

Summary:
Phospholipase C (PLC) enzymes hydrolyse phosphoinositide lipids to inositol phosphates and diacylglycerol. Direct activation of PLCβ by Gαq and/or Gβγ subunits mediates signalling by Gq and some Gi coupled G protein-coupled receptors (GPCRs), respectively. PLCβ isoforms contain a unique C-terminal extension, consisting of proximal and distal C-terminal domains (CTD) separated by a flexible linker. The structure of PLCβ3 bound to Gαq is known, however, for both Gαq and Gβγ, the mechanism for PLCβ activation on membranes is unknown. We examined PLCβ2 dynamics on membranes using hydrogen deuterium exchange mass spectrometry (HDX-MS). Gβγ caused a robust increase in dynamics of the distal C-terminal domain (CTD). Gαq showed decreased deuterium incorporation at the Gαq binding site on PLCβ. In vitro Gβγ-dependent activation of PLC is inhibited by the distal CTD. The results suggest that disruption of auto-inhibitory interactions with the CTD leads to increased PLCβ hydrolase activity.
Keywords: G protein, G protein-coupled receptor, GPCR, signal transduction, membrane interactions, phospholipase C, Hydrogen-deuterium exchange, HDX-MS, Gβγ, Gαq, Lipids, protein dynamics
Introduction:
Receptor-dependent activation of phospholipase C (PLC) enzymes stimulates hydrolysis of the lipid substrate phosphatidylinositol 4,5 bisphosphate to the second messengers, diacylglycerol (DAG), and inositol 1,4,5 trisphosphate (IP3), resulting in downstream activation of protein kinase C (PKC), and calcium release, respectively (Berridge, 1987; Kadamur and Ross, 2013). There are 6 families of PLC isoforms: β, δ, γ, ε, ζ and η (Harden et al., 2011; Kadamur and Ross, 2013; Smrcka et al., 2012). All share a common core domain structure consisting of four EF hand repeats, a TIM barrel catalytic domain, separated into X and Y domains connected by a flexible “X/Y” insert linker, and a C2 domain (Kadamur and Ross, 2013). Most PLC isozymes have a pleckstrin homology (PH) domain at the amino terminus. The individual PLC isotypes differ in their presence or absence of other regulatory domains.
Members of the PLCβ family are among the most well studied PLC isoforms. They are important for a myriad of biological functions including inflammation (Li et al., 2000), opioid analgesia (Bianchi et al., 2009; Xie et al., 1999), and cardiovascular function (Filtz et al., 2009; Howes et al., 2003; Woodcock et al., 2009; Woodcock et al., 1995) downstream of G protein-coupled receptor (GPCR) activation. There are 4 isoforms of the PLCβ family; 1,2,3 and 4 that are all directly activated by binding Gαq subunits (Lee et al., 1994; Singer et al., 2002; Smrcka et al., 1991; Smrcka and Sternweis, 1993; Taylor et al., 1991). PLCβ2 and β3 are also activated by Gβγ (Lee et al., 1994; Smrcka and Sternweis, 1993). A unique feature of the PLCβ family is a ~400 amino acid C terminal extension comprised of a well conserved proximal C-terminal domain helix-loop-helix domain, a flexible linker region, followed by a less well conserved distal C-terminal domain (CTD) (Figure 1). The distal CTD has Bin, Amphiphysin and Rvs. (BAR) domain homology and folds into a helical bundle (Lyon et al., 2013; Singer et al., 2002). The first structure of PLCβ3 bound to Gαq shows direct interactions of Gαq with the proximal CTD and the EF hands (Waldo et al., 2010). A more recent structure of full length PLCβ3 includes the extended C-terminus and can serve as a model for the domain organization of all PLCβ isoforms (Fig. 1) (Lyon et al., 2011). Both the proximal and distal CTD are required for activation by Gαq, however the distal CTD is dispensable for Gβγ activation (Lee et al., 1993; Wu et al., 1993).
Figure 1: Domain Architecture and model of PLCβ2 architecture.
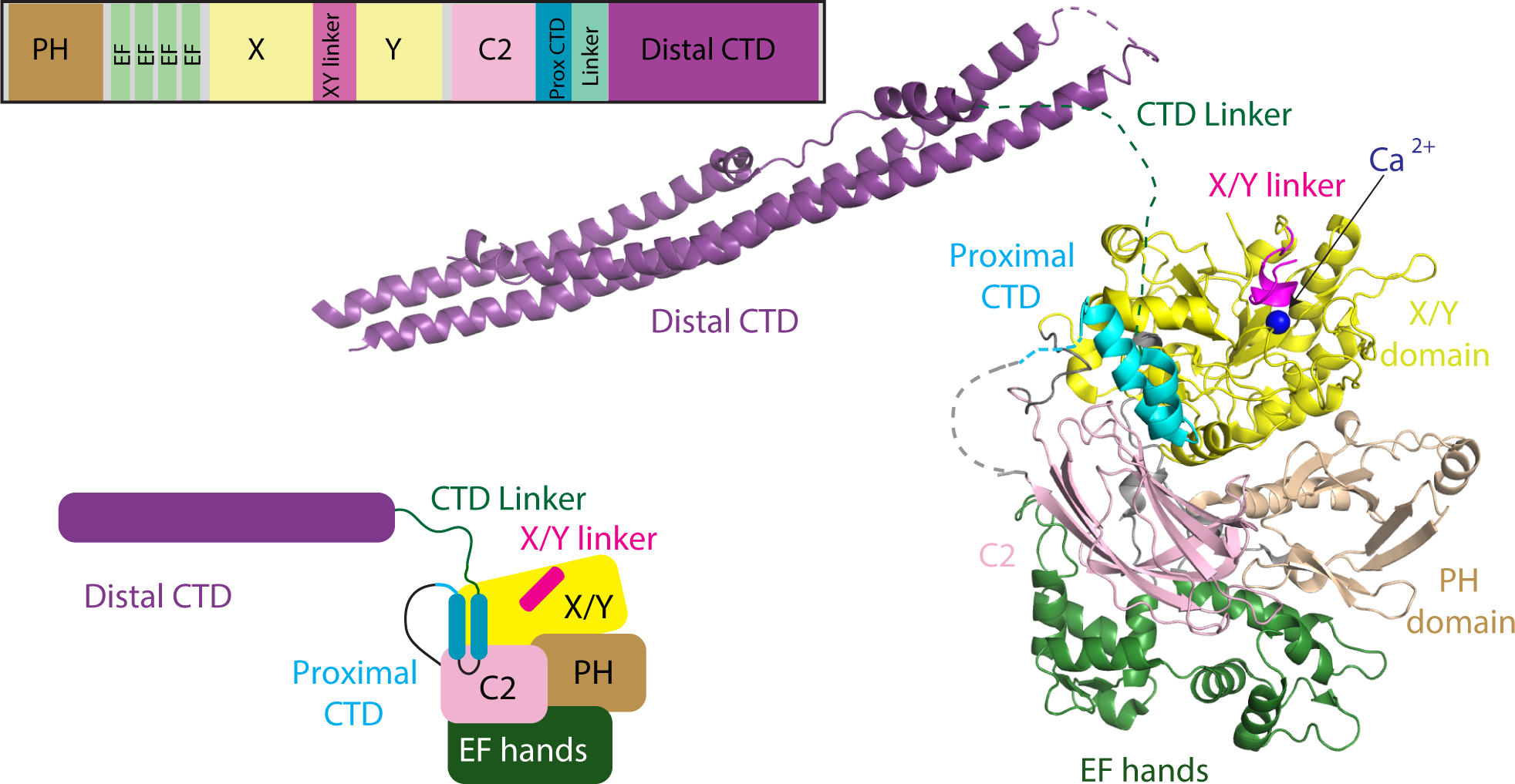
A model of PLCβ2 based on a monomer of PLCβ3 from the co-crystal structure of Gαq-PLCβ3 (PDB:4GNK), and the resolved portion of the proximal CTD from the structure of S. officinalis PLC21 (PDB: 3QR0). This model is oriented such that the active site (as indicated by the catalytic Ca2+ ion) is oriented towards the top of the figure. Domains are colored as in the domain schematics in the top and bottom left and the domain boundaries are (PLCβ2 numbering system) PH domain: 15–135; EF hands: 143–296; X: 321–466; XY Linker: 466–537; Y: 537–662; C2: 672–803; Prox CTD: 809–837; CTD Linker: 838–874; Distal CTD: 875–1150. The Gαq binding Helix-loop-Helix (amino acids: 810–827) is disordered in the structures solved in the apo PLCβ models with no bound Gαq. A model of the structure is shown on the right, with this used as a template for describing all HDX-MS data.
There are two proposed general mechanisms of PLCβ activation, with multiple models proposed for how GPCRs mediate this activation. The binding site for Gαq bound to PLCβ is well defined, but no clear binding site has been identified for Gβγ. Proposed mechanisms of activation involve either the X/Y linker or the proximal CTD (also referred to as the helix loop helix region). The negatively charged X/Y linker acts as a lid on the active site occluding access to PIP2 substrate. It has been proposed that negative charges on the flexible region of the linker are repelled upon membrane binding, or reorientation on the membrane surface, and this acts to remove the “lid” allowing for substrate (PIP2) to bind the catalytic site (Hicks et al., 2008). Genetic deletion of the flexible X/Y linker leads to an increase in basal activity, but this activity can be further stimulated by G-proteins, suggesting there is more to G-protein regulation of PLC activity than removal of the X/Y linker steric occlusion of the active site. A second proposed mechanism for activation involves the proximal CTD being autoinhibitory when packed against the catalytic core of PLC. In an inactive structure of invertebrate PLC (PLC21) (PDB: 3QR0 and 3QR1) the proximal CTD interacts with the core of the enzyme (Lyon et al., 2013; Lyon et al., 2011). In Gαq-PLCβ3 co-crystal structures the proximal CTD is pulled away from the core via binding to Gα, leading to disruption of the autoinhibitory interface (Lyon et al., 2011; Waldo et al., 2010). Supporting this hypothesis, mutations that interfere with the proximal CTD-core interactions cause a robust increase in activity that is not further stimulated by Gαq.
There are two proposed binding sites of Gβγ on PLCβ. One of the proposed binding sites for Gβγ on PLCβ is in the PH domain. Indeed, the isolated PH domain has been shown to inhibit Gβγ stimulated PLC activity by competing for binding to Gβγ, and chimeras containing the PH domain of PLCβ2 fused to the catalytic core of PLCδ confers the ability to be activated by Gβγ subunits (Han et al., 2011; Kadamur and Ross, 2016; Wang et al., 2000). Interestingly, one proposed PH domain binding site is not surface exposed in structural models of PLC, with binding of Gβγ proposed to be mediated by dynamic dissociation from the PLC core, leading to exposure of the Gβγ-binding surface on the PH domain (Kadamur and Ross, 2016). Covalent cross-linking to restrict this putative PH-domain movement inhibits both Gβy binding to, and activation of, PLCβ. The other proposed binding site of Gβγ on PLCβ is in the Y-domain. Mutational analysis of this region decreases Gβγ activation (Bonacci et al., 2005), and peptides corresponding to this region inhibits Gβγ-dependent activation of PLCβ2, and can be directly crosslinked to Gβγ (Kuang et al., 1996; Sankaran et al., 1998). Complicating analysis of the molecular basis for G protein-dependent activation of PLC, is that the activation occurs on a membrane surface, and studying these interactions by standard biophysical methods is extremely challenging.
To further define the molecular mechanisms that mediate activation of PLCβ2 by GPCRs on membranes we utilized hydrogen deuterium exchange mass spectrometry (HDX-MS). This technique has provided key insight into the molecular mechanisms of activation of lipid signalling enzymes downstream of GPCRs (Dbouk et al., 2012; Vadas et al., 2013). Our analysis reveals unique insights into the role of C-terminal domains of PLCβ2 in activation by both Gαq and Gβγ. The results show that Gβγ causes large scale allosteric rearrangement of the distal CTD domain, and that Gαq interacts with the distal CTD as well as the proximal CTD. Biochemical assays reveal a direct CTD-catalytic core interaction that inhibits PLC activation. Overall, this work provides fundamental insight into unique regulatory mechanisms that control PLCβ activation by GPCRs, and indicates a unifying role for the C-terminal extension in mediating G-protein activation.
Results:
Activation of PLC by G proteins requires a membrane surface (Charpentier et al., 2014). High resolution structural studies of peripheral membrane proteins are generally not compatible with, or are very difficult to perform on membrane surfaces. We used hydrogen-deuterium exchange mass spectrometry (HDX-MS) to examine the dynamics of PLCβ2 in the presence and absence of membranes, Gβγ, and activated Gαq. HDX-MS measures the exchange rate of amide hydrogens with deuterated solvent, and as the main determinant of exchange is the stability of secondary structure, it is an excellent probe of conformational dynamics. Deuterium incorporation is localised at peptide level resolution through the generation of pepsin generated peptides. The HDX-MS coverage of PLCβ2 was composed of 192 identified peptides that spanned 82% of the primary sequence of PLCβ2 (Supplemental Figure 1A), including multiple charge states for a total of 232 monitored isotopic peaks (complete HDX-MS data is found in Data S1). HDX-MS experiments compared the dynamics of PLCβ2 on and off lipid vesicles composed of phosphatidylethanolamine(PE), phosphatidylserine (PS), and phosphatidylinositol 4,5 bisphosphate (PIP2), and in the presence or absence of either Gβγ or activated Gαq.
HDX-MS reveals conformational changes in the C-terminus and PLCβ core that occur upon membrane binding
Coincubation of PLCβ2 with PS/PE/PIP2 containing lipid vesicles revealed multiple regions of significantly decreased deuterium exchange (defined as changes greater than both 4% and 0.4 Da at any timepoint with a student t-test value less than p<0.05) that occurred across multiple domains of PLCβ2 (Figure 2A–C). There were decreases in exchange that occurred on all helices of the distal CTD, spanning regions 878–931, 1020–1109 and 1169–1185. This is consistent with direct interactions of lipid with the distal CTD, which has been shown to be a primary determinant of membrane binding in PLCβ (Adjobo-Hermans et al., 2013; Kim et al., 1996; Wu et al., 1993). It is possible that some of these changes also may represent conformational changes mediated by interactions of the distal CTD with the PLC core that occur upon membrane binding, since changes were seen on multiple sides of the CTD domain (Lyon et al., 2013). There were also decreases in exchange in regions spanning the PLCβ2 core, including in the EF hand domain (residues 255–290) and C2 domain (661–681). These changes were only at the earliest time point of exchange, with these regions all being highly dynamic, as indicated by their very high percentage of deuterium exchange (Supplemental figure 1B). This indicates that the amides being protected either have no, or very limited, secondary structure. These changes were surprising since these regions would not be predicted to participate in membrane binding based on previous crystallographic studies. Intriguingly, both of these regions are very close to the C-terminus of the C2 domain, which is immediately followed by the proximal CTD domain (Figure 1, Figure 2A). This suggests that these regions are involved in allosteric changes in PLC associated with membrane binding. The region in the C2 domain with decreases in exchange would be predicted to be directly in contact with the putative autoinhibitory proximal CTD binding site based on the PLC21 structure (Lyon et al., 2011), suggesting a possible change in the interaction of the proximal CTD with the catalytic core upon membrane binding.
Figure 2. Membrane dependent changes in PLCβ2 dynamics.
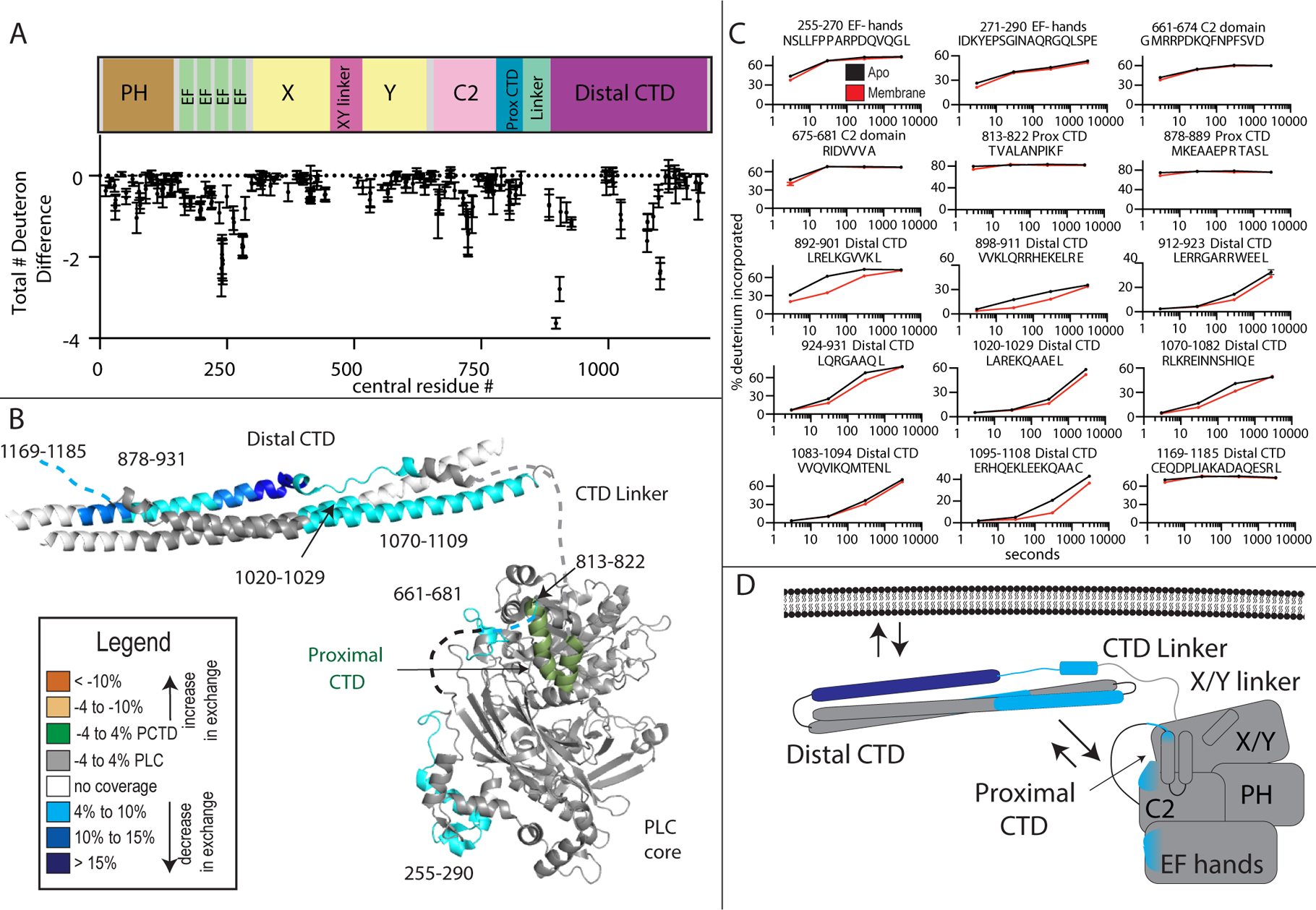
(A) The total number of deuteron difference between apo and membrane bound states for all peptides analyzed over the entire deuterium exchange time course for in PLCβ2. Every point represents the centroid of an individual peptide, with the domain schematic present above. Error bars represent S.D. (n=3). (See Table S1 for HDX data characterization)
(B) Peptides with significant changes in deuterium incorporation (both >0.4 Da and >4% and T-test p < 0.05 at time point) in the presence of membrane are mapped on a structural model of PLCβ2 as described in Fig. 1. Differences are mapped according to the legend.
(C) Representative PLCβ2 peptides displaying significant increases or decreases in exchange in the presence of membrane are shown. For all panels, error bars show SD (n=3), with most smaller than the size of the point. The full list of all peptides and their deuterium incorporation is shown in Data S1. For dynamic peptides with changes in exchange at only the earliest time point (813–822, 878–889, and 1169–1185) there are additional graphs in Supplemental Fig 1B that show these differences more clearly.
(D) Cartoon schematic displaying deuterium exchange differences, and potential mechanisms for altered protein dynamics.
We saw no changes in deuterium exchange consistent with the X/Y linker displacement by the membrane surface, but exchange rates in this area were extremely rapid indicating a high degree of flexibility of this region of the protein. We observed decreased deuterium incorporation in the proximal CTD, including residues 813–822 of PLCβ2, which are disordered in apo PLC crystal structures and missing from the electron density. Similar to the C2 and EF hand domains this change was only seen at very early time points of exchange. This may reflect an altered conformation of unstructured regions upon binding to membrane, or direct interaction with membranes. In the Gαq-PLCβ3 complex models this region forms a structured helix-loop-helix (HLH) that is bound directly to Gαq (Figure 3B). It is possible that the altered conformation induced by membranes may prime the proximal CTD for interaction with Gαq.
Figure 3. Gαq dependent changes in PLCβ2 dynamics.
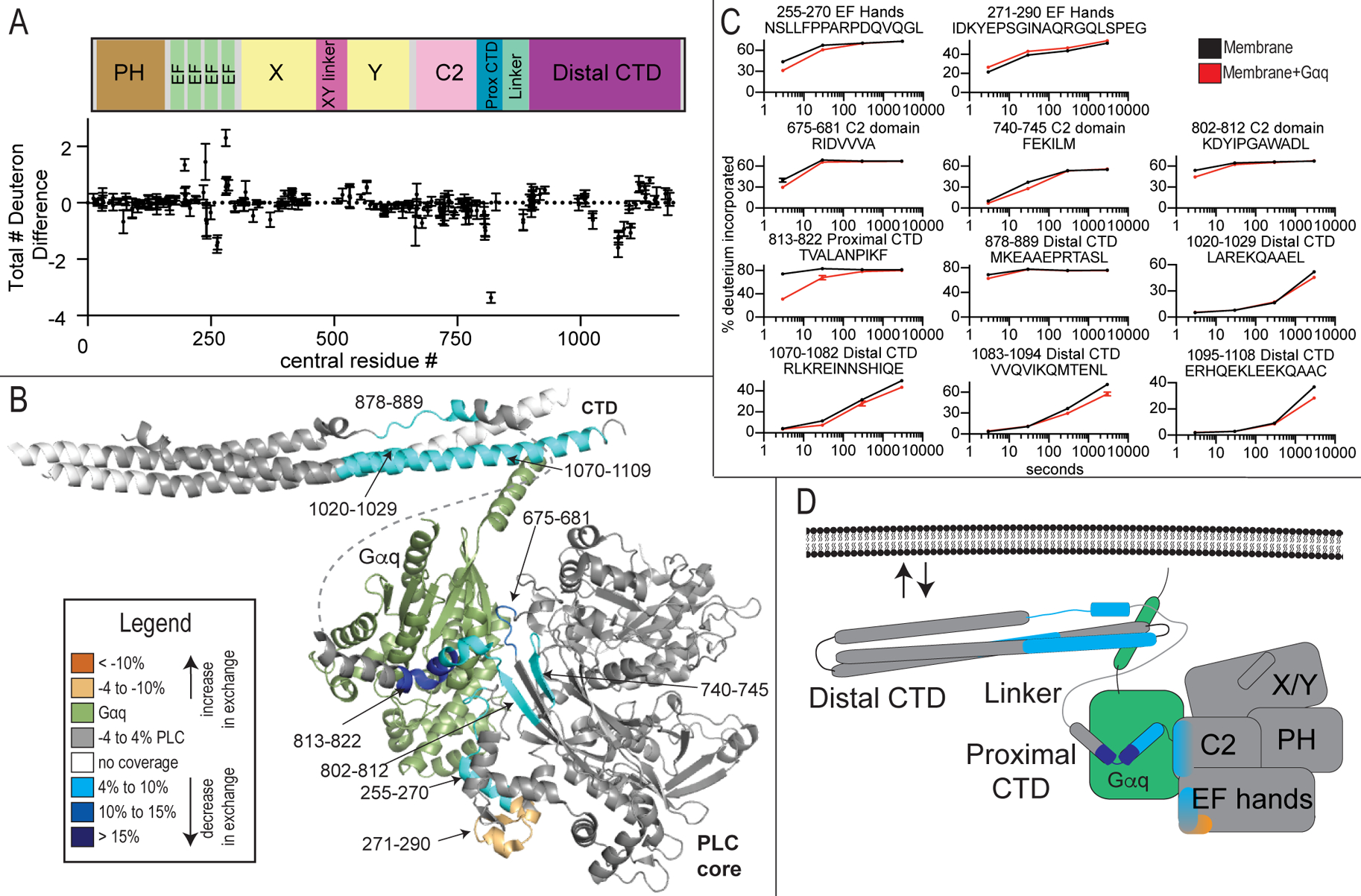
(A) The total number of deuteron difference between membrane and membrane-Gαq states for all peptides analyzed over the entire deuterium exchange time course for PLCβ2. Every point represents the centroid of an individual peptide. Error bars represent S.D. (n=3).
(B) Peptides with significant changes in deuterium incorporation (both >0.4 Da and >4% and T-test p < 0.05 at time point) in the presence of Gαq are mapped on a structural model of PLCβ2 as described in Fig. 1. Differences are mapped according to the legend.
(C) Representative PLCβ2 peptides displaying significant increases or decreases in exchange in the presence of Gαq are shown. For all peptides, error bars show SD (n=3), with most smaller than the size of the point. The full list of all peptides and their deuterium incorporation is shown in Data S1.
(D) Cartoon schematic displaying deuterium exchange differences on a model of Gαq binding.
Conformational changes that result from Gαq binding to PLCβ on membranes
Next, we compared HDX rate differences between PLC-liposome complexes to PLC-Gαq-liposome complexes. There were significant decreases in exchange at regions that correspond directly to the known binding site of Gαq on PLC in solution as determined by X-ray crystallography (Lyon et al., 2013; Lyon et al., 2011), which revealed that the complex of PLC with Gαq on membranes is consistent with the structure in solution. This included the portion of the proximal CTD (813–822) that forms part of the principle binding site for Gαq (Lyon et al., 2013; Waldo et al., 2010) showing a >40% decrease in deuterium incorporation (Figure 3A–D) at the earliest time points of exchange. In addition, regions of the EF hand (residues 255–272), and C2 domain (675–681, 740–745, and 802–812), either at the interface with Gαq, or directly adjacent, also showed small but significant decreases in exchange upon binding to Gαq on membranes. This region of the EF hand directly corresponds to structural elements directly involved activation of the GTPase activity of Gαq (Waldo et al., 2010). There was also an increase in exchange in a region of the EF hands (271–290) adjacent to the Gαq binding interface, revealing allosteric conformational changes that occur upon Gαq binding. The distal CTD showed decreases in exchange directly at the contact site with the Gαq N-terminal region (CTD region 1070–1109), as well as other regions directly adjacent (878–889 and 1020–1029). Gαq appears to contact the N-terminal helix in the Gαq-full length PLCβ3 co-crystal structure but there was some concern that this may represent a crystal packing artefact (Lyon et al., 2013). The results from the HDX experiments are consistent with contacts observed in the static X-ray crystal structures, and confirms that these contacts are retained outside of the crystal lattice and in the presence of lipid membranes. Additional conformational alterations are revealed that may be critical for activation.
Gβγ binding to PLCβ on membranes reveals significant conformational changes in the distal CTD
Next, we compared changes in deuterium incorporation between PLC-membrane complexes and PLC-Gβγ-membrane complexes. The most striking and unexpected effect of Gβγ binding to PLCβ was increased deuterium exchange on almost the entire distal CTD of PLCβ2 (multiple peptides spanning 892–1114) (Figure 4A–D). This is not due to competition for membrane binding because the other membrane associated changes in dynamics i.e. EF hand and C2 domain do not show this increase. (Figure 4A–D). Additionally, even under the high protein concentrations used for HDX experiments, Gβγ still increased PLC activity 3–4-fold (Supplemental figure 2). This indicates that Gβγ causes PLCβ2 to undergo a conformational change that rearranges the distal CTD. One possibility is that Gβγ binding leads to disruption of an auto-inhibitory interaction of the CTD with the PLCβ2 core, but no increases in exchange in the PLCβ2 core were observed that would be consistent with this. However; disruption of intramolecular interactions of the CTD with the core, with concomitant binding of Gβγ to the core would result in no net change in deuterium incorporation in the core.
Figure 4. Gβγ binding to PLCβ2 causes conformational changes in the distal CTD domain.
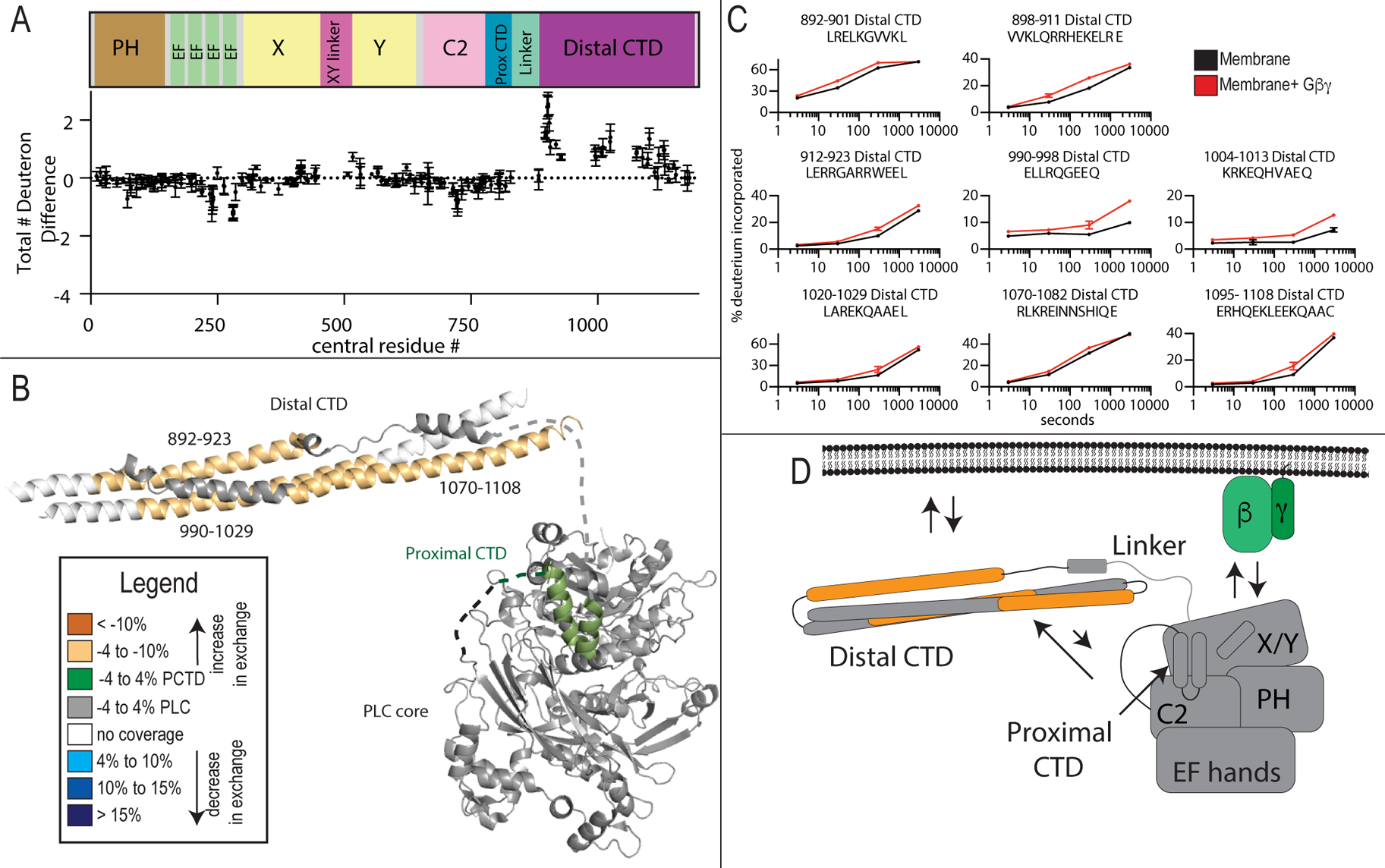
(A)The total number of deuteron difference between membrane and membrane- Gβγ states for all peptides analyzed over the entire deuterium exchange time course for PLCβ2. Every point represents the centroid of an individual peptide. Error bars represent S.D. (n=3). (See also Figure S2)
(B) Peptides with significant changes in deuterium incorporation (both >0.4 Da and >4% T-test p < 0.05 at any time point) in the presence of membrane are mapped on a structural model of PLCβ2 as described in Fig. 1. Differences are mapped according to the legend.
(C) Representative PLCβ2 peptides displaying significant increases or decreases in exchange in the presence of Gβγ are shown. For all panels, error bars show SD (n=3), with most smaller than the size of the point.
(D) Cartoon schematic displaying deuterium exchange differences upon Gβγ, and potential mechanisms for altered protein dynamics.
The distal CTD is autoinhibitory
To test the hypothesis that the distal CTD was autoinhibitory, we generated a PLCβ2 lacking the distal CTD by truncating the protein at amino acid 856 (PLCβ2-ΔCTD). A diagram of this truncation is shown in Figure 5A. We then examined the ability of PLCβ2-ΔCTD to be activated by Gβγ. We incubated varying concentrations of Gβγ with PLCβ2-ΔCTD or full-length PLCβ2 in a PLC enzyme activity assay using phospholipid vesicles containing PIP2 substrate. We predicted that the distal CTD-deleted PLC would have increased responsiveness to Gβγ stimulation as it would lack one of the autoinhibitory constraints opposing activation by Gβγ. Gβγ activated PLCβ2-ΔCTD with increased efficacy but no change in the EC50 (Figure 5B). Basal Ca2+ stimulated activity of PLCβ2-ΔCTD was similar to that of full-length PLCβ2 (supplemental figure 3). These data suggest that the distal CTD acts an autoinhibitory domain.
Figure 5: The Distal CTD inhibits Gβγ activation of PLCβ2.
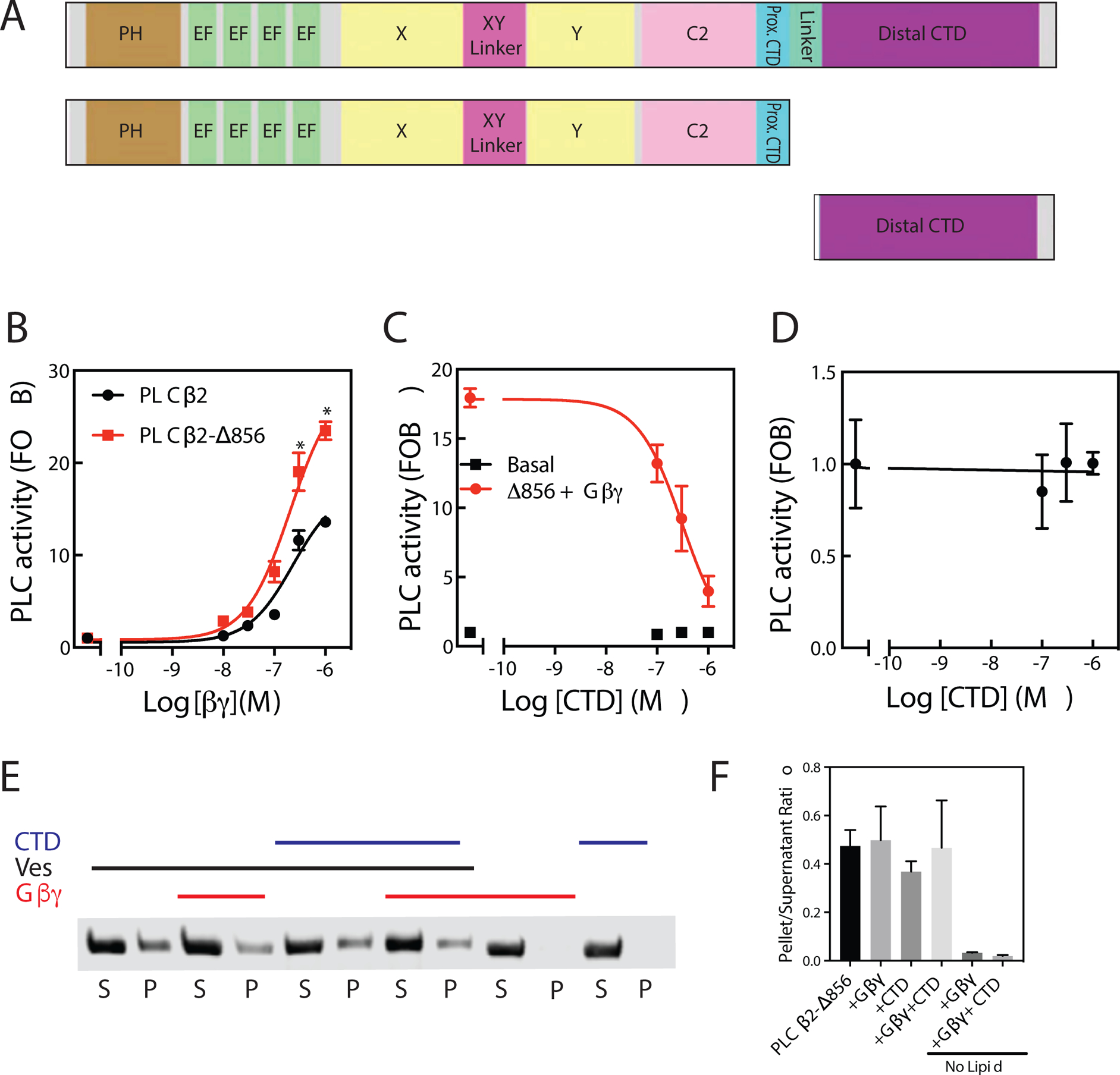
(A) Schematic of PLCβ2 constructs. (B) Assay of PLC enzymatic activity for the indicated purified PLC protein with the indicated concentrations of purified Gβγ subunits performed at least 3 times in duplicate. The data shown are mean ± S.D. and analysed by 2-way ANOVA with multiple comparisons post-test. *, p < 0.05 versus Full length PLCβ2 at equal concentration of Gβγ. (C) Assay of PLC enzymatic activity for the PLCβ2-ΔCTD with 250 nM Gβγ subunits and incubated with the indicated concentrations of purified CTD performed at least 3 times in duplicate. The data shown are mean ± S.D. (D) Expanded version of panel C to emphasize the PLCβ2-ΔCTD basal activity. The data shown are mean ± S.D. (Basal CPM = 234±15, total CPM = 6700 in the assay) (See also Figure S3). (E) Representative western blot image of PLCβ following ultracentrifugation in the presence (+) or absence (−) of PE/PIP2 liposomes, 1 μM CTD and/or purified 500ng Gβγ as indicated. (F) The ratio of protein present in the supernatant samples versus the respective total protein control samples is shown. No significant differences in lipid binding for PLCβ2-ΔCTD in the presence of CTD or Gβγ was detected. Data represent at least three independent experiments. The data shown are mean ± S.D.
To provide further evidence that the distal CTD is autoinhibitory, we tested whether the addition of purified distal CTD could inhibit Gβγ stimulated PLCβ activity when incubated with PLCβ2-ΔCTD in trans. We expressed and purified the triple helical bundle of the distal CTD (residues 864–1184) and incubated varying concentrations of purified CTD with Gβγ and PLCβ2-ΔCTD and measured PLC enzyme activity. We predicted that the distal CTD would inhibit Gβγ-dependent PLC activation. The purified CTD inhibited Gβγ stimulated activity in a concentration-dependent manner while having no appreciable effect on basal activity (Figure 5C and D).
One possible explanation of these results is that, since the CTD binds lipids, the CTD added in trans could compete for binding of PLC to the vesicle surface and thereby inhibit PLC activity. The observation that CTD addition did not affect PLC basal activity argues against this hypothesis, but to further test this idea, we measured binding of purified PLCβ2 binding to lipid vesicles, in the presence or absence of purified CTD, using a centrifugation-based lipid binding assay. At the highest concentration of CTD tested in the PLC activity reconstitution assay, the CTD had no significant effect on lipid binding of PLCβ2-ΔCTD (Figure 5 E, F). Taken together, these data suggest the CTD is inhibitory to PLC activity, and this inhibition is through direct binding of the distal CTD to the catalytic core of PLCβ2.
CTD movement is important for activation of PLCβ
To determine if the movement of the distal CTD upon Gβγ binding observed by HDX-MS is important for activation we generated a construct lacking the flexible linker between the proximal CTD and distal CTD (PLCβ2-Δ-linker) (Figure 6A). We hypothesized that deleting this linker would prevent the distal CTD from forming auto-inhibitory interactions with the catalytic core due to decreased conformational flexibility (Figure 6B). If movement of the distal CTD was important for activation, it would be expected that PLCβ2-Δ-linker would have increased basal activity, and would not be further activated by Gβγ. We examined activation of PLCβ2-Δ-linker by G-proteins in a cos-7 cell co-transfection assay. As expected, PLCβ2-Δ-linker had increased basal activity that was not increased in the presence of either Gβγ or Gαq (Figure 6 C and D). Since no further increase was seen in activity in the presence of either G-protein, this suggests a unified mechanism where the orientation of the proximal and distal CTD domains is critical for PLCβ2 activation by heterotrimeric G proteins. These data also agree with the changes in dynamics we see in HDX with Gβγ suggesting that the rearrangement of the distal CTD is an integral step in Gβγ activation.
Figure 6: Restriction of Distal CTD movement increases PLC activity.
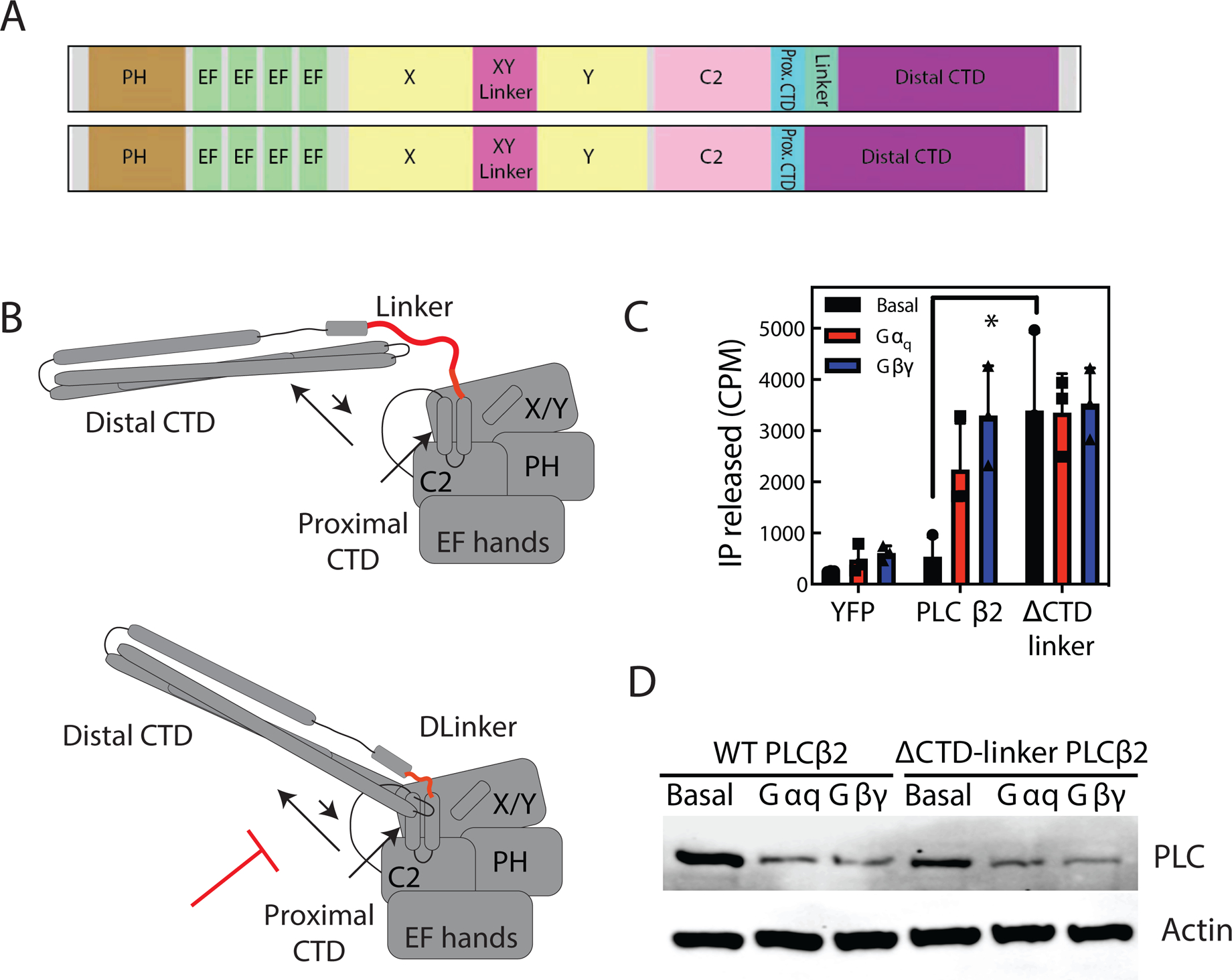
(A) Schematic of CTD-linker deletion (amino acids 839–872 deleted). (B) Cartoon diagram of model of CTD-linker deletion effects. By preventing C-terminal domain rearrangement, CTD linker would prevent autoinhibition. (C) Total inositol phosphate assay. COS-7 cells were transfected with 250 ng PLCβ2 WT or PLCβ2-ΔCTD-linker in the presence or absence of 200 ng Gβ1 and 200 ng Gγ2 or 200 ng Gαq, and total [3H] inositol phosphate accumulation was measured. The data shown are mean ± S.D. for at least three independent experiments and analysed by 2-way ANOVA with multiple comparisons. *, p < 0.05. (D) Western blot for PLCβ2 wt and PLCβ2-ΔCTD-linker mutant expressed in C, and actin as a loading control.
Proximal CTD inhibits PLC activation
As the mechanism of activation of PLCβ by Gαq has been established to use both the distal and proximal CTD, and these two domains are covalently tethered, we hypothesized that distal CTD rearrangement could destabilize the proximal CTD, preventing it from adopting an autoinhibited state. We tested PLCβ2 with a c-terminal deletion of both the proximal CTD and distal CTD (PLCβ2-Δ818) and a mutation (PLCβ2-P819A) predicted to disrupt formation of the helix loop helix conformation of the proximal CTD that is stabilized upon Gαq binding (Figure 7A). Both of these mutations greatly reduced basal PLC activity, but also prevented activation by wild type Gαq, and, surprisingly, constitutively active Rac1(G12V), seen as a significant reduction in the stimulated activity relative to basal activity when Gαq and Rac1 are cotransfected. (Figure 7B, C). This indicates this region is important for activation of PLCβ2 by these G proteins. While the Gβγ-stimulated activity was reduced (probably as a result of the overall reduction in the PLC activity of these mutants), there was little effect on the ability of Gβγ to increase PLC activity relative to the basal activity of that mutant. This suggests less reliance on proximal CTD interactions with the core for the mechanism for activation by Gβγ.
Figure 7: The Proximal CTD is involved PLC activation by Gαq and Rac1.
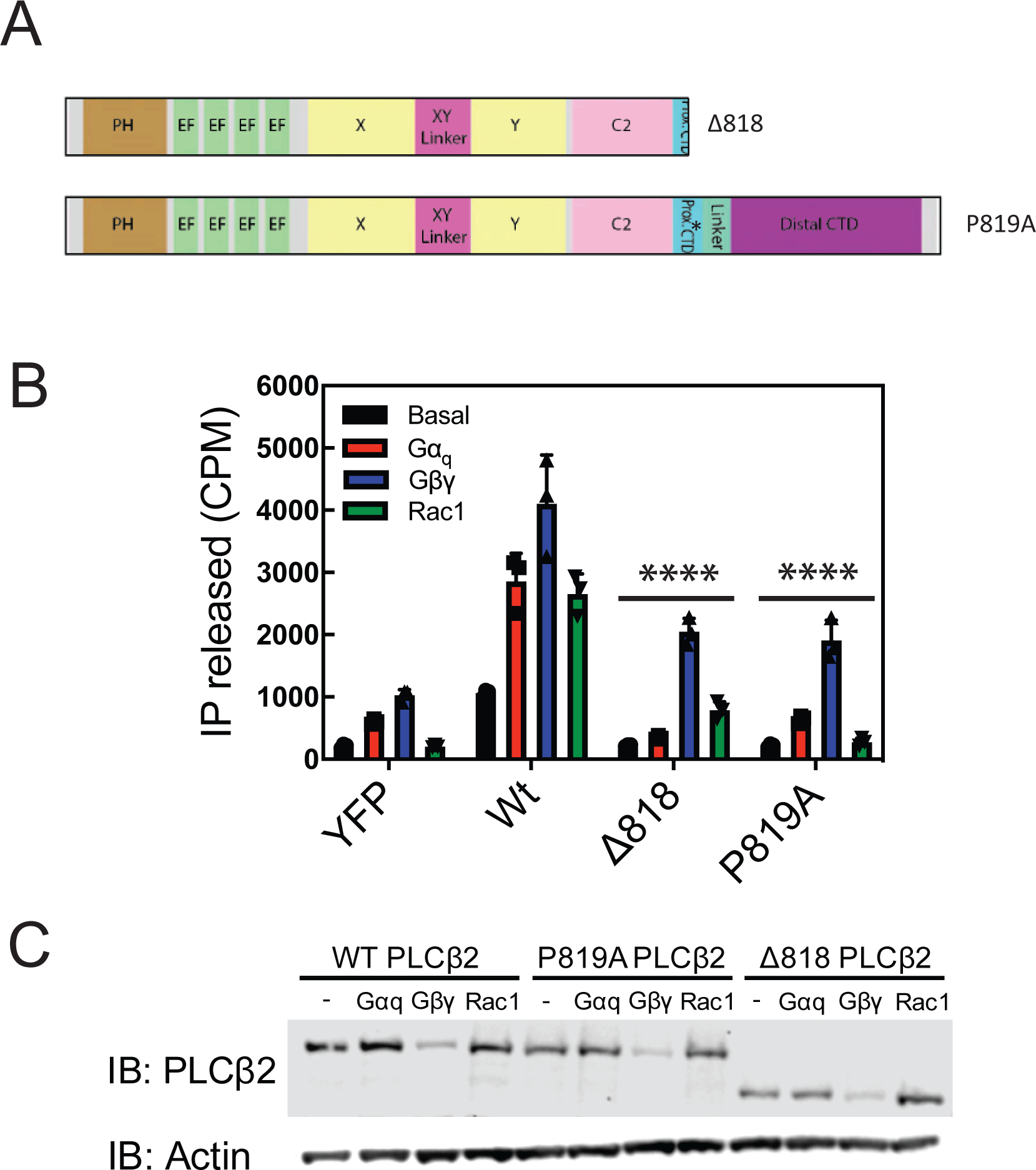
(A) Schematic of mutants and deletions of PLCβ2. (B) COS-7 cells were transfected with 250ng of WT PLCβ2, P819A PLCβ2, or Δ818-PLCβ2 in the presence or absence of 200ng Gβ1 and 200 ng Gγ2 or 300 ng Rac1G12V or 200ng wild type Gαq and total [3H] inositol phosphate accumulation was measured. The data shown are mean ± S.D. for at least three independent experiments and analysed by 2-way ANOVA with multiple comparisons. ****, p < 0.001 statistically different that wt PLC transfection. (C) Western blot for PLCβ2 wild type and mutants in B and C with an actin loading control.
Discussion:
Here we used HDX-MS to examine the molecular mechanisms that mediate PLCβ3 activation by GPCR subunits Gαq/Gβγ on membranes. Intriguingly, we find that Gβγ binding causes dramatic conformational rearrangement of the distal CTD, suggesting that the CTD is involved in activation by Gβγ. As part of our model for PLCβ activation, we propose that rearrangement of the distal CTD breaks autoinhibitory contacts between the CTD and the catalytic core as part of the mechanism for enzyme activation by Gβγ. To provide support for this idea we show 1) that the CTD can the inhibit the activity of the catalytic core when added in-trans, and 2) restricting interactions of the CTD with the catalytic core through removal of the flexible linker strongly increased PLCβ activity that was not further activated by G proteins. This model is consistent with prior CryoEM analysis where 30% of the particles used to create the Cryo-EM projections were in a conformation with the distal CTD contacting the catalytic core (Lyon et al., 2013). That PLCβ2 lacking the CTD was still activated by Gβγ indicates that other mechanisms, beyond disruption of CTD-core interactions, may also be involved in Gβγ activation. In prior work, and in the work presented here, deleting the CTD linker to restrict CTD domain interactions with the core, and perhaps other conformational alterations involving the CTD, led to increased basal activity and that could not be further increased by Gαq (Lyon et al., 2013), or Gβγ (this work Figure 6). This indicates that conformational reorientation of the distal CTD, likely involving, at least in part, disruption of CTD-core interactions, is important for activation by both Gβγ and Gαq.
Deletion or mutation of the proximal CTD did not dramatically inhibit Gβγ-dependent activation of PLC, suggesting that rearrangement of the proximal CTD is not required for Gβγ activation. These data indicate that rearrangement of either the proximal or distal CTDs are generally involved in all activation processes, while proximal CTD rearrangement is critical for regulation of activity Gαq and Rac but not Gβγ.
It is interesting to note incubation with phospholipid vesicles caused decreased deuterium exchange in several domains not thought to be involved in membrane binding. Domains far from the active site and the C-terminus that show decreased dynamics on lipid vesicles include the EF hands and the proximal CTD. This suggests that there are conformational changes in PLC that occur upon membrane binding, and this might be due to either changed inter-domain interfaces or conformational changes in PLC upon membrane binding. Since membranes are critical for the allosteric activation of PLCβ enzymes by G-proteins, these conformational changes could be essential for activation by Gβγ or Gα, as predicted for G-protein activation of class I PI3Ks (Dbouk et al., 2012; Vadas et al., 2013). It has been shown that conformational flexibility involving EF hands 3 and 4, and the PH domain are important for activity of multiple PLC variants (Garland-Kuntz et al., 2018).
Surprisingly, we did not observe G-protein or membrane dependent changes in deuterium exchange in either the X/Y linker, PH domain, or the Y domain. There were no changes in the dynamics of the interface between the PH domain and EF hand in the presence of membranes or G proteins. Overall our data does not provide evidence to support rearrangement of the X/Y linker by the membrane, a dynamic PH-EF hand interface for Gβγ binding, or binding of Gβγ to the Y domain, but the HDX-MS data are not incompatible with these processes.
A surprise from our data is that activation of PLCβ by different G-proteins converge on a unifying mechanism involving disruption of inhibition by the c-terminal extension, with the different proximal and distal CTD domains participating in different ways. On membrane surfaces, Gαq interfaces directly with the proximal CTD and distal CTD, with these interactions disrupting all auto-inhibitory interactions. On membranes Gβγ disrupts auto-inhibition by the distal CTD, most likely due to it forming a direct interaction with a portion of the PLCβ core. The binding interface for Gβγ on the PLC core is unknown. It is unclear why no regions were protected from exchange by Gβγ binding. It is possible that the region exposed upon disruption of the distal CTD domain inhibitory interface is what is responsible for Gβγ binding, and this would lead to no overall difference in deuterium incorporation.
Our data, taken together, suggest an activation mechanism of PLC by Gβγ where Gβγ causes a conformational rearrangement of the distal CTD through an allosteric mechanism, breaking the interaction between the catalytic core and the distal CTD, while Gαq binds directly to both the proximal and distal CTD in order to break autoinhibitory interactions of the C-terminal domain, in this way activation of PLCβ by either G-protein involves C-terminal domain rearrangement but each G-protein engages this regulatory region of PLC differently, Gαq utilizing direct interaction while Gβγ somehow coordinates a conformational change involving this region. Further studies will be required to conclusively define the exact molecular mechanisms by which Gβγ both directly binds to PLCβ, and releases auto-inhibition.
STAR Methods:
Resource Availability
Lead contact:
Further information and requests for resources and reagents will be fulfilled by the lead contact Alan Smrcka (avsmrcka@umich.edu).
Materials availability:
All unique reagents generated in this study are available from the lead contact.
Data and code availability:
The published article includes all datasets generated or analysed during this study.
Experimental Model and Subject Details
Cell Lines: COS-7 cells were obtained from ATCC and High Five (Trichoplusia ni) and and Sf9 (Spodoptera frugiperda) insect cells were obtained from Thermo Fisher and were authenticated by the suppliers. Both cells were used to a maximum of 20 passages. The sex of these cell lines is unknown.
Method Details
Plasmid constructs and baculoviruses
Variations of the wild -type human PLCβ2 in either pFastbac1 or pMT2 were generated using Quick Change XL II site -directed mutagenesis kit (Agilent Genomics) following manufacturers protocol then verified by sequencing. Baculoviruses were generated using the Fastbac recombinant baculovirus system (Invitrogen/Thermo Fisher Scientific, Inc.) and Sf9 (Spodoptera frugiperda) insect cells were used for virus production. For expression in mammalian cells Gβ1, Gγ2 and EYFP were in pCI-Neo (Madukwe et al., 2018). Rac1(G12V) and Gαq were in pCDNA 3.1+.
Co-Transfection of COS-7 cells with Gβ1, γ2, Rac1 or Gαq with PLCβ and quantitation of phospholipase C activity
All cell culture reagents were obtained from Invitrogen. COS-7 cells were seeded in 12-well culture dishes at a density of 100,000 cells per well and maintained in high-glucose Dulbecco’s modified Eagle’s medium containing 5% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C. The following day indicated plasmids were transfected using Lipofectamine 2000 (Invitrogen) transfection reagent (2.1 μl Lipofectamine per 1 μg of DNA). Total DNA varied from 700 to 900 ng per well and included EYFP control vector as necessary to maintain equal amount of DNA per well within individual experiments (see figure legends for experiment-specific conditions). Approximately 18–24 h after transfection, the culture medium was changed to low-inositol Ham’s F-10 medium (Gibco) containing 1.5 μ Ci/well Myo-[2-3H(N)]inositol (Perkin Elmer) for 16–18h. Accumulation of [3H]inositol phosphate was quantitated after the addition of 10 mM LiCl for 1 h to inhibit inositol monophosphate phosphatases. Media were aspirated and cells washed with 1× PBS, followed by the addition of ice-cold 50 mM formic acid to lyse cells. Soluble cell lysates containing [3H] inositol phosphate were transferred onto Dowex AGX8 chromatography columns to separate total IP by anion exchange chromatography. Columns were washed with 50 mM formic acid, then 100 mM formic acid and eluted with 1.2 M ammonium formate into scintillation vials containing scintillation fluid and counted. Basal activity is defined as the IP released of the transfected PLC construct relative to the YFP control and in the absence of transfection of any protein activators (G proteins).
Purification of Gβγ and Gαq
Purification of Gβ1γ2 (Gβγ) was performed by co-expressing Gβγ with His6 Gαi1 in High Five insect cells and nickel-agarose chromatography as described previously (Davis et al., 2005; Kozasa and Gilman, 1995). One liter cultures of High Five cells were triply infected with baculovirus encoding G protein His6-αi1, γ2 and β1 subunits. Cells were harvested 60 h postinfection, harvested by centrifugation at 2500g, frozen in liquid N2 and stored at −80 °C. All procedures were at 4 °C unless otherwise indicated. Cells pellets were thawed in 15 ml of lysis buffer (50 mM HEPES, pH8.0, 3 mM MgCl2, 10 mM β-mercaptoethanol, 0.1 mM EDTA, 100 mM NaCl, 10 μM GDP, and protease inhibitors (133 μM PMSF, 21 μg/ml TLCK and TPCK, 0.5 μg/ml) by freeze-thawing 4x in liquid nitrogen. The lysate was diluted to 100 ml with lysis buffer, distributed into multiple centrifuge tubes and membranes were harvested by centrifugation at 100,000g for 30 min. Each membrane pellet was suspended by douncing in 5 mL extraction buffer (50 mM HEPES, pH8.0, 3 mM MgCl2, 50 mM NaCl, 10 mM β-mercaptoethanol, 10 μM GDP, and protease inhibitors) and combined and diluted to 60 ml. Cholate was added to 1% and the mixture was stirred for 1 h at 4C to extract membrane proteins. Detergent extracts were clarified by centrifugation at 100,000 g for 45 min and pellets were discarded. The supernatant was diluted 5-fold with buffer A1 (50 mM HEPES, pH8.0, 3 mM MgCl2, 10 mM β-mercaptoethanol, 100 mM NaCl, 10 μM GDP, 0.5% polyoxyethylene(10) lauryl ether (C12E10), and protease inhibitors) and applied to a packed 4 mL column of Ni-NTA agarose pre-equilibrated with buffer A. The column washed a 4C with 100 mL of buffer A1+300mM NaCl+5 mM imidazole. The column was brought to room temperature and washed with 12 mL buffer A1+300 mM NaCl+5 mM imidazole at 30 C. The Gβ1γ2 subunits were eluted from bound (His)6-Gαi1 using 6 successive 4 ml elutions with RT elution buffer (bufferA+150 mM NaCl, 5 mM imidazole, 50 mM MgCl2, 10 mM NaF, 10 μM AlCl3, and 1% cholate) separately collected on ice. Elutions were analysed by SDS-PAGE on 12% acrylamide gels and Coomassie staining to assess purity. To concentrate the protein and exchange buffers and detergents, fractions containing 95% pure Gβγ were pooled and loaded onto a column packed with 0.5 mL ceramic hydroxyapatite preequilibrated with 20 mM HEPES, pH 8, 1 mM DTT, 100 mM NaCl and 1% octyl-β-D-glucopyranoside. The column was washed with 2 ml of equilibration buffer and eluted with equilibration buffer+200 mM K-phosphate, pH 8.0. Protein concentrations were determined by an Amido Black protein assay(Schaffner and Weissmann, 1973), fractions were snap frozen in liquid N2 and stored at −80 °C. The final protein concentration was 20–40 μM.
Gαq was purified according to established methods with minor modifications (Kozasa and Gilman, 1995). Gαq, His6Gβ1, Gγ2, and Ric-8A (Tall and Gilman, 2004)were co-expressed from baculoviruses in 4L of High-Five cells. The cells were collected by centrifugation and lysed by nitrogen cavitation using a disruption vessel (Parr Industries). Membranes were collected by high speed centrifugation. The membranes were washed and Dounce homogenized and extracted with 1% sodium cholate detergent for 1 hour at 4 °C. The sodium cholate detergent extract was diluted to 0.2% sodium cholate and 0.5% C12E10 detergent before application to the Ni-NTA column. The Ni-NTA column was washed while exchanging detergents to 11 mM (3-(3-Cholamidopropyl)dimethylammonio)-1-propanesulfonate) (CHAPS) and eluted with AlF4− -containing buffer. The eluate was applied to a Hi-trap Q HP column using an FPLC and eluted with a linear NaCl gradient. Fractions containing Gαq were concentrated in storage buffer (20 mM Hepes pH 8.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 11 mM CHAPs, 10 μM GDP) using an Amicon ultracentrifugal device with 30,000 MWCO. Elutions were analysed by SDS-PAGE on 12% acrylamide gels and Coomassie staining to assess purity. Protein concentrations were determined by an Amido Black protein assay (Schaffner and Weissmann, 1973), fractions were snap frozen in liquid N2 and stored at −80 °C. The final protein concentrations was 60 μM.
Purification of His6 PLCβ2, PLCβ2-ΔCTD, PLCβ2 C-terminal domain.
PLCs were purified based on modification of previously published methods (Ghosh et al., 2004). Sf9 insect cells were cultured in S6900 II serum free media and infected with baculovirus at a density of 2×106 cells/mL at an MOI of 1. After 48h cells were harvested by centrifugation, frozen in liquid N2 and stored at −80°C. Frozen insect cell pellets expressing His6 PLCβ2 and His6 PLCβ2-ΔCTD, or His6 C-terminal domain of PLCβ2 were rapidly thawed and lysed by Dounce on ice in lysis buffer containing 20 mM HEPES, pH 8, 50 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM EDTA, 0.1 M EGTA, 133 μM PMSF, 21 μg/ml TLCK and TPCK, 0.5 μg/ml, Aprotonin, 0.2 μg/ml Leupeptin, 1 μg/ml Pepstatin A, 42 μg/ml TAME, 10 μg/ml SBTI. The lysate was centrifuged at 100,000 × g, and the supernatant was collected and diluted with lysis buffer with NaCl to a final concentration of 800 mM. The diluted supernatant was then centrifuged at 100,000 × g and the supernatant was loaded onto a Ni-NTA column pre-equilibrated with buffer A (20 mM HEPES, pH 8, 100 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM EDTA, and 0.1 M EGTA). The column was washed with 3 column volumes (CVs) of buffer A, followed by 3 CVs of buffer A supplemented with 300 mM NaCl and 10 mM imidazole. The protein was eluted from the column with 3–10 CVs of buffer A, supplemented with 200 mM imidazole. Proteins were concentrated and loaded onto tandem Superdex 200 columns (10/300 GL; GE Healthcare) equilibrated with SEC buffer (20 mM HEPES, pH 8, 200 mM NaCl, 2 mM DTT, 0.1 mM EDTA, and 0.1 M EGTA). Fractions of interest at greater than 95% purity were confirmed by SDS-PAGE and Coomassie staining, pooled, concentrated, and snap frozen in liquid nitrogen. Protein concentrations were determined by an Amido Black protein assay.
Hydrogen deuterium exchange mass spectrometry (HDX-MS)
HDX experiments were performed with or without liposomes (final concentration 0.1 mg/mL) containing brain porcine phosphatidylethanolamine, brain porcine phosphatidylserine and brain porcine phosphatidylinositol 4,5-bisphosphate in a 10:5:1 molar ratio (Avanti Polar Lipids) in a total reaction volume of 50 μl with a final concentration of 400 nM PLC, 1mM NaF, 30μM AlCl3 and 600 nM Gαq or Gβγ where appropriate. Deuterium incorporation was initiated by the addition of 40 μl of deuterium oxide buffer (75 mM HEPES pH 7.5, 50 mM NaCl, and 69% (v/v) final deuterium oxide). The constructs were allowed to exchange over three time courses: 3, 30, 300, and 3000s at 16 °C. Exchange was terminated by the addition of 25 μl of ice-cold quench buffer (3% formic acid, 2M guanidine), and samples were immediately frozen in liquid nitrogen.
MS experiments were carried out as described previously (Vadas et al., 2017). In brief, samples were quickly thawed and injected onto an ultra-performance LC system (Dionex Ultimate 3000 RSLC nano system coupled to a leap technologies PAL RTC) at 2 °C. The samples were subjected to two immobilized pepsin columns (Applied Biosystems, Poroszyme, 2-3131-00) at 10 and 2 °C, respectively, at a flow of 200 μl/min for 3 min. Peptides were subsequently collected and desalted on a VanGuard precolumn Trap column (Waters) and were eluted onto an Acquity 1.7 μM particle, 100 × 1 mm2 C18 ultra-performance LC column (Waters). Separation and elution of peptides from the analytical column was achieved using a gradient ranging from 3 to 70% mobile phase B (buffer A: 0.1% formic acid, LC/MS grade; buffer B: 100% acetonitrile, LC/MS grade) over 16 min. Mass spectrometry analyses were performed using an Impact II TOF (Bruker) with an electrospray ionization source operated at a temperature of 200 °C and a spray voltage of 4.5 kV, and data were acquired over a range of 150–2200 m/z. Peptides were identified using a target decoy database search in the software program PEAKS 7. The database was composed of common affinity purification protein contaminants, pepsin, and PLC. The search parameters were set with a precursor tolerance of 15 ppm, fragment mass error 0.015 Da, charge states from 1–7, a false discovery rate of 1%, leading to a selection criteria of peptides that had a −10logP score of >23.3.
Levels of deuterium incorporation were calculated using the HD-Examiner Software (Sierra Analytics), and data from each individual peptide were inspected for correct charge state, quality of spectra, and the presence of overlapping peptides. Levels of deuteration were computed by HD-Examiner using the centroid of the isotopic cluster corresponding to each peptide at each time point. The deuteration levels calculated are presented as relative levels of deuterium incorporation, with the only correction for back exchange being for the amount of deuterium present in the buffer (69%). The back exchange of the fluidics system was 20–45% as validated through the use of a fully deuterated model protein. The data were further analyzed and curated using Excel, and changes in deuteration levels above 4% and 0.4 Da with a paired t test value of p < 0.05 were deemed to be significant. All deuterium incorporation values for all peptides can be found in the xls file in the Data S1. For figures data is shown both with percent and number of deuterium incorporation/difference to not bias towards data from short or long peptides. The full analysis parameters as required by the IC-HDX-MS (Masson et al., 2019) for all experiments are found in supplemental table 1.
Liposome Pulldown:
200 μM hen egg white phosphatidylethanolamine and 50 μM brain phosphatidylinositol 4,5-bisphosphate (Avanti Polar Lipids) were mixed and dried under N2. Lipids were resuspended in sonication buffer (50 mM HEPES, pH 7, 80 mMKCl, 3 mM EGTA, and 2 mM DTT) and sonicated for 2 minutes. Liposomes (130 μl) were combined with 200 ng of PLCβ2 and sonication buffer to a final volume of 200 μl in the presence or absence of 1 μM CTD and/or 500 ng Gβγ. All samples were incubated for 1 h on ice. Samples were then centrifuged at 100,000 × g for 1 h. The supernatant was removed, and the pellet was resuspended in 200 μl of sonication buffer. For analysis by SDS-PAGE and Image Studio, 16 μl of the supernatant, or resuspended pellet was denatured with 4 μl of 5× SDS loading dye, and 10 μl of the total sample was analyzed by SDS-PAGE using anti-PLCβ2 antibody (Invitrogen Ca#PA5–75551).
Phospholipase C activity assay
The assay was performed as described previously (Bonacci et al., 2005), with minor modification. Briefly, lipids containing ~5000 cpm of [3H-inositol]PIP2, 37.5 μM PIP2, and 150 μM phosphatidylethanolamine were mixed with 1.4 ng of PLCβ2 and equimolar amounts of PLCβ2-ΔCTD (1 ng). Varying concentrations of Gβγ were added to the PLC and lipid mixture. The reaction was set at 30 °C for 30 min in the presence of 1.5 mM CaCl2 and 3 mM EGTA (Free Ca2+=100nM), and quenched using ice cold 10% TCA. Released soluble [3H] IP3 was separated through the addition of 5% BSA and centrifugation then measured by liquid scintillation counting. Basal activity is defined as the activity in the presence of Ca2+ and no other protein activators.
Western blotting
Cells were lysed in 1× SDS sample buffer, boiled, and loaded onto a 10% (w/v) SDS–polyacrylamide gel electrophoresis (PAGE). After SDS-PAGE, proteins were transferred to PDVF for 16 hours at 25 V, which was followed by incubation with an antibody against either PLCβ2 (Thermo Fisher PA5–75551) (1:2000) or Actin (Novus 8H10D10) (1:2000). Goat anti-rabbit, or goat anti-mouse HRP secondary antibody (1:10,000) was added respectively. Western blots were imaged and quantified with a GeneGnome imaging system.
Quantification and Statistical Analysis
Mass spectrometry
Experiments were performed with two separate preparations of protein with triplicate mass spectrometry runs for each. Changes in deuteration levels above 4% and 0.4 Da with a paired t test value of p < 0.05 were deemed to be significant and reported only if they were seen in both experiments.
Cell biology and biochemistry
All transfections were performed in triplicate technical replicates with at least three independent biological replicate experiments. All biochemical assays were performed in at least three independent experiments. All biological replicates (1 biological replicate is N=1) were analyzed with a two way ANOVA with a multiple comparisons post test for statistical significance. P values are defined in the figure legends.
Supplementary Material
Data S1. Related to figures 2–4 and STAR Methods “Hydrogen deuterium exchange mass spectrometry (HDX-MS)”: Source HDX-MS data in Excel format.
Acknowledgements.
Work in the AVS laboratory was supported by a grant from the National Institutes of Health R35GM127303. Work in the JEB laboratory was supported by NSERC through a discovery grant (NSERC-2014–05218) and NSERC Canada Graduate Scholarship-Masters to MLJ.
Footnotes
Declaration of Interests.
The authors declare no competing interests.
References
- Adjobo-Hermans MJ, Crosby KC, Putyrski M, Bhageloe A, van Weeren L, Schultz C, Goedhart J, and Gadella TW Jr. (2013). PLCβ isoforms differ in their subcellular location and their CT-domain dependent interaction with Gαq. Cell Signal 25, 255–263. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1987). Inositol tisphosphate and diacylglycerol: two interacting second messengers. Ann Rev Biochem 56, 159–193. [DOI] [PubMed] [Google Scholar]
- Bianchi E, Norcini M, Smrcka A, and Ghelardini C (2009). Supraspinal Gβγ-dependent stimulation of PLCβ originating from G inhibitory protein-μ opioid receptor-coupling is necessary for morphine induced acute hyperalgesia. J.Neurochem 111, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacci TM, Ghosh M, Malik S, and Smrcka AV (2005). Regulatory interactions between the amino terminus of G-protein βγ subunits and the catalytic domain of PLCβ2. J Biol Chem 280, 10174–10181. [DOI] [PubMed] [Google Scholar]
- Charpentier TH, Waldo GL, Barrett MO, Huang W, Zhang Q, Harden TK, and Sondek J (2014). Membrane-induced Allosteric Control of Phospholipase C-β Isozymes. J Biol Chem 289, 29545–29557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TL, Bonacci TM, Sprang SR, and Smrcka AV (2005). Structural Definition of a Preferred Protein Interaction Site in the G protein β1γ2 heterodimer. Biochemistry 44, 10593–10604. [DOI] [PubMed] [Google Scholar]
- Dbouk HA, Vadas O, Shymanets A, Burke JE, Salamon RS, Khalil BD, Barrett MO, Waldo GL, Surve C, Hsueh C, et al. (2012). G protein-coupled receptor-mediated activation of p110bby Gβγ is required for cellular transformation and invasiveness. Sci Signal 5, ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filtz TM, Grubb DR, McLeod-Dryden TJ, Luo J, and Woodcock EA (2009). Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J 23, 3564–3570. [DOI] [PubMed] [Google Scholar]
- Garland-Kuntz EE, Vago FS, Sieng M, Van Camp M, Chakravarthy S, Blaine A, Corpstein C, Jiang W, and Lyon AM (2018). Direct observation of conformational dynamics of the PH domain in phospholipases C and β may contribute to subfamily-specific roles in regulation. J Biol Chem 293, 17477–17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Wang H, Kelley GG, and Smrcka AV (2004). Purification of phospholipase C β and phospholipase C ε from Sf9 cells. Met Mol Biol 237. [DOI] [PubMed] [Google Scholar]
- Han DS, Golebiewska U, Stolzenberg S, Scarlata SF, and Weinstein H (2011). A dynamic model of membrane-bound phospholipase Cβ2 activation by Gβγ subunits. Mol Pharmacol 80, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden TK, Waldo GL, Hicks SN, and Sondek J (2011). Mechanism of Activation and Inactivation of Gq/Phospholipase C-β Signaling Nodes. Chem Rev 111, 6120–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks SN, Jezyk MR, Gershburg S, Seifert JP, Harden TK, and Sondek J (2008). General and versatile autoinhibition of PLC isozymes. Mol Cell 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes AL, Arthur JF, Zhang T, Miyamoto S, Adams JW, Dorn GW 2nd, Woodcock EA, and Brown JH (2003). Akt-mediated cardiomyocyte survival pathways are compromised by G α q-induced phosphoinositide 4,5-bisphosphate depletion. J Biol Chem 278, 40343–40351. [DOI] [PubMed] [Google Scholar]
- Kadamur G, and Ross EM (2013). Mammalian phospholipase C. Annu Rev Physiol 75, 127–154. [DOI] [PubMed] [Google Scholar]
- Kadamur G, and Ross EM (2016). Intrinsic Pleckstrin Homology (PH) Domain Motion in Phospholipase C-β Exposes a Gβγ Protein Binding Site. J Biol Chem 291, 11394–11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CG, Park D, and Rhee SG (1996). The role of carboxyl-terminal basic amino acids in Gqα- dependent activation, particulate association, and nuclear localization of phospholipase C-β1. J Biol Chem 271, 21187–21192. [DOI] [PubMed] [Google Scholar]
- Kozasa T, and Gilman AG (1995). Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α 12 and inhibition of adenylyl cyclase by α z. J Biol Chem 270, 1734–1741. [DOI] [PubMed] [Google Scholar]
- Kuang Y, Wu Y, Smrcka A, Jiang H, and Wu D (1996). Identification of a phospholipase C β2 region that interacts with Gβγ. Proc Natl Acad Sci U S A 93, 2964–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Lee KH, Lee SB, Park D, and Rhee SG (1994). Regulation of phospholipase C-β4 by ribonucleotides and the α subunit of G q J Biol Chem 269, 25335–25338. [PubMed] [Google Scholar]
- Lee SB, Shin SH, Hepler JR, Gilman AG, and Rhee SG (1993). Activation of phospholipase C-·2 mutants by G protein α q and βγ subunits. J Biol Chem 268, 25952–25957. [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, and Wu D (2000). Roles of PLCβ2 and β3 and PI3K in Chemoattractant-Mediated Signal Transduction. Science 287, 1046–1049. [DOI] [PubMed] [Google Scholar]
- Lyon AM, Dutta S, Boguth CA, Skiniotis G, and Tesmer JJ (2013). Full-length Gα(q)-phospholipase C-β3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat Struct Mol Biol 20, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon AM, Tesmer VM, Dhamsania VD, Thal DM, Gutierrez J, Chowdhury S, Suddala KC, Northup JK, and Tesmer JJG (2011). An autoinhibitory helix in the C-terminal region of phospholipase C-β mediates Gαq activation. Nat Struct Mol Biol 18, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madukwe JC, Garland-Kuntz EE, Lyon AM, and Smrcka AV (2018). G protein βγ subunits directly interact with and activate phospholipase C ε. J Biol Chem 293, 6387–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson GR, Burke JE, Ahn NG, Anand GS, Borchers C, Brier S, Bou-Assaf GM, Engen JR, Englander SW, Faber J, et al. (2019). Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Methods 16, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran B, Osterhout J, Wu D, and Smrcka AV (1998). Identification of a structural element in phospholipase C β2 that interacts with G protein βγ subunits. J Biol Chem 273, 7148–7154. [DOI] [PubMed] [Google Scholar]
- Schaffner W, and Weissmann C (1973). A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 56, 502–514. [DOI] [PubMed] [Google Scholar]
- Singer AU, Waldo GL, Harden TK, and Sondek J (2002). A unique fold of phospholipase Cβ mediates dimerization and interaction with G αq. Nat Struct Biol. 9, 32–36. [DOI] [PubMed] [Google Scholar]
- Smrcka AV, Brown JH, and Holz GG (2012). Role of phospholipase Cε in physiological phosphoinositide signaling networks. Cell Signal 24, 1333–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Hepler JR, Brown KO, and Sternweis PC (1991). Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 251, 804–807. [DOI] [PubMed] [Google Scholar]
- Smrcka AV, and Sternweis PC (1993). Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C β by G protein α and βγ subunits. J Biol Chem 268, 9667–9674. [PubMed] [Google Scholar]
- Tall GG, and Gilman AG (2004). Purification and functional analysis of Ric-8A: a guanine nucleotide exchange factor for G-protein α subunits. Methods in enzymology 390, 377–388. [DOI] [PubMed] [Google Scholar]
- Taylor SJ, Chae HZ, Rhee SG, and Exton JH (1991). Activation of the β1 isozyme of phospholipase C by α subunits of the G q class of G proteins. Nature 350, 516–518. [DOI] [PubMed] [Google Scholar]
- Vadas O, Dbouk HA, Shymanets A, Perisic O, Burke JE, Abi Saab WF, Khalil BD, Harteneck C, Bresnick AR, Nurnberg B, et al. (2013). Molecular determinants of PI3Kgamma-mediated activation downstream of G-protein-coupled receptors (GPCRs). Proc Natl Acad Sci U S A 110, 18862–18867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadas O, Jenkins ML, Dornan GL, and Burke JE (2017). Using Hydrogen-Deuterium Exchange Mass Spectrometry to Examine Protein-Membrane Interactions. Methods Enzymol 583, 143–172. [DOI] [PubMed] [Google Scholar]
- Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, et al. (2010). Kinetic Scaffolding Mediated by a Phospholipase C–β and Gq Signaling Complex. Science 330, 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Dowal L, El-Maghrabi MR, Rebecchi M, and Scarlata S (2000). The pleckstrin homology domain of phospholipase C-β(2) links the binding of Gβγ to activation of the catalytic core. J Biol Chem 275, 7466–7469. [DOI] [PubMed] [Google Scholar]
- Woodcock EA, Kistler PM, and Ju YK (2009). Phosphoinositide signalling and cardiac arrhythmias. Cardiovascular Res 82, 286–295. [DOI] [PubMed] [Google Scholar]
- Woodcock EA, Suss MB, and Anderson KE (1995). Inositol Phosphate Release and Metabolism in Rat Left Atria. Circ Res 76, 252–260. [DOI] [PubMed] [Google Scholar]
- Wu D, Jiang H, Katz A, and Simon MI (1993). Identification of critical regions on phospholipase C-β1 required for activation by G-proteins. J Biol Chem 268, 3704–3709. [PubMed] [Google Scholar]
- Xie W, Samoriski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, and Wu D (1999). Genetic alteration of phospholipase C beta3 expression modulates behavioral and cellular responses to mu opioids. Proc Natl Acad Sci U S A 96, 10385–10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Related to figures 2–4 and STAR Methods “Hydrogen deuterium exchange mass spectrometry (HDX-MS)”: Source HDX-MS data in Excel format.
Data Availability Statement
The published article includes all datasets generated or analysed during this study.