

Abstract
Brugia malayi is a human filarial nematode parasite that causes lymphatic filariasis or ‘elephantiasis’ a disfiguring neglected tropical disease. This parasite is a more tractable nematode parasite for the experimental study of anthelmintic drugs and has been studied with patch-clamp and RNAi techniques. Unlike in C. elegans however, calcium signaling in B. malayi or other nematode parasites has not been achieved, limiting the studies of the mode of action of anthelmintic drugs. We describe here the development of calcium imaging methods that allow us to characterize changes in cellular calcium in the muscles of B. malayi. This is a powerful technique that can help in elucidating the mode of action of selected anthelmintics. We developed two approaches that allow the recording of calcium signals in the muscles of adult B. malayi: (a) soaking the muscles with Fluo-3AM, promoting large-scale imaging of multiple cells simultaneously and, (b) direct insertion of Fluo-3 using microinjection, providing the possibility of performing dual calcium and electrophysiological recordings. Here, we describe the techniques used to optimize dye entry into the muscle cells and demonstrate that detectable increases in Fluo-3 fluorescence to elevated calcium concentrations can be achieved in B. malayi using both techniques.
Keywords: Calcium imaging, Fluo-3, B. malayi, Muscle
Introduction
Neglected tropical diseases (NTDs) caused by parasitic nematode infections affect over a billion people globally. Lymphatic filariasis, commonly known as elephantiasis, is an illness caused by the clade III filarial nematodes Brugia malayi, Wuchereria bancrofti and Brugia timori, which become lodged in the lymphatic vessels, impairing the drainage and producing swelling of the tissues including the limbs. Although the symptoms are not life threatening, heavy infections can lead to abnormal enlargement of body parts resulting in pain and severe disability. Approximately over 850 million people are at risk of suffering from lymphatic filariasis. There are no vaccines available to help treat filarial parasitic infections and prevention relies on the use anthelmintics as chemotherapeutic agents. To prevent the spread of filarial parasites, the World Health Organization recommends Mass Drug Administration (MDA), using albendazole in combination with both ivermectin and diethylcarbamazine (DEC) to kill the microfilariae (King et al. 2018). However, MDA does not kill the adult parasites and although treatment is effective at reducing transmission, there are concerns that resistance to the drugs may occur. Therefore, the development of new or improved use of existing anthelmintics is urgently required to ensure the effective killing of both the adult parasites and microfilariae.
Studies observing effects of anthelmintics on different receptors have often been conducted in the model organism C. elegans, due to the ease of maintenance and genetic manipulation. However, this organism is free living and does not possess many of the features of parasitic nematodes. Many of the ‘parasite genes’ are not present in the animal’s genome (Coghlan 2005; Gilleard 2004). Additionally, the composition of C. elegans receptors can be different compared to various parasitic species of nematode, for example, the nicotinic acetylcholine receptors (nAChRs) (Holden-Dye et al. 2013; Williamson et al. 2007). To overcome these limitations, we focus on the human filarial nematode Brugia malayi. Previously, we have studied the effect of anthelmintics on B. malayi muscles and their target receptors, including levamisole, pyrantel, morantel, bephenium, tribendimidine and emodepside (Kashyap et al. 2019; Martin et al. 2015; Robertson et al. 2013; Verma et al. 2017). Our overall aim is to dissect further, how anthelmintics manipulate or interfere with muscular signaling in B. malayi and to identify mechanisms that promote increased sensitivity. A major signal that can be measured to determine the effects of anthelmintics on muscular signaling is the calcium signal. Calcium is a key secondary messenger that promotes multiple downstream effects including contraction, transcription and opening of channels. Detectable increases of calcium in the muscle by anthelmintic manipulation may be generated either through the activation and opening of ion channels promoting extracellular calcium entry or by release from intracellular stores.
Calcium imaging has been an important technique for recording the activity of neurons and muscles in both invertebrates and vertebrates. For nematodes, C. elegans has been the most tractable model organism as transgenic animals expressing different types of genetically encoded calcium indicators (GECIs) in specific cells and tissues can be generated (Kerr 2006). Calcium imaging techniques have yet to be adapted for parasitic species including B. malayi, due to difficulties generating transgenic lines expressing GECIs and propagating viable isolates (Lok and Unnasch 2013). Therefore, we turned the older calcium indicators such as the synthetic organic fluorescent dyes (Fluo-3, Fura-2). Although older, these single-wavelength-activated dyes demonstrate fast binding and disassociation kinetics, photostability and calcium specificity (Badura et al. 2014; Mank and Griesbeck 2008).
In this study, we focused on using the fluorescent calcium indicator dyes, particularly Fluo-3 and Fluo-3AM and adapted two approaches to perform calcium imaging in B. malayi muscles. Fluo-3AM is a fluorescent dye, which has an acetoxymethyl group (AM) added to prevent degradation and activation by light and allows the dye to pass across the cellular membrane. Once inside the cell, non-specific endogenous esterases cleave the AM group trapping and activating the dye in the muscle (Tsien 1981). Using this approach, we loaded and recorded calcium signals from multiple muscle cells simultaneously. The second technique we adapted involved directly injecting Fluo-3 into a single muscle via a patch pipette, with aim of recording calcium and electrophysiological signals simultaneously. Using these adapted techniques, we can insert Fluo-3 into B. malayi muscles and successfully record changes in Fluo-3 fluorescence by increasing the calcium concentration.
Materials and methods
Brugia malayi supply and maintenance
Only female Brugia malayi worms were used for the study due to their large size. Live adult female B. malayi were sourced from the NIH/NIAID Filariasis Research Reagent Resource Centre (FR3; College of Veterinary Medicine, University of Georgia, Athens, GA, USA). B. malayi were maintained in non-phenol red Roswell Park Memorial Institute (RPMI) 1640 media (Life Technologies, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Fisher-Scientific, USA) and 1% penicillin–streptomycin (Life Technologies, USA). Parasites were separated individually into a 24 well microtiter plate containing 2 mL of the RPMI media. Parasites were held in an incubator set to 37 °C with 5% CO2. All parasites were used up to 7 days post-delivery.
Dissection of B. malayi
Dissection of B. malayi was performed as previously described (Robertson et al. 2011, 2013; Verma et al. 2017). All dissections were performed at room temperature. Briefly, worms were cut into 1 cm pieces from the anterior region and placed into a recording chamber filled with B. malayi bath solution (23 mM NaCl, 110 mM sodium acetate, 5 mM KCl, 6 mM CaCl2, 4 mM MgCl2, 5 mM HEPES, 10 mM d-glucose, 11 mM sucrose, pH 7.2 adjusted with NaOH, ~ 320 mOsmol). The base of the chamber was a coverslip (24 × 50 mm) coated with a thin layer of Sylgard. The body piece was immobilized by gluing each end to the Sylgard pad using Glushield cyanoacrylate glue (Glustitch) and pinned by creating a wall down one side. The body piece was cut longitudinally using a tungsten needle, and the resulting ‘muscle flap’ was glued to the coverslip along the cut edge exposing the muscle cells. Intestines and the uterus were removed using fine forceps and the preparation was washed with bath solution to remove any eggs or debris (Fig. 1). All dissections were performed under DIC optics (400×) on an inverted light microscope (SMZ-U, Nikon, USA).
Fig. 1.
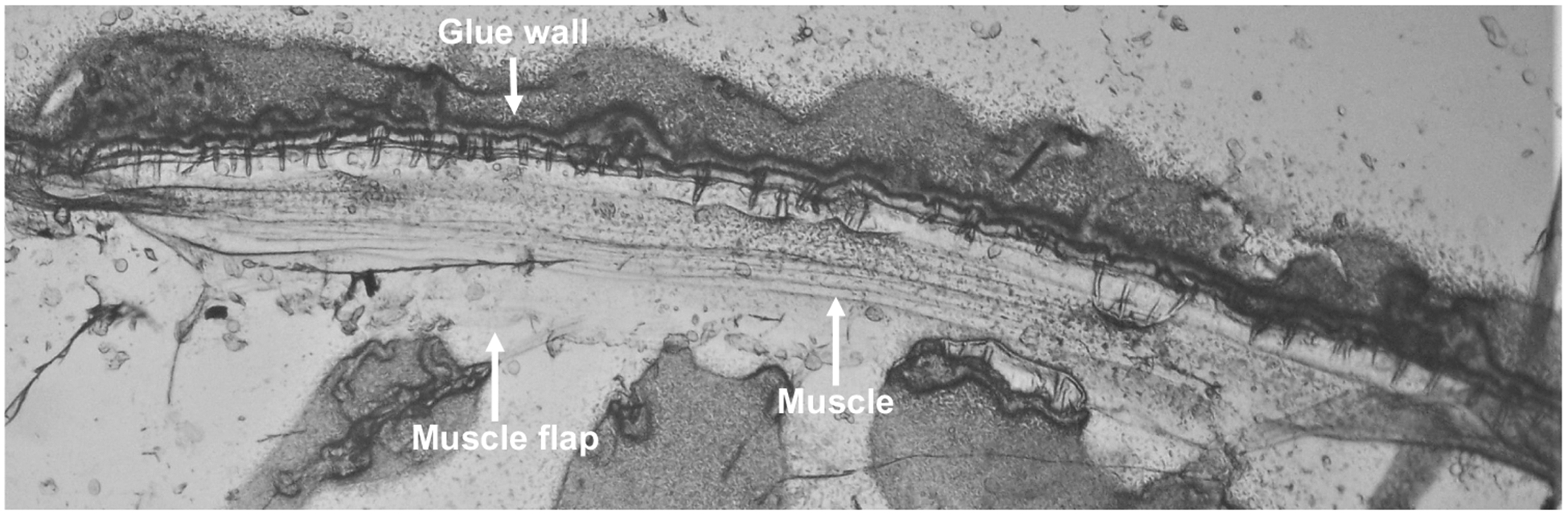
Micrograph of a dissected body piece of B. malayi: micrograph of a suitable dissected B. malayi body piece with intestine and uterus removed used for calcium and electrophysiological experiments. Key structures glue wall, muscle and muscle flap indicated
Fluo-3AM soaking
After dissection, the recording chamber was connected to a Dual Automatic Temperature Controller (Warner Instruments, Hamden, CT) and the temperature was maintained between 34 and 36 °C. The muscles were incubated in 5 μM Fluo-3AM (Sigma-Aldrich, USA) diluted in bath solution with no added Ca2+ (total [Ca2+] is < 100 μM) for 60 min, allowing for Fluo-3AM uptake and the cleavage of the ester group by endogenous non-specific esterases. For samples treated with Pluronic F-127 (Invitrogen, USA), 10% (v/v) of PF-127 diluted in DMSO was added to the 5 μM Fluo-3AM solution. For samples treated with collagenase, muscles were treated with 2 mg/mL collagenase (type 1A, Sigma-Aldrich, USA) in bath solution for 15–20 s and washed several times before Fluo-3AM was added. Fluo-3AM, with or without PF-127, solutions were prepared in Ca2+-free bath solution daily before every experiment. After incubation, the sample was continuously perfused with regular bath solution containing 6 mM CaCl2 or low calcium bath solution containing 1 mM CaCl2. All solutions were delivered to the chamber under gravity feed through solenoid valves controlled using a VC-6 Six-Channel Valve Controller (Warner Instruments, Hamden, CT, USA) through an inline heater set at 37 °C (Warner Instruments, Hamden, CT, USA) at a rate of 1.5 mL/min. To measure changes in calcium, all samples were subjected to 10 mM CaCl2. Muscles that elicited Ca2+ signals were considered viable while those that failed to elicit response were classified as non-viable. Absence of Ca2+ signals in the muscles were also monitored and achieved by the continuous perfusion of bath solution containing 1 mM CaCl2.
Fluo-3 injections
For microinjections, dissected muscles were treated with 2 mg/mL collagenase (Type 1A) for 30 s and were washed in buffer several times to remove excess collagenase. Patch pipettes were pulled from capillary glass (G85150T; Warner Instruments Inc., Hamden, CT, USA) and fire polished. The pipettes were filled with either pipette solution (120 mM KCl, 20 mM KOH, 4 mM MgCl2, 5 mM TRIS, 0.25 mM CaCl2, 4 mM NaATP, 5 mM EGTA and 36 mM sucrose (pH 7.2 with KOH, ~ 315–330 mOsmol), TRIS-HCl solution (1 M TRIS-HCl pH 7.4 with KOH) or K-gluconate recording solution (125 mM K-Glu, 2 mM KCl, 5 mM MgATP, 0.3 mM NaGTP, 5 mM HEPES (pH 7.4 with KOH)). All solutions had 5 μM Fluo-3 diluted in DMSO added before each experiment and were kept in a dark environment to prevent degradation of the dye. Pipettes with resistances of 1.8–3 MΩ were used. Giga ohm seals were formed before breaking the membrane by suction. After breaking in cells were left for at least 10 min to allow Fluo-3 to diffuse into the muscle cell before being subjected to 10 mM CaCl2. Cells were consistently perfused with bath solution containing 1 mM CaCl2 at a rate of 1.5 mL/min as described previously.
Calcium imaging
All recordings were performed on a Nikon Eclipse TE3000 microscope (20×/0.45 Nikon PlanFluor objective), fitted with a Photometrics Retiga R1 Camera (Photometrics, Surrey, BC, Canada). Light control was achieved using a Lambda 10–2 two-filter wheel system with a shutter controller (Lambda Instruments, Switzerland). Filter wheel one was set on a green filter (525–530 mm) between the microscope and camera. Filter wheel two set on the blue filter (490 mm) between a Lambda LS Xenon bulb light box, which delivered light via a fiber optic cable to the microscope (Lambda Instruments, Switzerland), to activate Fluo-3. Blue light emission was controlled using a shutter. Minimal exposure to blue light during recording setup was used to prevent reduction in signal strength.
Calcium signal recordings were acquired and analyzed using MetaFluor 7.10.2 (MDS Analytical Technologies, Sunnyvale, CA, USA) with exposure settings at 250 ms with 2× binning. Maximal Ca2+ signal amplitudes (ΔF) were calculated using the equation F1 − F0/F0 × 100, where F1 is the fluorescent value and F0 is the baseline value. All F0 values were determined as being the value at the time the stimulus was applied to the sample for all recordings analyzed. All pictures were taken using Ocular 2.0.1.496 (Digital optics, Auckland, New Zealand). Exposure settings were 150 ms with 2× binning. Statistical analysis was performed using two-tailed unpaired student t tests with P values < 0.05 were considered significant and two-way ANOVA where P values < 0.0001 were considered significant. All statistical analyses were performed in GraphPad Prism 5.0 (Graphpad Software, Inc., La Jolla, CA, USA).
Results
Fluo-3: a suitable candidate for calcium imaging in B. malayi
We used the fluorescent dye calcium reporter Fluo-3. Insertion of Fluo-3 into the B. malayi muscle can be carried out by two methods: invasively by microinjection or non-invasively as a permeable acetoxymethyl (AM) ester (Gobel and Helmchen 2007; Mank and Griesbeck 2008; Tsien 1981). A major benefit of using Fluo-3AM is that more absorption into tissues can be achieved, allowing the calcium signal to be measured in multiple cells simultaneously.
To ensure that Fluo-3 fluorescence was able to be detected, we looked for any autofluorescence present in untreated tissues and in the glue used to immobilize the worm that may hinder our recordings. The bright-field Fig. 2a shows the main structural features of the muscle cell including the soma, the muscle arms, the nucleus, lateral line, nerve cord and the edge of the glue wall. We exposed untreated muscles to blue light and observed no autofluorescence within the muscle cell. However, some autofluorescence was visible from the glue prompting us to avoid imaging near the glue wall (Fig. 2b). Muscle flaps were incubated in 5 μM Fluo-3AM for 60 min and then thoroughly washed. Fluo-3 fluorescence was observed under blue light with the muscle cells being visible (Fig. 2c). These observations showed that Fluo-3AM can be absorbed and activated in the muscles.
Fig. 2.
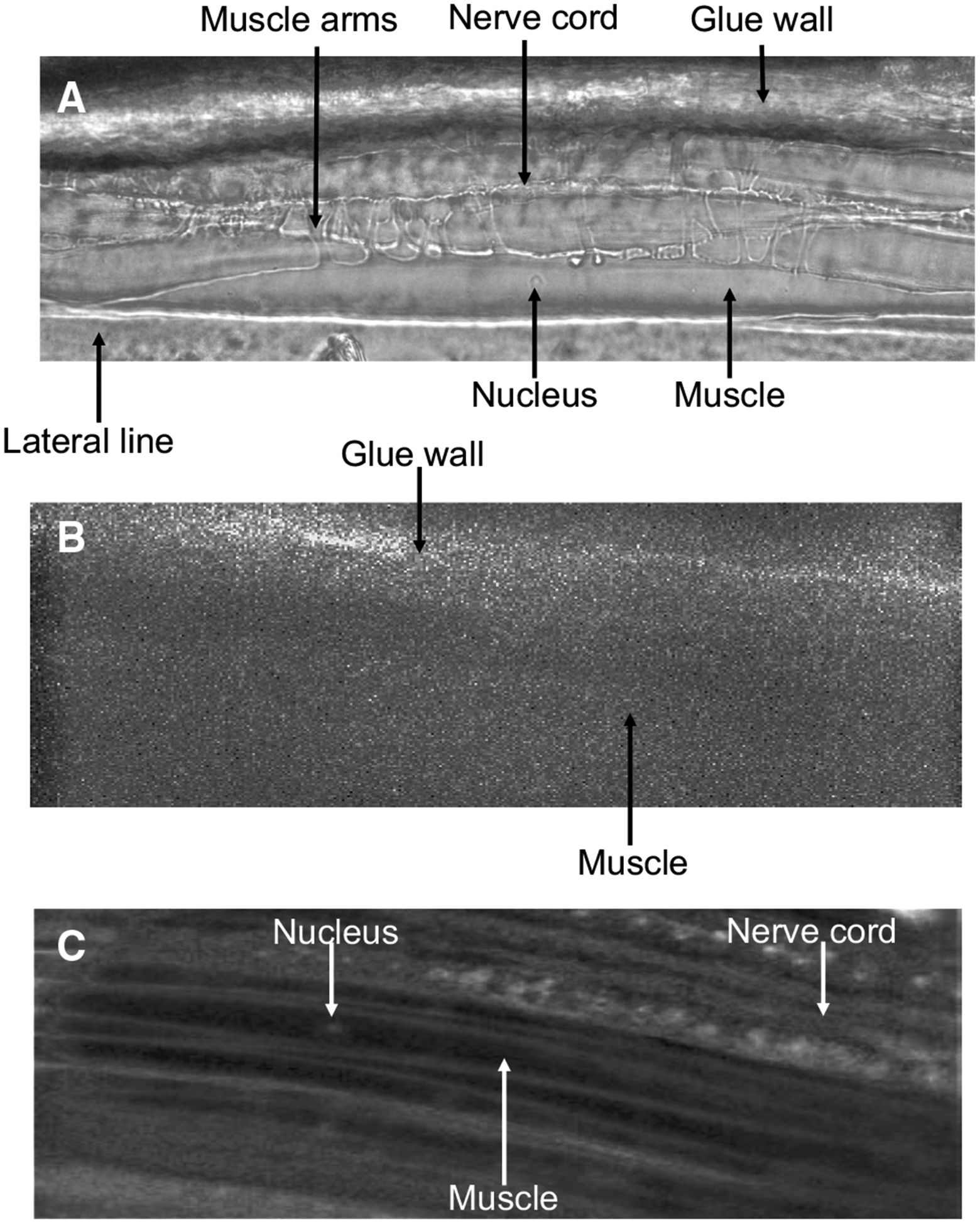
Micrographs of B. malayi muscles: a micrograph of B. malayi muscles under white light. b Untreated muscle exposed to blue light. c Muscles after being treated with 5 μM Fluo-3AM for 60 min at 34–36 °C. Key structures, muscle arms, muscle, nucleus, nerve cord, lateral line and glue wall are highlighted
Optimizing Fluo-3AM uptake
Although Fig. 2c shows that Fluo-3AM can enter B. malayi muscle and become active, the fluorescence level of Fluo-3 was modest and much of the dye was localized in the nucleus or outer edge of the muscle. To increase the level of Fluo-3 inside the muscle cells, we tested different incubation conditions to increase the uptake of the dye. We tested the fluorescence every 10 min in muscles treated with Fluo-3AM alone for 60 min (Fig. 3 black bars) and observed the slow uptake of the dye. To improve dye uptake, we added 10% (v/v) Pluronic F-127 to the Fluo-3AM solution because low levels of Pluronic F-127 in DMSO have been shown to improve uptake of AM dyes including Fluo-3AM (Hamad et al. 2015; Maruyama et al. 1989). We observed no improvement in dye uptake and overall fluorescence (Fig. 3 white bars) as addition of PF-127 alone did not significantly improve overall fluorescence compared to untreated samples (Table 1). One explanation for the limited uptake of Fluo-3 AM dye was the presence of the extracellular collagen matrix covering the muscle cells. We pretreated the muscle preparations with Type 1A collagenase, to remove the extracellular material (Robertson et al. 2013; Verma et al. 2017). We applied 2 mg/mL concentration of Type 1A collagenase to the muscles for 15–20 s and thoroughly washed the preparation. We again incubated the sample in 5 μM Fluo-3AM for 60 min at 34–36 °C and measured the fluorescence intensity every 10 min. Muscles treated with collagenase appeared to have increased dye uptake and fluorescence compared to the untreated samples (Fig. 3 gray bars). Although dye uptake and fluorescence were improved, there was no overall significance between collagenase treated and untreated samples after 60 min (Table 1). We hypothesized the addition of PF-127 would further improve uptake. Accordingly, we treated muscles cells with collagenase and added 10% (v/v) PF-127 to the Fluo-3AM solution, which significantly improved dye uptake compared to untreated samples (Fig. 3 dashed bar, Table 1). Based on our observations, we suggest that the optimum method for B. malayi muscle preparations is to use 2 mg/mL collagenase for 20 s before applying 5 μM Fluo-3AM with 10% PF-127 (v/v) for 60 min to maximize dye uptake and overall fluorescence.
Fig. 3.
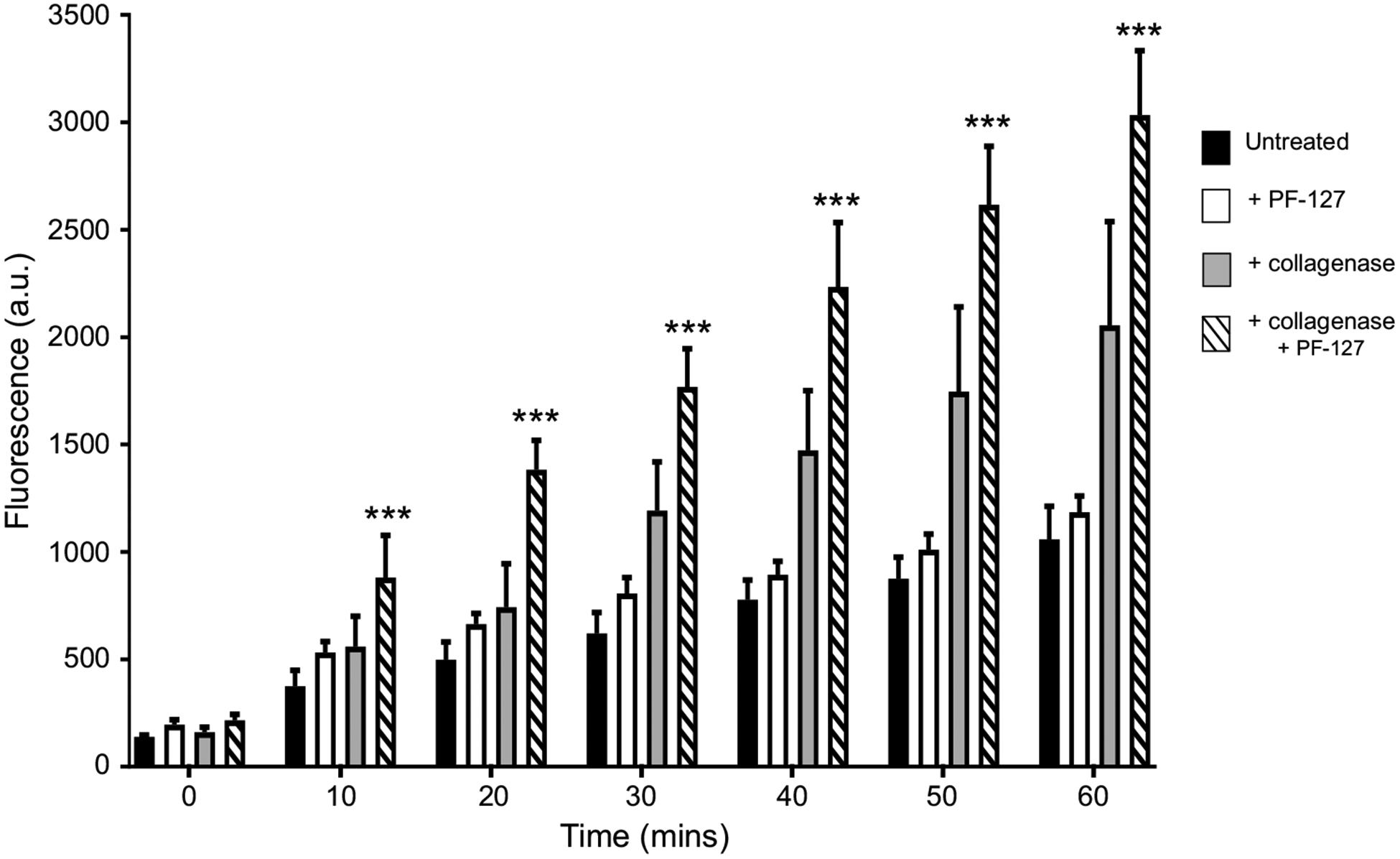
Collagenase and Pluronic F-127 improve Fluo-3AM absorption: 60 min time course of overall Fluo-3 fluorescencein B. malayi muscle cells for tissues treated with Fluo-3AM alone (black bars), Fluo-3AM and 10% (v/v) Pluronic F-127 (white bars), Fluo-3AM and 2 mg/mL collagenase (gray bars) and Fluo-3AM, collagenase and Pluronic F-127 (dashed bars). ***Significantly different to untreated (Fluo-3AM alone) (P = < 0.001; two-way ANOVA). For all treatments, n = 5. All values are represented as mean ± SEM
Table 1.
Summary of two-way ANOVA results for treatment and incubation time: summary of statistical results comparing effects of treatment and time on Fluo-3 fluorescence in B. malayi muscle cells between untreated, PF-127 treated, collagenase treated and collagenase + PF-127 treated sampless
| Untreated versus PF-127 | Untreated versus collagenase | Untreated versus collagenase + PF-127 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Source | df | MS | F | P | Source | df | MS | F | P | Source | df | MS | F | P |
| Treatment | 6 | 4,649,000 | 4.189 | 0.0749 | Treatment | 6 | 4,649,000 | 4.189 | 0.0749 | Treatment | 6 | 21,770,000 | 56.15 | <0.0001 |
| Time | 6 | 2,485,000 | 30.09 | <0.0001 | Time | 6 | 2,485,000 | 30.09 | <0.0001 | Time | 6 | 4,276,000 | 38.84 | <0.0001 |
| Treatment × time | 6 | 333,500 | 4.039 | 0.0024 | Treatment × time | 6 | 333,500 | 4.039 | 0.0024 | Treatment × time | 6 | 1,129,000 | 14.83 | <0.0001 |
Fluo-3AM treated muscles elicit calcium transients
We challenged muscles with 10 mM CaCl2 to test for a successful recording of Ca2+ signals in B. malayi muscles treated with Fluo-3AM. Our initial experiments met with modest success when we applied 10 mM CaCl2 to the muscle preparations maintained in our regular bath solution which contains 6 mM CaCl2 (Verma et al. 2017). We observed a small change in fluorescence intensity that had an average amplitude of 1.75% (Fig. 4a, c). We hypothesized that the small difference in the overall CaCl2 concentration between the bath solution and stimulus (from 6 to 10 mM) was the cause of the low Fluo-3 amplitudes. Therefore, we reduced the amount of CaCl2 in the bath solution to 1 mM providing a larger change in CaCl2 concentration when the stimulus was applied. As expected, application of 10 mM CaCl2 elicited a stereotypical Ca2+ signal in muscles when maintained in 1 mM CaCl2 bath solution with an average change in signal fluorescence of 14%. This change is significantly larger than our initial observations regular bath solution (Fig. 4b, c). These observations show that detectable Ca2+ signals can be recorded in B. malayi muscles soaked in Fluo-3AM when the methods are optimized using PF-127, collagenase and bathing solution with 1 mM CaCl2.
Fig. 4.
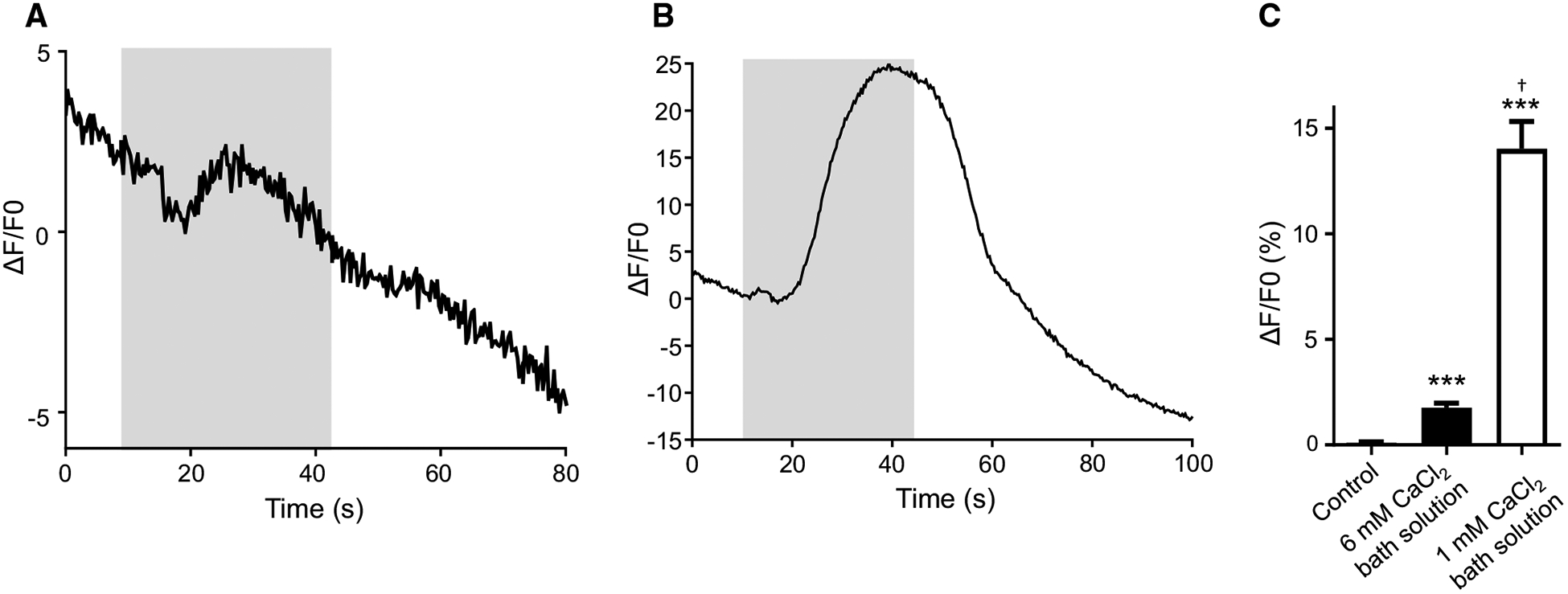
Fluo-3AM treated muscles cells display Ca2+ signals: a representative calcium trace of a muscle exposed to 10 mM CaCl2 that was maintained in regular bath solution before stimulus application. Gray box represents stimulus application. b Representative calcium trace in response to 10 mM CaCl2 in a muscle maintained in low CaCl2 (1 mM) bath solution. Gray box represents stimulus application. c Amplitudes of calcium signals in muscles perfused with low calcium bath solution, 10 mM CaCl2 in regular bath solution and 10 mM CaCl2 in low CaCl2 bath solution. ***Significantly different from untreated (untreated vs. 10 mM CaCl2 regular buffer; P < 0.0001, t = 6.792, df = 25, unpaired t test), (untreated vs. 10 mM CaCl2 low CaCl2 buffer; P < 0.0001, t = 8.273, df = 30, unpaired t test). † = significantly different to 10 mM CaCl2 in regular buffer (10 mM CaCl2 in regular buffer vs. 10 mM CaCl2 in low CaCl2 buffer P = < 0.0001, t = 8.108, df = 33, unpaired t test). Untreated n = 12; 10 mM CaCl2 in regular buffer n = 15, 10 mM CaCl2 in low CaCl2 buffer n = 20. All values are represented as mean ± SEM
Microinjection of Fluo-3 into B. malayi muscles
Although we have had success soaking muscles in Fluo-3AM, another goal was to achieve dual recordings of calcium and electrophysiological signals. Unfortunately, soaking limits this approach due to the delicate and fragile nature of the muscles after an hour incubation resulting in loss of patched cells before recordings can be achieved. Therefore, we patched onto muscle cells using a patch pipette containing the cell-impermeant indicator Fluo-3, which can only enter the muscle cell either by diffusion or driven by charge. Microinjection is a technique that has been used in earlier calcium imaging studies in different organisms (Jospin et al. 2002; Klauke and Plattner 1997; Lindsay et al. 1992). Various solutions have been used to promote dye entry into cells. We chose to test the two most common solutions: internal pipette solution (Jospin et al. 2002) and TRIS-HCl (Klauke and Plattner 1997). Additionally, we tested K-gluconate recording solutions because of the potential benefit of gluconate acting as a calcium buffer to prevent dye activation and degradation during patching. We added 5 μM Fluo-3 to each different pipette solution before recording and maintained the solutions in a dark environment to prevent the photobleaching and degradation of the fluorescent dye. Muscle cells were treated with collagenase, washed with buffer and patched immediately after washing, while being maintained in bath solution containing 1 mM CaCl2 (see ‘Materials and methods’ section). Upon successfully patching and breaking into a cell, the cells were left for 10 min while being perfused with 1 mM CaCl2 bath solution to allow the dye to enter and diffuse throughout the muscle cell. After 10 min, the muscle cells were visualized under blue light to determine if the dye entered the cell. As shown in Fig. 5, each of the three solutions had different effects on how well the dye diffused inside the muscle. The solution, which appeared to promote the best loading, was our regular electrophysiology internal pipette solution (Fig. 5a). After 10 min, the entire cell was filled with cell having a high level of fluorescence. The second-best pipette solution was TRIS-HCl, which filled the cell but not as effectively as the internal pipette solution and was not as fluorescent (Fig. 5b). Finally, cells patched with pipettes containing K-gluconate were characterized by most of the dye and fluorescence remaining near the pipette tip, suggesting that this solution prevented Fluo-3 diffusion in the cell (Fig. 5c).
Fig. 5.
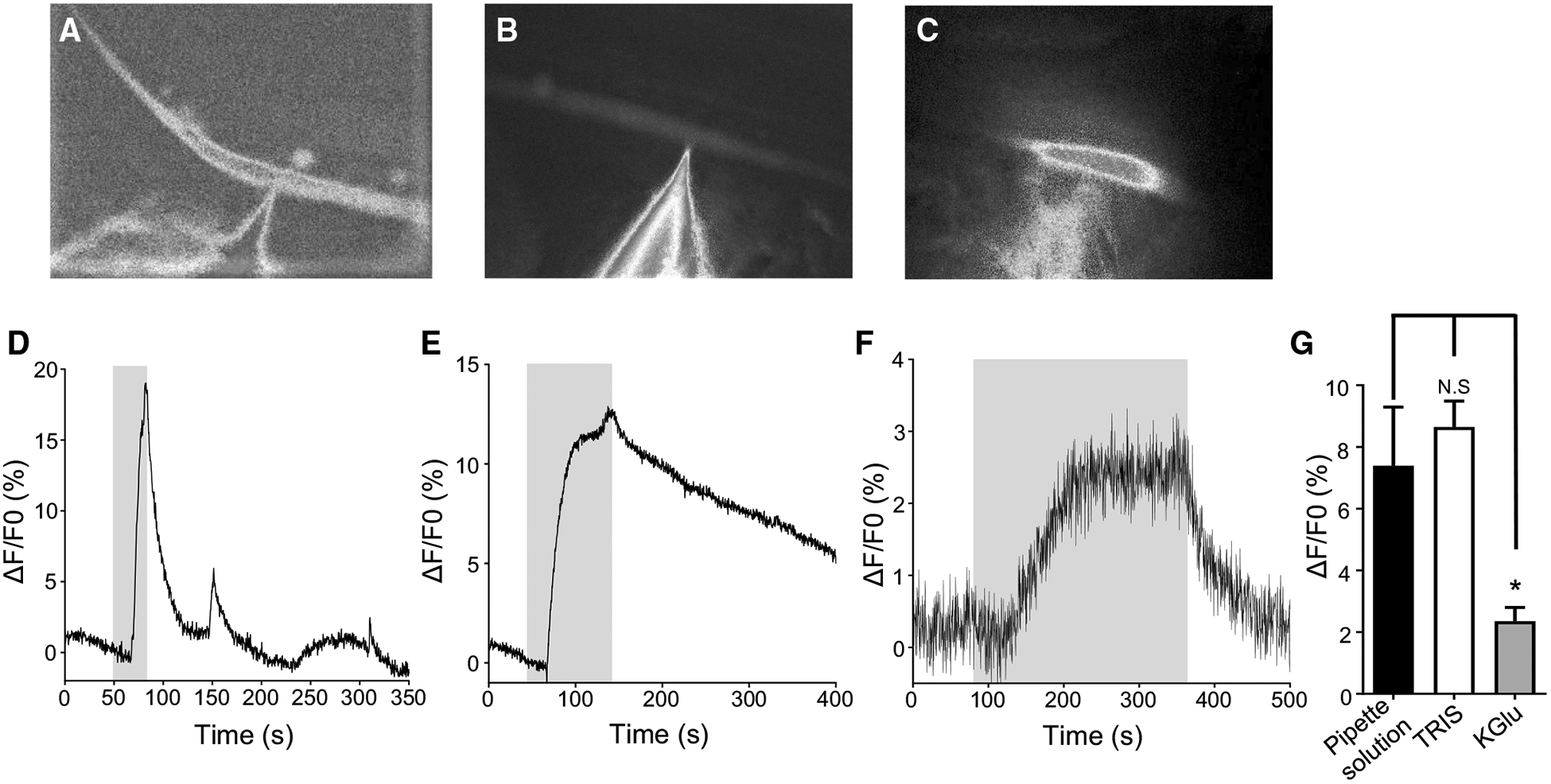
Microinjection of Fluo-3: a micrograph of muscle under blue light injected with electrophysiology pipette solution containing 5 μM Fluo-3. b Micrograph of Fluo-3 injection using TRIS-HCl. c Micrograph of Fluo-3 injection using K-gluconate solution. d Representative trace of a calcium response to 10 mM CaCl2 in a muscle injected with pipette solution containing Fluo-3. Gray box indicates stimulus application. e Representative trace of TRIS-HCl Fluo-3 inject muscles to 10 mM CaCl2. Gray box indicates stimulus application. f Representative trace of calcium signal in muscle inject with Fluo-3 using K-gluconate solution. Gray box indicates stimulus application. g Quantification of calcium amplitudes to 10 mM CaCl2 in muscles injected using pipette solution (black bar), TRIS-HCl (white bar) and K-gluconate (gray bar). N.S. = not significant pipette solution versus TRIS-HCl (P = 0.5526, t = 0.6031, df = 22, unpaired t test). *Significantly different pipette solutions versus K-Glu (P = 0.0220, t = 2.473, df = 21, unpaired t test). Pipette solution n = 12; TRIS-HCl n = 12, K-Glu n = 11. All values are represented as mean ± SEM
To determine if the muscles were viable after patching and injected with the dye, we tested muscle cells with a 10 mM CaCl2 stimulus. Similar to the Fluo-3AM treated samples, cells were perfused with bath solution containing 1 mM CaCl2, before being subjected to an increase in CaCl2 concentration. As seen with the level of dye loading, cells treated with regular internal pipette solution had strong changes in calcium, which quickly returned to baseline when the stimulus was removed (Fig. 5d, g). TRIS-HCl treated cells also had stereotypical Ca2+ responses and on average had a similar peak amplitude as the cells injected with the internal pipette solution (Fig. 5e, g). However, after removal of the 10 mM CaCl2, the signal took longer to return to baseline. Lastly, cells that were injected using K-gluconate had the lowest response: they were low amplitude and were very noisy (Fig. 5f, g). The observations showed that we are able to achieve measurable changes in calcium with two of the three pipette solutions, but due to the ease of patching and overall level of dye entry into the cell, we decided to pursue further experiments using the internal pipette solution, with aim of performing dual calcium imaging and electrophysiology experiments.
Discussion
The measurement of calcium signals by calcium imaging is a powerful technique to track and follow effects of drugs on cellular signaling. In this paper, we show how calcium imaging may be performed in muscle cells of the parasitic nematode Brugia malayi. We have successfully recorded rises in Fluo-3 fluorescence produced by increasing the CaCl2 concentrations. We have adapted two techniques to promote calcium imaging of B. malayi muscles using two Fluo-3 variants: Fluo-3AM that allows the recording from multiple muscle cells and the direct injection of Fluo-3 into a single muscle. The use of the dye Fluo-3 to measure Ca2+ signals has many advantages including: fast binding and disassociation kinetics, allowing the detection of rapid Ca2+ fluctuations; large fluorescence changes; near-linear response properties; high photostability, preventing rapid bleaching and high pH-resistance (Badura et al. 2014; Mank and Griesbeck 2008; Russell 2011).
Fluo-3AM incubation promotes large-scale calcium imaging
Incubation using Fluo-3AM permits experiments that will seek to record calcium signals from multiple cells simultaneously (Paredes et al. 2008; Roe et al. 2006). The addition of AM esters makes the dye membrane permeable, inactive and avoids the need for ‘invasive’ microinjection, which is labor intensive, prevents the study of larger populations of cells and can result in cellular damage (Paredes et al. 2008; Roe et al. 2006; Takahashi et al. 1999). Once inside the cell, non-specific esterases cleave the AM group and trap the dye inside the cell (Kao et al. 2010; Tsien 1981). This method effectively concentrates the Ca2+ indicator in cells, meaning a extracellular low concentration of dye (1–5 μM) is sufficient to result in a higher intracellular concentration of ~ 100 μM by ion-trapping (Kao et al. 2010; Paredes et al. 2008). Our aim here was to develop techniques to promote insertion of Fluo-3AM into multiple B. malayi muscles and successfully record changes in fluorescence from several muscle cells at the same time.
We found that incubating muscles in 5 μM Fluo-3AM in regular bath solution produced a poor level of fluorescence and very few muscles could be observed under blue light. We hypothesize that the presence of Ca2+ in the solution may have resulted in the activation of non-specific endogenous esterases present in the parasite, cleaving the Fluo-3AM before it was able to enter the muscles. We modified the protocol and found that for best uptake of Fluo-3 AM the preparation needed to be exposed to collagenase to remove the basement membrane, and to Pluronic F-127 to facilitate the dispersal and solubility of fluorescent dyes with AM groups (Fig. 3). To demonstrate that a positive change in Fluo-3 fluorescence was achievable, we exposed Fluo-3AM treated muscles to elevated concentrations of CaCl2. We found that to demonstrate the strong calcium signals with a test 10 mM stimulus of CaCl2, it was necessary to reduce the calcium concentration in the normal bathing solution to 1 mM from 6 mM (Fig. 4).
We point out that there are some disadvantages when using Fluo-3AM. Firstly, incubating the entire preparation can lead to expression of the dye in neighboring cells and tissues of non-interest, resulting in a decrease in signal quality from muscles, and may prevent the detection of smaller, subtler signals that may be triggered by receptor manipulation (Badura et al. 2014; O’Donovan et al. 1993). In addition, for absorption and activation of Fluo-3 AM, a long-term incubation is required (60 min), but this may give rise to the dye being compartmentalized and accumulated in intracellular organelles, including the nucleus (Fig. 2c). The compartmentalization reduces signal strength and quality (Cobbold and Rink 1987; Gee et al. 2000; Qu et al. 2016). Furthermore, fluorescent dyes may be extruded from the cell relatively quickly and high concentrations of the dye inside the cell can lead to toxicity (Badura et al. 2014; Kao et al. 2010; Mank and Griesbeck 2008; O’Donovan et al. 1993; Roe et al. 2006). With further optimization of dye loading, we hope to achieve suitable levels of fluorescence after a shorter period of incubation to avoid the problems inherent with using Fluo-3AM.
Fluo-3 microinjection a technique for dual calcium and electrophysiology recordings
Fluo-3AM incubation allows recording of calcium changes in a number of muscle cells simultaneously, but there may be reduced signal clarity and the longer incubation time can degrade the health of the cells. By inserting Fluo-3 directly into a cell by diffusion from a patch pipette, a single cell can be loaded with a controlled amount of the probe, to allow the cell to be analyzed with high contrast (Qu et al. 2016; Russell 2011). Once inside the cell, salt forms of Fluo-3 equilibrate quickly, allowing Ca2+ signals to be recorded within minutes (Paredes et al. 2008). This was one of the reasons we pursued the development of the second technique to allow for the recording of calcium from a single muscle cell.
Direct injection of dye by patching typically uses two solutions, an internal pipette solution or TRIS-HCl (Jospin et al. 2002; Klauke and Plattner 1997). We also used a third electrophysiology patching solution that contained K-gluconate, to act as a calcium buffer to reduce dye activation and degradation during patching. Use of all three solutions allowed for direct patching on the muscle cell, and after 10 min, cells showed Fluo-3 loading in the muscle, with patch pipette solution loading cells more effectively than TRIS-HCl and K-gluconate having very poor cellular loading (Fig. 5a, b, c). When challenged with an increase in CaCl2 concentration, cells injected using pipette solution and TRIS-HCl had similar overall amplitudes in Fluo-3 fluorescence, 7.4% for pipette solution and 8.6% for TRIS-HCl (Fig. 5g). However, cells injected using K-gluconate and had very low and noisy signals, 2.3% (Fig. 5f, g), suggesting that this solution is very poor for dye injection.
With the success of generating recordable Ca2+ signals using direct injection methods, we observed that after removal of CaCl2, a second or sometimes a third increase in fluorescence was detected (Fig. 5d). This spontaneous increase in Ca2+ fluorescence suggests that other cellular mechanisms may be occurring within the cell, either through an increase in intracellular Ca2+ release or by the activation of stretch receptors promoting extracellular calcium entry, which have been implicated in C. elegans (Fieseler et al. 2018; Li et al. 2006; Yeon et al. 2018).
Direct injection also has some disadvantages. Firstly, this technique is invasive and as such there is high risk of cellular damage reducing the overall data output. Additionally, this technique requires specialized equipment, is labor intensive and requires a high skill level. The incubation in Fluo-3AM allows for the recording of multiple muscles simultaneously; direct injection only permits one or perhaps a few cells in a single preparation to be recorded (Russell 2011).
In conclusion, our long-term goal is to understand how anthelmintics manipulate cellular signaling in B. malayi. We have previously demonstrated how anthelmintics manipulate muscle signaling using electrophysiology (Kashyap et al. 2019; Verma et al. 2017), but these studies do not allow us to record changes in intracellular signaling and to measure the effects on downstream secondary messengers. By developing two methods to record calcium signals in B. malayi muscles, we hypothesize we will be able to measure the effects of anthelmintics in numerous ways to achieve a greater understanding how these compounds interfere with cellular signaling in parasitic nematodes.
Acknowledgements
The authors would like to thank the Filariasis Research Reagent Resource Centre (FR3; College of Veterinary Medicine, University of Georgia, USA) for the supply of live adult B. malayi.
Funding This study was funded by National Institutes of Health NIAID (NIH R01 AI047194 and R21AI138967 to RJM) and the EA Benbrook Endowed Chair of Pathology and Parasitology. The funding agencies had no role in the design, execution or publication of this study. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases.
Footnotes
Availability of data and materials All data generated or analyzed during this study are included in this published article.
Conflict of interest The authors declare that they have no conflict of interests.
References
- Badura A, Sun XR, Giovannucci A, Lynch LA, Wang SS (2014) Fast calcium sensor proteins for monitoring neural activity. Neurophotonics 1(2):025008 10.1117/1.NPh.1.2.025008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbold PH, Rink TJ (1987) Fluorescence and bioluminescence measurement of cytoplasmic free calcium. Biochem J 248(2):313–328. 10.1042/bj2480313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A (2005) Nematode genome evolution. WormBook. 10.1895/wormbook.1.15.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieseler C, Kunert-Graf J, Kutz JN (2018) The control structure of the nematode Caenorhabditis elegans: neuro-sensory integration and proprioceptive feedback. J Biomech 74:1–8. 10.1016/j.jbiomech.2018.03.046 [DOI] [PubMed] [Google Scholar]
- Gee KR, Brown KA, Chen WN, Bishop-Stewart J, Gray D, Johnson I (2000) Chemical and physiological characterization of fluo-4 Ca(2 +)-indicator dyes. Cell Calcium 27(2):97–106. 10.1054/ceca.1999.0095 [DOI] [PubMed] [Google Scholar]
- Gilleard JS (2004) The use of Caenorhabditis elegans in parasitic nematode research. Parasitology 128(Suppl 1):S49–S70. 10.1017/S003118200400647X [DOI] [PubMed] [Google Scholar]
- Gobel W, Helmchen F (2007) In vivo calcium imaging of neural network function. Physiology (Bethesda) 22:358–365. 10.1152/physiol.00032.2007 [DOI] [PubMed] [Google Scholar]
- Hamad MI, Krause M, Wahle P (2015) Improving AM ester calcium dye loading efficiency. J Neurosci Methods 240:48–60. 10.1016/j.jneumeth.2014.11.010 [DOI] [PubMed] [Google Scholar]
- Holden-Dye L, Joyner M, O’Connor V, Walker RJ (2013) Nicotinic acetylcholine receptors: a comparison of the nAChRs of Caenorhabditis elegans and parasitic nematodes. Parasitol Int 62(6):606–615. 10.1016/j.parint.2013.03.004 [DOI] [PubMed] [Google Scholar]
- Jospin M, Jacquemond V, Mariol MC, Segalat L, Allard B (2002) The L-type voltage-dependent Ca2+ channel EGL-19 controls body wall muscle function in Caenorhabditis elegans. J Cell Biol 159(2):337–348. 10.1083/jcb.200203055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JP, Li G, Auston DA (2010) Practical aspects of measuring intracellular calcium signals with fluorescent indicators. Methods Cell Biol 99:113–152. 10.1016/B978-0-12374841-6.00005-0 [DOI] [PubMed] [Google Scholar]
- Kashyap SS, Verma S, Voronin D, Lustigman S, Kulke D, Robertson AP et al. (2019) Emodepside has sex-dependent immobilizing effects on adult Brugia malayi due to a differentially spliced binding pocket in the RCK1 region of the SLO-1K channel. PLoS Pathog 15(9):e1008041 10.1371/journal.ppat.1008041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr RA (2006) Imaging the activity of neurons and muscles. Worm-Book 10.1895/wormbook.1.113.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CL, Suamani J, Sanuku N, Cheng YC, Satofan S, Mancuso B et al. (2018) A trial of a triple-drug treatment for lymphatic filariasis. N Engl J Med 379(19):1801–1810. 10.1056/NEJMoa1706854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauke N, Plattner H (1997) Imaging of Ca2+ transients induced in paramecium cells by a polyamine secretagogue. J Cell Sci 110(Pt 8):975–983 [DOI] [PubMed] [Google Scholar]
- Li W, Feng Z, Sternberg PW, Xu XZ (2006) A C. elegans stretch receptor neuron revealed by a mechanosensitive TRP channel homologue. Nature 440(7084):684–687. 10.1038/nature04538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay LL, Hertzler PL, Clark WH Jr (1992) Extracellular Mg2+ induces an intracellular Ca2+ wave during oocyte activation in the marine shrimp Sicyonia ingentis. Dev Biol 152(1):94–102. 10.1016/0012-1606(92)90159-e [DOI] [PubMed] [Google Scholar]
- Lok JB, Unnasch TR (2013) Transgenesis in animal parasitic nematodes: Strongyloides spp. and Brugia spp. In: WormBook (ed) The C. elegans research community, wormBook 10.1895/wormbook.1.163.1. http://www.wormbook.org [DOI] [Google Scholar]
- Mank M, Griesbeck O (2008) Genetically encoded calcium indicators. Chem Rev 108(5):1550–1564. 10.1021/cr078213v [DOI] [PubMed] [Google Scholar]
- Martin RJ, Puttachary S, Buxton SK, Verma S, Robertson AP (2015) The Conqueror Worm: recent advances with cholinergic anthelmintics and techniques excite research for better therapeutic drugs. J Helminthol 89(4):387–397. 10.1017/S0022149X1400039X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama I, Hasegawa T, Yamamoto T, Momose K (1989) Effects of pluronic F-127 on loading of fura 2/AM into single smooth muscle cells isolated from guinea pig taenia coli. J Toxicol Sci 14(3):153–163. 10.2131/jts.14.153 [DOI] [PubMed] [Google Scholar]
- O’Donovan MJ, Ho S, Sholomenko G, Yee W (1993) Real-time imaging of neurons retrogradely and anterogradely labelled with calcium-sensitive dyes. J Neurosci Methods 46(2):91–106. 10.1016/0165-0270(93)90145-h [DOI] [PubMed] [Google Scholar]
- Paredes RM, Etzler JC, Watts LT, Zheng W, Lechleiter JD (2008) Chemical calcium indicators. Methods 46(3):143–151. 10.1016/j.ymeth.2008.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu H, Xing W, Wu F, Wang Y (2016) Rapid and inexpensive method of loading fluorescent dye into pollen tubes and root hairs. PLoS ONE 11(4):e0152320 10.1371/journal.pone.0152320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AP, Puttachary S, Martin RJ (2011) Single-channel recording from adult Brugia malayi. Invert Neurosci 11(1):53–57. 10.1007/s10158-011-0118-1 [DOI] [PubMed] [Google Scholar]
- Robertson AP, Buxton SK, Martin RJ (2013) Whole-cell patch-clamp recording of nicotinic acetylcholine receptors in adult Brugia malayi muscle. Parasitol Int 62(6):616–618. 10.1016/j.parint.2013.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe MW, Fiekers JF, Philipson LH, Bindokas VP (2006) Visualizing calcium signaling in cells by digitized wide-field and confocal fluorescent microscopy. Methods Mol Biol 319:37–66. 10.1007/978-1-59259-993-6_3 [DOI] [PubMed] [Google Scholar]
- Russell JT (2011) Imaging calcium signals in vivo: a powerful tool in physiology and pharmacology. Br J Pharmacol 163(8):1605–1625. 10.1111/j.1476-5381.2010.00988.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Camacho P, Lechleiter JD, Herman B (1999) Measurement of intracellular calcium. Physiol Rev 79(4):1089–1125. 10.1152/physrev.1999.79.4.1089 [DOI] [PubMed] [Google Scholar]
- Tsien RY (1981) A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 290(5806):527–528. 10.1038/290527a0 [DOI] [PubMed] [Google Scholar]
- Verma S, Kashyap SS, Robertson AP, Martin RJ (2017) Functional genomics in Brugia malayi reveal diverse muscle nAChRs and differences between cholinergic anthelmintics. Proc Natl Acad Sci U S A 114(21):5539–5544. 10.1073/pnas.1619820114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SM, Walsh TK, Wolstenholme AJ (2007) The cys-loop ligand-gated ion channel gene family of Brugia malayi and Trichinella spiralis: a comparison with Caenorhabditis elegans. Invert Neurosci 7(4):219–226. 10.1007/s10158-007-0056-0 [DOI] [PubMed] [Google Scholar]
- Yeon J, Kim J, Kim DY, Kim H, Kim J, Du EJ et al. (2018) A sensory-motor neuron type mediates proprioceptive coordination of steering in C. elegans via two TRPC channels. PLoS Biol 16(6):e2004929 10.1371/journal.pbio.2004929 [DOI] [PMC free article] [PubMed] [Google Scholar]