

Graphical abstract
Keywords: Virtual screening, SARS CoV-2, Boceprevir, MD simulation, MM-PBSA
Abstract
SARS-CoV-2 has posed global challenge for healthcare due to COVID-19. The main protease (Mpro) of this virus is considered as a major target for drug development efforts. In this work, we have used virtual screening approach with molecular dynamics simulations to identify high affinity and low molecular weight alternatives of boceprevir, a repurposed drug currently being evaluated against Mpro. Out of 180 compounds screened, two boceprevir analogs (PubChem ID: 57841991 and 58606278) were reported as potential alternatives with comparable predicted protease inhibitor potential and pharmacological properties. Further experimental validation of the reported compounds may contribute to the ongoing investigation of boceprevir.
1. Introduction
As the world keeps on battling the COVID-19 pandemic, worldwide endeavors are progressing for the rapid identification of effective vaccines as well as antivirals. At present there are 63 vaccines under clinical trials (https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines). Antivirals such as Ivermectin and Danoprevir (clinical trial ID: NCT04435587 and NCT04291729 respectively) are under phase IV of clinical trial among others. Albeit multiple vaccines and inhibitors have entered clinical trials, endeavors to develop inhibitors and antibodies are as yet critical to treat this disease on account of clinical need and mutations in the virus that could influence the efficacy of the antiviral agents [1], [2].
The single stranded positive-sense RNA genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) comprises of ~ 30,000 nucleotides [3], [4]. The replicase gene of the virus, encompassing more than 21,000 nucleotides, encodes two overlapping polyproteins, pp1a (~486 kDa) and pp1ab (~790 kDa). These polyproteins are essential for the replication and transcription of coronaviruses. The main protease, a 33.8-kDa cysteine protease, and papain-like protease (PLpro) are essential for proteolytic processing of these polyproteins into functional peptides in a multistep process [5], [6]. As a drug target, PLpro is not so much preferred as it recognizes the C-terminal sequence of ubiquitin, and compounds inhibiting PLpro may also inhibit host cell deubiquitinases. However, the Mpro cleaves at a very specific cleave site of the polypeptide sequences and prefers a glutamine residue at the P1 site. The cleavage site of Mpro is [Leu-Gln↓ (Ser, Ala, Gly)] (↓: cleavage site). Such cleavage specificity is not reported in any human proteases [7], [8]. These functional attributes make Mpro an attractive target for drug development studies. The determination of multiple experimental structures of Mpro in its apo form and with co-crystallized ligands dramatically facilitates the process of drug discovery. The usual drug development process typically costs a lot of time and money. Hence, multiple efforts are going on for computational screening of large chemical libraries as well as re-purposing antivirals for the rapid identification of lead compounds with the potential to be translated into effective and safe drugs.
The main protease (Mpro) from SARS-CoV-2 shows structural similarity with other main proteases and has three domains. The two domains, domains I (residues 8–101) and II (residues 102–184) have mainly antiparallel β-barrel structures while domain III covering amino acid residues 201–303 contain five α- helices [6], [9]. The substrate-binding site (Met49, Gly143, Ser144, His163, His164, Met165, Glu166, Leu167, Asp187, Arg188, Gln189, Thr190, Ala191, and Gln192), located in the cleft between domain I and II and partly in the loop joining the domain II and III, harbors the catalytic dyad formed by Cys145 and His41 [10].
Boceprevir (M.W.: 519.68 g/mol,) is an FDA approved drug against hepatitis C virus (HCV) that targets serine protease NS3 for inhibitor. It is reported to be well tolerated with few drug-drug interactions, and no major hepatic or renal toxicity [11]. A recent report suggests that boceprevir inhibits the enzymatic activity of Mpro with an IC50 value of 4.13 µM [12]. The cellular cytotoxicity testing was also found to be well tolerated with a CC50 value of over 100 µM for all the cell lines tested. The same study also reported that boceprevir has potent antiviral activity against SARS-CoV-2. The crystal structure of Mpro complexed with boceprevir (PDB ID: 6WNP) also shows that it effectively binds to the catalytic residues.
Since boceprevir is a promising candidate to be an effective drug, we have used virtual screening in combination with molecular dynamics (MD) simulations to find a possible candidate with better binding from a library of compounds similar to boceprevir. Among the 180 low molecular weight boceprevir analogs, two compounds (PubChem ID: 57841991 and 58606278) formed stronger and stable interactions to the binding site than boceprevir. We hope this study will aid the rapid discovery of inhibitors against Mpro with more efficiency.
2. Materials and methods
2.1. Structure-based virtual screening
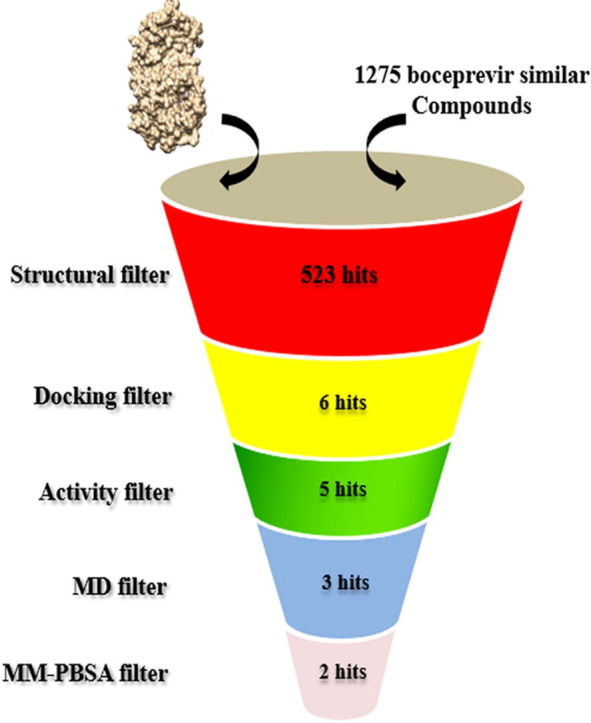
Out of 1,275 similar compounds of boceprevir available in PubChem database [13], we retrieved a total of 180 compounds having molecular weight less than 519.68 g/mol with available 3D conformers. Compounds having a Tanimoto score of 0.9 or greater were considered as similar compounds based on 2D similarity[14]. The Mpro structure at room temperature (PDB ID: 6WQF) [9] was selected for docking since it had the suggested protonation states of histidine residues, and the structure was solved at room temperature. It has been suggested that this structure solved at room temperature is more appropriate for molecular docking studies as it provides more relevant information at physiological temperatures [9]. The compounds were energy minimized with the steepest descent algorithm prior to docking using UCSF Chimera [15]. The selected compounds, along with boceprevir as control were docked by centering the grid box centered on the substrate-binding site of Mpro (x, y and z center coordinates :36.56, 46.43 and 56.04) with the AutoDock vina program [16]. The screening was performed with a exhaustiveness value of 8 and ten runs for each compound, and the best pose was selected based on the lowest binding free energy (kcal/mol). The 2D ligand interactions were visualized with the Discovery studio visualizer [17].
2.2. Biological activity and pharmacokinetic property prediction
The PASS (Prediction of Activity Spectra for Substances) server (http://www.pharmaexpert.ru/passonline/) [18] was utilized to predict the protease inhibition probability of the selected compounds. The server can predict pharmacological effects and biochemical mechanisms of a compound based on the structural formula and the prediction may be analyzed based on the ratio of probability to be active (Pa) and probability to be inactive (Pi). The absorption (A), distribution (D), metabolism (M), and toxicity (T) profiles of the compounds were calculated based on human intestinal absorption (HIA), blood–brain barrier permeability, CYP2D6 inhibitor/substrate, and Ames mutagenicity and mouse carcinogenicity parameters respectively. The excretion parameter is represented by HIA, as it is a sum of bioavailability and absorption evaluated from the ratio of excretion or cumulative excretion in urine, bile, and feces [19]. The pharmacokinetic parameters were predicted by using the pre-ADMET server (https://preadmet.bmdrc.kr/) [20].
2.3. Molecular dynamics simulation
MD Simulation is a routinely applied method to decipher the dynamic interactions between biomolecules with small molecules, peptides, or nanoparticles [21], [22], [23], [24]. For exploring the binding dynamics, the apo-protein, and docked complexes were simulated using GROMACS v.2018 with the GROMOS 54a7 force field [25]. PRODRG server was used to calculate the ligand parameters [26]. The catalytic dyad is suggested to be activated by a proton transfer from Cys145 to His41 possibly activated by substrate binding [9]. Therefore, the His41 and Cys145 were prepared in their neutral state, with His41 protonated at Nδ. Based on previous studies protonation states of other histidine residues were assigned at the Nδ (HISA) or Nε (HISB) sites as follows: HISA64, HISA80, HISB163, HISB164, HISB172, and HISB246 [27], [28]. The system was solvated in a cubic box of 1.2 nm containing SPC water molecules. The addition of NA+ ions neutralized the charge of the systems. The steepest descent method was utilized for energy minimization for 50,000 steps for all systems with a tolerance of 1000 kJ mol-1nm−1. NVT and NPT equilibrations were carried out at 300 K and 1 bar using the V-rescale thermostat and Berendsen barostat [9]. The final production of 130 ns was performed using the leap-frog integration with Nose-Hoover and Parinello-Rahman couplings [29]. Bond-lengths were constrained using the Linear Constraint Solver (LINCS) algorithm [30]. The trajectories were analyzed using GROMACS inbuilt utilities.
2.4. MM-PBSA analysis
To estimate the post-simulation binding free energy of the complexes, the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) approach was utilized. This approach has been successfully applied to various protein–ligand/peptide complexes [31], [32]. The binding free energy, and per residue energy contributions were calculated using the g_mmpbsa tool [33]. This GROMACS tool estimates Gibb's free energy of binding using the MM-PBSA method as described by the equation below. The SASA model was used for calculation of nonpolar solvation energy in this study.
Gbind = (ΔEelec + ΔEvdw) + (ΔGpolar + ΔGSASA)
The g_mmpbsa tool also calculates the energy contribution of each residue by decomposing the total binding free energy at the residue level [33].
3. Results
3.1. Interaction of the compounds with Mpro
The control compound boceprevir (PubChem ID: 10324367) scored −6.8 kcal/mol while docked with AutoDock vina. The top six compounds 58606278, 58605188, 59115738, 58908774, 58605473, and 57841991 were selected based on the binding energy (Fig. 1 and Table 1 ). All the compounds interacted within the binding pocket and formed interactions with the vital substrate-binding residues and the catalytic residues His41 and Cys145 (Table 1 and Fig. 2 ). Boceprevir formed 5 hydrogen bonds (H-bonds) with the binding site residues Thr26, Asn142, Gly143, Cys145 and Gln189. The compound 58606278 formed 6H-bonds with the residues Thr26, Asn142, Gly143, Cys145 and Glu166. The compound 58605188 formed 4H-bonds with the residues Asn142, Gly143, Cys145, and Glu166.The compound 59115738 formed 5H-bonds with the residues. The compound 58908774 formed 6H-bonds with the residues Thr26, Asn142, Gly143, Cys145, Glu166 and Gln189. The compound 58605473 formed 6H-bonds with the residues Thr26, Asn142, Gly143, Cys145, Glu166 and Gln189. The compound 57841991 formed 5H-bonds with the residues Gly143, His163, Glu166 and Arg188. In all the compounds, Glu166 formed H-bonds consistently. The catalytic residue His41 formed either van der Waals or akyl bond with all the compounds. Other substrate-binding residues formed van der Waals, akyl and carbon hydrogen bonds. Details of the docking results of the rest of the compounds were shown in supplementary table 1.
Fig. 1.

2D chemical structures of boceprevir and top scoring compounds.
Table 1.
Details of the top six compounds including boceprevir (10324367) from the docking analysis with Mpro substrate binding site. Residues forming H-bonds were shown in bold.
| SL No | Ligand | MW. g/mol |
Binding Energy (kcal/mol) | H-bond interactions P-L [Length in Å] |
Other interactions |
|---|---|---|---|---|---|
| 1 | 10324367 |
519.68 |
−6.8 |
NH(Thr26)-O [2.57] HD22(Asn142)-O [2.52] NH(Gly143)-O [2.12] HG(Cys145)-O [2.71] HE21(Gln189)-O [2.53] |
Thr25#, Leu27#, His41#, Cys44#, Thr45#, Ser46#, Met49*, Phe140#, Leu141#, Ser144#, His163*, His164#, Met165*, Glu166#, Arg188# |
| 2 | 58606278 |
517.66 |
−7.5 | O(Thr26)–NH [2.85] O(Thr26)–NH [2.66] HD22(Asn142)-O [2.81] NH(Gly143)-O [1.89] HG(Cys145)-O [2.88] O(Glu166)–NH [2.14] |
Thr25#, Leu27#, His41#, Cys44#, Ser46#, Met49*, Leu141#, His163#, Met165*, Leu167*, Pro168*, Arg188#, Gln189#, Thr190#, Gln192# |
| 3 | 58605188 |
515.64 | −7.4 | HD22(Asn142)-O [2.51] NH(Gly143)-O [2.41] HG(Cys145)-O [2.81] O(Glu166)–NH [2.52] |
Thr25#, Thr26#, Leu27#, His41*, Cys44#, Ser46#, Met49*, Phe140#, Leu141#, His163*, Met165*, Leu167#, Pro168*, Arg188$, Gln189#, Thr190#, Gln192# |
| 4 | 59115738 | 517.66 | −7.3 |
O(Thr26)–NH [2.73] HD22(Asn142)-O [2.43] NH(Gly143)-O [2.36] HG(Cys145)-O [2.89] O(Glu166)–NH [2.22] |
Thr25#, Leu27#, His41#, Cys44#, Ser46#, Met49#, Phe140#, Leu141#, His163#, Met165*, Leu167*, Pro168*, Arg188#, Gln189#, Thr190#, Gln192# |
| 5 | 58908774 | 477.6 | −7.2 |
O(Thr26)–NH [2.60] HD22(Asn142)-O [2.53] NH(Gly143)-O [2.32] HG(Cys145)-O [2.84] O(Glu166)–NH [2.63] HE21(Gln189)-O [2.30] |
Thr25#, Leu27#, His41#, Cys44#, Ser46#, Met49#, Phe140#, Leu141#, His163*, Met165*, Leu167#, Pro168*, Arg188$, Thr190#, Gln192# |
| 6 | 58605473 | 477.60 | −7.2 |
O(Thr26)–NH [2.65] HD22(Asn142)-O [2.54] NH(Gly143)-O [2.33] HG(Cys145)-O [2.82] O(Glu166)–NH [2.46] HE21(Gln189)-O[2.34] |
Thr25#, Leu27#, His41*#, Cys44#, Ser46#, Met49#, Phe140#, Leu141#, His163*, Met165*, Leu167#, Pro168*, Arg188$, Thr190#, Gln192# |
| 7 | 57841991 | 505.6 | −7.2 |
NH(Gly143)-O [2.62] HE2(His163)-O [2.22] O(Glu166)–NH [2.33] NH(Glu166)-O [2.43] O(Arg188)–NH [2.20] |
His41*, Met49*, Phe140#, Leu14#1, Asn142$, Ser144#, Cys145#, Met165$, Leu167$, Pro168$, His172#, Gln189$, Thr190#, Gln192# |
#van der Waals, *Akyl and $ carbon-hydrogen bond.
Fig. 2.

Interactions of the docked compounds to Mpro: a) Surface representation of SARS-CoV-2 Mpro showing the mode of binding by the compounds (domain I: brown, domain II: cyan, domain III: pink and loop connecting domain I and II: silver). 2D representations of the interactions: b) boceprevir, c) 58606278, d) 58605188, e) 59115738, f) 58908774, g) 58605473 and h) 57841991.
3.2. Biological activity and pharmacokinetic property prediction
The compounds were analyzed in the PASS prediction server to explore the possible protease inhibitor potential (Table 2 ). The protease inhibitory activity of the compounds was compared with boceprevir. It was observed the protease inhibitor activity predictions of the compounds are comparable to boceprevir except the compound 59115738, which scored the lowest value of 0.214. The compound 57841991 had the closest protease inhibition value to boceprevir. Although the Pa values are less than 0.5, this could mean the descriptors of the compounds to the particular activity are new to the PASS training set, as the protease inhibition capacity of boceprevir has been proved experimentally for both HCV and SARS CoV-2[12], [18], [34]. The absorption (A), distribution (D), metabolism (M), and toxicity (T) prediction values of all the compounds were found comparable to the control compound boceprevir (Table 3 ). All the compounds showed good absorption values having either well absorbed values (70–100%) or moderately absorption values (20–70%). The low distribution values (less than 0.1) of the compounds indicate that they have low absorption to central nervous system (CNS). The compounds also demonstrated no CYP2D6 inhibitor/substrate potential, Ames mutagenicity and mouse carcinogenicity similar to boceprevir. Since 59115738 had a very low probability of being a protease inhibitor and was therefore excluded from further analysis.
Table 2.
Protease inhibitor potential along with ADMT properties of the selected compounds.
| Sl no | Compound ID (PubChem) |
Protease inhibitor Activity | Antiviral (Hepatitis) | A | D | M | T | ||
|---|---|---|---|---|---|---|---|---|---|
| Pa | Pi | Pa | Pi | ||||||
| 1 | 10324367 |
0.347 | 0.006 | 0.729 | 0.002 | 75.07 | 0.07 | Non | Non |
| 2 | 58606278 | 0.287 | 0.012 | 0.714 | 0.002 | 75.53 | 0.07 | Non | Non |
| 3 | 58605188 | 0.250 | 0.019 | 0.701 | 0.002 | 75.86 | 0.06 | Non | Non |
| 4 | 59115738 | 0.214 | 0.027 | 0.731 | 0.002 | 75.47 | 0.07 | Non | Non |
| 5 | 58908774 | 0.268 | 0.015 | 0.726 | 0.002 | 66.30 | 0.05 | Non | Non |
| 6 | 58605473 | 0.268 | 0.015 | 0.726 | 0.002 | 66.30 | 0.05 | Non | Non |
| 7 | 57841991 | 0.324 | 0.008 | 0.736 | 0.002 | 72.44 | 0.06 | Non | Non |
Table 3.
The components of ligand binding free energy with Mpro. The values are in (kcal/mol).
| Ligands | Binding Energy | van der Waal Energy | Electrostatic Energy | Polar Solvation Energy | SASA Energy |
|---|---|---|---|---|---|
| Boceprevir | −5.83 ± 0.50 | −15.52 ± 1.00 | −13.54 ± 0.36 | 25.35 ± 0.57 | −2.12 ± 0.03 |
| 57841991 | −14.32 ± 0.27 | −31.88 ± 0.25 | −13.85 ± 0.99 | 35.52 ± 0.28 | −4.11 ± 0.22 |
| 58605473 | −9.01 ± 0.43 | –23.64 ± 0.21 | −2.83 ± 0.19 | 20.83 ± 0.41 | −3.37 ± 0.33 |
| 58606278 | −6.48 ± 1.93 | −20.25 ± 0.23 | −17.78 ± 0.24 | 34.65 ± 0.50 | −3.10 ± 0.03 |
3.3. MD simulation of the complexes
The binding stability of the compounds boceprevir, 58605188, 58605473, 58606278, 57841991, and 58908774 in complex with Mpro was submitted to 130 ns MD simulation each. The evolution of the backbone RMSD over time suggests a stable behavior for the boceprevir complex and the complexes made by the compounds except for the 58605188, 58605473, and 58908774 complexes which displayed comparatively inconsistent trend (Fig. 3 a). To observe the residue level fluctuations in each complex, the average RMSF of all residues and substrate-binding residues were calculated after 100 ns. High values of fluctuations were observed in the complexes made by 58605188 and 58908774. The complex made by the compound 57841991 displayed the lowest RMSF among the simulated complexes followed by 58606278 and 58605473 complexes (Fig. 3b). A similar pattern of RMSF was observed for the substrate-binding region as well (Fig. 3c), where the complexes formed by compounds 57841991 and 58606278 displayed comparatively lower RMSF, while the average RMSF of 58605473 complex was found to be close to boceprevir complex. The RMSF of residues His41, Met49, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, Glu166, and Leu167 were found to be lower than the apo-protein and boceprevir complex in the case of 57841991 complex. The RMSF of substrate binding residues for the 58605188 complex was found to highest among the complexes.
Fig. 3.

Comparative analysis of trajectories: a) RMSD analysis of the apo-protein and the complexes calculated over 130 ns trajectories, b) Average RMSF of protein with simulated complexes, c) RMSF of substrate binding site residues [inset: average RMSF].
The interactions of the compounds with Mpro were mapped by hydrogen bond (H-bond) graphs (Fig. 4 ). Boceprevir initially formed a maximum of 8H-bonds but the number was reduced after 50 ns (Fig. 4a). The compound 57,841,991 (Fig. 4b) formed higher H-bonds up to 10 and maintained overall 8H-bonds from 20 ns. The compound 58605188 maintained 5H-bonds consistently from 70 ns (Fig. 4c) while the compound 58605473 formed 4H-bonds in most of the simulation period (Fig. 4d). The compound 58606278 formed 6H-bonds in a comparatively consistent way throughout the simulation period (Fig. 4e). Among the complexes studied, 58,908,774 formed the lowest number of H-bonds which was found to 2 on average (Fig. 4f). These trends of interactions can be correlated with changes in the RMSF of the complexes. The higher RMSF values of the 58605188 and 58908774 complexes may lead to loss of interactions found during the docking calculations.
Fig. 4.

The H-bond graphs of the complexes with the compounds a) Boceprevir, b) 57814991, c) 58605188, d) 586054743, e) 58606278 and f) 58908774.
3.4. Binding free energy calculations
Based on the RMSD, RMSF, and H-bond interaction analysis, the complexes formed by the compounds 57841991, 58605473, and 58606278 were selected for MM-PBSA analysis along with boceprevir. Energy components of each complex were calculated for 101 snapshots that were extracted at every 0.1 ns from the final 10 ns. Among the complexes, 57841991 scored the highest binding free energy −14.32 kcal/mol, which is higher than the boceprevir complex (-5.83 kcal/mol). The other two complexes formed by 58605473 and 58606278 scored −9.01 and −6.48 kcal/mol respectively. In the case of the 57841991 complex the contributions of van der Waals, polar solvation energy, and SASA energy to the binding free energy were found to be higher among the complexes. The binding energy differences in the 58605473 and 58606278 complexes were contributed by the energy contributions from van der Waals, electrostatic energy, and polar solvation energy.
To evaluate the binding pattern, the strong energy contribution of the substrate-binding residues for these complexes were calculated (Fig. 5 ). From the residue contribution analysis, it was observed that for 58605473 complex, there is more negative contribution of substrate binding residues on average (-0.16 kcal/mol). Whereas the average energy contribution of the residues in the complexes 57841991 and 58606278 were found to be more positively contributing −0.47 kcal/mol and −0.28 kcal/mol respectively, which is better than boceprevir complex (-0.24 kcal/mol). The energy contribution from the critical catalytic residues His41 and Cys145 were also found to be stronger in these two complexes, which were found to be contributing poorly in the 58605473 complex. Other highly contributing residues in the 57841991 complex are Met165, Asp187, Arg188, Gln189, and Thr190 which were higher among the complexes.
Fig. 5.

MM/PBSA per residue energy contribution to the binding energy for the complexes formed by the compounds boceprevir, 57841991, 58605473 and 58606278.
4. Discussion
Understanding protein structure is of fundamental importance [35]. Moreover, with the growing number of daily cases of COVID-19 infections, subsequent efforts to combat the disease are also rising. Apart from the efforts to the development of a successful vaccine candidate, there are also multiple computational and experimental efforts to identify effective drugs/inhibitors either by repurposing the existing drugs or designing novel compounds [36], [37], [38], [39], [40]. Multiple studies also proposed natural compounds as potential inhibitors of SARS CoV-2 main protease[41], [42], [43], [44].
In this work, we have computationally investigated possible high affinity and low molecular weight alternative of Boceprevir, a repurposed drug currently being evaluated against SARS-CoV-2 main protease. Among the 180 boceprevir similar compounds studied, we have identified 57,841,991 and 58606278 as high affinity and lower molecular weight alternatives. The probabilities of being a protease inhibitor for the compounds 57841991 and 58606278 were also found to be very close to the investigational inhibitor boceprevir together with the predicted absorption, distribution, metabolism, and toxicity profiles. The reported compounds formed stable complexes with Mpro as suggested by RMSD and RMSF analysis and formed a consistently higher number of hydrogen bonds than boceprevir during the MD simulation analysis. These two complexes also scored higher binding energy than the boceprevir complex during the post-simulation binding energy analysis. Although the 58605473 complex scored better binding free energy than 58606278, per residue contribution to the free energy of binding revealed that the average energy contribution from the substrate-binding region was poor in the 58605473 complex. The important catalytic residues His41 and Cys145 were found to be highly contributing in the case of 57841991 and 58606278 complexes.
In conclusion, the overall observations from this computational study suggest that the compounds 57841991 and 58606278 could be investigated as low molecular weight alternatives of boceprevir. Experimental validation and optimization of the reported compounds might contribute to the development of a more potent analog of boceprevir against SARS-CoV-2.
CRediT authorship contribution statement
Subhomoi Borkotoky: . Manidipa Banerjee: . Gyan Prakash Modi: Conceptualization. Vikash Kumar Dubey: Conceptualization.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The infrastructure facilities of IIT (BHU) Varanasi and DST funded I-DAPT Hub Foundation, IIT BHU [DST/NMICPS/TIH11/IIT(BHU)2020/02] are sincerely acknowledged. The support and resources provided by PARAM Shivay Facility under the National Supercomputing Mission, Government of India at the Indian Institute of Technology, Varanasi are gratefully acknowledged. MB acknowledges the facilities of IIT Delhi. Project grant support through SERB funding [CVD/2020/000031] to VKD and GPM is acknowledged.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cplett.2021.138446.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Tang X., Wu C., Li X., Song Y., Yao X., Wu X., Duan Y., Zhang H., Wang Y., Qian Z., et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020;7(6):1012–1023. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Q., Kang C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms. 2020;8(8) doi: 10.3390/microorganisms8081250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakagawa S., Miyazawa T. Genome evolution of SARS-CoV-2 and its virological characteristics. Inflamm Regen. 2020;40:17. doi: 10.1186/s41232-020-00126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rastogi M., Pandey N., Shukla A., Singh S.K. SARS coronavirus 2: from genome to infectome. Respir. Res. 2020;21(1):318. doi: 10.1186/s12931-020-01581-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 6.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. PNAS. 2003;100(23):13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30(17) doi: 10.1016/j.bmcl.2020.127377. 127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kneller D.W., Phillips G., O'Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL M(pro) active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11(1):3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goyal B., Goyal D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Comb. Sci. 2020;22(6):297–305. doi: 10.1021/acscombsci.0c00058. [DOI] [PubMed] [Google Scholar]
- 11.Rizza S.A., Talwani R., Nehra V., Temesgen Z. Boceprevir. Drugs Today (Barc) 2011;47(10):743–751. doi: 10.1358/dot.2011.47.10.1656503. [DOI] [PubMed] [Google Scholar]
- 12.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B.A., et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016;44(D1):D1202–D1213. doi: 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S., Bolton E.E., Bryant S.H. Similar compounds versus similar conformers: complementarity between PubChem 2-D and 3-D neighboring sets. J Cheminform. 2016;8:62. doi: 10.1186/s13321-016-0163-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 16.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biovia D.S. CA, USA; San Diego: 2017. Discovery studio visualizer; p. 936. [Google Scholar]
- 18.Lagunin A., Stepanchikova A., Filimonov D., Poroikov V. PASS: prediction of activity spectra for biologically active substances. Bioinformatics. 2000;16(8):747–748. doi: 10.1093/bioinformatics/16.8.747. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y.H., Le J., Abraham M.H., Hersey A., Eddershaw P.J., Luscombe C.N., Butina D., Beck G., Sherborne B., Cooper I., et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure-activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 2001;90(6):749–784. doi: 10.1002/jps.1031. [DOI] [PubMed] [Google Scholar]
- 20.Lee SK, Lee IH, Kim HJ, Chang GS, Chung JE, No KT: The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions (Blackwell Publishing) 2003:418-420.
- 21.Jimenez-Alberto A., Ribas-Aparicio R.M., Aparicio-Ozores G., Castelan-Vega J.A. Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors. Comput. Biol. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107325. 107325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma T., Siddiqi M.I. In silico identification and design of potent peptide inhibitors against PDZ-3 domain of Postsynaptic Density Protein (PSD-95) J. Biomol. Struct. Dyn. 2019;37(5):1241–1253. doi: 10.1080/07391102.2018.1454851. [DOI] [PubMed] [Google Scholar]
- 23.Hazarika Z., Jha A.N. Computational Analysis of the Silver Nanoparticle-Human Serum Albumin Complex. ACS Omega. 2020;5(1):170–178. doi: 10.1021/acsomega.9b02340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das S., Hazarika Z., Sarmah S., Baruah K., Rohman M.A., Paul D., Jha A.N., Singha Roy A. Exploring the interaction of bioactive kaempferol with serum albumin, lysozyme and hemoglobin: A biophysical investigation using multi-spectroscopic, docking and molecular dynamics simulation studies. J. Photochem. Photobiol., B. 2020;205 doi: 10.1016/j.jphotobiol.2020.111825. 111825. [DOI] [PubMed] [Google Scholar]
- 25.Schmid N., Eichenberger A.P., Choutko A., Riniker S., Winger M., Mark A.E., van Gunsteren W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. European Biophysics J. : EBJ. 2011;40(7):843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 26.Schuttelkopf A.W., van Aalten D.M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 27.Acharya A, Agarwal R, Baker M, Baudry J, Bhowmik D, Boehm S, Byler K, Coates L, Chen SY-C, Cooper CJ: Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. 2020. [DOI] [PMC free article] [PubMed]
- 28.Paasche A., Zipper A., Schafer S., Ziebuhr J., Schirmeister T., Engels B. Evidence for substrate binding-induced zwitterion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochemistry. 2014;53(37):5930–5946. doi: 10.1021/bi400604t. [DOI] [PubMed] [Google Scholar]
- 29.Martonak R., Laio A., Parrinello M. Predicting crystal structures: the Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003;90(7) doi: 10.1103/PhysRevLett.90.075503. 075503. [DOI] [PubMed] [Google Scholar]
- 30.Hess B., Bekker H., Berendsen H.J., Fraaije J.G. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18(12):1463–1472. [Google Scholar]
- 31.Chen J-g, Wu S-f, Zhang Q-f, Yin Z-p. Zhang L: α-Glucosidase inhibitory effect of anthocyanins from Cinnamomum camphora fruit: Inhibition kinetics and mechanistic insights through in vitro and in silico studies. Int. J. Biol. Macromol. 2020;143:696–703. doi: 10.1016/j.ijbiomac.2019.09.091. [DOI] [PubMed] [Google Scholar]
- 32.Ma R., Wong S.W., Ge L., Shaw C., Siu S.W., Kwok H.F. In vitro and MD simulation study to explore physicochemical parameters for antibacterial peptide to become potent anticancer peptide. Molecular Therapy-Oncolytics. 2020;16:7–19. doi: 10.1016/j.omto.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumari R., Kumar R., Lynn A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 34.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P., et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11(1):4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J., Dubey V.K., Longo L.M., Blaber M. A logical OR redundancy within the Asx-Pro-Asx-Gly type I β-turn motif. J. Mol. Biol. 2008;377(4):1251–1264. doi: 10.1016/j.jmb.2008.01.055. [DOI] [PubMed] [Google Scholar]
- 36.Hussain I., Hussain A., Alajmi M.F., Rehman M.T., Amir S. Impact of repurposed drugs on the symptomatic COVID-19 patients. J Infect Public Health. 2020;14(1):24–38. doi: 10.1016/j.jiph.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z., Li X., Huang Y.Y., Wu Y., Liu R., Zhou L., Lin Y., Wu D., Zhang L., Liu H., et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad Sci. USA. 2020;117(44):27381–27387. doi: 10.1073/pnas.2010470117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiorucci D., Milletti E., Orofino F., Brizzi A., Mugnaini C., Corelli F. Computational drug repurposing for the identification of SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020:1–7. doi: 10.1080/07391102.2020.1796805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43(17):4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 40.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004;11(10):1445–1453. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muteeb G., Alshoaibi A., Aatif M., Rehman M.T., Qayyum M.Z. Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic. Appl. Biol. Chem. 2020;63(1):79. doi: 10.1186/s13765-020-00564-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rehman M.T., AlAjmi M.F., Hussain A. Natural Compounds as Inhibitors of SARS-CoV-2 Main Protease (3CLpro): A Molecular Docking and Simulation Approach to Combat COVID-19. Curr. Pharm. Des. 2020 doi: 10.2174/1381612826999201116195851. [DOI] [PubMed] [Google Scholar]
- 43.Verma S., Twilley D., Esmear T., Oosthuizen C.B., Reid A.M., Nel M., Lall N. Anti-SARS-CoV Natural Products With the Potential to Inhibit SARS-CoV-2 (COVID-19) Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.561334. 561334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z.R., Zhang Y.N., Li X.D., Zhang H.Q., Xiao S.Q., Deng F., Yuan Z.M., Ye H.Q., Zhang B. A cell-based large-scale screening of natural compounds for inhibitors of SARS-CoV-2. Signal Transduct. Target Ther. 2020;5(1):218. doi: 10.1038/s41392-020-00343-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.