

We present the genomes of two isolated bacteriophages infecting Pelagibacter ubique HTCC1062. Pelagibacter phage Mosig EXVC030M (Myoviridae) and Pelagibacter phage Lederberg EXVC029P (Podoviridae) were isolated by dilution-to-extinction culturing from the oxygen minimum zone at Devil’s Hole (Harrington Sound, Bermuda).
ABSTRACT
We present the genomes of two isolated bacteriophages infecting Pelagibacter ubique HTCC1062. Pelagibacter phage Mosig EXVC030M (Myoviridae) and Pelagibacter phage Lederberg EXVC029P (Podoviridae) were isolated by dilution-to-extinction culturing from the oxygen minimum zone at Devil’s Hole (Harrington Sound, Bermuda).
ANNOUNCEMENT
Viruses infecting the heterotrophic bacterial clade of Pelagibacterales are an important component of marine microbial communities throughout global oceans (1). Since the discovery and first isolation of four pelagiphages in 2013 (2), 38 more have been isolated and sequenced (3–5). Out of the 38 isolated pelagiphages, 36 belong to the Podoviridae family, with only one species each of Myoviridae and Siphoviridae. Here, we report the draft genome sequences of a novel pelagimyophage and a novel pelagipodophage, both isolated on Pelagibacter ubique HTCC1062.
A 2-liter water sample was taken (12 July 2019) using a hand-held Niskin bottle, fired at a 20-m depth at Devil’s Hole, Bermuda, a seasonal oxygen minimum zone in Bermuda (6) (latitude 32.32421, longitude −64.71849). The water sample was taken to the Bermuda Institute of Ocean Sciences for processing, where planktonic cells were removed with 0.1-μm polyethersulfone filters. Viruses were concentrated by tangential flow filtration (50R VivaFlow 100-kDa Hydrosart filter; Sartorius Lab Instruments, Göttingen, Germany). We used previously described dilution-to-extinction-based methods (4) with HTCC1062 as a bait host (grown in artificial seawater medium ASM1 [7]) in 96-well Teflon plates, which does not rely on plaque formation, because the host does not grow on solid medium. The purification process was repeated five times; nonetheless, final sequence data contained two genomes, suggesting an impure culture.
For DNA isolation, a 50-ml HTCC1062 culture (106 cells/ml), amended with 5 ml of 0.1-μm-filtered lysate, was grown in ASM1 (18°C) until cell death (detected via flow cytometry). Debris was removed using 0.1-μm-pore polyvinylidene difluoride (PVDF) filters, and lysate was subjected to PEG8000/NaCl DNA isolation (modified from https://doi.org/10.17504/protocols.io.c36yrd, as described previously (4).
DNA libraries (Nextera XT) were prepared and sequenced by the Exeter Sequencing Service (Illumina paired end [2 × 250 bp], NovaSeq S Prime [SP], targeting 30-fold coverage). Raw reads (13.18 million) were trimmed, quality controlled, and error corrected using tadpole (default settings [8] within BBMap v38.22 [https://sourceforge.net/projects/bbmap/]) and assembled with SPAdes v3.13 (7). Viral contigs were confirmed and gene called with VirSorter v1.05 (9) and imported into DNA Master v5.23.3 (10) for manual curation with additional gene calls using GenMark v2.5 (11), GeneMarkS v4.28 (12), GeneMarkS-2 v1.14 (13), GeneMark.hmm v3.25 (14), Glimmer v3.02 (15), and Prodigal v2.6.3 (16). Open reading frames were annotated with NCBI’s nonredundant protein database (17) and phmmer v2.41.1 (18) against the UniProtKB, uniprotrefprot (19), SWISS PROT (20), and Pfam (21) databases (accessed May 2020) and were evaluated using a previously described scoring system (10). Genome completion was verified with CheckV (22). Sequences similar to our isolates were identified with ClusterGenomes v5.1 (https://github.com/simroux/ClusterGenomes) and vConTACT 2 v0.9.19 (23) using previously isolated Pelagiphages (2, 3, 5), fosmid-derived contigs from Mediterranean metagenomes (uvMed) (24), and putative pelagimyophages from genome-resolved metagenomics (PMP-MAVG) (25). Conserved genes were identified (GET_HOMOLOGUES v09072020 [22]), aligned (MUSCLE v3.8.1551 [26]), curated (Gblocks v0.91b [27]), and concatenated manually (all with default settings). Bayesian inference trees were generated via Phylogeny.fr (28) using MRBAYES v3.2.7 (29) (100,000 generations, sampled every 10 generations, 5,000 tree burn-in) (Fig. 1).
FIG 1.
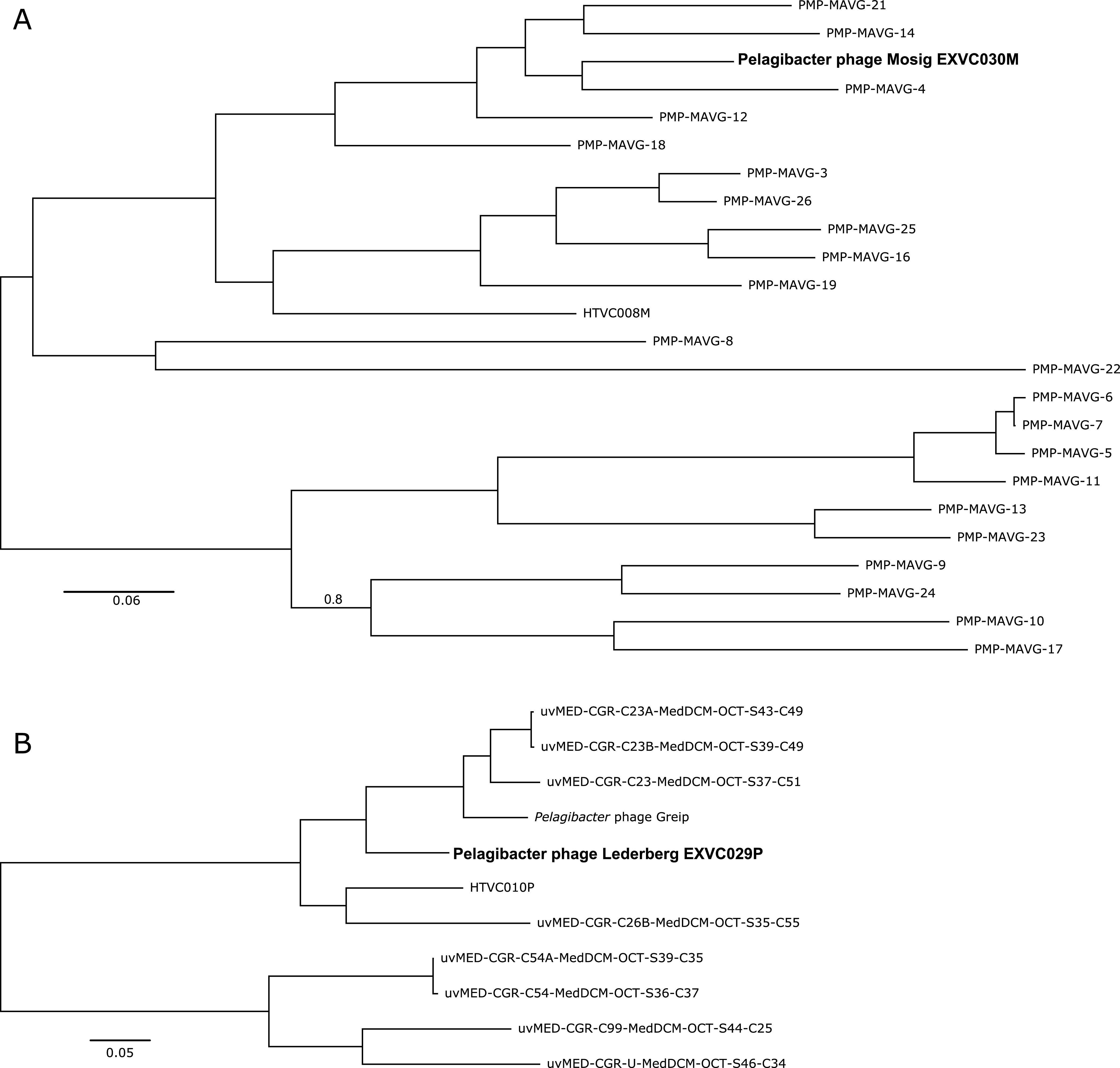
Bayesian inference tree from conserved genes found in pelagiphages (2, 3, 5), contigs from Mediterranean metagenomes (uvMed) (24), and putative pelagimyophages from genome-resolved metagenomics (PMP-MAVG) (25). (A) Terminase large subunit, tail sheath protein, and tail tube protein; (B) head-tail connector protein, capsid assembly protein, major capsid protein, tail tubular protein A, and putative acetyltransferase. Branch support values of 1 were omitted for clarity. The scale bar represents the estimated substitution per site.
Pelagimyophage Mosig (named after microbiologist Gisela Mosig in recognition of her work on Escherichia coli phage T4) was 141,462 bp long (348× coverage; GC content, 30.01%), linear, and 75.73% complete (CheckV [22]). Out of 208 genes, 98 were putative, 3 were tRNAs, 30 were structural, and 77 were associated with DNA replication.
Pelagibacter phage Lederberg (named after microbiologist Esther Lederberg in recognition of her work on the E. coli phage λ) was 33,623 bp long (5,849× coverage; GC content, 33.13%) and predicted as circularly permuted/complete. Lederberg had a total of 71 genes, out of which 9 were structural, 8 were associated with DNA replication, and 54 were without known function.
Data availability.
The complete genome sequences were deposited under GenBank accession numbers MT647605 (Lederberg) and MT647606 (Mosig). The corresponding read data were deposited in the Sequence Read Archive (SRA) under BioProject number PRJNA625644 and SRA accession number SRR12024324.
ACKNOWLEDGMENTS
The efforts of H.H.B. in this work were funded by the Natural Environment Research Council (NERC) GW4+ Doctoral Training Program. R.J.P., N.R.B., and B.T. were funded by the Simons Foundation BIOS-SCOPE program. M.M. and B.T. were part-funded by NERC (NE/R010935/1). This project used equipment funded by the Wellcome Trust Institutional Strategic Support Fund (WT097835MF), Wellcome Trust Multi-User Equipment Award (WT101650MA), and BBSRC LOLA award (BB/K003240/1).
We acknowledge the use of the University of Exeter High-Performance Computing (HPC) facility in carrying out this work.
REFERENCES
- 1.Weitz JS, Stock CA, Wilhelm SW, Bourouiba L, Coleman ML, Buchan A, Follows MJ, Fuhrman JA, Jover LF, Lennon JT, Middelboe M, Sonderegger DL, Suttle CA, Taylor BP, Frede Thingstad T, Wilson WH, Eric Wommack K. 2015. A multitrophic model to quantify the effects of marine viruses on microbial food webs and ecosystem processes. ISME J 9:1352–1364. doi: 10.1038/ismej.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao Y, Temperton B, Thrash JC, Schwalbach MS, Vergin KL, Landry ZC, Ellisman M, Deerinck T, Sullivan MB, Giovannoni SJ. 2013. Abundant SAR11 viruses in the ocean. Nature 494:357–360. doi: 10.1038/nature11921. [DOI] [PubMed] [Google Scholar]
- 3.Zhao Y, Qin F, Zhang R, Giovannoni SJ, Zhang Z, Sun J, Du S, Rensing C. 2019. Pelagiphages in the Podoviridae family integrate into host genomes. Environ Microbiol 21:1989–2001. doi: 10.1111/1462-2920.14487. [DOI] [PubMed] [Google Scholar]
- 4.Buchholz HH, Michelsen ML, Bolaños LM, Brown E, Allen MJ, Temperton B. 2021. Efficient dilution-to-extinction isolation of novel virus–host model systems for fastidious heterotrophic bacteria. ISME J doi: 10.1038/s41396-020-00872-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Qin F, Chen F, Chu X, Luo H, Zhang R, Du S, Tian Z, Zhao Y. 2020. Culturing novel and abundant pelagiphages in the ocean. Environ Microbiol doi: 10.1111/1462-2920.15272. [DOI] [PubMed] [Google Scholar]
- 6.Parsons RJ, Nelson CE, Carlson CA, Denman CC, Andersson AJ, Kledzik AL, Vergin KL, McNally SP, Treusch AH, Giovannoni SJ. 2015. Marine bacterioplankton community turnover within seasonally hypoxic waters of a subtropical sound: Devil’s Hole, Bermuda. Environ Microbiol 17:3481–3499. doi: 10.1111/1462-2920.12445. [DOI] [PubMed] [Google Scholar]
- 7.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bushnell B, Rood J, Singer E. 2017. BBMerge: accurate paired shotgun read merging via overlap. PLoS One 12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roux S, Enault F, Hurwitz BL, Sullivan MB. 2015. VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salisbury A, Tsourkas PK. 2019. A method for improving the accuracy and efficiency of bacteriophage genome annotation. Int J Mol Sci 20:3391. doi: 10.3390/ijms20143391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borodovsky M, McIninch J. 1993. GenMark: parallel gene recognition for both DNA strands. Comput Chem 17:123–133. doi: 10.1016/0097-8485(93)85004-V. [DOI] [Google Scholar]
- 12.Besemer J, Lomsadze A, Borodovsky M. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomsadze A, Gemayel K, Tang S, Borodovsky M. 2018. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res 28:1079–1089. doi: 10.1101/gr.230615.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu W, Lomsadze A, Borodovsky M. 2010. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. 2018. HMMER Web server: 2018 update. Nucleic Acids Res 46:W200–W204. doi: 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.UniProt Consortium. 2019. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bairoch A, Apweiler R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Contreras-Moreira B, Vinuesa P. 2013. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol 79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolduc B, Jang HB, Doulcier G, You Z-Q, Roux S, Sullivan MB. 2017. vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 5:e3243. doi: 10.7717/peerj.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizuno CM, Rodriguez-Valera F, Kimes NE, Ghai R. 2013. Expanding the marine virosphere using metagenomics. PLoS Genet 9:e1003987. doi: 10.1371/journal.pgen.1003987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaragoza-Solas A, Rodriguez-Valera F, López-Pérez M. 2020. Metagenome mining reveals hidden genomic diversity of pelagimyophages in aquatic environments. mSystems 5:e00905-19. doi: 10.1128/mSystems.00905-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar RC 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 28.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard J-F, Guindon S, Lefort V, Lescot M, Claverie J-M, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The complete genome sequences were deposited under GenBank accession numbers MT647605 (Lederberg) and MT647606 (Mosig). The corresponding read data were deposited in the Sequence Read Archive (SRA) under BioProject number PRJNA625644 and SRA accession number SRR12024324.