

Abstract
To signal, Ras isoforms must be enriched at the plasma membrane (PM). It was suggested that phosphodiesterase-δ (PDEδ) can bind and shuttle some farnesylated Ras isoforms to the PM, but not all. Among these, interest focused on K-Ras4B, the most abundant oncogenic Ras isoform. To study PDEδ/Ras interactions, we modeled and simulated the PDEδ/K-Ras4B complex. We obtained structures, which were similar to two subsequently determined crystal structures. We next modeled and simulated complexes of PDEδ with the farnesylated hypervariable regions of K-Ras4A and N-Ras. Earlier data suggested that PDEδ extracts K-Ras4B and N-Ras from the PM, but surprisingly not K-Ras4A. Earlier analysis of the crystal structures advanced that the presence of large/charged residues adjacent to the farnesylated site precludes the PDEδ interaction. Here, we show that PDEδ can bind to farnesylated K-Ras4A and N-Ras like K-Ras4B, albeit not as strongly. This weaker binding, coupled with the stronger anchoring of K-Ras4A in the membrane (but not of electrostatically neutral N-Ras), can explain the observation why PDEδ is unable to effectively extract K-Ras4A. We thus propose that farnesylated Ras isoforms can bind PDEδ to fulfill the required PM enrichment, and argue that the different environments, PM versus solution, can resolve apparently puzzling Ras observations. These are novel insights that would not be expected based on the crystal structures alone, which provide an elegant rationale for previously puzzling observations of the differential effects of PDEδ on farnesylated Ras family proteins.
Graphical Abstract
INTRODUCTION
The Ras superfamily of small GTPases, including Ras isoforms KRAS (K-Ras4A, and K-Ras4B), HRAS, and NRAS, as well as the Rho and Rab subfamilies, regulate an astonishing diversity of cellular functions such as proliferation, differentiation, cell morphology, motility, intracellular trafficking, and gene expression.1–3 It has been known for many years that the attachment of Ras superfamily proteins to plasma membranes (PMs) is essential for their function.4 To gain full biological activity, following their synthesis as soluble precursors, the Ras superfamily proteins undergo a series of post-translational modifications (PTMs).5–9 Ras prenylation involving farnesylation and geranylgeranylation is crucial for membrane signaling and trafficking.10
Ras and Rho family proteins contain a CAAX sequence (where “C” is a Cys, “A” is an aliphatic residue, and “X” denotes any amino acid) at their C-termini that undergoes post-translational farnesylation. Signaling by these prenylated proteins relies on their subcellular distribution and localization.11 Guanosine nucleotide dissociation inhibitors (GDIs), such as RhoGDI and RabGDI, are responsible for the intracellular trafficking of Rho and Rab proteins, respectively.12,13 Similarly, the delta subunit of the cGMP PDE from retinal rod cells (phosphodiesterase-δ (PDEδ)) can interact with prenylated proteins, such as K-Ras, H-Ras, and Rheb, and assist in their trafficking through the cytosol between different membrane compartments.14–18 PDEδ shares a significant structural similarity with RhoGDI. It features a hydrophobic prenyl-binding pocket into which prenylated small G proteins insert their farnesylated C-terminus. This facilitates the intracellular diffusion of these proteins by shielding the hydrophobic moiety from the cytosol. However, the prenyl-binding pocket of PDEδ is much shorter than the RhoGDI binding pocket resulting in a weaker interaction between PDEδ and geranylgeranylated proteins.14,15,17 PDEδ also lacks an N-terminal helix−loop−helix that can interact with the GTPase switch regions. Consequently, unlike RhoGDI binding, PDEδ binding to prenylated proteins is nucleotide independent.15,16 PDEδ appears more promiscuous in terms of ligand specificity and can associate with various prenylated proteins including protein kinases, PDE subunits, and GTPases. In addition to these proteins, PDEδ associates with two nonprenylated small G proteins, Arl2 and Arl3, from the Arf family. These PTM-independent interactions involve the switch regions and are specific to the GTP-bound state, hence classifying PDEδ as Arl2 and Arl3 effectors. 15,19,20
Biochemical studies indicate that PDEδ binds more strongly to farnesylated proteins, such as Rheb and K-Ras4B, than to other prenylated proteins and the binding stoichiometry is 1:1.14,17 Binding of Rheb to PDEδ occurs mainly through the farnesylated C-terminus. Unlike the Rho/RhoGDI interaction, the interaction between Rheb and PDEδ does not involve the switch regions of Rheb.13,21,22 Additionally, the farnesyl group together with the last three residues penetrates much more deeply into the hydrophobic pocket than RhoGDI. This presumably compensates for the weaker contacts with the rest of the protein. Binding of K-Ras4B to PDEδ also occurs mainly through the farnesylated and methylated C-terminus of the hypervariable region (HVR) and is enhanced by the negatively charged surface of PDEδ and the positively charged C-terminus of K-Ras4B HVR, with no additional contacts between PDEδ and the catalytic domain of K-Ras4B.18,23
The frequent mutations of Ras in human cancers24–26 and its significant role in cell proliferation25,27 have led to attempts to pharmacologically inhibit oncogenic Ras.26,28 Ras localization at the PM is required for its biological activity;29 thus interfering with Ras localization is one of the approaches to target Ras.30,31 However, despite its significance in drug development, the transport of Ras proteins to the PM is not fully understood.32,33 There is evidence that farnesylated and palmitoylated H-Ras, N-Ras, and K-Ras4A proteins, but not K-Ras4B, are vesicle-transported from the Golgi complex to the PM.11,34 On the other hand, K-Ras4B is transported to the PM through a nonvesicular, diffusional mechanism. This intracellular diffusion is a slow process and the inward membrane flow of endocytic vesicles counteracts this movement to randomize the Ras distribution over all membranes.35 Studies involving inhibition or knock down of PDEδ indicated that PDEδ is essential for the dynamic shuttling of the polybasic-stretch-containing K-Ras4B, raising the question of whether K-Ras4A can also be shuttled in a similar way.36 PDEδ increases the kinetics of trapping and thereby counteracts the entropic tendency of the membranes to randomly distribute Ras over all membranes and maintains K-Ras4B localized enrichment at the PM.11,14,37,38
Until recently, the experimental structure of K-Ras4B in complex with PDEδ was unavailable. Dharmaiah et al.18 crystallized the human farnesylated-methylated K-Ras4B/PDEδ complex in two different forms. Here, we modeled and simulated the K-Ras4B/PDEδ complex to gain structural and conformational dynamics insight into the interaction of the HVR/farnesyl (and geranylgeranyl) moiety with the PDEδ prenyl-binding pocket. We asked whether the catalytic domain of K-Ras4B also interacts with PDEδ, and what are the implications for other Ras isoforms, splice isoform K-Ras4A, and N-Ras. This question is significant because experiments indicated that both K-Ras4B and N-Ras bind PDEδ,36–38 as well as depalmitoylated H-Ras, but surprisingly, apparently not K-Ras4A; at least it was not extracted from the PM by PDEδ, whereas N-Ras was.36 We also investigated the similarities and differences between the interaction of PDEδ with farnesylated and geranylgeranylated K-Ras4B. When we took on this project, the crystal structures were unavailable. Thus, we modeled the K-Ras4B/PDEδ structures and performed explicit-solvent molecular dynamics (MD) simulations using six different initial conformations. Our results are in good agreement with recent crystal structures18 and earlier experimental observations.11,38 Not surprisingly, they indicate that the HVR and the prenyl moiety are the main players of the K-Ras4B/PDEδ interaction, and that the conformation that the HVR adopts affects the K-Ras4B/PDEδ interaction strength. To address the question of whether in its farnesylated but depalmitoylated form K-Ras4A39 can interact with PDEδ as well, we modeled and simulated the farnesylated HVRs of K-Ras4A, and for comparison with the Tsai et al.36 and Chandra et al.37 experiments, also N-Ras complexed with the PDEδ in its two major states. The sequence properties of the HVR of K-Ras4A (167RLKKISKEEKTPGCVKIKKC185) resemble those of K-Ras4B (167KEKMSKDGKKKKKKSKTKC186), presenting a highly positively charged HVR (+6e) albeit not to the same extent as K-Ras4B’s (+9e),29,39 and we wondered whether conformational dynamics will resolve the steric clash that otherwise appears to take place with K-Ras4A’s two HVR lysine residues. Noteworthily, the sequence properties of the HVR of N-Ras (167RMKKLNSSDDGTQGCMGLPC186) differ from K-Ras4B’s, being weakly charged (+1e) with a neutral anchor region (residues 177−86). Further support for isoform-PDEδ binding comes from the observation that the key stabilizing factor in the Ras/PDEδ complex is the docking of the farnesyl (or geranylgeranyl) into its partner’s pocket. Additionally, Chandra et al.’s comparison of the palmitoylation levels indicated that at least half of the N-Ras molecules were depalmitoylated (as compared with less than a quarter of the H-Ras), leading the authors to suggest that a substantial fraction of N-Ras was already in the depalmitoylated state once it reached the PM through the secretory pathway.37 Thus, a certain fraction of K-Ras4A can be in the depalmitoylated state as well.
Here, we show that farnesylated and depalmitoylated K-Ras4A can bind PDEδ, as can N-Ras. Why then was K-Ras4A not extracted from the PM by PDEδ,36 when simulations suggest that formation of a K-Ras4A/PDEδ complex is possible? Here we argue that we need to consider the different environments. The Philips group36 probed isoform extraction; they did not test the binding of depalmitoylated K-Ras4A to PDEδ in solution. At the same time, analysis of the crystal structures18 and our simulations probe the interactions in a solution environment. To understand Ras isoform behavior, we need to consider the relative stabilities in the PM versus with PDEδ in solution. If the interactions of the two isoforms with PDEδ are comparable, but with the PM they are not, the outcome will differ. In our case here, K-Ras4A and N-Ras have comparable interactions with PDEδ, but not with the PM, thus explaining the seemingly surprising K-Ras4A observations.
METHODS
Generating Initial Configurations.
The simulation inputs were built using CHARMM40 c39b2 with the CHARMM 36 force field.41 Bonded parameters for the GTP and the prenylated and methylated cysteine were adopted from previous works.42–45 Farnesylated and geranylgeranylated K-Ras4B-GTP proteins were constructed using the crystal structures (PDB ID: 3GFT and 4DSO) as described earlier.24,42,44 The former is GNP-bound K-Ras4B1−167 with Q61H mutation, and the latter is GSP-bound K-Ras4B1−180 with G12D mutation. Both GTP analogues were converted to GTP, and their sequences were modified into human. The missing HVR coordinates, HVR168−185 for the former and HVR181−185 for the latter, were modeled as a flexible loop. At the C-terminus, Cys185 was modified with the farnesyl/geranylgeranyl and methyl groups. Thus, we obtained two K-Ras4B-GTP models, denoted KM1 and KM2. The HVR of KM1 is a flexible chain, whereas the HVR of KM2 partially folds into an α-helix extending from α5 of the catalytic domain (Figure 1A). To model the interactions of PDEδ with farnesyl and geranylgeranyl moieties, two crystal structures were used (PDB ID: 5F2U and 5E8F). The former PDEδ crystal structure contained the farnesyl group in the hydrophobic cavity, and the latter involved the geranylgeranyl group in the cavity. Due to the relatively longer geranylgeranyl moiety, the latter PDEδ has a deeper hydrophobic cavity resulting from the Phe133 aromatic ring flipped downward. In contrast, the former PDEδ embedding the farnesyl has a shallower cavity resulting from the Phe133 aromatic ring flipped upward. Depending on the orientations of the Phe133 side chain, we designated PDEδ into two different states; state 1 is for the downward side chain, and state 2 is for the upward side chain (Figure 1B). In the initial model construction, with farnesylated K-Ras4B-GTP, we took these two PDEδ states. For state 1, the originally embedded geranylgeranyl group was replaced with the farnesyl group. As a result, a couple of HVR backbone residues slipped into the cavity. For geranylgeranylated K-Ras4B-GTP, only state 1 of PDEδ was modeled. The combinations of two K-Ras4B models, KM1 and KM2, and PDEδ in states 1 and 2, obtained six model systems; four with farnesylated and two with geranylgeranylated K-Ras4B-GTP (Table 1). For comparison, we also simulated another six PDEδ systems containing the HVR peptide only, without the catalytic domain. The HVR peptides of three Ras isoforms, K-Ras4A, K-Ras4B, and N-Ras, were modeled with PDEδ in states 1 and 2. Two modeled systems for each Ras isoform’s HVR with PDEδ in states 1 and 2 are designated: system A1 and system A2 for K-Ras4A, system B1 and system B2 for K-Ras4B, and system N1 and system N2 for N-Ras (Table 2). In the HVR/PDEδ complex, the HVR peptides of both K-Ras4A and N-Ras were modified with depalmitoylation, but with a farnesyl modification as the HVR peptide of K-Ras4B.
Figure 1.

Crystal structures used to model the K-Ras4B-GTP/PDEδ interaction. (A) Two K-Ras4B-GTP models (KM1 and KM2) constructed for modeling the K-Ras4B-GTP/PDEδ complex. Two crystal structures of the K-Ras4B catalytic domain (PDB ID: 3GFT and 4DSO) were used. The GTP analogues in the crystal structures were converted to GTP (red). The missing HVR (dark blue) coordinates were modeled as a flexible loop. The prenyl (yellow) and methyl groups were added to Cys185, at the C-terminus. (B) PDEδ models used for modeling the K-Ras4B-GTP/PDEδ complex. To model the interactions of PDEδ with farnesyl and geranylgeranyl moieties, two crystal structures were used (PDB ID: 5F2U and 5E8F). The former PDEδ crystal structure contained the farnesyl group in the hydrophobic cavity, and the latter involved the geranylgeranyl group in the cavity. Depending on the orientations of the Phe133 side chain resulting from the deep/shallow insertion of the prenyl groups, we designated PDEδ into two different states; state 1 is for the downward side chain, and state 2 is for the upward side chain.
Table 1.
Initial Configurations of the Prenylated K-Ras4B-GTP/PDEδ Systems
| K-Ras4B-GTP |
PDEδ |
|||||||
|---|---|---|---|---|---|---|---|---|
| simulation system | PDB | HVR | prenylation | model | PDB | Phe133 side chain | state | prenyl insertion |
| system 1 | 3GFT | flexible loop | farnesyl | KM1 | 5E8F | down | 1 | deep |
| system 2 | 4DSO | partial α-helix | farnesyl | KM2 | 5E8F | down | 1 | deep |
| system 3 | 3GFT | flexible loop | farnesyl | KM1 | 5F2U | up | 2 | shallow |
| system 4 | 4DSO | partial α-helix | farnesyl | KM2 | 5F2U | up | 2 | shallow |
| system 5 | 3GFT | flexible loop | geranylgeranyl | KM1 | 5E8F | down | 1 | deep |
| system 6 | 4DSO | partial α-helix | geranylgeranyl | KM2 | 5E8F | down | 1 | deep |
Table 2.
Initial Configurations of the Farnesylated HVR/PDEδ Systems
| HVR |
PDEδ |
|||||
|---|---|---|---|---|---|---|
| simulation system | Ras | prenylation | PDB | Phe133 side chain | state | prenyl insertion |
| system A1 | K-Ras4A | farnesyl | 5E8F | down | 1 | deep |
| system A2 | K-Ras4A | farnesyl | 5F2U | up | 2 | shallow |
| system B1 | K-Ras4B | farnesyl | 5E8F | down | 1 | deep |
| system B2 | K-Ras4B | farnesyl | 5F2U | up | 2 | shallow |
| system N1 | N-Ras | farnesyl | 5E8F | down | 1 | deep |
| system N2 | N-Ras | farnesyl | 5F2U | up | 2 | shallow |
Atomistic MD Simulations.
The crystal structures of PDEδ provided a short peptide with 4−5 residues along with the prenyl group at the gate of the hydrophobic pocket. The sequence of the prenylated peptide was converted to that of the anchor portion of the HVR at the C-terminal region of K-Ras4B. In the initial construction toward the starting point, the backbone of the peptide and the prenyl group were kept intact during the pre-equilibration cycles. The rest of the HVR backbone was modeled as a flexible loop and then covalently connected to both the catalytic domains using the crystal structures and the HVR anchor portions converted from the crystal peptides. To align the two proteins well with each other, the K-Ras4B-GTP/PDEδ systems were minimized with rigid body motions for the K-Ras4B catalytic domain and PDEδ to enhance the interaction between them. A number of rigid body minimization steps were performed for the systems with the modeled HVR portion the only held unrestraint. The CHARMM40 program was used to construct six initial configurations of the K-Ras4B-GTP/PDEδ system (Figure S1). A total of 13 MD simulations including three K-Ras4BHVR/PDEδ, two K-Ras4A-HVR/PDEδ, and two N-Ras-HVR/PDEδ were performed in a water environment. The initial K-Ras4B-GTP/PDEδ and HVR/PDEδ configurations were solvated by the modified TIP3P water model46 and gradually relaxed with the proteins held rigid. For both the farnesylated and geranylgeranylated K-Ras4B-GTP/PDEδ systems, the unit cell box of 100 Å × 100 Å × 100 Å contains around 102 000 atoms with the ions 31 Na+, 1 Mg2+, and 30 Cl−, to satisfy a total cation concentration near 100 mM. For the farnesylated HVR/PDEδ systems, the unit cell box of 80 Å × 80 Å × 80 Å contains around 51 900 atoms with ions, 20 Na+ and 20 Cl−, for systems with N-Ras, 20 Na+ and 25 Cl−, for systems with K-Ras4A, and 20 Na+ and 28 Cl− for systems with K-Ras4B. In the dynamics, long-range electrostatic interactions were calculated by using the particle mesh Ewald method. The Langevin temperature control was used to maintain the constant temperature at 310 K, and the Nosé−Hoover−Langevin piston pressure control was used to maintain the pressure at 1 atm. A total of 1.8 μs simulations was performed for the six K-Ras4B-GTP/PDEδ systems; each has 300 ns simulation with the constant temperature at 310 K. The HVR/PDEδ systems were simulated for 200 ns. All simulations were performed in the NPT (constant number of atoms, pressure, and temperature) ensemble. The nanoscale molecular dynamics (NAMD)47 parallel computing code was employed in the production runs on a Biowulf cluster at the National Institute of Health.
Molecular Mechanics-Generalized Born Surface Area (MM/GBSA) Calculations.
To compare the interaction strengths of the systems, binding energy calculations have been performed. To this end, the sum of gas-phase energies (including Coulombic energy, van der Waals energy, as determined by a Lennard−Jones potential and internal energy), and solvation free energies (including a nonpolar part and an electrostatic part obtained from GB calculations) were calculated using the molecular mechanics energies combined with GB and surface area continuum solvation (MM/GBSA) method. The gas-phase and solvation free energy values were averaged over 3000 snapshots taken at 100 ps intervals from the 300 ns of MD simulations of K-Ras4B-GTP/PDEδ and 2000 snapshots taken at 100 ps intervals from the 200 ns of MD simulations of HVR/PDEδ systems. The calculations were performed with Amber v14 software using a modified GB model developed by Onufriev et al.48 To this end, the NAMD PSF and coordinate files were converted into Amber format using the chamber program in Amber.49 The change in binding energy due to the dimerization is calculated by using the equation
| (1) |
The binding energy was calculated as a sum of the gas-phase contribution and the solvation energy contribution 〈ΔGgas〉 = 〈ΔGsol〉 + 〈ΔGb〉, where 〈〉 denotes an average along the MD trajectory.
RESULTS
Interaction of the Catalytic Domain of K-Ras4B-GTP with PDEδ.
We carried out standard Rosetta50,51 global docking to build putative models for the K-Ras4B catalytic domain interacting with PDEδ. We selected the top four models from the prediction for further evaluation (Figure S2). Because the interaction of K-Ras4B with PDEδ is mediated mainly through the contacts between the HVR/farnesyl group and the prenyl-binding pocket, we investigated whether the orientation of the catalytic domain with respect to PDEδ in the selected models allows the penetration of the farnesyl group into the prenyl-binding pocket. However, none of the models are compatible with this scenario because the predicted models are almost identical. The models show that the K-Ras4B catalytic domain locates at the opposite side of the gate of the prenyl-binding pocket. The distance between the C-terminus of the K-Ras4B catalytic domain and the PDEδ prenyl-binding pocket is larger than the length of the HVR. The catalytic domain and the PDEδ are away from each other in the initial structures, suggesting that the interaction between these two proteins occurs only through the prenylated C-terminus.18
Interaction of Prenylated K-Ras4B-GTP with PDEδ.
We carried out 300 ns MD simulations on the six systems described in the Methods section to analyze the interactions between the prenylated K-Ras4B-GTP and the PDEδ hydrophobic pocket in detail. During the simulations, no immediate dissociation of the prenyl groups of K-Ras4B-GTP from the hydrophobic pocket of PDEδ was observed (Figure S3). The K-Ras4B-GTP/PDEδ complex was highly preserved, although there were large fluctuations between these two proteins connected by the HVR (Figure 2). The fluctuations were prominent when we superimposed the systems with respect to the K-Ras4B catalytic domain (Figure S4). To monitor and compare the stability of the systems, the root-mean-squared deviations (RMSDs) with respect to the initial structures of the six K-Ras4B/PDEδ systems were calculated (Figure S5 and Table S1). As expected, K-Ras4B has a larger RMSD than PDEδ due to the HVR. The results also suggest that the averaged RMSDs of the complexes of systems 4 and 6 are smaller than those for the other four systems. We also computed the center of mass distance between K-Ras4B and PDEδ (Figure S6). The results indicate that unlike the other systems, the distance between the two proteins remains larger in system 1 and lower in systems 4 and 6, which explains why system 1 has the least and systems 4 and 6 have the best binding energies. The dynamics of both the catalytic domain and PDEδ show a rigid body motion with respect to each other, which is supported by the flexible HVR. The fluctuations of the prenyl groups cause a conformational rearrangement of the β-sandwiched pocket in PDEδ. For systems 1, 2, 5, and 6, the conformational transition of PDEδ from state 1 to 2 can be observed. PDEδ shifts the equilibrium toward the Phe133 aromatic ring flipping upward, suggesting that both farnesyl and geranylgeranyl favor the upward state of the aromatic ring. However, System 3 shows the transition from state 2 to 1 during the simulations, indicating that interconversion between the states of the Phe133 side chain is possible, depending on the dynamics of the prenyl groups. We noted that the pseudo-helical conformation of the HVR in KM2 gradually unfolds, suggesting that the HVR is intrinsically disordered.
Figure 2.

Superimposed snapshots of the final conformation (colored cartoon) and the starting point (white transparent cartoon) of these six K-Ras4B-GTP/PDEδ systems (systems 1−6). All coordinates were reoriented with respect to the PDEδ coordinates.
The major interaction of K-Ras4B with PDEδ derives from the HVR. In contrast, the catalytic domain interaction is transient, marginally contacting the PDEδ in some system configurations. To corroborate the K-Ras4B interaction with PDEδ, we analyzed key residues involved in dimer formation (Figure 3 and Table S2). The results indicate that the anchor HVR residues, Lys182, Thr183, and Lys184, play a significant role in PDEδ binding. They form hydrogen bonds (H-bonds) and salt bridges with Glu88 from PDEδ. In addition, the linker HVR residues, Lys169, Lys172, and Lys177, also participate in K-Ras4B/PDEδ association. Lys169 forms a salt bridge with Glu93 from PDEδ. Lys172 forms multiple H-bonds and salt bridges with residues Asp136 and Asp137. Lys177 forms a H-bond and a salt bridge with Glu89. In particular, Ser181 makes stronger associations with Glu88 in geranylgeranylated systems via main and side-chain oxygen atoms. Comparison of the interacting residues between the farnesylated and geranylgeranylated systems also suggests that the catalytic domain of geranylgeranylated K-Ras4B forms some interactions with PDEδ. The catalytic domain residues, Arg73 and Asp69, on the α2 helix form H-bonds with Glu77 and Lys79 from PDEδ, respectively. However, no catalytic domain interaction with PDEδ was observed in the farnesylated systems, suggesting that the interactions between the catalytic domain of farnesylated K-Ras4B and PDEδ are highly transient.
Figure 3.

Key residues involved in K-Ras4B-GTP/PDEδ complex formation. (A) The prominent H-bonds and (B) salt bridges formed between K-Ras4B and PDEδ during the simulations. The residues involved in hydrogen bonding are represented as ball-and-stick with parental colors.
At the starting point of farnesylated systems with PDEδ in state 1, the farnesyl group inserted deeply into the pocket. Subsequently, a couple of the HVR backbone residues initially slipped into the hydrophobic cavity. However during the simulations, the HVR residues bounced from the pocket due to unfavorable interactions taking place at the gate of the pocket. As a result, PDEδ allowed the farnesyl to reside in the shallow region of the cavity. In the pocket, the farnesyl favors hydrophobic interactions with Trp32, Ile53, Val59, Leu87, and Val145 from PDEδ (Figure 4A). At the starting point of the farnesylated systems with PDEδ in state 2, the farnesyl group inserted shallowly into the pocket. The shallow farnesyl insertion was favorable for PDEδ, which provides many hydrophobic residues, Met20, Leu22, Trp32, Leu38, Ile53, Trp90, Ile109, Phe133, and Leu147, to interact with the farnesyl moiety (Figure 4B). Evidently, the farnesyl group makes more contacts with the PDEδ hydrophobic pocket. The geranylgeranylated systems have only PDEδ in state 1 due to the longer hydrocarbon chain. In the pocket, geranylgeranyl favors the hydrophobic interactions with Trp32, Leu38, Val59, Ala111, Phe133, Val145, and Leu147 from PDEδ (Figure 4C).
Figure 4.

Hydrophobic interactions involved in K-Ras4B-GTP/PDEδ complex formation. PDEδ residues that participate in hydrophobic interactions with the K-Ras4B prenyl group for (A) system 2, (B) system 4, and (C) system 5.
Comparison of the crystal with the simulated states indicates that the long helix observed in state 2 in the crystal structure becomes partially disordered; in state 1 about a third of the helix becomes disordered as well. Inspection of the crystal lattice indicates that the disordered part of the helix in form 1 (state 1) is stabilized by crystal packing in form 2 (state 2).
Binding Energy in Dimer Formation.
To quantify the six K-Ras4B-GTP/PDEδ complex systems, we calculated the binding energy, ΔGb, using the MM/GBSA method (Table 3). With an entropic contribution, the binding energy is directly related to the binding free energy of dimer formation. For the farnesylated systems, shallowly inserted configurations yield stronger binding for the K-Ras4B-GTP/PDEδ complex, which suggests that the farnesyl interaction in the pocket is significant for high-affinity binding. For the geranylgeranylated systems, the binding energies are comparable to those of the farnesylated systems. Overall, the binding energy results suggest that PDEδ can transport the K-Ras4B protein, whether it is modified with farnesyl or geranylgeranyl. In general, prenylated K-Ras4B interacts more strongly with the PDEδ hydrophobic pocket when the HVR is folded because under these circumstances additional catalytic domain interactions with PDEδ partially contribute to the binding energy as well. We expect that the simulation times were not long enough to achieve comparable energies for the two minima (systems 1 and 2 vs 3 and 4).
Table 3.
Binding Energy in Dimer Formation Using the MM/GBSA Method for the Prenylated K-Ras4B-GTP/PDEδ Systems
| K-Ras4B/PDEδ | 〈ΔGgas〉 (kcal/mol) | 〈ΔGsol〉 (kcal/mol) | 〈ΔGb〉 (kcal/mol) |
|---|---|---|---|
| system 1 | −411.3 ± 162.0 | 368.8 ± 158.6 | −42.5 ± 8.5 |
| system 2 | −694.5 ± 235.4 | 635.4 ± 217.0 | −59.2 ± 23.6 |
| system 3 | −769.5 ± 266.0 | 705.2 ± 247.8 | −64.2 ± 21.2 |
| system 4 | −820.6 ± 96.4 | 736.7 ± 86.0 | −83.9 ± 13.3 |
| system 5 | −621.4 ± 74.9 | 548.5 ± 68.3 | −71.9 ± 11.0 |
| system 6 | −570.9 ± 112.4 | 487.9 ± 105.8 | −83.0 ± 17.2 |
Interactions of Farnesylated HVRs of K-Ras4A, K-Ras4B, and N-Ras with PDEδ.
We carried out 200 ns MD simulations on the HVR/PDEδ systems described in the Methods section to investigate the isoform-specific interactions between the farnesylated HVR peptides and the PDEδ hydrophobic pocket. During the simulations, even without the catalytic domain, no immediate dissociation of the HVR peptide from PDEδ was observed (Figure 5). The HVR/PDEδ complex was highly preserved for all farnesylated Ras isoforms, supporting the observation that the catalytic domain interaction with PDEδ is transient. Similar to the K-Ras4B-GTP/PDEδ systems, for all HVR/PDEδ systems including K-Ras4A and N-Ras, the conformational transition of PDEδ from state 1 to 2 with the Phe133 aromatic ring flipping upward can also be observed, verifying that the farnesyl favors the upward state of the aromatic ring of PDEδ.
Figure 5.

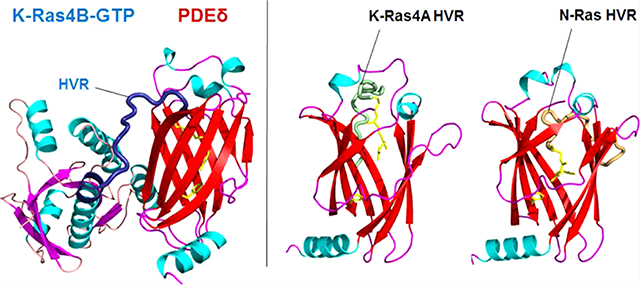
Snapshots of the farnesylated HVR/PDEδ complex for K-Ras4A (systems A1 and A2), K-Ras4B (systems B1 and B2), and N-Ras (systems N1 and N2) at the start, 100 ns, and 200 ns points. In the cartoon, green, blue, and orange tubes represent the K-Ras4A, K Ras4B, and N-Ras HVRs, respectively.
To identify the key binding residues in the HVR/PDEδ complex, we performed a detailed interface analysis. Because the K-Ras4B HVR interactions with PDEδ are similar to those defined for the K-Ras4B-GTP/PDEδ complexes, we closely monitored the atomic pair interactions of the HVRs of both K-Ras4A and N-Ras with PDEδ (Figure 6 and Table S3). We observed that the residues at the anchor region of K-Ras4A HVR, Lys182, Lys184, and Lys185 are important for PDEδ binding. They form H-bonds and salt bridges with Glu88 from PDEδ via main and side-chain oxygen atoms. For the N-Ras HVR/PDEδ complex, we observed that the anchor residues of N-Ras HVR, Cys181, Met182, and Gly183 interact with Glu88, and Glu110 from PDEδ via H-bonds and salt bridges. For both K-Ras4A and N-Ras, the linker residue Arg167 at the HVR commonly participates in the HVR/PDEδ association, suggesting that the linker interaction occurs due to the peptide effect, in the absence of the catalytic domain. Compared with K-Ras4A and K-Ras4B, the N-Ras HVRs do not form a salt bridge with PDEδ because the anchor region of the N-Ras HVR is electrostatically neutral. Overall, the residue Glu88 on the β6 of PDEδ constitutes the most important residue in the interactions of both farnesylated K-Ras4A and N-Ras HVRs with PDEδ.
Figure 6.

Key residues involved in HVR/PDEδ complex formation. (A) The prominent H-bonds and (B) salt bridges formed between the farnesylated HVR and PDEδ during the simulations. The residues involved in hydrogen bonding are represented as ball-and-stick with parental colors.
A detailed analysis of the resulting structures reveals that the stabilizing interactions of the farnesyl with the hydrophobic residues in the pocket of PDEδ support the conformation of the HVR/PDEδ complex. In system A1, the farnesyl favors the hydrophobic interactions with Met20, Trp32, Leu38, Val49, Val59, Ile109, and Ile129 from PDEδ (Figure 7A). In system A2, the farnesyl favors the hydrophobic interactions with Trp32, Ala47, and Met117 from PDEδ (Figure 7B). For the K-Ras4A HVR/PDEδ complex, the deep farnesyl insertion results in increased contacts between the farnesyl and PDEδ. In contrast, for the N-Ras HVR/PDEδ complex, there are more hydrophobic contacts of the farnesyl with PDEδ, when the farnesyl is inserted shallowly into the pocket. This is most likely to compensate for the lack of H-bonds and salt bridges. In system N1, the farnesyl favors the hydrophobic interactions with Trp32 and Trp90 from PDEδ (Figure 7C). In system N2, the farnesyl favors the hydrophobic interactions with Met20, Trp32, Leu63, Ile109, Ala111, Val145, and Leu147 from PDEδ (Figure 7D). Although the catalytic domains were not included in these simulations, we expect that their fluctuations would weaken the associations, both in K-Ras4A and N-Ras.
Figure 7.

Hydrophobic interactions involved in HVR/PDEδ complex formation. PDEδ residues that participate in hydrophobic interactions with the farnesylated HVR for (A) system A1, (B) system A2, (C) system N1, and (D) system N2.
DISCUSSION
Over the years, numerous attempts have been made to pharmacologically inhibit oncogenic Ras. Despite intensive efforts, Ras is still widely considered as “undruggable”. The prenyl-binding protein PDEδ has been shown to augment (oncogenic) Ras signaling by enriching Ras at the PM.11,37,38 Ras localization at the PM profoundly influences its activation and signaling capacity. This localization increases its effective local concentration through dimerization52–54 and nanocluster formation,55–57 as well as acts in the signaling selectivity of Ras isoforms.43–45,58–62 Importantly, it also locates Ras in the vicinity of receptors that receive the signaling cues, such as epidermal growth factor receptor, its nucleotide exchange factors, and membrane-recruited kinase effectors,63 such as Raf and PI3K, and nonkinase effectors, such as RASSF5, which links Ras with the MST1/2 kinase and the Hippo pathway.64,65 Therefore, interfering with Ras localization by inhibiting its interaction with PDEδ can be an alternative approach to target oncogenic Ras.66 Here, we simulated six full-length prenylated K-Ras4B-GTP/PDEδ complex systems and six prenylated HVR/PDEδ systems. Structural analysis of snapshots of the interfaces of K-Ras4B-GTP/PDEδ reveals a remarkable similarity to the recently solved crystal structures as well as their behavior in solution.18 Our results indicate that the HVR/prenyl moiety of K-Ras4B is responsible for the interaction with PDEδ and the “SKTK” motif plays a significant role in PDEδ binding. They further suggest that glutamate residues at positions 88, 89, 110, 136, and 137 are the key binding residues of PDEδ. The conserved aspartates, 136 and 137, are also involved in the interaction of RhoGDI and Cdc42. They help stabilizing the interaction of the N-terminal helix−loop−helix region of RhoGDI and the switch II domain of Cdc42.13 Thus, the electrostatically negative surface of PDEδ plays an important role in the interaction with the positively charged K-Ras4B HVR. They also point to two dynamic states.
Comparison of the interfaces of farnesylated and geranylgeranylated systems reveals that a few residues on the catalytic domain of geranylgeranylated but not farnesylated K-Ras4B interact with PDEδ. Our results are in accordance with the previous findings showing that the interaction between the catalytic domain of farnesylated K-Ras4B and PDEδ is very weak and transient.18,38 The absence of any specific interaction between the catalytic domain and PDEδ makes this association nucleotide independent. MM/GBSA calculations of the full-length prenylated K-Ras-4B-GTP/PDEδ systems indicate that the interaction between farnesylated K-Ras4B and PDEδ is stronger when the farnesyl inserts properly into the pocket. To understand the contribution of the catalytic domain, if any, to the binding, we also studied the prenylated K-Ras4B HVR/ PDEδ systems. The results corroborate other studies, pointing out that the HVR and the prenyl moiety are the main players of PDEδ binding.18,38 Overall, the simulations indicate that factors such as the prenylation status and the HVR conformation affect the K-Ras4B/PDEδ interaction, and that K-Ras4B has the highest affinity for PDEδ when it is farnesylated.
Experimental data verify that PDEδ binds to K-Ras4B and to N-Ras;37 but not to K-Ras4A, a splice variant of K-Ras4B.36 K-Ras4B is only farnesylated; K-Ras4A and N-Ras are farnesylated and palmitoylated, however, palmitoylation is reversible. Sequence-wise, the HVR of K-Ras4A is very similar to that of K-Ras4B, with both being highly positively charged; on the other hand, the anchor region of the N-Ras HVR is electrostatically neutral. With respect to the catalytic domain, the K-Ras isoforms are highly similar: K-Ras4A differs from K-Ras4B by only four residues. This raises the significant question of how to resolve these apparently puzzling experimental observations.
Oncogenic K-Ras4A was observed to be involved in K-Ras4B colorectal cancer, which is a K-Ras4B cancer, in addition to involvement in N-Ras cancer, such as melanoma, which led us to propose that K-Ras4A can exist in two states, depalmitoylated K-Ras4B-like, which we called state 1, and palmitoylated N-Ras-like, which we called state 2.29,39 Thus, to address the question of why K-Ras4B and in particular also N-Ras were observed to bind to PDEδ but apparently not K-Ras4A, we simulated PDEδ with the HVRs of K-Ras4A and N-Ras isoforms. We observed that even though steric clashes appear to exist with the two K-Ras4A lysines located upstream of the farnesylated Cys, conformational dynamics can accommodate these, permitting the interaction of a depalmityolated K-Ras4A molecule with PDEδ. This was not surprising, as along similar lines, calmodulin, which has been suggested to be K-Ras4B-specific, appears to also interact with depalmitoylated K-Ras4A.67,68 Moreover, the proposition that depalmitoylated K-Ras4A can interact with PDEδ is supported by experimental N-Ras observations. Coexpression of N-Ras and PDEδ showed that a marked fraction of N-Ras in the cytoplasm interacts with the PDEδ. Comparison of the level of palmitoylation indicated that at least half the N-Ras molecules were depalmitoylated at the steady state. We observe that the interaction of K-Ras4A with PDEδ is comparable to that of N-Ras. Further in line with this, H-Ras (with a farnesyl and two palmitoyls) experiments pointed to less than a quarter of the molecules at the steady state.37 This led the authors to conclude that a substantial fraction of N-Ras is already depalmitoylated when it reaches the PM through the secretory pathway and the relative palmitoylation level (H-Ras > N-Ras) determines the PDEδ binding tendencies of Ras isoforms.
To explain why PDEδ can extract from the PM, K-Ras4B and N-Ras but not K-Ras4A,36 it was proposed that Ras family proteins that bind to PDEδ avoid large/charged residues adjacent to the prenylated site. However, our conformational dynamics indicate that the disordered fluctuating K-Ras4A HVR can accommodate the two adjacent lysine residues and bind PDEδ.
Here, we argue that PDEδ can bind to all farnesylated but nonpalmitoylated Ras isoforms, with affinities that may reflect the electrostatic properties of the HVR and the adjacent residues. A key factor in understanding why K-Ras4B and N-Ras are extracted from the PM by PDEδ but not K-Ras4A is the relative stabilities of the interactions with the PM versus in solution with PDEδ. The stabilities of the interactions with PDEδ of the electrostatically neutral N-Ras and the electrostatically positively charged K-Ras4A, but with an apparent steric hindrance that needs to be accommodated, are similar; however, that is not the case with respect to the PM. There, K-Ras4A and H-Ras are more stably lodged. This emphasizes the need to consider both environments when seeking to understand physical observations. The Tsai et al. experiment36 considered extraction from the membrane; not direct PDEδ binding in solution. The consideration of steric hindrance based on the crystal and our modeling and simulations are in solution, and do not consider the competing PM interactions. Thus, PDEδ can shuttle isoforms that have farnesyl (but not palmitoyl), positively charged HVR, and preferentially lack bulky residues adjacent to the prenylated sites, which could weaken the interaction, even though these can be accommodated.
The implications of our observations are significant. In a recent remarkable paper, Schmick et al.11 posited that PDEδ acts to counteract an otherwise entropy-driven localization of Ras isoforms in endomembranes. The large total surface area that they present would result in randomized Ras distributions. PDEδ solubilizes the Ras molecules and shuttles Ras to the PM, opposing the inward cytoplasmic flow driven by endocytosis. Schmick et al. argued that cytosolic PDEδs constantly sequester mislocalized K-Ras from endomembranes, shuttling it to the recycling endosome, where it is trapped, and subsequently vesicle-transported to restore its enrichment on the PM. Here we show that in principle, all Ras isoforms can be shuttled. This emphasizes the potential of small molecules interfering with this action. The conformational details of the two states provided here may help in this venture.
CONCLUSIONS
Our results further emphasize the importance of considering and comparing the interactions in the respective environments. We already showed that apparently conflicting experimental results can be reconciled.64 There too, considering that some experiments were performed in vivo, whereas others in vitro, resolves the question of whether RASSF5 (NORE1A), a Ras effector that links Ras (and the MAPK pathway) with the Hippo pathway, is a tumor suppressor (observed in vivo) or an activator (in vitro).64,69 In the cell, RASSF5 is a tumor suppressor. In vitro, in the absence of the membrane, it is a tumor activator. There, the PM acts in the localization of Ras and RASSF5.
Finally, the crystal structures and the K-Ras4B simulations underscore PDEδ evolution to pre-exist in two major states, which can accept farnesyl and geranylgeranyl. These two minima are critical in permitting enrichment of Ras family GTPases at the PM, which is required for Ras to fulfill its function. Within this framework, it would be of interest to explore the structure and interactions of SmgGDS, a chaperone that interacts with Ras family proteins entering the prenylation pathway by recognizing the last amino acid in the CAAX motif (-CAAL more than -CAAM (in wild type K-Ras) via the ARM A and B domains, as inferred by computational modeling).
This also highlights the challenges facing Ras pharmacology. Farnesyltransferase inhibitors increase the binding of K-Ras to SmgGDS-607, suggesting that pharmacological intervention in the geranylgeranylation of K-Ras also promotes the interaction of K-Ras with SmgGDS-607.70
This work emphasizes the notion that fundamental concepts from physical chemistry and chemical physics dominate the structure−function paradigm.71 Here, they elucidate K-Ras4A behavior and explain why PDEδ binding to farnesylated Ras family proteins can provide a route to proper membrane localization.11
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by TUBITAK Research Grant No: 114M196. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mentioning of trade names, commercial products, or organizations imply endorsement by the U.S. Government. All simulations were performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.7b03035.
Supporting figures include the stating points of the K-Ras4B-GTP/PDEδ systems, the predicted models of the K-Ras4B catalytic domain interacting with PDEδ, the snapshots during the simulations, the RMSD plots, and the center of mass distance in the six K-Ras4B/PDEδ systems (Figures S1–S6); supporting tables include the average RMSD values and the types of atomic interactions in the prenylated K-Ras4B-GTP/PDEδ and HVR/PDEδ complexes (Tables S1–S3) (PDF)
REFERENCES
- (1).Hall A G Proteins and Small GTPases: Distant Relatives Keep in Touch. Science 1998, 280, 2074–2075. [DOI] [PubMed] [Google Scholar]
- (2).Vojtek AB; Der CJ Increasing Complexity of the Ras Signaling Pathway. J. Biol. Chem 1998, 273, 19925–19928. [DOI] [PubMed] [Google Scholar]
- (3).Chavrier P; Goud B The Role of ARF and Rab GTPases in Membrane Transport. Curr. Opin. Cell Biol 1999, 11, 466–475. [DOI] [PubMed] [Google Scholar]
- (4).Willumsen BM; Norris K; Papageorge AG; Hubbert NL; Lowy DR Harvey Murine Sarcoma Virus p21 Ras Protein: Biological and Biochemical Significance of the Cysteine Nearest the Carboxy Terminus. EMBO J. 1984, 3, 2581–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Trahey M; McCormick F A Cytoplasmic Protein Stimulates Normal N-Ras p21 GTPase, but Does Not Affect Oncogenic Mutants. Science 1987, 238, 542–545. [DOI] [PubMed] [Google Scholar]
- (6).Seabra MC Membrane Association and Targeting of Prenylated Ras-Like GTPases. Cell. Signalling 1998, 10, 167–172. [DOI] [PubMed] [Google Scholar]
- (7).Lowy DR; Willumsen BM Function and Regulation of Ras. Annu. Rev. Biochem 1993, 62, 851–891. [DOI] [PubMed] [Google Scholar]
- (8).Sprang SR How Ras Works: Structure of a Rap-Raf Complex. Structure 1995, 3, 641–643. [DOI] [PubMed] [Google Scholar]
- (9).Vetter IR; Wittinghofer A The Guanine Nucleotide-Binding Switch in Three Dimensions. Science 2001, 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- (10).Ray A; Jatana N; Thukral L Lipidated Proteins: Spotlight on Protein-Membrane Binding Interfaces Prog. Biophys. Mol. Biol 2017. 10.1016/j.pbiomolbio.2017.01.002. [DOI] [PubMed] [Google Scholar]
- (11).Schmick M; Vartak N; Papke B; Kovacevic M; Truxius DC; Rossmannek L; Bastiaens PI KRas Localizes to the Plasma Membrane by Spatial Cycles of Solubilization, Trapping and Vesicular Transport. Cell 2014, 157, 459–471. [DOI] [PubMed] [Google Scholar]
- (12).Wu SK; Zeng K; Wilson IA; Balch WE Structural Insights into the Function of the Rab GDI Superfamily. Trends Biochem. Sci 1996, 21, 472–476. [DOI] [PubMed] [Google Scholar]
- (13).Hoffman GR; Nassar N; Cerione RA Structure of the Rho Family GTP-Binding Protein Cdc42 in Complex with the Multifunctional Regulator RhoGDI. Cell 2000, 100, 345–356. [DOI] [PubMed] [Google Scholar]
- (14).Zhang H; Liu XH; Zhang K; Chen CK; Frederick JM; Prestwich GD; Baehr W Photoreceptor cGMP Phosphodiesterase Delta Subunit (PDEδ) Functions as a Prenyl-Binding Protein. J. Biol. Chem 2004, 279, 407–413. [DOI] [PubMed] [Google Scholar]
- (15).Hanzal-Bayer M; Renault L; Roversi P; Wittinghofer A; Hillig RC The Complex of Arl2-GTP and PDE Delta: From Structure to Function. EMBO J. 2002, 21, 2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nancy V; Callebaut I; El Marjou A; de Gunzburg J The Delta Subunit of Retinal Rod cGMP Phosphodiesterase Regulates the Membrane Association of Ras and Rap GTPases. J. Biol. Chem 2002, 277, 15076–15084. [DOI] [PubMed] [Google Scholar]
- (17).Chen YX; Koch S; Uhlenbrock K; Weise K; Das D; Gremer L; Brunsveld L; Wittinghofer A; Winter R; Triola G; et al. Synthesis of the Rheb and K-Ras4B GTPases. Angew. Chem., Int. Ed 2010, 49, 6090–6095. [DOI] [PubMed] [Google Scholar]
- (18).Dharmaiah S; Bindu L; Tran TH; Gillette WK; Frank PH; Ghirlando R; Nissley DV; Esposito D; McCormick F; Stephen AG; et al. Structural Basis of Recognition of Farnesylated and Methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. U.S.A 2016, 113, E6766–E6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Linari M; Hanzal-Bayer M; Becker J The Delta Subunit of Rod Specific Cyclic GMP Phosphodiesterase, PDE Delta, Interacts with the Arf-like Protein Arl3 in a GTP Specific Manner. FEBS Lett. 1999, 458, 55–59. [DOI] [PubMed] [Google Scholar]
- (20).Veltel S; Kravchenko A; Ismail S; Wittinghofer A Specificity of Arl2/Arl3 Signaling Is Mediated by a Ternary Arl3-Effector-GAP Complex. FEBS Lett. 2008, 582, 2501–2507. [DOI] [PubMed] [Google Scholar]
- (21).Gosser YQ; Nomanbhoy TK; Aghazadeh B; Manor D; Combs C; Cerione RA; Rosen MK C-Terminal Binding Domain of Rho GDP-dissociation Inhibitor Directs N-terminal Inhibitory Peptide to GTPases. Nature 1997, 387, 814–819. [DOI] [PubMed] [Google Scholar]
- (22).Scheffzek K; Stephan I; Jensen ON; Illenberger D; Gierschik P The Rac-RhoGDI Complex and the Structural Basis for the Regulation of Rho Proteins by RhoGDI. Nat. Struct. Biol 2000, 7, 122–126. [DOI] [PubMed] [Google Scholar]
- (23).Ismail SA; Chen YX; Rusinova A; Chandra A; Bierbaum M; Gremer L; Triola G; Waldmann H; Bastiaens PI; Wittinghofer A Arl2-GTP and Arl3-GTP Regulate a GDI-like Transport System for Farnesylated Cargo. Nat. Chem. Biol 2011, 7, 942–949. [DOI] [PubMed] [Google Scholar]
- (24).Lu S; Banerjee A; Jang H; Zhang J; Gaponenko V; Nussinov R GTP Binding and Oncogenic Mutations May Attenuate Hypervariable Region (HVR)-Catalytic Domain Interactions in Small GTPase K-Ras4B, Exposing the Effector Binding Site. J. Biol. Chem 2015, 290, 28887–28900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lu S; Jang H; Nussinov R; Zhang J The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep 2016, 6, No. 21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lu S; Jang H; Gu S; Zhang J; Nussinov R Drugging Ras GTPase: A Comprehensive Mechanistic and Signaling Structural View. Chem. Soc. Rev 2016, 45, 4929–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nussinov R; Tsai CJ; Jang H; Korcsmaros T; Csermely P Oncogenic KRAS Signaling and YAP1/β-catenin: Similar Cell Cycle Control in Tumor Initiation. Semin. Cell Dev. Biol 2016, 58, 79–85. [DOI] [PubMed] [Google Scholar]
- (28).Lu S; Jang H; Muratcioglu S; Gursoy A; Keskin O; Nussinov R; Zhang J Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev 2016, 116, 6607–6665. [DOI] [PubMed] [Google Scholar]
- (29).Chakrabarti M; Jang H; Nussinov R Comparison of the Conformations of KRAS Isoforms, K-Ras4A and K-Ras4B, Points to Similarities and Significant Differences. J. Phys. Chem. B 2016, 120, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gysin S; Salt M; Young A; McCormick F Therapeutic Strategies for Targeting Ras Proteins. Genes Cancer 2011, 2, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cox AD; Der CJ; Philips MR Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin. Cancer Res 2015, 21, 1819–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ahearn IM; Haigis K; Bar-Sagi D; Philips MR Regulating the Regulator: Post-Translational Modification of RAS. Nat. Rev. Mol. Cell Biol 2011, 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhou M; Wiener H; Su W; Zhou Y; Liot C; Ahearn I; Hancock JF; Philips MR VPS35 Binds Farnesylated N-Ras in the Cytosol to Regulate N-Ras Trafficking. J. Cell Biol 2016, 214, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lynch SJ; Snitkin H; Gumper I; Philips MR; Sabatini D; Pellicer A The differential palmitoylation states of N-Ras and H-Ras determine their distinct Golgi subcompartment localizations. J. Cell. Physiol 2015, 230, 610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Apolloni A; Prior IA; Lindsay M; Parton RG; Hancock JF H-Ras but Not K-Ras Traffics to the Plasma Membrane Through the Exocytic Pathway. Mol. Cell. Biol 2000, 20, 2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tsai FD; Lopes MS; Zhou M; Court H; Ponce O; Fiordalisi JJ; Gierut JJ; Cox AD; Haigis KM; Philips MR K-Ras4A Splice Variant Is Widely Expressed in Cancer and Uses a Hybrid Membrane-Targeting Motif. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chandra A; Grecco HE; Pisupati V; Perera D; Cassidy L; Skoulidis F; Ismail SA; Hedberg C; Hanzal-Bayer M; Venkitaraman AR; et al. The GDI-Like Solubilizing Factor PDEδ Sustains the Spatial Organization and Signalling of Ras Family Proteins. Nat. Cell Biol 2011, 14, 148–158. [DOI] [PubMed] [Google Scholar]
- (38).Weise K; Kapoor S; Werkmuller A; Mobitz S; Zimmermann G; Triola G; Waldmann H; Winter R Dissociation of the K-Ras4B/PDEδ Complex upon Contact with Lipid Membranes: Membrane Delivery Instead of Extraction. J. Am. Chem. Soc 2012, 134, 11503–11510. [DOI] [PubMed] [Google Scholar]
- (39).Nussinov R; Tsai CJ; Chakrabarti M; Jang H A New View of Ras Isoforms in Cancers. Cancer Res. 2016, 76, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Brooks BR; Brooks CL 3rd; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; et al. CHARMM: the Biomolecular Simulation Program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chavan TS; Jang H; Khavrutskii L; Abraham SJ; Banerjee A; Freed BC; Johannessen L; Tarasov SG; Gaponenko V; Nussinov R; et al. High-Affinity Interaction of the K-Ras4B Hypervariable Region with the Ras Active Site. Biophys. J 2015, 109, 2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jang H; Abraham SJ; Chavan TS; Hitchinson B; Khavrutskii L; Tarasova NI; Nussinov R; Gaponenko V Mechanisms of Membrane Binding of Small GTPase K-Ras4B Farnesylated Hypervariable Region. J. Biol. Chem 2015, 290, 9465–9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jang H; Banerjee A; Chavan TS; Lu S; Zhang J; Gaponenko V; Nussinov R The Higher Level of Complexity of K-Ras4B Activation at the Membrane. FASEB J. 2016, 30, 1643–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jang H; Muratcioglu S; Gursoy A; Keskin O; Nussinov R Membrane-Associated Ras Dimers Are Isoform-Specific: K-Ras Dimers Differ from H-Ras Dimers. Biochem. J 2016, 473, 1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Durell SR; Brooks BR; Bennaim A Solvent-Induced Forces Between Two Hydrophilic Groups. J. Phys. Chem 1994, 98, 2198–2202. [Google Scholar]
- (47).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Onufriev A; Bashford D; Case DA Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins 2004, 55, 383–394. [DOI] [PubMed] [Google Scholar]
- (49).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of Multiple Amber Force Fields and Development of Improved Protein Backbone Parameters. Proteins 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kilambi KP; Pacella MS; Xu J; Labonte JW; Porter JR; Muthu P; Drew K; Kuroda D; Schueler-Furman O; Bonneau R; et al. Extending RosettaDock with Water, Sugar, and pH for Prediction of Complex Structures and Affinities for CAPRI Rounds 20–27. Proteins 2013, 81, 2201–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Adams PD; Baker D; Brunger AT; Das R; DiMaio F; Read RJ; Richardson DC; Richardson JS; Terwilliger TC Advances, Interactions, and Future Developments in the CNS, Phenix, and Rosetta Structural Biology Software Systems. Annu. Rev. Biophys 2013, 42, 265–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Muratcioglu S; Chavan TS; Freed BC; Jang H; Khavrutskii L; Freed RN; Dyba MA; Stefanisko K; Tarasov SG; Gursoy A; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Chen M; Peters A; Huang T; Nan X Ras Dimer Formation as a New Signaling Mechanism and Potential Cancer Therapeutic Target. Mini-Rev. Med. Chem 2016, 16, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nan X; Tamguney TM; Collisson EA; Lin LJ; Pitt C; Galeas J; Lewis S; Gray JW; McCormick F; Chu S Ras-GTP Dimers Activate the Mitogen-Activated Protein Kinase (MAPK) Pathway. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 7996–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhou Y; Hancock JF Ras Nanoclusters: Versatile Lipid-Based Signaling Platforms. Biochim. Biophys. Acta 2015, 1853, 841–849. [DOI] [PubMed] [Google Scholar]
- (56).Zhou Y; Liang H; Rodkey T; Ariotti N; Parton RG; Hancock JF Signal Integration by Lipid-Mediated Spatial Cross Talk Between Ras Nanoclusters. Mol. Cell. Biol 2014, 34, 862–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Nussinov R; Jang H; Tsai CJ Oligomerization and Nanocluster Organization Render Specificity. Biol. Rev. Cambridge Philos. Soc 2015, 90, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Janosi L; Li Z; Hancock JF; Gorfe AA Organization, Dynamics, and Segregation of Ras Nanoclusters in Membrane Domains. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 8097–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Prakash P; Zhou Y; Liang H; Hancock JF; Gorfe AA Oncogenic K-Ras Binds to an Anionic Membrane in Two Distinct Orientations: A Molecular Dynamics Analysis. Biophys. J 2016, 110, 1125–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Banerjee A; Jang H; Nussinov R; Gaponenko V The Disordered Hypervariable Region and the Folded Catalytic Domain of Oncogenic K-Ras4B Partner in Phospholipid Binding. Curr. Opin. Struct. Biol 2016, 36, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Li ZL; Buck M Computational Modeling Reveals that Signaling Lipids Modulate the Orientation of K-Ras4A at the Membrane Reflecting Protein Topology. Structure 2017, 25, No. e679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Erwin N; Patra S; Dwivedi M; Weise K; Winter R Influence of Isoform-Specific Ras Lipidation Motifs on Protein Partitioning and Dynamics in Model Membrane Systems of Various Complexity. Biol. Chem 2017, 398, 547–563. [DOI] [PubMed] [Google Scholar]
- (63).Nussinov R; Tsai CJ; Muratcioglu S; Jang H; Gursoy A; Keskin O Principles of K-Ras Effector Organization and the Role of Oncogenic K-Ras in Cancer Initiation Through G1 Cell Cycle Deregulation. Expert Rev. Proteomics 2015, 12, 669–682. [DOI] [PubMed] [Google Scholar]
- (64).Liao TJ; Tsai CJ; Jang H; Fushman D; Nussinov R RASSF5: An MST Activator and Tumor Suppressor in vivo but Opposite in vitro. Curr. Opin. Struct. Biol 2016, 41, 217–224. [DOI] [PubMed] [Google Scholar]
- (65).Donninger H; Schmidt ML; Mezzanotte J; Barnoud T; Clark GJ Ras Signaling Through RASSF Proteins. Semin. Cell Dev. Biol 2016, 58, 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Spiegel J; Cromm PM; Zimmermann G; Grossmann TN; Waldmann H Small-Molecule Modulation of Ras Signaling. Nat. Chem. Biol 2014, 10, 613–622. [DOI] [PubMed] [Google Scholar]
- (67).Nussinov R; Muratcioglu S; Tsai CJ; Jang H; Gursoy A; Keskin O The Key Role of Calmodulin in KRAS-Driven Adenocarcinomas. Mol. Cancer Res 2015, 13, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Nussinov R; Wang G; Tsai CJ; Jang H; Lu S; Banerjee A; Zhang J; Gaponenko V Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 2017, 3, 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Liao TJ; Jang H; Tsai CJ; Fushman D; Nussinov R The Dynamic Mechanism of RASSF5 and MST Kinase Activation by Ras. Phys. Chem. Chem. Phys 2017, 19, 6470–6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Schuld NJ; Vervacke JS; Lorimer EL; Simon NC; Hauser AD; Barbieri JT; Distefano MD; Williams CL The chaperone protein SmgGDS interacts with small GTPases entering the prenylation pathway by recognizing the last amino acid in the CAAX motif. J. Biol. Chem 2014, 289, 6862–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Nussinov R; Wolynes PG A Second Molecular Biology Revolution? The Energy Landscapes of Biomolecular Function. Phys. Chem. Chem. Phys 2014, 16, 6321–6322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.