12V mutants (>3 hours) [156]. Because of this fast cycling time, such mutants remain sensitive to upstream signals rendering ARS-853 more effective when coupled with EGFR inhibitors like erlotinib that increase the cellular fraction of GDP-KRAS. In contrast, combination treatment with MEK1/2 inhibitors renders ARS-853 treatment less effective due to loss of negative feedback inhibition of ERK1/2 on SOS1 (a RAS GEF), increasing the amount of GTP-KRAS that cannot be bound by ARS-853 [154]. These in vitro studies led to the development of ARS-1620 by structure-based design, a highly potent and selective KRAS G12C inhibitor that induced tumor regression in vivo [151].
The first-in-human clinical trials with Amgen’s G12C inhibitor AMG 510 are underway with promising preliminary results in lung cancer patients (clinicaltrials.gov identifier NCT03600883, phase1/2), but not colorectal cancer patients. Although 47% of lung cancer patients (11 out of 23) experienced tumor shrinkage, this occurred in only 3% (1 out of 29) of colorectal cancer patients [149, 157, 158]. Clinical trials studying the effects of combining AMG 510 with MEK or PD1 inhibitors in solid KRAS G12C tumors (clinicaltrials.gov identifier NCT04185883, phase 1b) and trials comparing AMG510 treatment versus Docetaxel in NSCLC (clinicaltrials.gov identifier NCT04303780, phase 3) are underway. Oher pharmaceutical companies have also manufactured KRAS G12C inhibitors that are currently in clinical trials such as Mirati Therapeutics MRTX894 (clinicaltrials.gov identifier NCT03785249, phase1/2) [159].
Recent work elucidated potential resistance mechanisms to AMG 510 in three models of KRASG12C lung cancer [160]. Single-cell RNAseq revealed that inhibitor treatment increased p21 and p27 expression levels indicative of a quiescent (G0) state [161] in 80% of the cell population while the remaining 20% were termed “adapting” cells. Adapting cells initially undergo growth inhibition upon treatment but this is followed by accumulation of GTP-KRAS and finally MAPK reactivation, a pattern seen in many cases of acquired drug resistance [162–164]. A genome-wide knock-out screen revealed that heparin-binding epidermal growth factor (HBEGF), aurora kinase A (AURKA) and KRAS were essential for the AMG 510-adapting cell population. Increased HBEGF in adapting cells led to activated EGFR signaling, which proved essential for escaping the G0 state. The role of AURKA in mitogenic signaling [165–167] and acquired resistance to PI3K or EGFR inhibitors has been well established [168, 169]. In this setting, AURKA stabilized the interaction between GTP-KRAS and CRAF; and co-treatment with AMG 510 and an AURKA inhibitor inhibited CRAF and ERK activation more extensively than treatment with either inhibitor alone. Cells expressing higher KRAS levels quickly convert GDP-KRAS to GTP-KRAS by increased upstream EGFR signaling and maintain the GTP-KRAS-CRAF interaction downstream by AURKA signaling, ultimately leading to cell cycle progression and escape from G0 [160]. These results likely explain why the majority of patients in the AMG 510 clinical trial have only had partial responses [157].
BI-2852 (available at https://opnme.com/molecules/kras-bi-2852) targets KRASG12C via a fundamentally different mechanism from other G12C inhibitors. BI-2852 binds to a pocket between switch I and switch II (SI/II-pocket), rather than above switch II (SII-pocket), which is unaffected by KRAS activation status [170]. Therefore, BI-2852 blocks interaction of both GDP- and GTP-KRAS with SOS1 [171] as well as GTP-KRAS with GAPs [172], CRAF and PI3Kα, inhibiting signaling and proliferation of KRAS-mutant cells in the low micromolar range [173, 174]. An obvious limitation of these inhibitors is that they are mutation specific, and most KRAS-mutant tumors have G12V or G12D mutations that have yet to be successfully targeted. However, this is a very active area of research [147, 175–177], with the SI/II pocket garnering most attention.
4.2. Downstream Effector Targeting
Identifying a suitable therapeutic window for agents that target KRAS or the MAPK pathway has been difficult as these pathways are essential for regulating cell growth and survival in normal cells. Efficacy has also been a recurring issue when trying to target the MAPK pathway, as doses required for efficient inhibition can be toxic [178–180]. Therefore, acquired drug resistance in tumor cells treated with these drugs is inevitable due to the strong selective pressure on cancer cells, which are inherently genetically unstable and able to adapt quite quickly. Downstream effector inhibitors have been extensively reviewed elsewhere [1, 3, 69, 99, 122, 181, 182] and so will only be considered briefly here to illustrate some mechanisms of resistance.
In the case of BRAF inhibitors for example, resistance is acquired through increased HGF secretion by stromal cells which activates both the MAPK and PI3K pathways via MET activation. Concordantly, BRAF-mutant melanoma patient data showed that those with stromal HGF had a significantly worse treatment response [183]. MET activation may also have a role in mitigating the action of gefitinib, an EGFR inhibitor, in non-small cell lung cancer [184–188]. HGF-induced resistance is immediate and innate as opposed to acquired resistance to drugs developed over time, but could be reduced by a combination of MEK and AKT inhibition [183]. BRAFV600E colorectal cancer cell lines treated with RAF inhibitors also develop resistance through EGFR activation [162, 164]. One recurring theme of resistance seen with many BRAF inhibitors is paradoxical activation of ERK due to alleviation of negative feedback mechanisms imposed by ERK on upstream components of the pathway [189–192], which is also seen with MEK1/2 inhibitors [99].
Rigosertib was developed as a RAS-mimetic that binds the RAS binding domain (RBD) of RAF [193]. This molecule inhibits RAS signaling but the mechanism of action remains unclear since Rigosertib binds the RAF-RBD with low affinity and cannot dissociate RAF from GTP-bound RAS. Rigosertib may induce mitotic stress and the generation of reactive oxygen species leading to JNK-mediated inhibition of SOS and RAF, to suppress the MAPK pathway in a KRAS non-specific manner [194].
SHP2 is a tyrosine phosphatase that acts upstream of KRAS to promote MAPK signaling and also mediates resistance to BRAF or MEK inhibitors in specific ERK-dependent tumors [195]. In addition, combination treatment with SHP2 inhibitors also abrogated the RTK reactivation observed with KRASG12C inhibitors, leading to sustained MAPK inhibition with better outcomes in vitro and in vivo. Several SHP2 inhibitors are currently under clinical evaluation since SHP2 inhibition has shown efficacy in RAS-mutant glioblastoma, pancreatic and lung cancers and in gastroesophageal cancers with WT KRAS amplification (ClinicalTrials.gov Identifiers: NCT03114319; NCT03565003; NCT03634982) [196–200]. Full details on all clinical trials involving RAS effector therapies can be found in a recent review [16].
4.3. Synthetic Lethality
Much work has been directed to identifying tumor-specific vulnerabilities that may be targeted to cause tumor cell death, a phenomenon termed synthetic lethality [201]. Such vulnerabilities may include non-oncogenic metabolic or genetic programs on which cancer cells have become dependent, so called non-oncogene addiction [202, 203]. Normal cellular processes that become critical for the maintenance of KRAS-mutant tumors include regulation of oxidative, genotoxic and ER stress, autophagy, macropinocytosis, glycolysis, and enhanced glutamine uptake in PDAC. Inhibitors of these pathways are currently in different stages of preclinical and clinical evaluation (reviewed in [204]).
For example, inhibiting KRAS function in PDAC cell lines leads to increased autophagy as a mechanism of cell survival [205–207]. Targeting both the MAPK and autophagy pathways has synergistic effects in vitro and in vivo in KRAS-mutant PDAC, NRAS-mutant melanoma and BRAF-mutant colorectal cancer. Moreover, coinhibition of MEK and autophagy using trametinib and hydroxychloroquine, respectively, in a single PDAC patient led to significant reduction in levels of CA19–9 tumor marker, overall tumor burden and cancer-associated pain [206].
KRAS G12R tumors exhibit a selective vulnerability to inhibitors of PI3K and macropinocytosis. Though quite rare in other cancers (~1% in NSCLC and CRC), G12R mutations comprise up to ~20% of KRAS mutations in PDAC. KRAS G12V and KRAS G12D expression stimulate macropinocytosis while KRAS G12R lacks this ability. The G12R substitution causes a structural change in switch II that impairs binding to the PI3K p110α subunit and hence activation of PI3K/AKT signaling, which in turn is essential for macropinocytosis. KRAS-independent upregulation of PI3Kγ signaling is therefore required to support macropinocytosis in KRAS G12R-PDAC. In consequence KRAS G12R-PDAC is more sensitive than G12D-PDAC and G12V-PDAC to ERK/MAPK and autophagy inhibition. Interestingly, simultaneously targeting both pathways was synergistic in G12D- and G12V-PDAC but additive in G12R-PDAC [208].
A genome-wide synthetic lethal RNAi screen in RAS-mutant tumors yielded multiple hits in the mitotic stress response pathway, including subunits of the APC/C complex and the mitotic kinase PLK1 [209]. Concordantly, mutant RAS positive lung cancer patients had increased overall survival if APC/C levels were low, whereas APC/C activity level had no prognostic bearing in mutant RAS negative patients. PLK1 is activated at the G2/M transition by Aurora A Kinase leading to mitotic progression. Activated KRAS may lead to oncogene-induced senescence and so KRAS-mutant cells seem to have a higher dependence on PLK1 to progress through mitosis, as evidenced by increased levels of total and activated (phospho-)PLK1 [209]. This compensatory mechanism was also seen with KRAS G12C inhibitor-resistant cells, which had increased Aurora A Kinase levels [160]. In this context the PLK1 inhibitor, BI-2536 significantly inhibited the growth of KRAS transformed HCT116 and DLD-1 colorectal cancer cell xenografts. Other pathways essential for RAS-mutant cell growth include ribosomal biogenesis, RNA splicing and mRNA translation, and posttranslational modifications such as neddylation and sumoylation [209]. An extensive list of RAS synthetic lethal functional genetic screens can be found in [204]. Unfortunately many of the early hits from these studies could not be validated or recapitulated pharmacologically [209–211], and to date none have advanced to the clinic for the treatment of mutant RAS cancers [212].
4.4. Inhibiting KRAS Posttranslational Modification and Plasma Membrane Recycling
A valid mechanism to inhibit KRAS function is to block PM localization [213]. As discussed previously, KRAS requires the posttranslational attachment of a membrane anchor to localize to the PM. The first step in this process can be blocked by Farnesyl Transferase inhibitors (FTIs). Highly potent and relatively non-toxic FTIs were developed in the early 1990s that blocked the growth of HRAS mutant tumors in mouse models [214–217]. Concordantly, an ongoing phase 2 clinical trial conducted by Kura Oncology for the FTI tipifarnib in patients with HRAS-mutant relapsed or refractory urothelial carcinomas has seen promising results: 5 of the 13 patients treated achieved objective responses and 4 experienced progression-free survival greater than 6 months (clinicaltrials.gov identifier NCT02535650) [218]. Kura oncology is conducting another phase 2 tipifarnib clinical trial in HRAS-mutant Head and Neck Squamous Cell Carcinoma (clinicaltrials.gov identifier NCT02383927) and have reported a 56% overall response rate with progression free survivals of 6.1 months compared to 2.1 months on previous therapies [219]. However, FTIs did not provide such promising results in earlier trials with KRAS-mutant tumors because in FTI-treated cells, KRAS and NRAS are alternatively prenylated by geranylgeranyltransferase I (GGTaseI) allowing normal PM localization [3, 220–224]. Attempts to circumvent this escape mechanism to date have been unsuccessful. A GTTase1 inhibitor (GGT1–2418) that was well tolerated in phase 1 studies had limited efficacy (https://drugs.ncats.io/drug/M67G28K74K) [16], and combining FTIs with GTTase1 inhibitors (GGTIs), whilst showing promising efficacy in inhibiting prenylation of all RAS isoforms in preclinical experiments, proved to be too cytotoxic, with a therapeutic index too low to be implemented as a treatment option [225, 226]. Probably because together, FTase and GGTase1 prenylate a large number of proteins important for normal cellular growth and function [227]. One dual prenyltransferase inhibitor, “L-778,123” that advanced to two different phase 1 clinical trials failed to inhibit KRAS prenylation [228, 229] despite some apparent efficacy in vitro [230].
A second approach to prevent PM KRAS localization involves blocking the function of PDEδ, which as reviewed above promotes recycling of cytosolic KRAS to maintain the fidelity of PM targeting [47, 49]. Inhibiting PDEδ leads to the accumulation of KRAS on endomembranes, whence it cannot signal. An early small molecule PDEδ inhibitor, Deltarasin, bound to the prenyl-binding pocket of PDEδ with nanomolar affinity and inhibited KRAS signaling in in vitro PDAC models but its effective in vivo dose was in the micromolar range and caused off-target cytotoxicity that limited efficacy [231]. A second generation PDEδ inhibitor, Deltazinone 1, to some extent circumvented these problems and was efficacious against PDAC [232]. Other compounds, such as Deltasonamide, 1 and 2 also inhibit KRAS-mutant colorectal cancer cell proliferation but suffer from low membrane permeability [233, 234].
4.5. Inhibiting KRAS Plasma Membrane Localization
An unbiased high content screen with a library of 1,120 FDA-approved small molecules was used to identify inhibitors of KRAS PM localization [235]. Some hits, also identified previously, included weak amphiphilic bases which only caused moderate dissociation of KRAS from the PM [236], but the top hit, fendiline, significantly mislocalized KRAS (IC50 =3.14 μM) as well as the PtdSer (IC50 =3.16 μM) from the PM in a dose-dependent manner. Importantly, fendiline had no effect on NRAS or HRAS localization or on CAAX processing [235, 237]. Fendiline is an L-type voltage-gated Ca2+ channel blocker originally used to treat angina [238]. Other Ca2+ channel blockers had no effect on KRAS, indicating a new function unrelated to Ca2+ channel activity and unrelated to changes in intracellular Ca2+ levels [235, 239]. Fendiline is a racemic compound; however, only the R-fendiline stereoisomer (hereafter fendiline, Figure 3B) significantly mislocalized KRAS. Fendiline treatment abolished KRAS-dependent MAPK and PI3K signaling at concentrations similar to those required for KRAS mislocalization, and more potently inhibited proliferation of KRAS-mutant cells in a panel of 21 pancreatic, lung, colorectal and endometrial cancer cell lines [235]. This selectivity was also observed in vivo in xenograft mouse models of KRAS-mutant or KRAS-WT pancreatic tumors with no observed organismal toxicity [240]. Because fendiline reduced KRAS PM levels and inhibited both the MAPK and PI3K/AKT pathways, it may have potential use as an adjuvant therapy with kinase inhibitors that lead to paradoxical re-activation of these pathways; such as mTOR and BRAF inhibitors that activate MAPK signaling [241, 242]. Fendiline derivatives have been synthesized that show increased potency in terms of KRAS mislocalization, inhibition of cell proliferation in vitro and tumor growth in vivo [243].
Fendiline operates as an indirect inhibitor of lysosomal acid sphingomyelinase (ASM) which converts sphingomyelin (SM) to ceramide (Cer), leading to SM accumulation in the lysosome [240, 244, 245]. Fendiline treatment reduces PM cholesterol levels, increases PM SM levels, and decreases PM PtdSer and cellular ceramide levels. Concordantly, since KRAS PM localization and nanoclustering depends on PM PtdSer levels, supplementation with exogenous PtdSer rescued fendiline-induced KRAS mislocalization and MAPK signaling [237]. ASM facilitates NPC2-mdiated cholesterol efflux from the lysosome in part explaining the decreased cholesterol content seen in the PM of fendiline-treated cells [240]. More broadly, SM accumulation destabilizes lysosomal membranes [246] and impedes membrane fusion events such as those occurring during autophagy [247].
Fendiline and other ASM inhibitors, such as siramesine and cationic amphiphilic drugs (CADs), are concentrated in the lysosome where they interfere with the binding of ASM to bis-monoacyl-glycerophosphate (BMP) on the inner lysosomal membrane [248]. Displacement of ASM from BMP results in relocation to the lysosomal lumen where it undergoes degradation [249]. Siramesine and other CADs such as desipramine and amlodipine selectively kill KRAS-mutant HCT116 colon cancer cells but not the KRAS-null isogenic line [248]. Cancer cells generally have destabilized lysosomes due to elevated proteolytic activity and dependency on autophagy [250] and siramesine has been shown to effectively inhibit autophagic flux [251, 252]; other clinically relevant CADs probably have the same effect. The additional effect of fendiline on KRAS PM localization would add loss of KRAS signaling to these detrimental shared effects on lysosomal function, perhaps accounting for the increased sensitivity of KRAS-mutant cancer cells to fendiline treatment [253, 254].
Three FDA-approved tricyclic antidepressants, desipramine, imipramine and amitriptyline, which also inhibit ASM, [244, 255] mislocalize KRAS and PtdSer at similar IC50 values in a dose-dependent manner and significantly decrease KRAS nanoclustering on the PM. These effects were translated to decreased MAPK signaling and preferential inhibition of KRAS-mutant pancreatic, colon, lung and endometrial cancer cells over KRAS-WT cells. ASM inhibitors also block mutant let-60 (KRAS homolog) signaling in C. elegans [240].
Sphingomyelin is generated by de novo synthesis from serine and palmitate by serine palmitoyl transferase (SPT) [256, 257], or by addition of a choline head group from phosphocholine to ceramide by sphingomyelin synthases (SMases) [258, 259]. Several targetable components of SM metabolism that decrease KRAS function were identified by an RNA interference screen against 18 enzymes of the Cer-SM biosynthetic pathway in the C. elegans system. Among the identified genes whose inhibition most potently suppressed let-60 signaling (~70%) were sphk-1 (sphingosine kinase 1), hyl-2 (ceramide synthase) and, unsurprisingly, asm-1 which encodes acid sphingomyelinase) [240]. SPHK1 is a kinase that phosphorylates sphingosine to sphingosine-1-phosphate (S1P), an extensively-studied active oncometabolite that promotes cell proliferation and survival [260, 261].
Validation of hits with pharmacological inhibitors in mammalian cells revealed that inhibition of ceramide synthase with fumonisin B1, dihydroceramide desaturase with GT11, or ceramide kinase with K1 potently misloclaized PM KRAS and PtdSer. These enzymes function in the Cer-SM de novo biosynthetic pathway and their inhibition in vitro significantly perturbed cellular SM levels or SM distribution and depleted PM PtdSer. Fumonisin B1, GT11 and K1 concordantly dose-dependently mislocalized KRAS from the PM at concentrations similar to those needed to inhibit their enzyme targets [240]. Additionally, inhibiting ORMDL3, a negative regulator of SPT, by very low concentrations of staurosporine (STS) [237] increased SM content by de novo synthesis [262].
G01 (3-O-methyl oxanthroquinone ethyl ester) is a derivative of oxanthroquinone, a polyketide structure discovered from a high content screen for inhibitors of KRAS PM binding (Figure 3C) [263]. G01 is an inhibitor of acylpeptide hydrolase (APEH), a prolyl-oligopeptidase that removes Nα-acylated amino acids from peptides and functions in protein degradation of oxidized and glycated proteins [264–270]. G01 treatment significantly inhibited the PM localization and nanoclustering of HRAS, KRAS4B (KRAS), KRAS4A and PtdSer with an IC50 of ~ 1μM [264, 271]. Further analysis suggests that this effect of G01 on multiple RAS isoforms [271] reflected a disruption of RE function which is crucial for maintaining RAS PM localization [49, 272]. The importance of the RE in maintaining RAS on the PM is echoed by studies showing that thioesterase inhibitors which prevent HRAS and NRAS depalmitoylation [273, 274], PDEδ inhibitors or RAB11 knockdown which prevent KRAS loading on to the RE [49, 231, 275] all lead to their redistribution to endomembranes. Similarly, a commercially available APEH inhibitor called ebelactone A inhibited APEH function at concentrations similar to that required to mislocalize KRAS from the PM [264, 269, 276]. Interestingly, in a cohort of patients encompassing 33 different types of cancer, APEH was expressed at significantly higher levels in KRAS-mutant as opposed to KRAS-WT tumors [264]. Precisely how APEH inhibition impairs RE function remains unclear, but may be related to aberrant proteasomal regulation [264].
G01 blocks MAPK signaling in cells expressing constitutively active KRAS or HRAS at the same concentrations that cause mislocalization from the PM. This translated to decreased proliferation of a panel of 14 cell lines comprising pancreatic, colon, endometrial and lung cancer cell lines treated with G01. Interestingly, cell lines harboring mutant KRAS were more sensitive to treatment than cell lines expressing WT KRAS in all but the lung cancer cell lines [271]. G01 also inhibited ASM and NSM, leading to elevated SM and cholesterol content [264] as seen with fendiline and fumonisin B1 [237, 240].
One unifying feature of these different mechanisms of lipid homeostasis is that they indirectly regulate PM PtdSer levels. Thus, their disruption decreases PM PtdSer content, and hence the capacity of KRAS to stably localize to and nanocluster on the PM. These observations in turn suggest that other approaches to disrupt PtdSer PM localization will also abrogate KRAS function.
4.5.1. Phosphatidylserine and KRAS Function
PtdSer accounts for 4–6% of total cellular lipids [277–279] but is the major anionic lipid species of the inner leaflet of the PM, accounting for ~25mol% [4, 280]. PtdSer is synthesized on the cytosolic leaflet of the ER by PtdSer synthase-1 (PSS1) and PtdSer synthase-2 (PSS2) [281] which, respectively, replace the choline or ethanolamine headgroups of phosphatidylcholine (PC) or phosphatidylethanolamine (PE) with serine [282–284]. Deletion of both PSS isoforms is lethal [285]; whereas cells with a non-functional PSS1 are viable [286–288]. From the ER, PtdSer is transported to mitochondria, PM, endosomes, lysosomes, and the trans-Golgi network by lipid transport proteins (LTPs) or vesicular transport [289–292]. Among intracellular organelles, PtdSer is most enriched in recycling endosomes (REs) [293]. PtdSer binding proteins such as evectin-2 mediate retrograde trafficking between the RE and Golgi and likely contribute to the maintenance of high PtdSer levels on the PM [282, 293, 294].
PtdSer is mainly transported from the ER to the PM by Oxysterol Related Binding Protein 5 and 8 (ORP5 and ORP8) encoded by OSBPL5 and OSBPL8, respectively [295]. ORP5 and ORP8 share 80% sequence identity [296] and are ER-resident proteins that function as lipid exchangers at membrane contact sites (MCSs) between the ER and PM, or mitochondrial outer membrane [297, 298]. MCSs are microdomains where two organelle membranes are arrayed within 10–30 nm of each other to facilitate molecule exchange [299]. At ER-PM MCSs, ORP5 and ORP8 couple PtdSer transport against its concentration gradient with PI4P transport down its concentration gradient. Dedicated modes of lipid transport between membranes generate membrane asymmetry which is then maintained by a variety of “trapping” mechanisms including asymmetric breakdown and synthesis. In the case of PI4P, it is synthesized on the PM by PM-localized PI4KIIIα and degraded immediately after transport to the ER by the ER-resident phosphatase SAC1P (Figure 4) [298–301].
Figure 4. ORP5 and ORP8 transport PtdSer to the plasma membrane in exchange for PI4P.
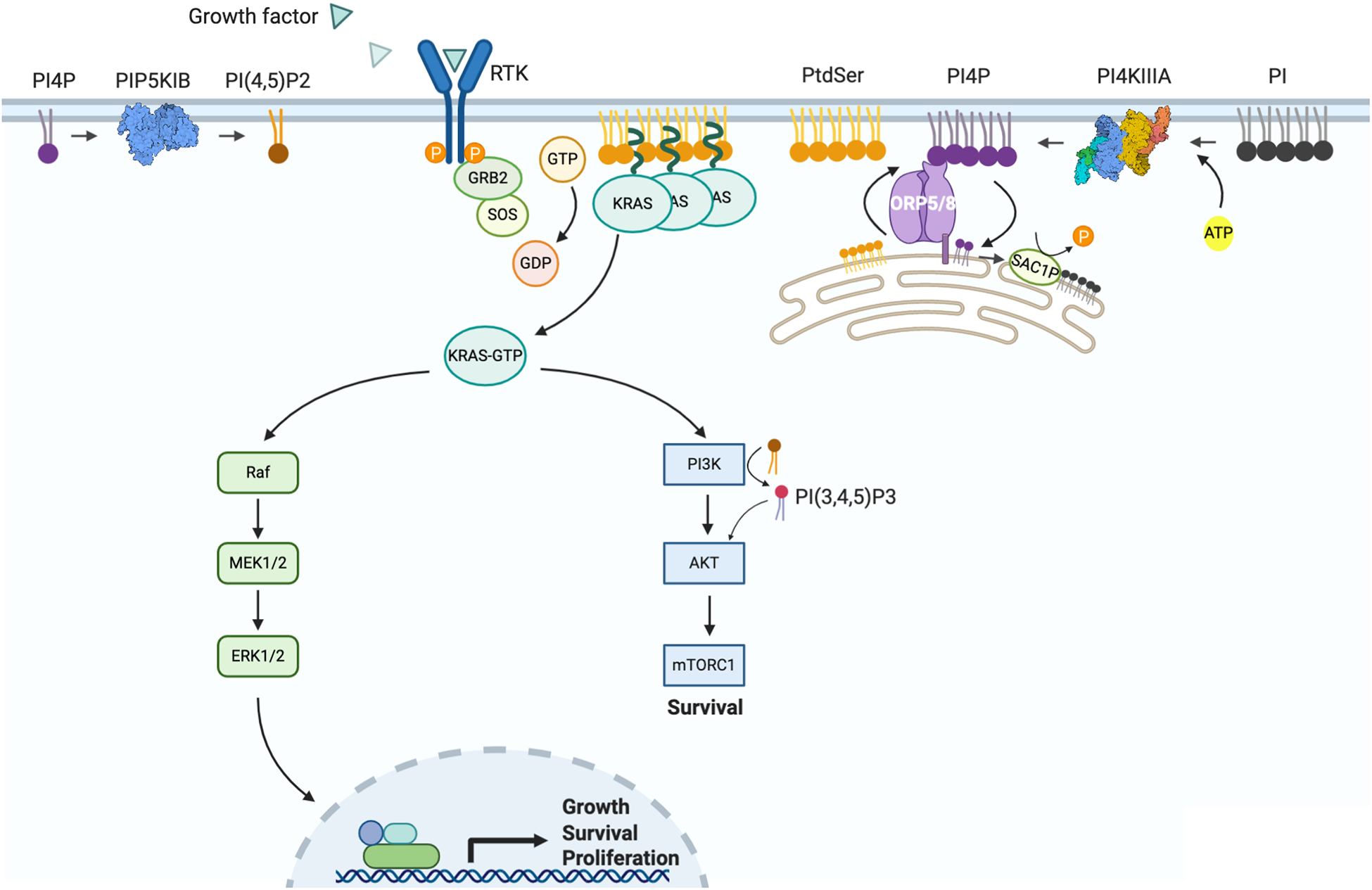
(A) ORP5 and ORP8 are lipid exchangers that transport PtdSer from the ER to the PM where they then bind to and transport PI4P to the ER. PI4P is immediately hydrolyzed to phosphoinositide (PI) by ER-resident SAC1P creating a PI4P concentration gradient that drives the counter transport process. PM PtdSer molecules are critical structural elements of KRAS nanoclusters that act as signaling platforms to activate many pathways, most importantly the RAF-MEK-ERK and PI3K-AKT-mTORC1 pathway which promote cell proliferation and survival. On the PM, PIP5K phosphorylates PI4P to PI(4,5)P2 which can subsequently be phosphorylated to PI(3,4,5)P3 by PI3K leading to the activation of the AKT pathway. ORP5/8: Oxysterol Related Binding Protein 5 and 8; SAC1P: SAC1 Like Phosphatidylinositide Phosphatase; PM, plasma membrane; ER, endoplasmic reticulum; PtdSer, phosphatidylserine; PI4P, phosphatidylinositol 4-phosphate; PI(4,5)P2, phosphatidylinositol-4,5-bisphosphate; PI(3,4,5)P3, phosphatidylinositol-3,4,5-trisphosphate; PI4KIIIA, Phosphatidylinositol 4-kinase III alpha; PIP5K1B, phosphatidylinositol-4-phosphate 5-kinase type 1 Beta; RAF, Rapidly Accelerated Fibrosarcoma; MEK, mitogen-activated protein kinase kinase; ERK, extracellular-signal-regulated kinase; PI3K, phosphoinositide 3-kinase; AKT, also known as protein kinase B (PKB); mTORC1, mammalian target of rapamycin complex 1.
PI4-kinases function mainly at the PM and Golgi to phosphorylate PI at position D4 of the inositol head group to synthesize PI4P, the precursor of PI(4,5)P2 (PIP2) and PI(3,4,5)P3 (PIP3) which are involved in phospholipase C (PLC) and PI3K/AKT signaling, respectively [302, 303]. There are three classes of PI4-kinases (PI4KI, PI4KII, PI4KIII) defined by sequence similarity and biochemical properties, each of which contain an α and a β isoform [302, 304]. The key isoform essential for PtdSer transport to the PM is PI4KIIIα which is recruited to the PM by a multiprotein complex containing the TTC7 scaffold protein and EFR3A, a palmitoylated protein that tethers the complex to the PM [304, 305].
Once at the PM, PtdSer is actively concentrated in the inner leaflet by ATP-dependent aminophospholipid flippases of the P4 subfamily of P-type ATPases [306–310]. In the PM, PtdSer has a multitude of functions involved in coagulation, signaling cascades, protein recruitment, phagocytosis and the apoptotic response [283, 311–314]. There are two pools of PtdSer, a reactive mobile pool and an immobile pool constrained by interactions with cortical actin [108, 279].
4.5.2. The ORP5/8 PtdSer Transport System as a Novel Targetable Pathway for KRAS Inhibition
The human ORP family contains 12 members in 6 subfamilies categorized by protein homology and gene organization [315, 316]. All members contain a highly conserved lipid binding domain that can transport cholesterol or different phospholipids [295, 317, 318]. The lipid transport domain in ORP5/8 is called the Oxysterol-binding-protein-Related-Domain (ORD) and binds PI4P or PtdSer; binding of these lipids is mutually exclusively because the hydrophobic pocket can only accommodate a single lipid molecule [299]. Both ORP5 and ORP8 contain an N-terminal Pleckstrin-Homology (PH) domain that binds to PI4P or PIP2 on the PM [297, 319–322] and a C-terminal hydrophobic transmembrane domain that tethers them to the ER [323–325]. Two splice variants of ORP8 exist: ORP8S and ORP8L which has an additional 42 amino acid acidic/negative stretch at the N-terminus. ORP5 is predominantly located at ER-PM MCSs, while the majority of ORP8L is ER localized, and ORP8S is intermediate between the two. The decreased levels of ORP8L at ER-PM contacts compared to ORP8S may be due to the ORP8L-specific negative N-terminus [297]. Additionally, ORP5 contains a cluster of basic/positive arginine residues that makes this protein especially favorable among the three for interaction with the negatively charged inner leaflet of the PM [326].
There is some debate about how ORP5 and ORP8 mechanistically interact. Some studies suggest that ORP5 helps recruit ORP8 to the PM when PIP2 levels rise due to elevated PI4P content that is subsequently phosphorylated to produce PIP2. Under these conditions ORP5 and ORP8 colocalize and directly interact with each other [297], consistent with studies showing an increased PI4P/PtdSer exchange rate upon dimerization [296]; by contrast other work suggests that ORP8 binding inhibits ORP5 recruitment to the PM [326]. There is also debate on which phosphoinositide ORP8 or ORP5 bind, with some studies accounting the difference to minor distinction in amino acid sequence and charge of each ORP protein [297]. More specifically, Sohn et al. showed that ORP8, and not ORP5, may preferentially bind PIP2 because it forms only 12 H-bonds with PI4P but 16 H-bonds with PIP2 and has additional electrostatic interactions with the two additional phosphates of PIP2 compared to PI4P. Therefore, while ORP5 activity at the PM depends on PI4P levels, ORP8 activity and recruitment depends on both PI4P and PIP2 levels [326]. It is yet to be determined if PI4P and PIP2 carry the same weight in terms of importance for PtdSer PM transport; however, it is clear that PIP2 levels can regulate PtdSer exchange by recruiting more ORP8 to the ER-PM MCSs, or by OPR8 using PIP2 for exchange, or a combination of both mechanisms [297, 327, 328].
Knockdown of ORP5 or ORP8 in C. elegans completely reverted the activated let-60 multi-vulva phenotype (MUV), indicative of pathway inhibition. In addition, and concordant with the concept that disrupting ER/PM PI4P concentration gradient will affect KRAS signaling, SAC1P knockdown also suppressed the MUV phenotype without affecting organismal viability. Similarly, knockdown of either ORP5 or ORP8 in human colorectal, breast and pancreatic cancer cell lines resulted in significant mislocalization of PtdSer and KRAS from the PM, along with decreased nanoclustering of any KRAS remaining on the PM and concomitant abrogation of KRAS-dependent MAPK activation [4]. Importantly, double knockdown of both homologs was not additive or synergistic, indicating that knockdown of either ORP protein alone is sufficient to mislocalize KRAS from the PM [4].
Knockdown of either ORP protein also decreased proliferation and anchorage-independent growth of KRAS-dependent but not KRAS-independent pancreatic cancer cells. More specifically, the KRAS-independent cell line PANC-1 was sensitive to ORP5 but not ORP8 knockdown, while KRAS-dependent lines were sensitive to either knockdown [4]. It has been reported that KRAS-independent cell lines depend on oxidative phosphorylation rather than glycolysis for metabolism, and hence have a higher amount of reactive oxygen species [329]. This observation is reconciled with studies showing ORP8 levels are higher at ER-mitochondria MCSs but ORP5 is the predominant homolog that interacts with mitochondrial outer membrane proteins [330]. Studies have shown that cells lacking either ORP have altered mitochondria morphology (reduced cristae) and reduced oxygen consumption. ORP5/8 interact with the VAPB-PTPIP51 protein complex on mitochondrial associated ER membranes (MAMs) and exchange mitochondrial PI4P with ER PtdSer for mitochondrial PE synthesis [330]. Interestingly, PANC-1 cells have a higher basal expression of ORP5 compared to other PDAC cell lines such as MiaPaCa-2 [331], which may indicate a more important role for this homolog in this cell line.
Corroborating evidence for a role of ORP5/8 in KRAS cancers comes from data showing that PDAC patients with increased ORP5 mRNA expression have poorer overall survival, with 1-year survival rates of high versus low ORP5 expression being 36.4% and 73.9%, respectively. The effect of high ORP5 levels on overall survival (OS) is seen across all stages of PDAC but is most pronounced for patients with stage I to stage III tumors with high ORP5 who have an OS of 6.7 months compared to 22.4 months for those with low ORP5 levels. High levels of ORP5 is also associated with increased invasion of pancreatic cancer cells in vitro and is manifested in patients as early relapse [332]. The consequences of increased ORP5 levels are not restricted to pancreatic cancer, as it is also highly expressed in metastasis-positive lung tumor samples [333]. In some pancreatic cancers, ORP5 expression correlates with upregulation of the cholesterol synthesis pathway and, although the mechanism is unclear, some have speculated that the effects of high ORP5 in these cancers can be tied back to reports that statins reduce invasion and metastasis in PDAC cells [331, 334, 335]. Ectopic expression of ORP5 also induced the expression of the efflux protein pump ABCB1 known as Multidrug Resistance gene MDR1 [331]. Others have shown that the lipid transferring function of ORP5 is absolutely required for cancer cell proliferation, invasion and migration in vitro and that when overexpressed, ORP5 interacts with mTORC1 by virtue of its ORD. Overexpression or down-regulation of ORP5 induced and repressed, respectively, mTORC1 activity in vitro. This may in part be due to the mislocalization of mTOR from lysosomes in ORP5 knockdown cells [336]. The most recent work; however, directly links both ORP5 and ORP8 to KRAS function and provides a straightforward explanation for the correlation of ORP5 levels with tumor progression. As such, in the context of what is now known, earlier models and speculations regarding ORP5 may not be accurate, moreover the link to KRAS function can also rationalize why high levels of ORP8 correlate negatively with patient survival in a variety of cancers including PDAC and NSCLC [4].
No inhibitors of ORP5 or ORP8 have been described but PtdSer-PI4P exchange can be inhibited by targeting PI4KIIIα upstream of this pathway. Overexpression of PI4KIIIα results in increased localization of ORP5/8 to PM-ER MCSs; conversely, inhibition of PI4KIIIα using the small molecule inhibitor A1 [337] leads to the dissociation of these complexes. In support of the rationale of targeting this protein in cancer, PI4KIIIα has been shown to contribute to the invasion and metastasis of pancreatic cancer in a difference analysis screen [338], and to the acquired resistance against gemcitabine in pancreatic cancer [339]. No activating mutations in PI4-kinases or inactivating deletions of their phosphatases have been identified in cancer [302]. Several pan and selective PI4K inhibitors have been synthesized (reviewed in [303]). The tool compound demonstrating the most selectivity towards PI4KIIIα is the small molecule C7 (IC50 against PI4KIIIα, PI4KIIIβ, PI3K: 6.3 nM, 1,250 nM, inactive, respectively) (Figure 3D). Treatment with C7 depletes PI4P from the PM [340, 341], which should lead to elimination of the PI4P concentration gradient required for ORP function, effectively phenocopying ORP5/8 knockdown. Concordantly, C7 treatment significantly mislocalized both PtdSer and KRAS from the PM in a dose-dependent manner, with the highest mislocalization occurring at concentrations previously reported to decrease PIP2 and PIP3 (generated from PI4P) levels [4, 341]. This translated to selective inhibition by C7 of the proliferation of KRAS-dependent PDAC cells, with minimal effects on KRAS-WT PDAC cells and no effect on non-transformed immortalized pancreatic cells. These results tie in nicely with the observations that ORP5 levels are specifically elevated in KRAS-mutant tumors across multiple cancer types, indicating the dependence of KRAS function, and hence KRAS-addicted cells, on ORP5 [4]. In addition to reducing the PI4P PM-to-ER gradient, PI4KIIIα inhibition may have other relevant mechanistic actions. For example, levels of PM-localized ORP5 and ORP8 are decreased upon PI4KIIIα inhibition [326]. Recent work has shown that knockdown of EFR3A, part of a protein complex that recruits PI4KIIIα to the PM, also leads to decreased PtdSer and KRAS PM content and reduced PDAC cell growth in vitro [342]. Others have shown that depleting PM PI4P by PI4KIIIα inhibition or knockout not only decreases PM PtdSer content, but also lowers overall PtdSer levels by 50% [297, 301, 326]. The overall reduction of PtdSer is due to its accumulation in the ER leading to product inhibition of PSSI and PSSII.
Prolonged overexpression of PIP5K, which converts PI4P to PIP2, also depletes PM PI4P, in turn decreasing PM PtdSer. Under physiological conditions, increased PIP5K activity increases PM PIP2 levels which leads to ORP5/8 redirecting more PI4P to the ER. Therefore, the ORP5/8 system also functions to regulate PIP2 levels on the PM. PIP2 dysregulation can be seen upon ORP5 knockdown, where PI4P accumulates on the PM leading to more PIP2 generation and consequent recruitment/activation of ORP8 to exchange PIP2 for PtdSer [4, 326]. In this context, some experiments suggest that ORP8 actually exhibits dual activity as a PtdSer/PI4P and PtdSer/PIP2 exchanger, as it has been reported that ORP5 knockdown leads to increased ORP8 protein expression which partially restores PtdSer PM levels, but that knocking down ORP8 did not lead to increased ORP5 expression [4]. This compensatory mechanism does not seem to operate or be relevant in KRAS-dependent cancer cells where loss of ORP5 or ORP8 is sufficient to significantly reduce cell proliferation [4]. Although PtdSer transport depends predominantly on ORP5 and ORP8 exchange with PI4P, ORP10 can also transport PtdSer from the ER to the trans-Golgi (TGN) from where it can be transported to the PM; however, knockdown of ORP10 did not alter PM PtdSer levels suggesting that this pathway is not a major contributor to PM PtdSer [343].
A critical question when considering PI4KIIIα as a drug target is toxicity. Indeed, one study claimed that pharmacological targeting of PI4KIIIα is not feasible since oral administration of PI4KIIIα inhibitors to mice induced gastric mucosal epithelial degeneration at high doses. Mice treated at lower doses remained alive but still exhibited differing degrees of GI toxicity probably due to the oral route of drug administration [337]. However, all of the PI4K inhibitors synthesized and tested on the mice variably inhibited multiple isoforms including PI4KIIIα, PI4KIIIβ, PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ [337, 344]. To date, the compound C7 seems to be the most selective PI4KIIIα inhibitor [303, 340] and at least in tissue culture experiments shows a high degree of selective toxicity for KRAS-mutant cancer cell lines with minimal effects on normal pancreatic cells [4]. PI4KIIIα inhibition also increases the radiosensitivity of diverse cancer cell lines in vitro as well as in immune-competent and nude mouse models of breast and brain cancer [345]. Thus, selective targeting of PI4KIIIα seems well tolerated in these animals at concentrations that result in anti-tumor effects.
Inhibition of MAPK signaling can lead to upregulation of PI3K-AKT-mTOR signaling as a compensatory mechanism and vice versa [346–348]. However, inhibiting PI4KIIIα can act in KRAS-mutant cells to simultaneously inhibit both the MAPK and PI3K pathways, which is an attractive notion given that 93% of PIK3CA mutations in PDAC co-occur with KRAS mutations [69, 349–353]. First, PI4KIIIα inhibition has the additional effect of decreasing pAKT activation by decreasing PIP3 PM levels (Figure 4) in breast cancer cells [345]. Secondly, PtdSer has been shown to be critical for the activation of PI3K/AKT signaling as it facilitates AKT-PIP3 binding and AKT conformational changes required for phosphorylation and downstream signaling [354]. Thirdly, one study showed that suppressing ORP5 increased levels of PTEN, a negative regulator of AKT/mTOR signaling [331].
Finally, PI4KIIIα as a target to inhibit KRAS function may circumvent the limitations seen with other inhibitors that show mutation-specific activity. For example, the EGFR inhibitor cetuximab, shows efficacy with KRAS G13 but not G12 mutants in advanced colorectal cancer [355]. In studies evaluating PtdSer mislocalization as anti-KRAS treatment, cell lines of different cancer types harboring different KRAS mutations were similarly affected [4, 235, 240, 271]. Additionally, PtdSer mislocalization most potently affected KRAS-dependent transformed cells, followed by KRAS-independent transformed cells and finally KRAS-WT cells. This demonstrates that levels of PM PtdSer are a selective vulnerability that may be exploited in KRAS-driven cancers.
5. Conclusion
RAS genes are the most frequently mutated oncogenes and drive of some of the deadliest human cancers, hence much effort has been expended to find anti-RAS therapies. The major clinical problem is KRAS. In addition to targeting classical downstream effectors, recent KRAS targeting strategies have expanded to include direct inhibition of KRAS, combination therapies exploiting KRAS-specific vulnerabilities, inhibiting KRAS post-translational processing and recycling, and preventing KRAS PM localization. The major benefit of targeting KRAS PM interactions has been the observed selectivity of these methods against oncogenic KRAS over WT KRAS irrespective of the mutation, and the ability to simultaneously downregulate two major KRAS signaling pathways without observable toxicities on organismal viability. One intriguing drug target is PI4KIIIα which has merit as a novel treatment target for KRAS-dependent tumors and warrants further research as inhibitors suppress the MAPK pathway by mislocalizing KRAS from the PM, and the PI3K/AKT/mTOR pathway by depleting both PM PtdSer and PIP3 required for the activation of this pathway.
6. Acknowledgements
Work in the authors’ laboratory is supported by grants from the NIH/NIGMS (R01 GM124233) and Cancer Prevention and Research Institute of Texas (CPRIT RP200047). WEK is additionally supported by the Andrew Sowell-Wade Huggins Fellowship/Professorship in Cancer Research and the Dr. John J. Kopchick Fellowship.
References
- 1.Downward J (2003) Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 3, 11–22 [DOI] [PubMed] [Google Scholar]
- 2.Wennerberg K, Rossman KL and Der CJ (2005) The Ras superfamily at a glance. J Cell Sci. 118, 843–846 [DOI] [PubMed] [Google Scholar]
- 3.Cox AD and Der CJ (2010) Ras history: The saga continues. Small GTPases. 1, 2–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kattan WE, Chen W, Ma X, Lan TH, van der Hoeven D, van der Hoeven R and Hancock JF (2019) Targeting plasma membrane phosphatidylserine content to inhibit oncogenic KRAS function. Life Sci Alliance. 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahearn I, Zhou M and Philips MR (2018) Posttranslational Modifications of RAS Proteins. Cold Spring Harbor perspectives in medicine. 8, a031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung HH, Benson DR, Cornish VW and Schultz PG (1993) Probing the role of loop 2 in Ras function with unnatural amino acids. Proc Natl Acad Sci U S A. 90, 10145–10149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibbs JB, Sigal IS, Poe M and Scolnick EM (1984) Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc Natl Acad Sci U S A. 81, 5704–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harvey J (1964) An unidentified virus which causes the rapid production of tumours in mice. Nature. 204, 1104–1105 [DOI] [PubMed] [Google Scholar]
- 9.Brown R, Marshall CJ, Pennie SG and Hall A (1984) Mechanism of activation of an N-ras gene in the human fibrosarcoma cell line HT1080. Embo j. 3, 1321–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bos JL (1989) Ras oncogenes in human cancer: a review. Cancer research. 49, 4682–4689 [PubMed] [Google Scholar]
- 11.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, Gierut JJ, Cox AD, Haigis KM and Philips MR (2015) K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 112, 779–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mo SP, Coulson JM and Prior IA (2018) RAS variant signalling. Biochem Soc Trans. 46, 1325–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prior IA, Lewis PD and Mattos C (2012) A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorfe AA, Hanzal-Bayer M, Abankwa D, Hancock JF and McCammon JA (2007) Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J Med Chem. 50, 674–684 [DOI] [PubMed] [Google Scholar]
- 15.Vetter IR and Wittinghofer A (2001) The guanine nucleotide-binding switch in three dimensions. Science. 294, 1299–1304 [DOI] [PubMed] [Google Scholar]
- 16.Khan I, Rhett JM and O’Bryan JP (2020) Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim Biophys Acta Mol Cell Res. 1867, 118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abankwa D, Gorfe AA and Hancock JF (2008) Mechanisms of Ras membrane organization and signalling: Ras on a rocker. Cell Cycle. 7, 2667–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parker JA and Mattos C (2018) The K-Ras, N-Ras, and H-Ras Isoforms: Unique Conformational Preferences and Implications for Targeting Oncogenic Mutants. Cold Spring Harb Perspect Med. 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henis YI, Hancock JF and Prior IA (2009) Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol Membr Biol. 26, 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG and Lowy DR (1984) The p21 ras C-terminus is required for transformation and membrane association. Nature. 310, 583–586 [DOI] [PubMed] [Google Scholar]
- 21.Reiss Y, Goldstein JL, Seabra MC, Casey PJ and Brown MS (1990) Inhibition of purified p21ras farnesyl:protein transferase by Cys-AAX tetrapeptides. Cell. 62, 81–88 [DOI] [PubMed] [Google Scholar]
- 22.Boyartchuk VL, Ashby MN and Rine J (1997) Modulation of Ras and a-factor function by carboxyl-terminal proteolysis. Science. 275, 1796–1800 [DOI] [PubMed] [Google Scholar]
- 23.Freije JM, Blay P, Pendas AM, Cadinanos J, Crespo P and Lopez-Otin C (1999) Identification and chromosomal location of two human genes encoding enzymes potentially involved in proteolytic maturation of farnesylated proteins. Genomics. 58, 270–280 [DOI] [PubMed] [Google Scholar]
- 24.Hancock JF (2003) Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 4, 373–384 [DOI] [PubMed] [Google Scholar]
- 25.Kim E, Ambroziak P, Otto JC, Taylor B, Ashby M, Shannon K, Casey PJ and Young SG (1999) Disruption of the mouse Rce1 gene results in defective Ras processing and mislocalization of Ras within cells. J Biol Chem. 274, 8383–8390 [DOI] [PubMed] [Google Scholar]
- 26.Otto JC, Kim E, Young SG and Casey PJ (1999) Cloning and characterization of a mammalian prenyl protein-specific protease. J Biol Chem. 274, 8379–8382 [DOI] [PubMed] [Google Scholar]
- 27.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, Ivanov IE and Philips MR (1999) Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 98, 69–80 [DOI] [PubMed] [Google Scholar]
- 28.Clarke S, Vogel JP, Deschenes RJ and Stock J (1988) Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases. Proc Natl Acad Sci U S A. 85, 4643–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai Q, Choy E, Chiu V, Romano J, Slivka SR, Steitz SA, Michaelis S and Philips MR (1998) Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulum. J Biol Chem. 273, 15030–15034 [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez L, Magee AI, Marshall CJ and Hancock JF (1989) Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. Embo j. 8, 1093–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hancock JF, Cadwallader K and Marshall CJ (1991) Methylation and proteolysis are essential for efficient membrane binding of prenylated p21K-ras(B). Embo j. 10, 641–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hrycyna CA, Sapperstein SK, Clarke S and Michaelis S (1991) The Saccharomyces cerevisiae STE14 gene encodes a methyltransferase that mediates C-terminal methylation of a-factor and RAS proteins. The EMBO journal. 10, 1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pillinger MH, Volker C, Stock JB, Weissmann G and Philips MR (1994) Characterization of a plasma membrane-associated prenylcysteine-directed alpha carboxyl methyltransferase in human neutrophils. J Biol Chem. 269, 1486–1492 [PubMed] [Google Scholar]
- 34.Hancock JF, Cadwallader K, Paterson H and Marshall CJ (1991) A CAAX or a CAAL motif and a second signal are sufficient for plasma membrane targeting of ras proteins. Embo j. 10, 4033–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swarthout JT, Lobo S, Farh L, Croke MR, Greentree WK, Deschenes RJ and Linder ME (2005) DHHC9 and GCP16 constitute a human protein fatty acyltransferase with specificity for H-and N-Ras. Journal of Biological Chemistry. 280, 31141–31148 [DOI] [PubMed] [Google Scholar]
- 36.Zhao L, Lobo S, Dong X, Ault AD and Deschenes RJ (2002) Erf4p and Erf2p form an endoplasmic reticulum-associated complex involved in the plasma membrane localization of yeast Ras proteins. Journal of Biological Chemistry. 277, 49352–49359 [DOI] [PubMed] [Google Scholar]
- 37.Apolloni A, Prior IA, Lindsay M, Parton RG and Hancock JF (2000) H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 20, 2475–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hancock JF, Paterson H and Marshall CJ (1990) A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 63, 133–139 [DOI] [PubMed] [Google Scholar]
- 39.Esteban LM, Vicario-Abejon C, Fernandez-Salguero P, Fernandez-Medarde A, Swaminathan N, Yienger K, Lopez E, Malumbres M, McKay R, Ward JM, Pellicer A and Santos E (2001) Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 21, 1444–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson RT, Umanoff H, Edelmann W, Kucherlapati R and Jacks T (1997) K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 11, 2468–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newlaczyl AU, Coulson JM and Prior IA (2017) Quantification of spatiotemporal patterns of Ras isoform expression during development. Sci Rep. 7, 41297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Omerovic J, Laude AJ and Prior IA (2007) Ras proteins: paradigms for compartmentalised and isoform-specific signalling. Cell Mol Life Sci. 64, 2575–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Potenza N, Vecchione C, Notte A, De Rienzo A, Rosica A, Bauer L, Affuso A, De Felice M, Russo T, Poulet R, Cifelli G, De Vita G, Lembo G and Di Lauro R (2005) Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 6, 432–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Umanoff H, Edelmann W, Pellicer A and Kucherlapati R (1995) The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci U S A. 92, 1709–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvarez-Moya B, Lopez-Alcala C, Drosten M, Bachs O and Agell N (2010) K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene. 29, 5911–5922 [DOI] [PubMed] [Google Scholar]
- 46.Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, Miura J, Wiener HH, Wright L, Saba SG, Yim D, Fein A, Perez de Castro I, Li C, Thompson CB, Cox AD and Philips MR (2006) PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 21, 481–493 [DOI] [PubMed] [Google Scholar]
- 47.Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M, Venkitaraman AR, Wittinghofer A and Bastiaens PI (2011) The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 14, 148–158 [DOI] [PubMed] [Google Scholar]
- 48.Cho KJ, Casteel DE, Prakash P, Tan L, van der Hoeven D, Salim AA, Kim C, Capon RJ, Lacey E, Cunha SR, Gorfe AA and Hancock JF (2016) AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol Cell Biol. 36, 3086–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L and Bastiaens PIH (2014) KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell. 157, 459–471 [DOI] [PubMed] [Google Scholar]
- 50.McMahon HT and Gallop JL (2005) Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 438, 590–596 [DOI] [PubMed] [Google Scholar]
- 51.Nir S, Bentz J, Wilschut J and Duzgunes N (1983) Aggregation and fusion of phospholipid vesicles. Progress in surface science. 13, 1–124 [Google Scholar]
- 52.Pomorski T, Holthuis JC, Herrmann A and van Meer G (2004) Tracking down lipid flippases and their biological functions. J Cell Sci. 117, 805–813 [DOI] [PubMed] [Google Scholar]
- 53.Ravoo BJ, Weringa WD and Engberts JB (1999) Membrane fusion in vesicles of oligomerizable lipids. Biophys J. 76, 374–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, Nussinov R and Gaponenko V (2015) Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. Journal of Biological Chemistry. 290, 9465–9477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLaughlin S and Aderem A (1995) The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 20, 272–276 [DOI] [PubMed] [Google Scholar]
- 56.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR and Futreal PA (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 39, D945–D950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maher J, Baker DA, Manning M, Dibb NJ and Roberts IA (1995) Evidence for cell-specific differences in transformation by N-, H- and K-ras. Oncogene. 11, 1639–1647 [PubMed] [Google Scholar]
- 58.Yeh JJ, Madigan JP, Campbell PM, Roberts PJ, DeGraffenreid L and Der CJ (2010) Chapter 329 - Targeting Ras for Anticancer Drug Discovery In Handbook of Cell Signaling (Second Edition) (Bradshaw RA and Dennis EA, eds.). pp. 2837–2857, Academic Press, San Diego [Google Scholar]
- 59.Hobbs GA, Der CJ and Rossman KL (2016) RAS isoforms and mutations in cancer at a glance. J Cell Sci. 129, 1287–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bryant KL, Mancias JD, Kimmelman AC and Der CJ (2014) KRAS: feeding pancreatic cancer proliferation. Trends in biochemical sciences. 39, 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheidig AJ, Burmester C and Goody RS (1999) The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure. 7, 1311–1324 [DOI] [PubMed] [Google Scholar]
- 62.Scheffzek K, Lautwein A, Kabsch W, Ahmadian MR and Wittinghofer A (1996) Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature. 384, 591–596 [DOI] [PubMed] [Google Scholar]
- 63.Manne V, Bekesi E and Kung HF (1985) Ha-ras proteins exhibit GTPase activity: point mutations that activate Ha-ras gene products result in decreased GTPase activity. Proc Natl Acad Sci U S A. 82, 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sweet RW, Yokoyama S, Kamata T, Feramisco JR, Rosenberg M and Gross M (1984) The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature. 311, 273–275 [DOI] [PubMed] [Google Scholar]
- 65.McGrath JP, Capon DJ, Goeddel DV and Levinson AD (1984) Comparative biochemical properties of normal and activated human ras p21 protein. Nature. 310, 644–649 [DOI] [PubMed] [Google Scholar]
- 66.Quinlan MP, Quatela SE, Philips MR and Settleman J (2008) Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol Cell Biol. 28, 2659–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quinlan MP and Settleman J (2008) Explaining the preponderance of Kras mutations in human cancer: An isoform-specific function in stem cell expansion. Cell Cycle. 7, 1332–1335 [DOI] [PubMed] [Google Scholar]
- 68.Quinlan MP and Settleman J (2009) Isoform-specific ras functions in development and cancer. Future Oncol. 5, 105–116 [DOI] [PubMed] [Google Scholar]
- 69.Waters AM and Der CJ (2018) KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med. 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marshall CJ (1996) Ras effectors. Curr Opin Cell Biol. 8, 197–204 [DOI] [PubMed] [Google Scholar]
- 71.Freeman AK, Ritt DA and Morrison DK (2013) The importance of Raf dimerization in cell signaling. Small GTPases. 4, 180–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morrison DK (2012) MAP kinase pathways. Cold Spring Harb Perspect Biol. 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khosravi-Far R and Der CJ (1994) The Ras signal transduction pathway. Cancer Metastasis Rev. 13, 67–89 [DOI] [PubMed] [Google Scholar]
- 74.McKay MM and Morrison DK (2007) Integrating signals from RTKs to ERK/MAPK. Oncogene. 26, 3113–3121 [DOI] [PubMed] [Google Scholar]
- 75.Pruitt K and Der CJ (2001) Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 171, 1–10 [DOI] [PubMed] [Google Scholar]
- 76.Shaul YD and Seger R (2007) The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 1773, 1213–1226 [DOI] [PubMed] [Google Scholar]
- 77.Yordy JS and Muise-Helmericks RC (2000) Signal transduction and the Ets family of transcription factors. Oncogene. 19, 6503–6513 [DOI] [PubMed] [Google Scholar]
- 78.Pacold ME, Suire S, Perisic O, Lara-Gonzalez S, Davis CT, Walker EH, Hawkins PT, Stephens L, Eccleston JF and Williams RL (2000) Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 103, 931–943 [DOI] [PubMed] [Google Scholar]
- 79.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD and Downward J (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 370, 527–532 [DOI] [PubMed] [Google Scholar]
- 80.Castellano E and Downward J (2011) RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2, 261–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kennedy SG, Wagner AJ, Conzen SD, Jordán J, Bellacosa A, Tsichlis PN and Hay N (1997) The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 11, 701–713 [DOI] [PubMed] [Google Scholar]
- 82.Bai D, Ueno L and Vogt PK (2009) Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer. 125, 2863–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Datta SR, Brunet A and Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev. 13, 2905–2927 [DOI] [PubMed] [Google Scholar]
- 84.Farhan M, Wang H, Gaur U, Little PJ, Xu J and Zheng W (2017) FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int J Biol Sci. 13, 815–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khwaja A, Rodriguez-Viciana P, Wennström S, Warne PH and Downward J (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. Embo j. 16, 2783–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marte BM and Downward J (1997) PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 22, 355–358 [DOI] [PubMed] [Google Scholar]
- 87.Rytömaa M, Lehmann K and Downward J (2000) Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: inhibition by PKB/Akt but not Raf signalling. Oncogene. 19, 4461–4468 [DOI] [PubMed] [Google Scholar]
- 88.Romashkova JA and Makarov SS (1999) NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 401, 86–90 [DOI] [PubMed] [Google Scholar]
- 89.Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T and Goldschmidt-Clermont PJ (1997) Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 275, 1649–1652 [DOI] [PubMed] [Google Scholar]
- 90.Joneson T and Bar-Sagi D (1999) Suppression of Ras-induced apoptosis by the Rac GTPase. Molecular and cellular biology. 19, 5892–5901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P and Finkel T (1996) rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol. 16, 7115–7121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cox AD and Der CJ (2003) The dark side of Ras: regulation of apoptosis. Oncogene. 22, 8999–9006 [DOI] [PubMed] [Google Scholar]
- 93.Mayo MW and Baldwin AS (2000) The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 1470, M55–62 [DOI] [PubMed] [Google Scholar]
- 94.Voice JK, Klemke RL, Le A and Jackson JH (1999) Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J Biol Chem. 274, 17164–17170 [DOI] [PubMed] [Google Scholar]
- 95.Yan J, Roy S, Apolloni A, Lane A and Hancock JF (1998) Ras isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 273, 24052–24056 [DOI] [PubMed] [Google Scholar]
- 96.Witkiewicz AK, Balaji U, Eslinger C, McMillan E, Conway W, Posner B, Mills GB, O’Reilly EM and Knudsen ES (2016) Integrated Patient-Derived Models Delineate Individualized Therapeutic Vulnerabilities of Pancreatic Cancer. Cell Rep. 16, 2017–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Foster SA, Whalen DM, Ozen A, Wongchenko MJ, Yin J, Yen I, Schaefer G, Mayfield JD, Chmielecki J, Stephens PJ, Albacker LA, Yan Y, Song K, Hatzivassiliou G, Eigenbrot C, Yu C, Shaw AS, Manning G, Skelton NJ, Hymowitz SG and Malek S (2016) Activation Mechanism of Oncogenic Deletion Mutations in BRAF, EGFR, and HER2. Cancer Cell. 29, 477–493 [DOI] [PubMed] [Google Scholar]
- 98.Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, Mochalkin I, Wong SS, Yue YG, Huber L, Conti I, Henry JR, Starling JJ, Plowman GD and Peng SB (2016) Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 6, 300–315 [DOI] [PubMed] [Google Scholar]
- 99.Papke B and Der CJ (2017) Drugging RAS: Know the enemy. Science. 355, 1158–1163 [DOI] [PubMed] [Google Scholar]
- 100.Guerra C and Barbacid M (2013) Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol. 7, 232–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hancock JF and Parton RG (2005) Ras plasma membrane signalling platforms. Biochemical Journal. 389, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hancock JF and Prior IA (2005) Electron microscopic imaging of Ras signaling domains. Methods. 37, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tian T, Harding A, Inder K, Plowman S, Parton RG and Hancock JF (2007) Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nature cell biology. 9, 905–914 [DOI] [PubMed] [Google Scholar]
- 104.Zhou Y and Hancock JF (2015) Ras nanoclusters: Versatile lipid-based signaling platforms. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 1853, 841–849 [DOI] [PubMed] [Google Scholar]
- 105.Zhou Y, Prakash P, Gorfe AA and Hancock JF (2017) Ras and the plasma membrane: a complicated relationship. Cold Spring Harbor perspectives in medicine, a031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou Y, Prakash P, Liang H, Cho K-J, Gorfe AA and Hancock JF (2017) Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell. 168, 239–251. e216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cho K. j., Park J-H, Piggott AM, Salim AA, Gorfe AA, Parton RG, Capon RJ, Lacey E and Hancock JF (2012) Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. The Journal of biological chemistry. 287, 43573–43584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou Y, Liang H, Rodkey T, Ariotti N, Parton RG and Hancock JF (2014) Signal integration by lipid-mediated spatial cross talk between Ras nanoclusters. Molecular and cellular biology. 34, 862–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prakash P, Sayyed-Ahmad A, Cho K-J, Dolino DM, Chen W, Li H, Grant BJ, Hancock JF and Gorfe AA (2017) Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Scientific reports. 7, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sarkar-Banerjee S, Sayyed-Ahmad A, Prakash P, Cho K-J, Waxham MN, Hancock JF and Gorfe AA (2017) Spatiotemporal analysis of K-Ras plasma membrane interactions reveals multiple high order homo-oligomeric complexes. Journal of the American Chemical Society. 139, 13466–13475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chung JK, Lee YK, Denson J-P, Gillette WK, Alvarez S, Stephen AG and Groves JT (2018) K-Ras4B remains monomeric on membranes over a wide range of surface densities and lipid compositions. Biophysical journal. 114, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weise K, Kapoor S, Denter C, Nikolaus J. r., Opitz N, Koch S, Triola G, Herrmann A, Waldmann H and Winter R (2011) Membrane-mediated induction and sorting of K-Ras microdomain signaling platforms. Journal of the American Chemical Society. 133, 880–887 [DOI] [PubMed] [Google Scholar]
- 113.Baass PC, Di Guglielmo G, Authier F, Posner BI and Bergeron JJ (1995) Compartmentalized signal transduction by receptor tyrosine kinases. Trends in cell biology. 5, 465–470 [DOI] [PubMed] [Google Scholar]
- 114.Di Guglielmo G, Baass P, Ou W, Posner B and Bergeron J (1994) Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf 1 by EGF but not insulin in liver parenchyma. The EMBO journal. 13, 4269–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen W-C, Zhou M, Court H, Shi J, Mendoza SL and Morten MJ (2019) KRAS4A directly regulates hexokinase 1. Nature. 576, 482–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rebollo A, Pérez-Sala D and Martínez-A C (1999) Bcl-2 differentially targets K-, N-, and H-Ras to mitochondria in IL-2 supplemented or deprived cells: implications in prevention of apoptosis. Oncogene. 18, 4930–4939 [DOI] [PubMed] [Google Scholar]
- 117.Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson II RL, Cox AD and Philips MR (2002) Ras signalling on the endoplasmic reticulum and the Golgi. Nature cell biology. 4, 343. [DOI] [PubMed] [Google Scholar]
- 118.Harding A and Hancock JF (2008) Ras nanoclusters: combining digital and analog signaling. Cell Cycle. 7, 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Harding A, Tian T, Westbury E, Frische E and Hancock JF (2005) Subcellular localization determines MAP kinase signal output. Curr Biol. 15, 869–873 [DOI] [PubMed] [Google Scholar]
- 120.Inder K, Harding A, Plowman SJ, Philips MR, Parton RG and Hancock JF (2008) Activation of the MAPK module from different spatial locations generates distinct system outputs. Mol Biol Cell. 19, 4776–4784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Daniels MA, Teixeiro E, Gill J, Hausmann B, Roubaty D, Holmberg K, Werlen G, Holländer GA, Gascoigne NR and Palmer E (2006) Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature. 444, 724–729 [DOI] [PubMed] [Google Scholar]
- 122.Cox AD, Fesik SW, Kimmelman AC, Luo J and Der CJ (2014) Drugging the undruggable RAS: mission possible? Nature reviews Drug discovery. 13, 828–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Santos E and Nebreda AR (1989) Structural and functional properties of ras proteins. Faseb j. 3, 2151–2163 [DOI] [PubMed] [Google Scholar]
- 124.Spiegel J, Cromm PM, Zimmermann G, Grossmann TN and Waldmann H (2014) Small-molecule modulation of Ras signaling. Nat Chem Biol. 10, 613–622 [DOI] [PubMed] [Google Scholar]
- 125.John J, Sohmen R, Feuerstein J, Linke R, Wittinghofer A and Goody RS (1990) Kinetics of interaction of nucleotides with nucleotide-free H-ras p21. Biochemistry. 29, 6058–6065 [DOI] [PubMed] [Google Scholar]
- 126.Becher I, Savitski MM, Savitski M. F. l., Hopf C, Bantscheff M and Drewes G (2013) Affinity profiling of the cellular kinome for the nucleotide cofactors ATP, ADP, and GTP. ACS chemical biology. 8, 599–607 [DOI] [PubMed] [Google Scholar]
- 127.Clark R, Wong G, Arnheim N, Nitecki D and McCormick F (1985) Antibodies specific for amino acid 12 of the ras oncogene product inhibit GTP binding. Proc Natl Acad Sci U S A. 82, 5280–5284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang J-L, Liu D-X, Zhen S-J, Zhou Y-G, Zhang D-J, Yang L-Y, Chen H-B and Feng Q (2016) A novel anti-p21Ras scFv antibody reacting specifically with human tumour cell lines and primary tumour tissues. BMC Cancer. 16, 131–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S and Frejd FY (2010) Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Letters. 584, 2670–2680 [DOI] [PubMed] [Google Scholar]
- 130.Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, Cobbert J, Lavoie H, Smith M, Rajakulendran T, Dowdell E, Okur MN, Dementieva I, Sicheri F, Therrien M, Hancock JF, Ikura M, Koide S and O’Bryan JP (2017) Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol. 13, 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Spencer-Smith R, Li L, Prasad S, Koide A, Koide S and O’Bryan JP (2019) Targeting the alpha4-alpha5 interface of RAS results in multiple levels of inhibition. Small GTPases. 10, 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guillard S, Kolasinska-Zwierz P, Debreczeni J, Breed J, Zhang J, Bery N, Marwood R, Tart J, Overman R, Stocki P, Mistry B, Phillips C, Rabbitts T, Jackson R and Minter R (2017) Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat Commun. 8, 16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sha F, Salzman G, Gupta A and Koide S (2017) Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 26, 910–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ambrogio C, Köhler J, Zhou Z-W, Wang H, Paranal R, Li J, Capelletti M, Caffarra C, Li S and Lv Q (2018) KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell. 172, 857–868. e815 [DOI] [PubMed] [Google Scholar]
- 135.Chen M, Peters A, Huang T and Nan X (2016) Ras dimer formation as a new signaling mechanism and potential cancer therapeutic target. Mini Reviews in Medicinal Chemistry. 16, 391–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Simanshu DK, Nissley DV and McCormick F (2017) RAS Proteins and Their Regulators in Human Disease. Cell. 170, 17–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Welsch ME, Kaplan A, Chambers JM, Stokes ME, Bos PH, Zask A, Zhang Y, Sanchez-Martin M, Badgley MA, Huang CS, Tran TH, Akkiraju H, Brown LM, Nandakumar R, Cremers S, Yang WS, Tong L, Olive KP, Ferrando A and Stockwell BR (2017) Multivalent Small-Molecule Pan-RAS Inhibitors. Cell. 168, 878–889.e829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Prakash P, Hancock JF and Gorfe AA (2015) Binding hotspots on K-ras: consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins. 83, 898–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Grant BJ, Lukman S, Hocker HJ, Sayyah J, Brown JH, McCammon JA and Gorfe AA (2011) Novel allosteric sites on Ras for lead generation. PLoS One. 6, e25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McCarthy MJ, Pagba CV, Prakash P, Naji AK, van der Hoeven D, Liang H, Gupta AK, Zhou Y, Cho KJ, Hancock JF and Gorfe AA (2019) Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega. 4, 2921–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, Salzberg AC and Huang ES (2007) Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol. 25, 71–75 [DOI] [PubMed] [Google Scholar]
- 142.Hopkins AL and Groom CR (2002) The druggable genome. Nat Rev Drug Discov. 1, 727–730 [DOI] [PubMed] [Google Scholar]
- 143.Smith RD, Hu L, Falkner JA, Benson ML, Nerothin JP and Carlson HA (2006) Exploring protein-ligand recognition with Binding MOAD. J Mol Graph Model. 24, 414–425 [DOI] [PubMed] [Google Scholar]
- 144.Tanaka T and Rabbitts TH (2010) Protocol for the selection of single-domain antibody fragments by third generation intracellular antibody capture. Nat Protoc. 5, 67–92 [DOI] [PubMed] [Google Scholar]
- 145.Visintin M, Tse E, Axelson H, Rabbitts TH and Cattaneo A (1999) Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc Natl Acad Sci U S A. 96, 11723–11728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tanaka T and Rabbitts TH (2010) Interfering with RAS-effector protein interactions prevent RAS-dependent tumour initiation and causes stop-start control of cancer growth. Oncogene. 29, 6064–6070 [DOI] [PubMed] [Google Scholar]
- 147.Quevedo CE, Cruz-Migoni A, Bery N, Miller A, Tanaka T, Petch D, Bataille CJR, Lee LYW, Fallon PS, Tulmin H, Ehebauer MT, Fernandez-Fuentes N, Russell AJ, Carr SB, Phillips SEV and Rabbitts TH (2018) Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat Commun. 9, 3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fang Z, Marshall CB, Nishikawa T, Gossert AD, Jansen JM, Jahnke W and Ikura M (2018) Inhibition of K-RAS4B by a Unique Mechanism of Action: Stabilizing Membrane-Dependent Occlusion of the Effector-Binding Site. Cell Chem Biol. 25, 1327–1336.e1324 [DOI] [PubMed] [Google Scholar]
- 149.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, Lanman BA, Werner J, Rapaport AS, San Miguel T, Ortiz R, Osgood T, Sun JR, Zhu X, McCarter JD, Volak LP, Houk BE, Fakih MG, O’Neil BH, Price TJ, Falchook GS, Desai J, Kuo J, Govindan R, Hong DS, Ouyang W, Henary H, Arvedson T, Cee VJ and Lipford JR (2019) The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 575, 217–223 [DOI] [PubMed] [Google Scholar]
- 150.Ostrem JM, Peters U, Sos ML, Wells JA and Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 503, 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Janes MR, Zhang J, Li L-S, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ and Darjania L (2018) Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 172, 578–589. e517 [DOI] [PubMed] [Google Scholar]
- 152.Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Janne PA, Meyerson M, Marto JA, Engen JR and Gray NS (2014) Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. 53, 199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lito P, Solomon M, Li LS, Hansen R and Rosen N (2016) Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 351, 604–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P and Liu Y (2016) Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 6, 316–329 [DOI] [PubMed] [Google Scholar]
- 155.Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, Ficarro SB, Xiong Y, Ambrogio C and Paranal RM (2017) Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell chemical biology. 24, 1005–1016. e1003 [DOI] [PubMed] [Google Scholar]
- 156.Trahey M and McCormick F (1987) A cytoplasmic protein stimulates normal N-ras p21 GTPase, but does not affect oncogenic mutants. Science. 238, 542–545 [DOI] [PubMed] [Google Scholar]
- 157.Govindan R, Fakih M, Price T, Falchook G, Desai J, Kuo J, Strickler J, Krauss J, Li B, Denlinger C, Durm G, Ngang J, Henary H, Ngarmchamnanrith G, Rasmussen E, Morrow P and Hong D (2019) OA02.02 Phase 1 Study of Safety, Tolerability, PK and Efficacy of AMG 510, a Novel KRASG12C Inhibitor, Evaluated in NSCLC. Journal of Thoracic Oncology. 14, S208 [Google Scholar]
- 158.Govindan R, F. MG, Price TJ, Falchook GS, Desai J, Kuo JC, Strickler JH, Krauss JC, Li BT, Denlinger CS, Durm G, Ngang J, Henary H, Ngarmchamnanrith G, Rasmussen E, Morrow PK, Hong DS. (2019) Phase 1 Study of AMG 510, a Novel Molecule Targeting KRAS G12C Mutant Solid Tumors. In Annals of Oncology ed.)^eds.) [Google Scholar]
- 159.Fell JB, Fischer JP, Baer BR, Ballard J, Blake JF, Bouhana K, Brandhuber BJ, Briere DM, Burgess LE, Burkard MR, Chiang H, Chicarelli MJ, Davidson K, Gaudino JJ, Hallin J, Hanson L, Hee K, Hicken EJ, Hinklin RJ, Marx MA, Mejia MJ, Olson P, Savechenkov P, Sudhakar N, Tang TP, Vigers GP, Zecca H and Christensen JG (2018) Discovery of Tetrahydropyridopyrimidines as Irreversible Covalent Inhibitors of KRAS-G12C with In Vivo Activity. ACS Med Chem Lett. 9, 1230–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, Kim D, Li C, de Stanchina E, Mazutis L, Risso D and Lito P (2020) Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 577, 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cheung TH and Rando TA (2013) Molecular regulation of stem cell quiescence. Nature reviews Molecular cell biology. 14, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D and Hung KE (2012) EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery. 2, 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lito P, Rosen N and Solit DB (2013) Tumor adaptation and resistance to RAF inhibitors. Nature medicine. 19, 1401. [DOI] [PubMed] [Google Scholar]
- 164.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A and Bernards R (2012) Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature. 483, 100–103 [DOI] [PubMed] [Google Scholar]
- 165.Gong X, Du J, Parsons SH, Merzoug FF, Webster Y, Iversen PW, Chio L-C, Van Horn RD, Lin X and Blosser W (2019) Aurora A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer discovery. 9, 248–263 [DOI] [PubMed] [Google Scholar]
- 166.Lim K-H, Brady DC, Kashatus DF, Ancrile BB, Der CJ, Cox AD and Counter CM (2010) Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Molecular and cellular biology. 30, 508–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Umstead M, Xiong J, Qi Q, Du Y and Fu H (2017) Aurora kinase A interacts with H-Ras and potentiates Ras-MAPK signaling. Oncotarget. 8, 28359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Donnella HJ, Webber JT, Levin RS, Camarda R, Momcilovic O, Bayani N, Shah KN, Korkola JE, Shokat KM and Goga A (2018) Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nature chemical biology. 14, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shah KN, Bhatt R, Rotow J, Rohrberg J, Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A and Kuhn J (2019) Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nature medicine. 25, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, Gollner A, Covini D, Fischer S, Gerstberger T, Gmaschitz T, Goodwin C, Greb P, Haring D, Hela W, Hoffmann J, Karolyi-Oezguer J, Knesl P, Kornigg S, Koegl M, Kousek R, Lamarre L, Moser F, Munico-Martinez S, Peinsipp C, Phan J, Rinnenthal J, Sai J, Salamon C, Scherbantin Y, Schipany K, Schnitzer R, Schrenk A, Sharps B, Siszler G, Sun Q, Waterson A, Wolkerstorfer B, Zeeb M, Pearson M, Fesik SW and McConnell DB (2019) Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A. 116, 15823–15829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D and Kuriyan J (1998) The structural basis of the activation of Ras by Sos. Nature. 394, 337–343 [DOI] [PubMed] [Google Scholar]
- 172.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F and Wittinghofer A (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 277, 333–338 [DOI] [PubMed] [Google Scholar]
- 173.Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R and Mattos C (2015) Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure. 23, 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Walker EH, Perisic O, Ried C, Stephens L and Williams RL (1999) Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 402, 313–320 [DOI] [PubMed] [Google Scholar]
- 175.Cruz-Migoni A, Canning P, Quevedo CE, Bataille CJR, Bery N, Miller A, Russell AJ, Phillips SEV, Carr SB and Rabbitts TH (2019) Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc Natl Acad Sci U S A. 116, 2545–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJ, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W and Fang G (2012) Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 109, 5299–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sun Q, Burke JP, Phan J, Burns MC, Olejniczak ET, Waterson AG, Lee T, Rossanese OW and Fesik SW (2012) Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 51, 6140–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fakih M and Vincent M (2010) Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Current Oncology. 17, S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jimeno A, Messersmith WA, Hirsch FR, Franklin WA and Eckhardt SG (2009) KRAS mutations and susceptibility to cetuximab and panitumumab in colorectal cancer. The Cancer Journal. 15, 110–113 [DOI] [PubMed] [Google Scholar]
- 180.Welsh SJ and Corrie PG (2015) Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Therapeutic advances in medical oncology. 7, 122–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stephen AG, Esposito D, Bagni RK and McCormick F (2014) Dragging ras back in the ring. Cancer Cell. 25, 272–281 [DOI] [PubMed] [Google Scholar]
- 182.Samatar AA and Poulikakos PI (2014) Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 13, 928–942 [DOI] [PubMed] [Google Scholar]
- 183.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J and Frederick DT (2012) Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 487, 500–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC and Janne PA (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 185.Lee YJ, Kim DH, Lee SH, Kim DW, Nam HS and Cho MK (2011) Expression of the c-Met Proteins in Malignant Skin Cancers. Ann Dermatol. 23, 33–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Puri N, Ahmed S, Janamanchi V, Tretiakova M, Zumba O, Krausz T, Jagadeeswaran R and Salgia R (2007) c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin Cancer Res. 13, 2246–2253 [DOI] [PubMed] [Google Scholar]
- 187.Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S and Yano S (2009) Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 15, 6630–6638 [DOI] [PubMed] [Google Scholar]
- 188.Williams RT, Roussel MF and Sherr CJ (2006) Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 103, 6688–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hall-Jackson CA, Eyers PA, Cohen P, Goedert M, Boyle FT, Hewitt N, Plant H and Hedge P (1999) Paradoxical activation of Raf by a novel Raf inhibitor. Chem Biol. 6, 559–568 [DOI] [PubMed] [Google Scholar]
- 190.Hall-Jackson CA, Goedert M, Hedge P and Cohen P (1999) Effect of SB 203580 on the activity of c-Raf in vitro and in vivo. Oncogene. 18, 2047–2054 [DOI] [PubMed] [Google Scholar]
- 191.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS and Malek S (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 464, 431–435 [DOI] [PubMed] [Google Scholar]
- 192.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C and Marais R (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 140, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, Gupta YK, Reddy MV, Ueno L, Hart JR, Vogt PK, Mulholland D, Guha C, Aggarwal AK and Reddy EP (2016) A Small Molecule RAS-Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell. 165, 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ritt DA, Abreu-Blanco MT, Bindu L, Durrant DE, Zhou M, Specht SI, Stephen AG, Holderfield M and Morrison DK (2016) Inhibition of Ras/Raf/MEK/ERK Pathway Signaling by a Stress-Induced Phospho-Regulatory Circuit. Molecular cell. 64, 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA and Poulikakos PI (2019) SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell reports. 26, 65–78. e65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, Raught B, Zhang Z-Y, Zadeh G and Ohh M (2015) Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nature communications. 6, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, Neel BG and Tang KH (2018) SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 8, 1237–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, de Wit N, Goncalves-Ribeiro S, Nadal E, Bardelli A, Villanueva A and Bernards R (2018) SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med. 24, 961–967 [DOI] [PubMed] [Google Scholar]
- 199.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Gorgulu K, Dantes Z, Wormann SM, Diakopoulos KN, Karpathaki AF, Kowalska M, Kaya-Aksoy E, Song L, van der Laan EAZ, Lopez-Alberca MP, Nazare M, Reichert M, Saur D, Erkan MM, Hopt UT, Sainz B Jr., Birchmeier W, Schmid RM, Lesina M and Algul H (2018) Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 24, 954–960 [DOI] [PubMed] [Google Scholar]
- 200.Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, Xu D, Schumacher SE, Puschhof J, McFarland J, Zou C, Dulak A, Henderson L, Xu P, O’Day E, Rendak R, Liao WL, Cecchi F, Hembrough T, Schwartz S, Szeto C, Rustgi AK, Wong KK, Diehl JA, Jensen K, Graziano F, Ruzzo A, Fereshetian S, Mertins P, Carr SA, Beroukhim R, Nakamura K, Oki E, Watanabe M, Baba H, Imamura Y, Catenacci D and Bass AJ (2018) Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 24, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nijman SMB (2011) Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS letters. 585, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Luo J, Solimini NL and Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 136, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Solimini NL, Luo J and Elledge SJ (2007) Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 130, 986–988 [DOI] [PubMed] [Google Scholar]
- 204.Aguirre AJ and Hahn WC (2018) Synthetic Lethal Vulnerabilities in KRAS-Mutant Cancers. Cold Spring Harb Perspect Med. 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, Gunda V, Pierobon M, Waters AM, George SD, Tomar G, Papke B, Hobbs GA, Yan L, Hayes TK, Diehl JN, Goode GD, Chaika NV, Wang Y, Zhang GF, Witkiewicz AK, Knudsen ES, Petricoin EF 3rd, Singh PK, Macdonald JM, Tran NL, Lyssiotis CA, Ying H, Kimmelman AC, Cox AD and Der CJ (2019) Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 25, 628–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT and Yap JT (2019) Protective autophagy elicited by RAF→ MEK→ ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nature medicine. 25, 620–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lee C-S, Lee LC, Yuan TL, Chakka S, Fellmann C, Lowe SW, Caplen NJ, McCormick F and Luo J (2019) MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proceedings of the National Academy of Sciences. 116, 4508–4517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN and Papke B (2020) Atypical KRASG12R mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer discovery. 10, 104–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK and Elledge SJ (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 137, 835–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, Fernando M, Quon K, Kassner PD, Ruefli-Brasse AA, Watson VJ, Fajardo F, Jackson A, Zondlo J, Sun Y, Ellison AR, Plewa CA, San MT, Robinson J, McCarter J, Schwandner R, Judd T, Carnahan J and Dussault I (2011) STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res. 71, 5818–5826 [DOI] [PubMed] [Google Scholar]
- 211.Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, Scherer CA, Scholl C, Frohling S, Carr SA, Stern AM, Schreiber SL and Golub TR (2012) STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc Natl Acad Sci U S A. 109, 2860–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Downward, J. (2015) RAS synthetic lethal screens revisited: still seeking the elusive prize? ed.)^eds.), AACR; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Willumsen BM, Christensen A, Hubbert NL, Papageorge AG and Lowy DR (1984) The p21 ras C-terminus is required for transformation and membrane association. Nature. 310, 583–586 [DOI] [PubMed] [Google Scholar]
- 214.Gibbs JB, Oliff A and Kohl NE (1994) Farnesyltransferase inhibitors: Ras research yields a potential cancer therapeutic. Cell. 77, 175–178 [DOI] [PubMed] [Google Scholar]
- 215.Gibbs JB and Oliff A (1997) The potential of farnesyltransferase inhibitors as cancer chemotherapeutics. Annual review of pharmacology and toxicology. 37, 143–166 [DOI] [PubMed] [Google Scholar]
- 216.Leonard DM (1997) Ras farnesyltransferase: a new therapeutic target. Journal of medicinal chemistry. 40, 2971–2990 [DOI] [PubMed] [Google Scholar]
- 217.Rowinsky EK, Windle JJ and Von Hoff DD (1999) Ras protein farnesyltransferase: a strategic target for anticancer therapeutic development. Journal of Clinical Oncology. 17, 3631–3652 [DOI] [PubMed] [Google Scholar]
- 218.Oncology K (2019) Kura Oncology Announces Positive Phase 2 Trial of Tipifarnib in HRAS Mutant Urothelial Carcinoma. ed.)^eds.)
- 219.Ho AL, Hanna GJ, Scholz CR, Gualberto A and Park SH (2020) Preliminary activity of tipifarnib in tumors of the head and neck, salivary gland and urothelial tract with HRAS mutations. In ASCO; 2020 ed.)^eds.) [Google Scholar]
- 220.Fiordalisi JJ, Johnson RL 2nd, Weinbaum CA, Sakabe K, Chen Z, Casey PJ and Cox AD (2003) High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J Biol Chem. 278, 41718–41727 [DOI] [PubMed] [Google Scholar]
- 221.James GL, Brown MS, Cobb MH and Goldstein JL (1994) Benzodiazepine peptidomimetic BZA-5B interrupts the MAP kinase activation pathway in H-Ras-transformed Rat-1 cells, but not in untransformed cells. J Biol Chem. 269, 27705–27714 [PubMed] [Google Scholar]
- 222.James GL, Goldstein JL and Brown MS (1995) Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J Biol Chem. 270, 6221–6226 [DOI] [PubMed] [Google Scholar]
- 223.Rowell CA, Kowalczyk JJ, Lewis MD and Garcia AM (1997) Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem. 272, 14093–14097 [DOI] [PubMed] [Google Scholar]
- 224.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR and Pai JK (1997) K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 272, 14459–14464 [DOI] [PubMed] [Google Scholar]
- 225.Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, Davide JP, deSolms SJ, Dinsmore CJ, Ellis-Hutchings MS, Kral AM, Liu D, Lumma WC, Machotka SV, Rands E, Williams TM, Graham SL, Hartman GD, Oliff AI, Heimbrook DC and Kohl NE (2001) Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 61, 8758–8768 [PubMed] [Google Scholar]
- 226.Sebti SM and Der CJ (2003) Opinion: Searching for the elusive targets of farnesyltransferase inhibitors. Nat Rev Cancer. 3, 945–951 [DOI] [PubMed] [Google Scholar]
- 227.Berndt N, Hamilton AD and Sebti SM (2011) Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 11, 775–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Britten CD, Rowinsky EK, Soignet S, Patnaik A, Yao SL, Deutsch P, Lee Y, Lobell RB, Mazina KE, McCreery H, Pezzuli S and Spriggs D (2001) A phase I and pharmacological study of the farnesyl protein transferase inhibitor L-778,123 in patients with solid malignancies. Clin Cancer Res. 7, 3894–3903 [PubMed] [Google Scholar]
- 229.Lobell RB, Liu D, Buser CA, Davide JP, DePuy E, Hamilton K, Koblan KS, Lee Y, Mosser S, Motzel SL, Abbruzzese JL, Fuchs CS, Rowinsky EK, Rubin EH, Sharma S, Deutsch PJ, Mazina KE, Morrison BW, Wildonger L, Yao SL and Kohl NE (2002) Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 1, 747–758 [PubMed] [Google Scholar]
- 230.Morgan MA, Onono FO, Spielmann HP, Subramanian T, Scherr M, Venturini L, Dallmann I, Ganser A and Reuter CW (2012) Modulation of anthracycline-induced cytotoxicity by targeting the prenylated proteome in myeloid leukemia cells. J Mol Med (Berl). 90, 149–161 [DOI] [PubMed] [Google Scholar]
- 231.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI and Waldmann H (2013) Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 497, 638–642 [DOI] [PubMed] [Google Scholar]
- 232.Papke B, Murarka S, Vogel HA, Martin-Gago P, Kovacevic M, Truxius DC, Fansa EK, Ismail S, Zimmermann G, Heinelt K, Schultz-Fademrecht C, Al Saabi A, Baumann M, Nussbaumer P, Wittinghofer A, Waldmann H and Bastiaens PI (2016) Identification of pyrazolopyridazinones as PDEdelta inhibitors. Nat Commun. 7, 11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Klein CH, Truxius DC, Vogel HA, Harizanova J, Murarka S, Martin-Gago P and Bastiaens PIH (2019) PDEdelta inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic KRas. Int J Cancer. 144, 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Martin-Gago P, Fansa EK, Klein CH, Murarka S, Janning P, Schurmann M, Metz M, Ismail S, Schultz-Fademrecht C, Baumann M, Bastiaens PI, Wittinghofer A and Waldmann H (2017) A PDE6delta-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew Chem Int Ed Engl. 56, 2423–2428 [DOI] [PubMed] [Google Scholar]
- 235.van der Hoeven D, Cho KJ, Ma X, Chigurupati S, Parton RG and Hancock JF (2013) Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 33, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Eisenberg S, Giehl K, Henis YI and Ehrlich M (2008) Differential interference of chlorpromazine with the membrane interactions of oncogenic K-Ras and its effects on cell growth. Journal of Biological Chemistry. 283, 27279–27288 [DOI] [PubMed] [Google Scholar]
- 237.Cho K. j., van der Hoeven D, Zhou Y, Maekawa M, Ma X, Chen W, Fairn GD and Hancock JF (2016) Inhibition of acid sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Molecular and cellular biology. 36, 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bayer R and Mannhold R (1987) Fendiline: a review of its basic pharmacological and clinical properties. Pharmatherapeutica. 5, 103–136 [PubMed] [Google Scholar]
- 239.Yeung T, Terebiznik M, Yu L, Silvius J, Abidi WM, Philips M, Levine T, Kapus A and Grinstein S (2006) Receptor activation alters inner surface potential during phagocytosis. Science. 313, 347–351 [DOI] [PubMed] [Google Scholar]
- 240.van der Hoeven D, Cho K. j., Zhou Y, Ma X, Chen W, Naji A, Montufar-Solis D, Zuo Y, Kovar SE and Levental KR (2018) Sphingomyelin metabolism is a regulator of K-Ras function. Molecular and cellular biology. 38, e00373–00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G and Kozma SC (2008) Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of clinical investigation. 118, 3065–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Cho K. j., Kasai RS, Park J-H, Chigurupati S, Heidorn SJ, van der Hoeven D, Plowman SJ, Kusumi A, Marais R and Hancock JF (2012) Raf inhibitors target ras spatiotemporal dynamics. Current biology. 22, 945–955 [DOI] [PubMed] [Google Scholar]
- 243.Wang P, Hoeven D. v. d., Ye N, Chen H, Liu Z, Ma X, Montufar-Solis D, Rehl KM, Cho K-J, Thapa S, Chen W, Hoeven R. v. d., Frost JA, Hancock JF and Zhou J (2020. (In Review)) Scaffold Repurposing of Fendiline: Identification of Potent KRAS Plasma Membrane Localization Inhibitors Journal of Medicinal Chemistry [DOI] [PubMed] [Google Scholar]
- 244.Gulbins E, Palmada M, Reichel M, Lüth A, Böhmer C, Amato D, Müller CP, Tischbirek CH, Groemer TW and Tabatabai G (2013) Acid sphingomyelinase–ceramide system mediates effects of antidepressant drugs. Nature medicine. 19, 934. [DOI] [PubMed] [Google Scholar]
- 245.Kornhuber J, Tripal P, Reichel M, Terfloth L, Bleich S, Wiltfang J and Gulbins E (2008) Identification of new functional inhibitors of acid sphingomyelinase using a structure– property– activity relation model. Journal of medicinal chemistry. 51, 219–237 [DOI] [PubMed] [Google Scholar]
- 246.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K and Arenz C (2010) Hsp70 stabilizes lysosomes and reverts Niemann–Pick disease-associated lysosomal pathology. Nature. 463, 549–553 [DOI] [PubMed] [Google Scholar]
- 247.Utermöhlen O, Herz J, Schramm M and Krönke M (2008) Fusogenicity of membranes: the impact of acid sphingomyelinase on innate immune responses. Immunobiology. 213, 307–314 [DOI] [PubMed] [Google Scholar]
- 248.Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, Ostenfeld MS, Ulanet D, Dovmark TH, Lonborg A, Vindelov SD, Hanahan D, Arenz C, Ejsing CS, Kirkegaard T, Rohde M, Nylandsted J and Jaattela M (2013) Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 24, 379–393 [DOI] [PubMed] [Google Scholar]
- 249.Kolter T and Sandhoff K (2010) Lysosomal degradation of membrane lipids. FEBS letters. 584, 1700–1712 [DOI] [PubMed] [Google Scholar]
- 250.Fehrenbacher N, Bastholm L, Kirkegaard-Sørensen T, Rafn B, Bøttzauw T, Nielsen C, Weber E, Shirasawa S, Kallunki T and Jäättelä M (2008) Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer research. 68, 6623–6633 [DOI] [PubMed] [Google Scholar]
- 251.Ostenfeld MS, Høyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, Puustinen P, Kirkegaard-Sørensen T, Nylandsted J and Farkas T (2008) Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 4, 487–499 [DOI] [PubMed] [Google Scholar]
- 252.Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra-Martin MA, Bampton ET, Glynn P, Bonanno G and Knight RA (2009) Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. Journal of cell science. 122, 3330–3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Barceló-Coblijn G, Martin ML, de Almeida RF, Noguera-Salvà MA, Marcilla-Etxenike A, Guardiola-Serrano F, Lüth A, Kleuser B, Halver JE and Escribá PV (2011) Sphingomyelin and sphingomyelin synthase (SMS) in the malignant transformation of glioma cells and in 2-hydroxyoleic acid therapy. Proceedings of the National Academy of Sciences. 108, 19569–19574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Dielschneider RF, Henson ES and Gibson SB (2017) Lysosomes as oxidative targets for cancer therapy. Oxidative medicine and cellular longevity. 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Beckmann N, Sharma D, Gulbins E, Becker KA and Edelmann B (2014) Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Frontiers in physiology. 5, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hannun YA and Obeid LM (2002) The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. Journal of Biological Chemistry. 277, 25847–25850 [DOI] [PubMed] [Google Scholar]
- 257.Linn S, Kim H, Keane E, Andras L, Wang E and Merrill A Jr (2001) Regulation of de novo sphingolipid biosynthesis and the toxic consequences of its disruption. ed.)^eds.), Portland Press Ltd. [DOI] [PubMed] [Google Scholar]
- 258.Tafesse FG, Ternes P and Holthuis JC (2006) The multigenic sphingomyelin synthase family. Journal of Biological Chemistry. 281, 29421–29425 [DOI] [PubMed] [Google Scholar]
- 259.Marchesini N and Hannun YA (2004) Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochemistry and cell biology. 82, 27–44 [DOI] [PubMed] [Google Scholar]
- 260.Kunkel GT, Maceyka M, Milstien S and Spiegel S (2013) Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nature reviews Drug discovery. 12, 688–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wang P, Yuan Y, Lin W, Zhong H, Xu K and Qi X (2019) Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell International. 19, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Maekawa M, Lee M, Wei K, Ridgway ND and Fairn GD (2016) Staurosporines decrease ORMDL proteins and enhance sphingomyelin synthesis resulting in depletion of plasmalemmal phosphatidylserine. Scientific reports. 6, 35762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Salim AA, Xiao X, Cho K-J, Piggott AM, Lacey E, Hancock JF and Capon RJ (2014) Rare Streptomyces sp. polyketides as modulators of K-Ras localisation. Organic & biomolecular chemistry. 12, 4872–4878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tan L, Cho K-J, Kattan WE, Garrido CM, Zhou Y, Neupane P, Capon RJ and Hancock JF (2019) Acylpeptide hydrolase is a novel regulator of KRAS plasma membrane localization and function. Journal of cell science. 132, jcs232132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Fujino T, Ando K, Beppu M and Kikugawa K (2000) Enzymatic removal of oxidized protein aggregates from erythrocyte membranes. The Journal of Biochemistry. 127, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 266.Fujino T, Watanabe K, Beppu M, Kikugawa K and Yasuda H (2000) Identification of oxidized protein hydrolase of human erythrocytes as acylpeptide hydrolase. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology. 1478, 102–112 [DOI] [PubMed] [Google Scholar]
- 267.Gonen H, Smith CE, Siegel NR, Kahana C, Merrick WC, Chakraburtty K, Schwartz AL and Ciechanover A (1994) Protein synthesis elongation factor EF-1 alpha is essential for ubiquitin-dependent degradation of certain N alpha-acetylated proteins and may be substituted for by the bacterial elongation factor EF-Tu. Proceedings of the National Academy of Sciences. 91, 7648–7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Hershko A, Heller H, Eytan E, Kaklij G and Rose IA (1984) Role of the alpha-amino group of protein in ubiquitin-mediated protein breakdown. Proceedings of the National Academy of Sciences. 81, 7021–7025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Palmieri G, Bergamo P, Luini A, Ruvo M, Gogliettino M, Langella E, Saviano M, Hegde RN, Sandomenico A and Rossi M (2011) Acylpeptide hydrolase inhibition as targeted strategy to induce proteasomal down-regulation. PloS one. 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zeng Z, Rulten SL, Breslin C, Zlatanou A, Coulthard V and Caldecott KW (2017) Acylpeptide hydrolase is a component of the cellular response to DNA damage. DNA repair. 58, 52–61 [DOI] [PubMed] [Google Scholar]
- 271.Tan L, Cho K-J, Neupane P, Capon RJ and Hancock JF (2018) An oxanthroquinone derivative that disrupts RAS plasma membrane localization inhibits cancer cell growth. Journal of Biological Chemistry. 293, 13696–13706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Misaki R, Morimatsu M, Uemura T, Waguri S, Miyoshi E, Taniguchi N, Matsuda M and Taguchi T (2010) Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. Journal of Cell Biology. 191, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M and Schölermann B (2010) Small-molecule inhibition of APT1 affects Ras localization and signaling. Nature chemical biology. 6, 449. [DOI] [PubMed] [Google Scholar]
- 274.Lin DTS and Conibear E (2015) ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife. 4, e11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Schmick M, Kraemer A and Bastiaens PI (2015) Ras moves to stay in place. Trends in cell biology. 25, 190–197 [DOI] [PubMed] [Google Scholar]
- 276.Sandomenico A, Russo A, Palmieri G, Bergamo P, Gogliettino M, Falcigno L and Ruvo M (2012) Small peptide inhibitors of acetyl-peptide hydrolase having an uncommon mechanism of inhibition and a stable bent conformation. Journal of medicinal chemistry. 55, 2102–2111 [DOI] [PubMed] [Google Scholar]
- 277.Junqueira L, Rothschild H and Fajer A (1957) Protein production by the rat pancreas. Experimental cell research. 12, 338–341 [DOI] [PubMed] [Google Scholar]
- 278.Kay JG and Grinstein S (2011) Sensing phosphatidylserine in cellular membranes. Sensors. 11, 1744–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kay JG, Koivusalo M, Ma X, Wohland T and Grinstein S (2012) Phosphatidylserine dynamics in cellular membranes. Mol Biol Cell. 23, 2198–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Van Meer G, Voelker DR and Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nature reviews Molecular cell biology. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Kuge O and Nishijima M (1997) Phosphatidylserine synthase I and II of mammalian cells. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism. 1348, 151–156 [DOI] [PubMed] [Google Scholar]
- 282.Lee S, Uchida Y, Emoto K, Umeda M, Kuge O, Taguchi T and Arai H (2012) Impaired retrograde membrane traffic through endosomes in a mutant CHO cell defective in phosphatidylserine synthesis. Genes to Cells. 17, 728–736 [DOI] [PubMed] [Google Scholar]
- 283.Leventis PA and Grinstein S (2010) The distribution and function of phosphatidylserine in cellular membranes. Annual review of biophysics. 39, 407–427 [DOI] [PubMed] [Google Scholar]
- 284.Stone SJ and Vance JE (2000) Phosphatidylserine synthase-1 and-2 are localized to mitochondria-associated membranes. Journal of Biological Chemistry. 275, 34534–34540 [DOI] [PubMed] [Google Scholar]
- 285.Arikketh D, Nelson R and Vance JE (2008) Defining the Importance of Phosphatidylserine Synthase-1 (PSS1) Unexpected Viability of PSS1-Deficient Mice. Journal of Biological Chemistry. 283, 12888–12897 [DOI] [PubMed] [Google Scholar]
- 286.Kuge O, Nishijima M and Akamatsu Y (1986) Phosphatidylserine biosynthesis in cultured Chinese hamster ovary cells. II. Isolation and characterization of phosphatidylserine auxotrophs. Journal of Biological Chemistry. 261, 5790–5794 [PubMed] [Google Scholar]
- 287.Kuge O, Nishijima M and Akamatsu Y (1991) A Chinese hamster cDNA encoding a protein essential for phosphatidylserine synthase I activity. Journal of Biological Chemistry. 266, 24184–24189 [PubMed] [Google Scholar]
- 288.Kuge O, Saito K and Nishijima M (1997) Cloning of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylserine synthase (PSS) II, overexpression of which suppresses the phosphatidylserine biosynthetic defect of a PSS I-lacking mutant of CHO-K1 cells. Journal of Biological Chemistry. 272, 19133–19139 [DOI] [PubMed] [Google Scholar]
- 289.Kay JG and Fairn GD (2019) Distribution, dynamics and functional roles of phosphatidylserine within the cell. Cell Commun Signal. 17, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Fairn GD, Schieber NL, Ariotti N, Murphy S, Kuerschner L, Webb RI, Grinstein S and Parton RG (2011) High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. Journal of Cell Biology. 194, 257–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Gagescu R, Demaurex N, Parton RG, Hunziker W, Huber LA and Gruenberg J (2000) The recycling endosome of Madin-Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol Biol Cell. 11, 2775–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A and Grinstein S (2008) Membrane phosphatidylserine regulates surface charge and protein localization. Science. 319, 210–213 [DOI] [PubMed] [Google Scholar]
- 293.Uchida Y, Hasegawa J, Chinnapen D, Inoue T, Okazaki S, Kato R, Wakatsuki S, Misaki R, Koike M and Uchiyama Y (2011) Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proceedings of the National Academy of Sciences. 108, 15846–15851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Matsudaira T, Mukai K, Noguchi T, Hasegawa J, Hatta T, Iemura S. i., Natsume T, Miyamura N, Nishina H and Nakayama J (2017) Endosomal phosphatidylserine is critical for the YAP signalling pathway in proliferating cells. Nature communications. 8, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Maeda K, Anand K, Chiapparino A, Kumar A, Poletto M, Kaksonen M and Gavin AC (2013) Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature. 501, 257–261 [DOI] [PubMed] [Google Scholar]
- 296.Ghai R, Du X, Wang H, Dong J, Ferguson C, Brown AJ, Parton RG, Wu J-W and Yang H (2017) ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns (4, 5) P 2) and regulate its level at the plasma membrane. Nature communications. 8, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F and De Camilli P (2015) INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science. 349, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Moser von Filseck J, Copic A, Delfosse V, Vanni S, Jackson CL, Bourguet W and Drin G (2015) INTRACELLULAR TRANSPORT. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science. 349, 432–436 [DOI] [PubMed] [Google Scholar]
- 299.Helle SC, Kanfer G, Kolar K, Lang A, Michel AH and Kornmann B (2013) Organization and function of membrane contact sites. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 1833, 2526–2541 [DOI] [PubMed] [Google Scholar]
- 300.Balla A, Tuymetova G, Tsiomenko A, Várnai P and Balla T (2005) A plasma membrane pool of phosphatidylinositol 4-phosphate is generated by phosphatidylinositol 4-kinase type-III alpha: studies with the PH domains of the oxysterol binding protein and FAPP1. Mol Biol Cell. 16, 1282–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Sohn M, Ivanova P, Brown HA, Toth DJ, Varnai P, Kim YJ and Balla T (2016) Lenz-Majewski mutations in PTDSS1 affect phosphatidylinositol 4-phosphate metabolism at ER-PM and ER-Golgi junctions. Proceedings of the National Academy of Sciences. 113, 4314–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Clayton EL, Minogue S and Waugh MG (2013) Mammalian phosphatidylinositol 4-kinases as modulators of membrane trafficking and lipid signaling networks. Progress in lipid research. 52, 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Boura E and Nencka R (2015) Phosphatidylinositol 4-kinases: Function, structure, and inhibition. Experimental cell research. 337, 136–145 [DOI] [PubMed] [Google Scholar]
- 304.Nakatsu F, Baskin JM, Chung J, Tanner LB, Shui G, Lee SY, Pirruccello M, Hao M, Ingolia NT and Wenk MR (2012) PtdIns4P synthesis by PI4KIIIα at the plasma membrane and its impact on plasma membrane identity. Journal of Cell Biology. 199, 1003–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Baskin JM, Wu X, Christiano R, Oh MS, Schauder CM, Gazzerro E, Messa M, Baldassari S, Assereto S and Biancheri R (2016) The leukodystrophy protein FAM126A (hyccin) regulates PtdIns (4) P synthesis at the plasma membrane. Nature cell biology. 18, 132–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Bai J and Pagano RE (1997) Measurement of spontaneous transfer and transbilayer movement of BODIPY-labeled lipids in lipid vesicles. Biochemistry. 36, 8840–8848 [DOI] [PubMed] [Google Scholar]
- 307.Daleke DL (2007) Phospholipid flippases. Journal of Biological Chemistry. 282, 821–825 [DOI] [PubMed] [Google Scholar]
- 308.Kornberg RD and McConnell HM (1971) Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry. 10, 1111–1120 [DOI] [PubMed] [Google Scholar]
- 309.Pomorski T and Menon A (2006) Lipid flippases and their biological functions. Cellular and Molecular Life Sciences CMLS. 63, 2908–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.van Meer G, Voelker DR and Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lentz BR (2003) Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Progress in lipid research. 42, 423–438 [DOI] [PubMed] [Google Scholar]
- 312.Williamson P and Schlegel RA (2002) Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 1585, 53–63 [DOI] [PubMed] [Google Scholar]
- 313.Wu Y, Tibrewal N and Birge RB (2006) Phosphatidylserine recognition by phagocytes: a view to a kill. Trends in cell biology. 16, 189–197 [DOI] [PubMed] [Google Scholar]
- 314.Zwaal RF, Comfurius P and Bevers EM (1998) Lipid–protein interactions in blood coagulation. Biochimica et Biophysica Acta (BBA)-Reviews on Biomembranes. 1376, 433–453 [DOI] [PubMed] [Google Scholar]
- 315.Jaworski CJ, Moreira E, Li A, Lee R and Rodriguez IR (2001) A family of 12 human genes containing oxysterol-binding domains. Genomics. 78, 185–196 [DOI] [PubMed] [Google Scholar]
- 316.Lehto M, Laitinen S, Chinetti G, Johansson M, Ehnholm C, Staels B, Ikonen E and Olkkonen VM (2001) The OSBP-related protein family in humans. Journal of lipid research. 42, 1203–1213 [PubMed] [Google Scholar]
- 317.Im YJ, Raychaudhuri S, Prinz WA and Hurley JH (2005) Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature. 437, 154–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Stefan CJ, Manford AG, Baird D, Yamada-Hanff J, Mao Y and Emr SD (2011) Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 144, 389–401 [DOI] [PubMed] [Google Scholar]
- 319.Haslam RJ, Koide HB and Hemmings BA (1993) Pleckstrin domain homology. Nature. 363, 309–310 [DOI] [PubMed] [Google Scholar]
- 320.Lemmon M and Ferguson K (2001) Molecular determinants in pleckstrin homology domains that allow specific recognition of phosphoinositides. ed.)^eds.), Portland Press Ltd. [DOI] [PubMed] [Google Scholar]
- 321.Mayer BJ, Ren R, Clark KL and Baltimore D (1993) A putative modular domain present in diverse signaling proteins. Cell. 73, 629–630 [DOI] [PubMed] [Google Scholar]
- 322.Rebecchi M and Scarlata S (1998) Pleckstrin homology domains: a common fold with diverse functions. Annual review of biophysics and biomolecular structure. 27, 503–528 [DOI] [PubMed] [Google Scholar]
- 323.Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ and Yang H (2011) A role for oxysterol-binding protein–related protein 5 in endosomal cholesterol trafficking. Journal of Cell Biology. 192, 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Olkkonen VM and Li S (2013) Oxysterol-binding proteins: sterol and phosphoinositide sensors coordinating transport, signaling and metabolism. Progress in lipid research. 52, 529–538 [DOI] [PubMed] [Google Scholar]
- 325.Yan D, Mäyränpää MI, Wong J, Perttilä J, Lehto M, Jauhiainen M, Kovanen PT, Ehnholm C, Brown AJ and Olkkonen VM (2008) OSBP-related protein 8 (ORP8) suppresses ABCA1 expression and cholesterol efflux from macrophages. Journal of Biological Chemistry. 283, 332–340 [DOI] [PubMed] [Google Scholar]
- 326.Sohn M, Korzeniowski M, Zewe JP, Wills RC, Hammond GR, Humpolickova J, Vrzal L, Chalupska D, Veverka V and Fairn GD (2018) PI (4, 5) P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER–PM contact sites. Journal of Cell Biology. 217, 1797–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B and Drin G (2011) Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. Journal of Cell Biology. 195, 965–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Tong J, Yang H, Yang H, Eom SH and Im YJ (2013) Structure of Osh3 reveals a conserved mode of phosphoinositide binding in oxysterol-binding proteins. Structure. 21, 1203–1213 [DOI] [PubMed] [Google Scholar]
- 329.Yuan TL, Amzallag A, Bagni R, Yi M, Afghani S, Burgan W, Fer N, Strathern LA, Powell K and Smith B (2018) Differential effector engagement by oncogenic KRAS. Cell reports. 22, 1889–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL and Giordano F (2016) ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO reports. 17, 800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Ishikawa S, Nagai Y, Masuda T, Koga Y, Nakamura T, Imamura Y, Takamori H, Hirota M, Funakosi A and Fukushima M (2010) The role of oxysterol binding protein-related protein 5 in pancreatic cancer. Cancer science. 101, 898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Koga Y, Ishikawa S, Nakamura T, Masuda T, Nagai Y, Takamori H, Hirota M, Kanemitsu K, Baba Y and Baba H (2008) Oxysterol binding protein-related protein-5 is related to invasion and poor prognosis in pancreatic cancer. Cancer science. 99, 2387–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Nagano K, Imai S, Zhao X, Yamashita T, Yoshioka Y, Abe Y, Mukai Y, Kamada H, Nakagawa S and Tsutsumi Y (2015) Identification and evaluation of metastasis-related proteins, oxysterol binding protein-like 5 and calumenin, in lung tumors. International journal of oncology. 47, 195–203 [DOI] [PubMed] [Google Scholar]
- 334.Bocci G, Fioravanti A, Orlandi P, Bernardini N, Collecchi P, Del Tacca M and Danesi R (2005) Fluvastatin synergistically enhances the antiproliferative effect of gemcitabine in human pancreatic cancer MIAPaCa-2 cells. British journal of cancer. 93, 319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Müller C, Bockhorn A, Klusmeier S, Kiehl M, Roeder C, Kalthoff H and Koch O (1998) Lovastatin inhibits proliferation of pancreatic cancer cell lines with mutant as well as with wild-type K-ras oncogene but has different effects on protein phosphorylation and induction of apoptosis. International journal of oncology. 12, 717–740 [DOI] [PubMed] [Google Scholar]
- 336.Du X, Zadoorian A, Lukmantara IE, Qi Y, Brown AJ and Yang H (2018) Oxysterol-binding protein–related protein 5 (ORP5) promotes cell proliferation by activation of mTORC1 signaling. Journal of Biological Chemistry. 293, 3806–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Bojjireddy N, Botyanszki J, Hammond G, Creech D, Peterson R, Kemp DC, Snead M, Brown R, Morrison A and Wilson S (2014) Pharmacological and genetic targeting of the PI4KA enzyme reveals its important role in maintaining plasma membrane phosphatidylinositol 4-phosphate and phosphatidylinositol 4, 5-bisphosphate levels. Journal of Biological Chemistry. 289, 6120–6132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Ishikawa S, Egami H, Kurizaki T, Akagi J, Tamori Y, Yoshida N, Tan X, Hayashi N and Ogawa M (2003) Identification of genes related to invasion and metastasis in pancreatic cancer by cDNA representational difference analysis. Journal of experimental & clinical cancer research: CR. 22, 299–306 [PubMed] [Google Scholar]
- 339.Giroux V, Iovanna J and Dagorn J-C (2006) Probing the human kinome for kinases involved in pancreatic cancer cell survival and gemcitabine resistance. The FASEB Journal. 20, 1982–1991 [DOI] [PubMed] [Google Scholar]
- 340.Raubo P, Andrews DM, McKelvie JC, Robb GR, Smith JM, Swarbrick ME and Waring MJ (2015) Discovery of potent, selective small molecule inhibitors of α-subtype of type III phosphatidylinositol-4-kinase (PI4KIIIα). Bioorganic & medicinal chemistry letters. 25, 3189–3193 [DOI] [PubMed] [Google Scholar]
- 341.Waring MJ, Andrews DM, Faulder PF, Flemington V, McKelvie JC, Maman S, Preston M, Raubo P, Robb GR and Roberts K (2014) Potent, selective small molecule inhibitors of type III phosphatidylinositol-4-kinase α-but not β-inhibit the phosphatidylinositol signaling cascade and cancer cell proliferation. Chemical Communications. 50, 5388–5390 [DOI] [PubMed] [Google Scholar]
- 342.Adhikari H, Kattan WE, Hancock JF and Counter CM (2020) Interrogating the RAS interactome identified EFR3A as a novel enhancer of RAS oncogenesis. In AACR Annual Meeting; ed.)^eds.) [Google Scholar]
- 343.Venditti R, Rega LR, Masone MC, Santoro M, Polishchuk E, Sarnataro D, Paladino S, D’Auria S, Varriale A and Olkkonen VM (2019) Molecular determinants of ER–Golgi contacts identified through a new FRET–FLIM system. Journal of Cell Biology. 218, 1055–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Vaillancourt FH, Brault M, Pilote L, Uyttersprot N, Gaillard ET, Stoltz JH, Knight BL, Pantages L, McFarland M, Breitfelder S, Chiu TT, Mahrouche L, Faucher AM, Cartier M, Cordingley MG, Bethell RC, Jiang H, White PW and Kukolj G (2012) Evaluation of phosphatidylinositol-4-kinase IIIalpha as a hepatitis C virus drug target. J Virol. 86, 11595–11607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Park Y, Park JM, Kim DH, Kwon J and Kim IA (2017) Inhibition of PI4K IIIalpha radiosensitizes in human tumor xenograft and immune-competent syngeneic murine tumor model. Oncotarget. 8, 110392–110405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, Spellman PT, Phillips WA, Gray JW and McMahon M (2012) A central role for RAF-->MEK-->ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2, 685–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Soares HP, Ming M, Mellon M, Young SH, Han L, Sinnet-Smith J and Rozengurt E (2015) Dual PI3K/mTOR Inhibitors Induce Rapid Overactivation of the MEK/ERK Pathway in Human Pancreatic Cancer Cells through Suppression of mTORC2. Mol Cancer Ther. 14, 1014–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Muzumdar MD, Chen PY, Dorans KJ, Chung KM, Bhutkar A, Hong E, Noll EM, Sprick MR, Trumpp A and Jacks T (2017) Survival of pancreatic cancer cells lacking KRAS function. Nat Commun. 8, 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, Chang DK, Cowley MJ, Gardiner BB, Song S, Harliwong I, Idrisoglu S, Nourse C, Nourbakhsh E, Manning S, Wani S, Gongora M, Pajic M, Scarlett CJ, Gill AJ, Pinho AV, Rooman I, Anderson M, Holmes O, Leonard C, Taylor D, Wood S, Xu Q, Nones K, Fink JL, Christ A, Bruxner T, Cloonan N, Kolle G, Newell F, Pinese M, Mead RS, Humphris JL, Kaplan W, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chou A, Chin VT, Chantrill LA, Mawson A, Samra JS, Kench JG, Lovell JA, Daly RJ, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Kakkar N, Zhao F, Wu YQ, Wang M, Muzny DM, Fisher WE, Brunicardi FC, Hodges SE, Reid JG, Drummond J, Chang K, Han Y, Lewis LR, Dinh H, Buhay CJ, Beck T, Timms L, Sam M, Begley K, Brown A, Pai D, Panchal A, Buchner N, De Borja R, Denroche RE, Yung CK, Serra S, Onetto N, Mukhopadhyay D, Tsao MS, Shaw PA, Petersen GM, Gallinger S, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Schulick RD, Wolfgang CL, Morgan RA, Lawlor RT, Capelli P, Corbo V, Scardoni M, Tortora G, Tempero MA, Mann KM, Jenkins NA, Perez-Mancera PA, Adams DJ, Largaespada DA, Wessels LF, Rust AG, Stein LD, Tuveson DA, Copeland NG, Musgrove EA, Scarpa A, Eshleman JR, Hudson TJ, Sutherland RL, Wheeler DA, Pearson JV, McPherson JD, Gibbs RA and Grimmond SM (2012) Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 491, 399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE and Kinzler KW (2008) Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 321, 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, Anagnostou V, Parpart-Li S, Murphy D, Kay Li Q, Hruban CA, Scharpf R, White JR, O’Dwyer PJ, Allen PJ, Eshleman JR, Thompson CB, Klimstra DS, Linehan DC, Maitra A, Hruban RH, Diaz LA Jr., Von Hoff DD, Johansen JS, Drebin JA and Velculescu VE (2015) Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 6, 7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, Bruxner TJ, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Wilson PJ, Markham E, Cloonan N, Anderson MJ, Fink JL, Holmes O, Kazakoff SH, Leonard C, Newell F, Poudel B, Song S, Taylor D, Waddell N, Wood S, Xu Q, Wu J, Pinese M, Cowley MJ, Lee HC, Jones MD, Nagrial AM, Humphris J, Chantrill LA, Chin V, Steinmann AM, Mawson A, Humphrey ES, Colvin EK, Chou A, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Pettitt JA, Merrett ND, Toon C, Epari K, Nguyen NQ, Barbour A, Zeps N, Jamieson NB, Graham JS, Niclou SP, Bjerkvig R, Grutzmann R, Aust D, Hruban RH, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Falconi M, Zamboni G, Tortora G, Tempero MA, Gill AJ, Eshleman JR, Pilarsky C, Scarpa A, Musgrove EA, Pearson JV, Biankin AV and Grimmond SM (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 518, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, White MA and Knudsen ES (2015) Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 6, 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Huang BX, Akbar M, Kevala K and Kim HY (2011) Phosphatidylserine is a critical modulator for Akt activation. J Cell Biol. 192, 979–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.De Roock W, Jonker DJ, Di Nicolantonio F, Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M, Piessevaux H, Van Cutsem E, O’Callaghan CJ, Khambata-Ford S, Zalcberg JR, Simes J, Karapetis CS, Bardelli A and Tejpar S (2010) Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. Jama. 304, 1812–1820 [DOI] [PubMed] [Google Scholar]
- 356.Grant BJ, Lukman S, Hocker HJ, Sayyah J, Brown JH, McCammon JA and Gorfe AA (2011) Novel allosteric sites on Ras for lead generation. PloS one. 6 [DOI] [PMC free article] [PubMed] [Google Scholar]