

Abstract
Medulloblastoma (MB) is a common yet highly heterogeneous childhood malignant brain tumor, however, clinically effective molecular targeted therapy is lacking. Modulation of hedgehog (HH) signaling by epigenetically targeting the transcriptional factors GLI through bromodomain-containing protein 4 (BRD4) has recently spurred new interest as potential treatment of HH-driven MB. Through screening of current clinical BRD4 inhibitors for their inhibitory potency against glioma-associated oncogene homolog (GLI) protein, the BRD4 inhibitor 2 was selected as the lead for further structural optimization, which led to the identification of compounds 25 and 35 as the high potency HH inhibitors. Mechanism profiling showed that both compounds suppressed HH signaling by interacting with the transcriptional factor GLI, and were equally potent against the clinical resistant mutants and the wild type of smoothened (SMO) receptor with IC50 values around 1 nmol/L. In the resistant MB allograft mice, compound 25 was well tolerated and markedly suppressed tumor growth at both 5 mg/kg (TGI = 83.3%) and 10 mg/kg (TGI = 87.6%) doses. Although further modification is needed to improve the pharmacokinetic (PK) parameters, compound 25 represents an efficacious lead compound of GLI inhibitors, possessing optimal safety and tolerance to fight against HH-driven MB.
Key words: Medulloblastoma, Hedgehog signaling pathway, Drug resistance, GLI, BRD4
Abbreviations: BCC, basal cell carcinoma; BET, bromo and extra C-terminal bromodomain proteins; BRD4, bromodomain-containing protein 4; hERG, human ether-a-go-go-related gene; HH, hedgehog; HTRF, homogeneous time-resolved fluorescence; i.v., intravenous injection; MB, medulloblastoma; PK, pharmacokinetic; p.o., per os; PTCH, patched; SAR, structure−activity relationship; SHH, Sonic hedgehog; SMO, smoothened; TGI, tumor growth inhibition; WNT, wingless
Graphical abstract
A systemic structure and hedgehog activity relationship study was conducted based on clinical BRD4 inhibitor ABBV-075. Compound 25 represents a new high potency hedgehog (HH) inhibitor both in vitro and in vivo, possessing optimal safety and tolerance to fight against HH-driven MB.

1. Introduction
Medulloblastoma (MB) is one of the most common childhood malignant brain tumors1,2, accounting for 6.6% of all pediatric central nervous system tumors (or 63.3% of all embryonal tumors) aging less than 19 years3. It is a small round blue cell tumor of the cerebellum with high heterogeneity, and biological characterization and molecular classification of MB remain as an unsettled challenge. Historically, MB is divided into four distinct molecular subgroups, namely: wingless (WNT), Sonic hedgehog (SHH), group 3 and group 44. Among which, the SHH contains 30% of all MB diagnosis and is the most studied MB subgroup5. The SHH MB is characterized by the somatic mutations in the SHH pathway genes (Fig. 1), including the 12-pass transmembrane receptor patched (PTCH1), G protein-coupled receptor smoothened (SMO), and the downstream transcriptional factors GLI (GLI1, GLI2, and GLI3)6,7. In addition to MB, aberrant activations of SHH have also been observed in many other cancers, such as basal cell carcinoma (BCC)8. Currently, three SHH signaling pathway inhibitors (vismodegib9,10, sonidegib11,12, and glasdegib13, Fig. 1) have been approved as SMO antagonists for clinical use of locally advanced or metastatic BCC, or acute leukemia. Unfortunately, clinical trials of these compounds with MB patients have not yet met success due to their transient effects as well as SMO mutation-induced drug resistance during treatment14, 15, 16. Therefore, the standard treatment for MB after surgery remains to be craniospinal irradiation, chemotherapy or their combinations, which are associated with many adverse effects17, 18, 19. Although more precise molecular classification of MB and the genomic and epigenetic alterations have been recently reported4, clinically effective treatment of MB remains to be an unmet medical need20.
Figure 1.

The hedgehog signaling pathway and representative inhibitors of SMO, GLI and BRD4. Ac, acetylation; BRD4, bromodomain-containing protein 4; GLI, glioma-associated oncogene homolog protein; GLIA, activated GLI; HH, hedgehog; PTCH, patched; SMO, smoothened; SUFU, suppressor of fused.
Since the transcriptional factor GLI is the final effector of the canonical HH signaling, acting downstream of SMO, inhibition of GLI function may overcome clinically observed drug resistance of SMO antagonist treatment20, 21, 22, 23. Meanwhile, Gli1 is the first identified HH pathway gene amplified in many HH-dependent cancers, and expression of Gli1 is a reliable marker of HH pathway activity20,24. Therefore, development of GLI-targeting inhibitors has become an attractive strategy to treat MB and many other cancers. Unfortunately, currently available GLI inhibitors such as GANT6125,26 and As2O327 (Fig. 1) suffer from either toxicity or poor druglikeness or both.
Recently, modulation of HH signaling by epigenetically targeting GLI through bromodomain-containing protein 4 (BRD4) has spurred new interest in the development of effective treatment for HH-dependent tumors, especially for MB28,29. BRD4, together with BRD2, BRD3 and the testis-specific BRDT constitutes the important family of bromo and extra C-terminal (BET) bromodomain proteins, all containing two bromodomains BD1 and BD2 at the N terminus30,31. As a serine kinase of RNA Pol II and an atypical histone acetyltransferase, BRD4 recruits transcriptional regulatory complexes to acetylate chromatin via recognition of acetylated lysines32. Global mRNA expression profiling of clinical primary MB and normal cerebellar samples indicate that BRD2, BRD3, and BRD4 are all up-expressed in primary MB, especially SHH MB subgroups, whereas only marginal expression of BRD4 expressed in normal cerebellar tissues33. These results indicate that BRD4 together with BRD2 and BRD3 is involved in the pathogenesis of MB.
To decode the underlying mechanism of BRD4 involved in MB, Tang et al.28 and Long et al.29 recently disclosed that the bromodomains of BRD4 interact with GLI by occupying the cancer-specific proximal promoters of Gli1 and Gli2 genes. Notably, knockdown of Brd4 did not abrogate GLI activity completely, and knockdown of either Brd2 or Brd3 also resulted in substantial GLI reduction, suggesting pan-BET inhibitors might be more efficacious than selective ones to modulate GLI28. Collectively, these pioneering studies provide the basis that inhibition of GLI can be achieved by suppression of BRD4, thus providing an alternative strategy for development of GLI inhibitors to treat HH-dependent MB. This strategy is especially appealing since development of highly potent GLI inhibitors is challenging due to the lack of targetable binding domains of GLI20. Therefore, in this report we first conducted a drug repurposing campaign by screening current clinically investigational BRD4 inhibitors bearing different chemotypes for their inhibitory potency against the HH pathway. The BRD4 inhibitor ABBV-07534, 35, 36, 37, 38 is then selected as the lead for further structural optimization leading to identification of compounds 25 and 35 as highly potent GLI inhibitors, especially compound 25 which was well tolerated and showed significant tumor growth inhibition in the resistant MB allograft mice. These results not only confirm the effectiveness of development of GLI inhibitors through optimization of BRD4 inhibitors, but also provide an alternative utility for BRD4 inhibitors in the treatment of many highly heterogeneous and untreatable pediatric brain tumors, such as MB33,39,40.
2. Results and discussions
2.1. Chemistry
The synthesis of carbamates 5 and 6 is outlined in Scheme 1. Pd-catalyzed coupling of 3-amino-5-bromo-1-methylpyridin-2(1H)-one (46) with borate 4735 provided compound 48 in 84% yield, which was then subjected to aromatic substitution to give 2,4-difluorophenyl ether 49. Substitution of 49 with ethyl carbonochloridate or 2-methoxyethyl carbonochloridate afforded corresponding carbamate 50a or 50b in 66%–71% yields. Subsequent reduction of 50a or 50b under iron powder followed by substitution with ethanesulfonyl chloride gave target compounds 5 and 6 in 54% and 70% yields, respectively.
Scheme 1.

Synthesis of compounds 5 and 6. Reagents and conditions: (a) 47, Pd(PPh3)4, 1,4-dioxane/2 mol/L Na2CO3 aq. (5:1), microwave 120 °C, 1 h, 84%; (b) 2,4-difluorophenol, Cs2CO3, DMF, r.t., 1 h; (c) ethyl carbonochloridate or 2-methoxyethyl carbonochloridate, NaH, THF, 0 °C to rt, 1 h, 66%–71%; (d) Fe, NH4Cl, EtOH/H2O (5:2), 70 °C, 3 h; (e) ethanesulfonyl chloride, pyridine, microwave 130 °C, 15 min, 54%–70%.
The preparation of 3-aminopyridinones 7–11 was described in Scheme 2. Compounds 52a−e were obtained from substitution reaction of aminopyridinone 46 or C–N coupling reaction of 51 with appropriate amines. Coupling of 52a−e with borate 5341 under Pd(PPh3)4 catalysis yielded compounds 54a−e in 59%–87% yields, which were readily converted to sulfonamides 7–11 by reacting with ethanesulfonyl chloride in the presence of Et3N.
Scheme 2.

Synthesis of compounds 7–11. Reagents and conditions: (a) RH, Et3N, DCM, r.t., 1 h; (b) RH, Pd2(dba)3, xantphos, Cs2CO3, toluene, 100 °C, 3 h, 19%–71%; (c) 10, Pd(PPh3)4, 1,4-dioxane/2 mol/L Na2CO3 aq. (5:1), 100 °C, 1 h, 59%–87%; (d) i) ethanesulfonyl chloride, Et3N, DCM, r.t., 1 h; ii) NaOH, 1,4-dioxane, 90 °C, 30 min, 33%–62%.
As shown in Scheme 3, substituted arylamine 55, which was prepared41 by following literature procedures, was reacted with sulfurisocyanatidic chloride followed by cyclization with 1,2-dibromoethane or 1,3-dibromopropane to deliver 56a and 56b in 90% and 88% yields, respectively. Subsequent reduction of the esteric moiety by NaBH4 followed by N-Ts deprotection with NaOH afforded compounds 12 and 13 in 72% and 78% yields, respectively.
Scheme 3.

Synthesis of compounds 12 and 13. Reagents and conditions: (a) methyl 2-(chlorosulfonyl)acetate, Et3N, DCM, r.t., 1 h, 88%; (b) 1,2-dibromoethane or 1-bromo-3-chloropropane, K2CO3, DMF, 70 °C, 88%–90%; (c) NaBH4, CaCl2, dry THF, 0 °C to r.t., 3 h; (d) NaOH, 1,4-dioxane, 90 °C, 30 min, 72%–78% over two steps.
The synthesis of sulfuric diamides 14–23 is illustrated in Scheme 4. Sulfuric diamide 57 was generated from substitution reaction of 55 with freshly made tert-butyl(chlorosulfonyl) carbamate in 74% yield. Cyclization of compound 57 with 1,2-dibromoethane or 1,3-dibromopropane delivered 1,2,5-thiadiazolidine 1,1-dioxides 58a and 58b in 54% and 75% yields, respectively. N-Boc deprotection of 58a or 58b with TFA and further substitution with different alkyl bromides afforded compounds 59a−d in 68%–98% yields. Subsequent N-Ts deprotection of compounds 59a−c in the presence of NaOH afforded corresponding compounds 14, 15 and 23 in 89%–93% overall yields. It should be noted that compounds 16–19 were prepared by N-Boc deprotection of 58b with TFA, subsequent N-Ts deprotection and further substitution with 1,1-difluoro-2-iodoethane or different alkyl bromides in 28%–55% overall yields. In addition, substitution of bromide 59d with different amines followed by N-Ts deprotection with NaOH delivered compounds 20–22 in 75%–78% yields.
Scheme 4.

Synthesis of compounds 14–23. Reagents and conditions: (a) sulfurisocyanatidic chloride, t-BuOH, Et3N, DCM, 0 °C to r.t., 2.5 h, 74%; (b) 1,2-dibromoethane or 1,3-dibromopropane, K2CO3, MeCN, reflux, overnight, 54%–75%; (c) TFA, DCM, r.t., 2 h; (d) RBr or (for 16) RI, K2CO3, DMF, 60 °C, 1–5 h; (e) NaOH, 1,4-dioxane, 90 °C, 30 min; (f) from 59h, amines, K2CO3, MeCN, 60 °C, 75%–78%.
As shown in Scheme 5, compounds 24 and 25 bearing the 3-fluoro-6-methyl-1,6-dihydro-7H-pyrrolo[2,3-c]pyridin-7-one scaffold were prepared. Starting from 4-fluoro-1H-pyrrole-2-carboxylic acid 6042, C-3 fluorinated bicyclic pyrrolopyridone core 62 was prepared in 68% overall yield by first condensation of 60 with 2,2-dimethoxyethanamine followed by self-cyclization in the presence of p-toluene sulfonic acid. Protection of pyrrolidine 62 with TsCl in the presence of NaH followed by bromination with NBS generated the intermediate 63 in 84% yield over two steps. Coupling of aryl bromide 63 with borate 53 under Pd(PPh3)4 catalysis afforded compound 64 in 85% yield. Treatment of compound 64 with 3-chloropropane-1-sulfonyl chloride or 4-chlorobutane-1-sulfonyl chloride yielded bis-substituted 65a and 65b in 64% and 68% yields, respectively. Subsequent N-Ts deprotection, N-sulfonyl removal and intramolecular cyclization of compounds 65a and 65b in one pot with NaOH afforded target compounds 24 and 25 in 95% and 91% yields, respectively.
Scheme 5.

Synthesis of compounds 24 and 25. Reagents and conditions: (a) 2,2-dimethoxyethanamine, DIPEA, propanephosphonic acid cyclic anhydride, THF, −5–60 °C, 20 h, 75%; (b) TsOH·H2O, THF, 60 °C, overnight, 90%; (c) TsCl, NaH, DMF, 0 °C, 1 h; (d) NBS, TsOH·H2O, THF, r.t., 1 h, 84% over 2 steps; (e) 53, Pd(PPh3)4, 1,4-dixone/2 mol/L Na2CO3 aq. (5:1), 100 °C, 1 h, 85%; (f) 3-chloropropane-1-sulfonyl chloride or 4-chlorobutane-1-sulfonyl chloride, Et3N, DCM, r.t., 1 h, 64%–68%; (g) NaOH, 1,4-dioxane, 90 °C, 0.5 h, 91%–95%.
As shown in Scheme 6, carboxylic acids 26 and 27 and amides 28–32 were obtained using 6-methyl-1-tosyl-1,6-dihydro-7H-pyrrolo[2,3-c]pyridin-7-one 66 as the starting material. Substitution of the pyrrole C-2 of 66 with ethyl chloroformate delivered ethyl ester 67, which was then converted to compounds 68a and 68b through a similar reaction as that for 65a and 65b. Subsequent N-Ts deprotection, N-sulfonyl removal, intramolecular cyclization and ethyl ester hydrolysis were conducted in one pot in the presence of NaOH leading to acids 26 and 27 in 83% and 89% yields, respectively. Condensation of 26 or 27 with NH4Cl, CH3CH2NH2 or 3-(4-methylpiperazin-1-yl)propan-1-amine in the presence of HOBT, EDCI and Et3N produced compunds 28–32 in 65%–78% yields.
Scheme 6.

Synthesis of compounds 26–32. Reagents and conditions: (a) n-BuLi, ethyl carbonochloridate, dry THF, N2, −10 °C, 1 h, 60%; (b) NBS, TsOH·H2O, THF, r.t., 1 h; (c) 53, Pd(PPh3)4, 1,4-dixone/2 mol/L Na2CO3 aq. (5:1), 100 °C, 1 h, 76%; (d) 3-chloropropane-1-sulfonyl chloride or 4-chlorobutane-1-sulfonyl chloride, Et3N, DCM, r.t., 1 h, 84%–88%; (e) NaOH, 1,4-dioxane, 90 °C, 0.5 h, 89%–92%; (f) HOBT, EDCI, Et3N, NH4Cl or CH3CH2NH2 or 3-(4-methylpiperazin-1-yl)propan-1-amine, DCM, r.t., 3 h, 65%–78%.
Compounds 33–39 bearing benzofuran or cycloalkyl amine moieties were synthesized as described in Scheme 7. Aromatic substitution of compound 6934 with benzofuran-5-ol or different amines furnished compounds 70a−e in 33%–98% yields, which were then reduced by hydrogen atmosphere under Pd/C or by iron powder in the presence of NH4Cl to afford corresponding anilines 71a−e. It is worth noting that N-Ts protecting group was partially taken off during substitution reaction with trans-4-methylcyclohexyl amine. In this case, additional N-Ts protection under NaH was necessary to afford compound 70b. Treatment of compounds 71a−e with 3-chloropropane-1-sulfonyl chloride or 4-chlorobutane-1-sulfonyl chloride provided di-substituted intermediate 72a and mono-substituted 72b−g in 34%–78% yields. It should be pointed out that for compound 71e, the two hydroxyl groups need to be protected with TMSCl before treatment with 4-chlorobutane-1-sulfonyl chloride. Finally, N-deprotection, N-sulfonyl removal and intramolecular cyclization of compounds 72a−g were conducted in one pot in the presence of NaOH to provide target compounds 33–39 in 85%–93% overall yields.
Scheme 7.

Synthesis of compounds 33–39. Reagents and conditions: (a) RH, DIPEA, DMSO, 80 °C, 6 h, 33%–51%; or (for 70a) RH, K2CO3, DMF, r.t., 30 min, 98%; or (for 70b): i) RH, DMSO, 80 °C, 6 h; ii) TsCl, NaH, DMF, 0 °C, 1 h, 72% over two steps. (b) Pd/C, H2, 45 °C, CHCl3/MeOH (1:1), 8 h; or (for 71a) Fe, NH4Cl, EtOH/H2O (5:2), 70 °C, 1 h. (c) 3-Chloropropane-1-sulfonyl chloride or 4-chlorobutane-1-sulfonyl chloride, Et3N, DCM, r.t., 1 h, 77%–88%; or (for 72g): i) TMSCl, Et3N, DMF, 0 °C, 0.5 h; ii) 4-chlorobutane-1-sulfonyl chloride, Et3N, DCM, r.t., 1 h; (d) NaOH, 1,4-dioxane, 90 °C, 0.5 h, 85%–93%.
The synthesis of C-2 or C-3 fluorinated pyrropyridinones 40–45 bearing a 4-methylcyclohexan-1-amino motif was described in Scheme 8. Fluorination of pyrrole C-2 of 66 with NFSI under n-BuLi delivered compound 73 in 45% yield, which was brominated to yield intermediate 74 through a similar reaction as described in Scheme 5. Borate 77 was obtained in 80% overall yield with 1-fluoro-4-nitrobenzene (75) as the starting material by substitution with trans-4-methylcyclohexyl amine followed by coupling with bis(pinacolato)diboron. The coupling of borate 77 with aryl bromide 63 or 74 delivered corresponding 78a and 78b in 57% and 68% yields, respectively, which were then converted to target compounds 40–45 in 65%–85% overall yields through a similar reaction procedure as described in Scheme 7.
Scheme 8.

Synthesis of compounds 40–45. Reagents and conditions: (a) n-BuLi, NFSI, dry THF, N2, −20 °C, 2.5 h, 45%; (b) NBS, TsOH·H2O, THF, r.t., 1 h; (c) trans-4-methylcyclohexyl amine, DMSO, 100 °C, 2 h; (d) bis(pinacolato)diboron, Pd(dppf)Cl2, AcOK, 1,4-dioxane, N2, 100, 5 h, 80% over two steps; (e) 63 or 73, Pd2(dba)3, meCgPPh, K3PO4, dioxane/H2O (4:1), 80 °C, 5 h, 57%–68%; (f) Pd/C, H2, 45 °C, CHCl3/MeOH (1:1), 8 h; (g) ethanesulfonyl chloride or 3-chloropropane-1-sulfonyl chloride or 4-chlorobutane-1-sulfonyl chloride, Et3N, DCM, r.t., 1 h, 63%–82%; (h) NaOH, 1,4-dioxane, 90 °C, 0.5 h, 65%–85%.
2.2. Screening of clinical BRD4 inhibitors against HH signaling pathway
To gain insights on the activity relationship between BRD4 and GLI, we selected seven clinically studied BRD4 inhibitors and tested their inhibitory activity in the HH signaling pathway in our dual luciferase reporter assays using light II cells. These cells are NIH-3T3 cells stably transfected with a GLI-responsive firefly luciferase reporter and renilla-luciferase expression vector21,43. As shown in Table 1, the seven clinically studied BRD4 inhibitors were found to show high but quite different potency against the HH signaling pathway with IC50 values ranging between 0.15 and 40.6 nmol/L. Among them, the phase II BRD4 inhibitor 1 (BMS-986158) is the most potent, whereas INCB-057643 is the least potent HH pathway inhibitor, with IC50 values of 0.15 and 40.6 nmol/L, respectively. The overall trend of activity is in agreement with the BRD4 activity where the phase II compound 1 remains the most potent BRD4 inhibitor, and compound 444 (AZD5153) is much less potent, with IC50 values of 0.16 and 18.2 nmol/L, respectively. This result indicates that development of high potency GLI-targeting HH pathway inhibitors might be realized by optimization of BRD4 activity through structural modification, since direct structural information on the transcriptional factor GLI is limited. Considering the potential toxicity liability and pharmacokinetic (PK) disadvantage of the tricyclic compounds (e.g., 1 and 3, Table 1)35,45, we decided to choose compound 2 (ABBV-075, Table 1) as our lead HH inhibitor for structural optimization. This compound shows high inhibitory activity (0.41 nmol/L) against HH pathway, but is nearly 10-fold less potent against BRD4 (3.4 nmol/L). Our objective is to identify a high potency and well tolerated HH pathway inhibitor for treatment of MB, especially those resistant to current SMO-targeting HH inhibitors.
Table 1.
Inhibition of clinical BRD4 inhibitors against BRD4 BD1 and the HH pathwaya.
| Compd. | Clinical stage | Structure | IC50 (nmol/L) |
|
|---|---|---|---|---|
| BRD4 (BD1) | GLI-luc | |||
| BMS-986158 (1) | I/II | 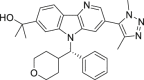 |
<0.16 | 0.15 ± 0.05 |
| ABBV-075 (2) | I | 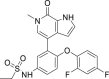 |
3.4 ± 0.2 | 0.41 ± 0.34 |
| PLX51107 | I | 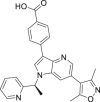 |
0.49 ± 0.14 | 5.5 ± 1.3 |
| OTX-015 | II | 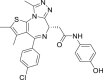 |
7.7 ± 0.21 | 0.64 ± 0.02 |
| JQ1 (3) | Discovery | 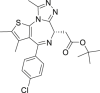 |
9.8 ± 2.2 | 2.6 ± 1.7 |
| INCB-057643 | I/II | 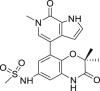 |
11.6 ± 1.8 | 40.6 ± 4.9 |
| AZD5153 (4) | I | 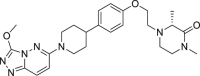 |
18.2 ± 2.7 | 32.1 ± 7.71 |
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments.
2.3. Structure-based drug design
4-Aryl-1,6-dihydro-7H-pyrrolo[2,3-c]pyridin-7-one 2 (ABBV-075, Fig. 2), developed by AbbVie, is a highly potent non-selective BRD4 inhibitor undergoing phase I clinical trials for the treatment of patients with acute myeloid leukemia, multiple myeloma and advanced solid tumors (NCT02391480)46. According to the reported co-crystal structure34 of its close analogue with BRD4 (BD2), the pyrrolopyridone core is located in the KAc binding pocket and provides critical interactions with Asn433 residue. The meta-difluorophenoxyl motif fills the WPF hydrophobic shelf (a hydrophobic region constructed by residues W81, P82 and F83), and the ethyl sulfonamide chain occupies the ZA channel of the BRD4 (BD2) protein. In addition, the pyrrole fragment and the ethyl sulfonamide chain are both exposed to the solvent interaction region, which may tolerate various substituents. Inspired by this analysis and in view of the lack of targetable binding domains of the transcriptional factors GLI20, we have designed several series of analogues by modifying these interaction sites through three approaches, including: (1) opening the pyrropyridinone bicyclic core to afford flexible 3-aminopyridiones; (2) cyclizing the ethanesulfonamide motif; (3) replacing the hydrophobic difluorophenyl with a water soluble moiety (Fig. 1).
Figure 2.

Structural analysis and design of new HH pathway inhibitors.
2.4. Structure−activity relationship (SAR) study
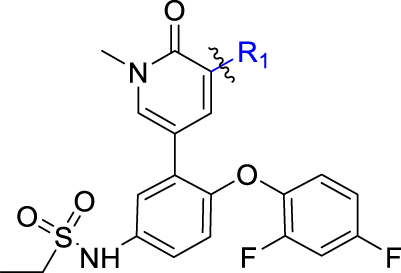
To investigate how the structural alterations impact activities of GLI, we first cleaved the bicyclic pyrrolopyridone core of compound 2 in 3-aminopyridinones 5–11. As shown in Table 2, all these compounds are much less potent than compound 2. Carbamate 5 has an IC50 value of 27.6 nmol/L against BRD4, which is 8-fold less potent than that of 2. Both carbamate 6 with a longer chain and the reverse amide 7 show more reduction of potency. 3-Ethanesulfonamido pyridinone 8 and 3-((1H-imidazol-2-yl)amino) pyridinone 9 display modest potency against BRD4 with IC50 value of 42.8 and 47.6 nmol/L, respectively. N-Heterocycle-substituted 3-aminopyridinones 10 and 11 show much reduced potency with IC50 values greater than 100 nmol/L. These results indicate that although the substituted 3-amino analogues may maintain the H-bonding with Asn433 residue in the KAc binding pocket of BRD4 (BD1), the electronic or steric property potentially affects the interaction with BRD4 as well. Disappointingly, all the compounds show markedly reduced potency against the HH pathway with IC50 values all greater than 100 nmol/L in the GLI-luciferase assay.
Table 2.
Inhibition of compounds against BRD4 (BD1) and the HH pathwaya.
| Compd. | R1 | IC50 (nmol/L) |
|
|---|---|---|---|
| BRD4 (BD1) | GLI-luc reporter | ||
| 5 | 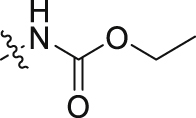 |
27.6 ± 5.7 | 157 ± 1.3 |
| 6 | 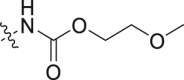 |
>100 | >100 |
| 7 | 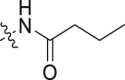 |
>100 | >100 |
| 8 | 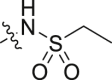 |
42.8 ± 9.6 | >100 |
| 9 | 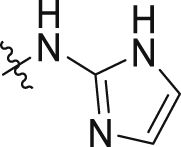 |
47.6 ± 2.9 | >100 |
| 10 | 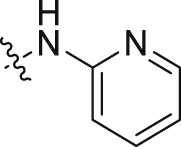 |
>100 | >100 |
| 11 | 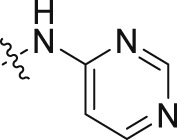 |
>100 | >100 |
| ABBV-075 (2) | – | 3.4 ± 0.2 | 0.41 ± 0.34 |
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments. –Not applicable.
Next, considering that there is a large space in the ZA channel interaction region in the binding mode of 2 with BRD4 (PDB code: 5uvx)34, we evaluated compounds derived from cyclization of the ethanesulfonamide. As shown in Table 3, the five- and six-membered cyclic sulfonamides 12 and 13 display much higher potency against BRD4 with IC50 values of 0.25 and 0.60 nmol/L, respectively. However, their potency against the HH pathway is significantly reduced with IC50 values of 3.29 and 20.5 nmol/L, respectively. In order to increase aqueous solubility (Table 7), we incorporated an additional N-atom moiety with various substituents leading to compounds 14–23. Most of these N-substituted cyclic aminosulfonamides except 16 and 22 were found much more potent than 2, by showing low- or sub-nanomolar potency against BRD4. However, all compounds are significantly less potent than 2 against the HH pathway with IC50 values between 17.7 and 100 nmol/L. Compound 22 bearing a substituted pyrrolidinylethyl moiety has the least potency in both assays. The five-membered analogues are slightly more potent than the six-membered congeners, especially against BRD4. These results indicate that the ZA channel of BRD4 BD1 can tolerate various substitutions, whereas larger substituents are not beneficial for interaction with the HH pathway.
Table 3.
Inhibition of compounds against BRD4 (BD1) and the HH pathwaya.
| Compd. | X | n | R2 | IC50 (nmol/L) |
|
|---|---|---|---|---|---|
| BRD4 (BD1) | GLI-luc reporter | ||||
| 12 | C | 1 | CH2OH | 0.25 ± 0.05 | 3.29 ± 0.14 |
| 13 | C | 2 | CH2OH | 0.60 ± 0.07 | 20.5 ± 7.74 |
| 14 | N | 1 |  |
0.20 ± 0.03 | 26.1 ± 5.17 |
| 15 | N | 1 | 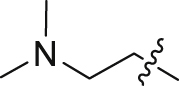 |
1.19 ± 0.10 | 71.9 ± 5.43 |
| 16 | N | 2 | 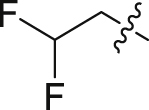 |
9.39 ± 0.80 | 28.9 ± 11.9 |
| 17 | N | 2 |  |
0.65 ± 0.36 | 33.9 ± 11.1 |
| 18 | N | 2 | 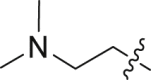 |
1.52 ± 0.72 | 56.1 ± 8.25 |
| 19 | N | 2 | 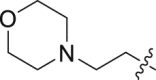 |
1.47 ± 0.03 | 17.7 ± 11.0 |
| 20 | N | 2 | 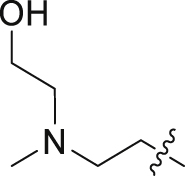 |
1.01 ± 0.34 | 42.8 ± 30.7 |
| 21 | N | 2 | 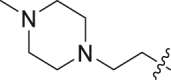 |
0.99 ± 0.19 | 39.4 ± 4.76 |
| 22 | N | 2 | 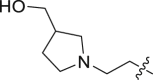 |
71.9 ± 5.43 | >100 |
| 23 | N | 2 | 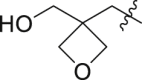 |
0.58 ± 0.10 | 45.8 ± 19.2 |
| 2 | – | – | – | 3.4 ± 0.2 | 0.41 ± 0.34 |
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments. –Not applicable.
Table 7.
hERG inhibition and predicted physico-chemical parameters of representative compoundsa.
| Compd. | GLI-luc reporter IC50 (nmol/L) | BRD4 IC50 (nmol/L) | hERG IC50 (μmol/L) | tPSAb (Å) | LogSb (μg/mL) | LogPappb (cm/s) | VDb (L/kg) |
|---|---|---|---|---|---|---|---|
| 43 | 0.062 ± 0.039 | 14.28 ± 1.27 | – | 87.2 | 17.88 | −5.228 | −0.24 |
| 30 | 0.05 ± 0.01 | 0.60 ± 0.41 | 6.4 | 113 | 21.35 | −5.135 | −0.795 |
| 34 | 0.12 ± 0.1 | 0.52 ± 0.03 | >40 | 87.2 | 17.60 | −5.194 | −0.034 |
| 35 | 0.33 ± 0.13 | 0.99 ± 0.01 | >40 | 87.2 | 17.94 | −5.235 | −0.078 |
| 25 | 0.48 ± 0.08 | 1.51 ± 0.43 | 27.3 | 84.4 | 9.14 | −4.79 | −0.551 |
| 24 | 0.93 ± 0.16 | 0.50 ± 0.06 | >40 | 84.4 | 18.14 | −4.773 | −0.518 |
| 31 | 0.81 ± 0.24 | 0.13 ± 0.005 | 11.7 | 113 | 30.16 | −5.164 | −0.799 |
| 33 | 1.03 ± 0.28 | 2.85 ± 0.50 | – | 97.5 | 9.35 | −5.312 | −0.345 |
| 28 | 1.5 ± 0.39 | 0.09 ± 0.04 | >40 | 127 | 51.80 | −5.189 | −0.754 |
| 2 | 0.41 ± 0.34 | 3.4 ± 0.2 | 35.5 | 93.2 | 19.60 | −5.01 | −0.547 |
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments.
Calculated through ADMET lab software viahttp://admet.scbdd.com; tPSA, topological polar surface area; logS, solubility; Papp, Caco-2 permeability; VD, volume distribution. –Not applicable.
As shown in Table 4, our next effort is to incorporate fluoro or other electron-withdrawing substituent on the C2- or C3-position of the pyrrolopyridone skeleton to the cyclic analogues of ethanesulfonamide motif. Compounds 24 and 25, both with a fluoro-substituent at the C3-position of the pyrrole component, display high potency against BRD4 and the HH pathway with IC50 values ranging between 0.48 and 1.5 nmol/L. However, compounds 26 and 27 bearing a carboxylic group at the C2-position of the pyrrole component show >4-fold decrease of potency against BRD4, together with markedly reduced potency (>100 nmol/L) against the HH pathway. Pleasantly, high potency was observed for compounds 28–32 containing an amide moiety at the C2-position of the pyrrole component. The unsubstituted carboxamides 28 and 29 show an identical IC50 value of 0.09 nmol/L against BRD4, whereas their potency against the HH pathway is slightly lower by showing IC50 values of 1.5 and 2.46 nmol/L, respectively. N-Ethyl substituted amide analogues 30 and 31 also display high potency, especially compound 30 having IC50 values of 0.6 and 0.05 nmol/L, respectively against BRD4 and the HH pathway, which are much more potent than compound 2. However, amide 32 with a longer N-substituent shows much reduced potency (>100 nmol/L) against the HH pathway, despite of the high potency for BRD4.
Table 4.
Inhibition of compounds against BRD4 (BD1) and the HH pathwaya.
| Compd. | n | R3 | R4 | IC50 (nmol/L) |
|
|---|---|---|---|---|---|
| BRD4 (BD1) | GLI-luc reporter | ||||
| 24 | 1 | H | F | 0.50 ± 0.06 | 0.93 ± 0.16 |
| 25 | 2 | H | F | 1.51 ± 0.43 | 0.48 ± 0.08 |
| 26 | 1 | COOH | H | 21.0 ± 11.0 | >100 |
| 27 | 2 | COOH | H | 14.3 ± 4.52 | >100 |
| 28 | 1 | CONH2 | H | 0.09 ± 0.04 | 1.5 ± 0.39 |
| 29 | 2 | CONH2 | H | 0.09 ± 0.08 | 2.46 ± 0.3 |
| 30 | 1 | CONHCH2CH3 | H | 0.60 ± 0.41 | 0.05 ± 0.01 |
| 31 | 2 | CONHCH2CH3 | H | 0.13 ± 0.005 | 0.81 ± 0.24 |
| 32 | 1 | 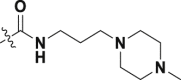 |
H | 0.75 ± 0.07 | >100 |
| 2 | – | – | – | 3.4 ± 0.2 | 0.41 ± 0.34 |
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments. –Not applicable.
Meanwhile, structural modification of the WPF interaction region of BRD4 (BD1) was conducted based on the ethanesulfonamide motif cyclized analogues. As shown in Table 5, compound 33 containing a benzofuran moiety to replace the original 2,4-difluorophenyl group maintains good potency against BRD4 (2.85 nmol/L), whereas the potency for the HH pathway was reduced (1.03 nmol/L). Compounds 34–39 all contain a water soluble substituent to replace the original phenylether motif. Compared to 2, these compounds generally retain good potency against BRD4, but the activity against the HH pathway is distinctly different. Notably, compounds 34 and 35 both bearing a 4-methylcyclohexyl amino substituent show extremely high potency in both assays, although the five-membered analogue 35 is slightly less potent than the six-membered analogue 34. Indeed, compound 34 is the most potent analogue among this series and shows IC50 values of 0.52 and 0.12 nmol/L, respectively against BRD4 and the HH pathway, and is 6.5- and 3.4-fold more potent than compound 2. Incorporation of an additional C4-hydroxyl group on the cyclohexyl (compounds 36 and 37) or reversing the chirality of the resulting quaternary carbon (compound 38) led to no improvement with IC50 values ranging between 1.2 and 3.4 nmol/L for both BRD4 and the HH pathway. In addition, compound 39 bearing a (3-hydroxy-4-hydroxymethyl) cyclopentylamino substituent lost the activity against the HH pathway significantly, despite remaining high potency for BRD4.
Table 5.
Inhibition of compounds against BRD4 (BD1) and the HH pathwaya.
| Compd. | n | R5 | IC50 (nmol/L) |
|
|---|---|---|---|---|
| BRD4 (BD1) | GLI-luc reporter | |||
| 33 | 1 | 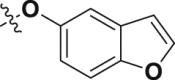 |
2.85 ± 0.50 | 1.03 ± 0.28 |
| 34 | 1 | 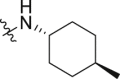 |
0.52 ± 0.03 | 0.12 ± 0.1 |
| 35 | 2 | 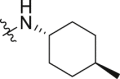 |
0.99 ± 0.01 | 0.33 ± 0.13 |
| 36 | 1 | 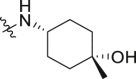 |
1.24 ± 0.09 | 1.58 ± 0.66 |
| 37 | 2 | 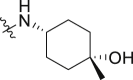 |
2.60 ± 056 | 1.47 ± 0.66 |
| 38 | 2 | 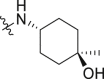 |
3.22 ± 0.11 | 1.34 ± 0.48 |
| 39 | 2 | 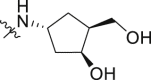 |
3.42 ± 0.25 | >100 |
| 2 | – | – | 3.4 ± 0.2 | 0.41 ± 0.34 |
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments. –Not applicable.
Based on the optimization results, we collected all the advantageous elements and incorporated them into a small series of multisubstituted pyrropyridinone analogues bearing a 4-methylcyclohexan-1-amino motif in the WPF binding site. As shown in Table 6, compared to the high potency of compound 34, all these compounds show reduced potency against BRD4 with IC50 values ranging between 9.51 and 20.54 nmol/L. The potency for the HH pathway is also decreased but distinctly different. Similarly modest potency was observed for compounds 40 and 41 in both assays indicating that there is no significant difference for the fluoro substituent on either the C2- or C3-position of the pyrrole component. Dramatically, compounds 42–45 with the ethanesulfonamido component cyclized to five- or six-membered sulfonamides show higher potency for the HH pathway, though their potency against BRD4 is modest, suggesting that the interaction mode for the HH signaling is different from that for BRD4. Among these, compound 43 is the most potent HH pathway inhibitor with IC50 values of 0.062 nmol/L, and shows 230-fold selectivity against BRD4.
Table 6.
Inhibition of compounds against BRD4 (BD1) and the HH pathwaya.
| Compd. | R2 | R3 | R4 | IC50 (nmol/L) |
|
|---|---|---|---|---|---|
| BRD4 (BD1) | GLI-luc reporter | ||||
| 40 | 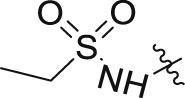 |
H | F | 14.25 ± 2.76 | 2.06 ± 1.16 |
| 41 | 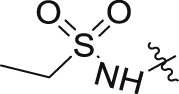 |
F | H | 17.57 ± 0.55 | 5.88 ± 2.72 |
| 42 | 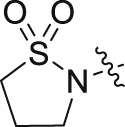 |
H | F | 9.51 ± 1.13 | 0.33 ± 0.056 |
| 43 | 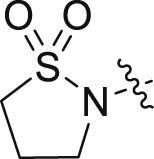 |
F | H | 14.28 ± 1.27 | 0.062 ± 0.039 |
| 44 | 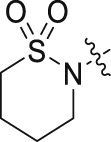 |
H | F | 19.10 ± 3.55 | 0.58 ± 0.35 |
| 45 | 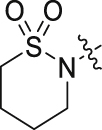 |
F | H | 20.52 ± 1.18 | 1.75 ± 0.63 |
| 2 | – | – | – | 3.4 ± 0.2 | 0.41 ± 0.34 |
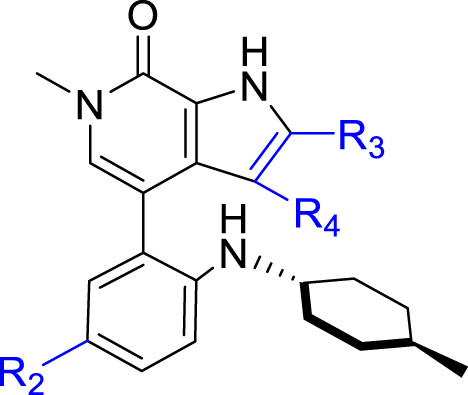
IC50 values are shown as the mean ± SD (nmol/L) from three separate experiments. –Not applicable.
2.5. hERG inhibition and predicted physico-chemical properties of potent compounds
From the SAR, we concluded that the inhibitory activity against BRD4 does not completely parallel with the potency of the HH pathway. Overall, BRD4 is well tolerant to various substitutions, whereas the activity for the HH pathway is much different and highly dependent on different substitution patterns. As listed in Table 7, Table 9 new compounds turn out as the high potency HH inhibitors with IC50 values less than 1.5 nmol/L. The most potent HH inhibitor 43 has an IC50 value of 0.062 nmol/L and is 230-fold more potent than against BRD4 (14.28 nmol/L), whereas compound 28, though highly potent against BRD4 (0.09 nmol/L), is 16-fold less potent for the HH pathway (1.5 nmol/L).
Table 9.
Antiproliferative inhibition of various cancer cells.
| GI50a (μmol/L) | H3132 | N87 | HT1080 | Colo680N | HSC-2 | KYSE450 | TOV-21G | CAL27 | SKM-1 | H1975 | DOHH-2 | MOLM-14 | MV4-11 | TMD8 | REC-1 | CHL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 25 | 0.14 | 0.13 | 1.0 | 2.3 | 0.77 | 1.6 | 0.023 | 0.11 | 0.019 | 0.034 | 0.0066 | 0.0081 | 0.0035 | 0.0093 | 0.015 | >10 |
| 35 | 0.33 | 0.46 | 0.88 | 4.0 | 0.32 | 1.4 | 0.008 | 0.12 | 0.0063 | 0.0054 | 0.0021 | 0.002 | 0.0013 | 0.0022 | 0.0022 | 1.1 |
Antiproliferative effects were initially tested in 10 μmol/L, and then diluted in eight diluted concentrations. GI50 values were obtained from two separate experiments.
To investigate the safety and druglikeness of these new HH inhibitors, we tested their inhibition on hERG channel and predicted their physico-chemical properties. As shown in Table 7, most compounds except 30 and 31 show negligible inhibition on the hERG with IC50 values greater than 20 μmol/L, indicating their low liability of cardiac toxicity. Compounds 30 and 31 have IC50 values of 6.4 and 11.7 μmol/L, indicating their slightly safety concerns on hERG, likely due to their more basic nature since both compounds contain a carboxamide substituent on the pyrrolopyridone component. Meanwhile, most compounds have similar predicated physico-chemical properties, including acceptable tPSA (<140 Å), modest aqueous solubility (logS), and low Caco-2 permeability (Table 7).
2.6. Pharmacokinetic parameters of potent compounds
To select optimal compound for further in vitro and in vivo antitumor study, the 7 highly potent HH inhibitors with IC50 values less than 1.0 nmol/L were selected for further evaluation for their pharmacokinetic (PK) parameters in Sprague–Dawley rats dosed intravenously (i.v., 1 mg/kg) and orally (p.o., 3 mg/kg). As shown in Table 8, these compounds generally have short half-life (t1/2) and modest plasma exposure (AUC). Compounds 24, 25 and 35 have relatively acceptable oral bioavailability (20%–30%) and modest clearance. Unfortunately, the most potent HH inhibitors 43 (0.062 nmol/L) and 30 (0.05 nmol/L) display poor PK parameters, especially with scarce oral bioavailability. Therefore, after consideration of the HH inhibitory potency and PK profile, we chose compounds 25 and 35 for further investigation.
Table 8.
PK parameters of potent compounds after p.o. (3 mg/kg) and i.v. (1 mg/kg) administration.
| Compd. | Route |
t1/2 (h) |
Tmax (h) |
Cmax (ng/mL) |
AUClast (h·ng/mL) |
AUCINF_obs (h·ng/mL) |
CL_obs (mL/min/kg) |
MRTINF_obs (h) |
Vss_obs (mL/kg) |
F |
|---|---|---|---|---|---|---|---|---|---|---|
| 24 | p.o. | 0.95 | 0.50 | 474 | 473 | 473 | – | 1.26 | – | 30.9 |
| i.v. | 0.56 | – | – | 509 | 509 | 33.2 | 0.76 | 1513 | – | |
| 25 | p.o. | 0.96 | 0.25 | 503 | 406 | 406 | – | 0.95 | – | 22.0 |
| i.v. | 0.54 | – | – | 614 | 614 | 27.2 | 0.77 | 1258 | – | |
| 30 | p.o. | 1.35 | 0.25 | 15.2 | 9.91 | 9.91 | – | 0.89 | – | 1.2 |
| i.v. | 0.68 | – | – | 278 | 278 | 61.5 | 0.794 | 2891 | – | |
| 31 | p.o. | 1.55 | 0.33 | 15.5 | 18.3 | 22.1 | – | 2.38 | – | 2.9 |
| i.v. | 0.69 | – | – | 213 | 217 | 77.6 | 0.57 | 3512 | – | |
| 34 | p.o. | 2.06 | 1.00 | 29.1 | 97.3 | 107 | – | 3.49 | – | 12.7 |
| i.v. | 1.09 | – | – | 255 | 261 | 76.4 | 1.66 | 8781 | – | |
| 35 | p.o. | 1.00 | 0.5 | 110 | 190 | 190 | – | 1.54 | – | 21.0 |
| i.v. | 1.06 | – | – | 300 | 300 | 59.2 | 1.20 | 4211 | – | |
| 43 | p.o. | 2.02 | 0.42 | 346 | 861 | 924 | – | 3.18 | – | 4.71 |
| i.v. | 1.05 | – | – | 6099 | 6135 | 2.78 | 1.46 | 239 | – | |
| 2 | p.o. | 3.34 | 2.75 | 401 | 2030 | 2052 | – | 5.45 | – | 57.0 |
| i.v. | 2.74 | – | – | 1186 | 1195 | 17.6 | 3.11 | 2772 | – |
aValues are the average of three runs. Vehicle: p.o., DMSO/0.5% HPMC (5/95, v/v); i.v., EtOH/PEG300/NaCl (10/40/50, v/v/v). CL, clearance; Vss, volume of distribution; t1/2, half-life; AUC, area under the plasma concentration time curve; F, oral bioavailability. –Not applicable.
2.7. Antiproliferative effects of compounds 25 and 35 against various cancer cells
Since the HH signaling pathway is a major regulator governing cell proliferation and differentiation, the selected HH inhibitors 25 and 35 were tested for their antiproliferative effects against a wide spectrum of cancer cell lines, including nine solid and six hematologic tumor cell lines, together with the CHL normal cell line. As shown in Table 9, no or low inhibition on the normal CHL cell lines was observed, especially for compound 25 with an GI50 value greater than 10 μmol/L, indicating the high selectivity against cancer cells. Notably, compared to the solid tumor cells, both compounds show markedly higher potency in the hematologic tumor cell lines, especially for SKM-1, DOHH-2, MOLM-14, MV4-11, MOLM-13 and TMD8 cell lines with GI50 value less than 10 nmol/L.
2.8. Inhibition of HH signaling pathway at the level of GLI
To investigate the exact mechanism of compounds 25 and 35 within the HH signaling pathway28, we investigated the suppressing effects of both compounds in the HH pathway by overexpressing GLI1 or GLI2. As shown in Fig. 3, compound 25 significantly inhibited GLI-luciferase activity provoked by forced expression of GLI1 or GLI2, with IC50 values of 0.97 and 2.2 nmol/L, respectively (Fig. 3A and B). Meanwhile, compound 25 dramatically reduced the abundance of endogenous GLI2 proteins in light2 cells at concentration of both 10 and 100 nmol/L, and the effect on GLI2 proteins of compound 25 (100 nmol/L) is superior to JQ1 (1 μmol/L), whereas the SMO inhibitor GDC-0449 (vismodegib) has no effect on the expression of endogenous GLI2 (Fig. 3C). In addition, compound 25 failed to inhibit the expression of exogenous GLI2-MYC (Fig. 3D).
Figure 3.

Compounds 25 and 35 inhibit HH activity at the level of GLI1/2. (A), (B) and (E), (F): GLI-luciferase activity in light2 cells treated with 25 or 35 after transfection with GLI1 or GLI2 construct; (C) and (G): Western blots evaluating endogenous GLI2 expression in light2 cells after treatment with 25, 35, JQ1 (1 μmol/L), and GDC-0449 (100 nmol/L); (D) and (H): GLI2-MYC protein expressions in light2 cells transfected with GLI2-MYC; (I): Western blots analysis of exogenous GLI1-FLAG.
Similarly, compound 35 also inhibited GLI-luciferase activity provoked by forced expression of GLI1 or GLI2, with IC50 values of 0.30 and 0.43 nmol/L, respectively (Fig. 3E and F). Meanwhile, compound 35 down-regulated endogenous GLI2 proteins in light2 cells (Fig. 3G), whereas no effect was observed on the expression of exogenous GLI1-FLAG and GLI2-MYC (Fig. 3H and I).
Given the fact that significant suppression on the HH activation provoked by forced expression of GLI1 or GLI2 was observed by compounds 25 and 35, but not by SMO inhibitor, as well as the robust inhibition of 25 and 35 on the activity of BRD proteins, we conclude that compounds 25 and 35 epigenetically inhibit HH signaling pathway at the level of GLI, not at the upstream targets of the HH signaling pathway, e.g., SMO.
2.9. Compounds 25 and 35 can circumvent drug resistance caused by SMO mutations
Having characterized that compounds 25 and 35 function as HH inhibitors by acting at the level of GLI, we continued to evaluate its in vitro ability of combating the resistance to SMO inhibitors caused by SMO mutation47. As expected, both compounds markedly blocked GLI-luciferase activity in light2 cells induced by ectopic expression of wild type SMO (SMOWT, Fig. 4A and E), as well as SMOD473H (Fig. 4B and F) and SMOW535L (Fig. 4C and G), two most predominant mutants47 of SMO responsible for the resistance to SMO inhibitors, with respective IC50 values of 1.16, 1.44 and 1.13 nmol/L for 25, and 0.39, 0.53 and 0.63 nmol/L for 35 (Fig. 4D and H). These findings indicate that compounds 25 and 35 are promising HH inhibitors with capacity of overcoming drug resistance caused by SMO mutations.
Figure 4.

Compounds 25 and 35 possess in vitro ability of circumventing the resistance to SMO inhibitors caused by SMO mutations. GLI-luciferase analysis of the inhibitory effects of compounds in light2 cells followed by transfection with SMOWT (A) and (E), SMOD473H (B) and (F), or SMOW535L (C) and (G), respectively. (D) and (H) IC50 values of compounds in light2 cells with forced expression of SMOWT, SMOD473H and SMOW535L.
2.10. In vivo antitumor effects in SMOA1 MB allograft model
To translate the in vitro ability of the GLI inhibitors 25 and 35 in combating the resistance caused by SMO inhibition into in vivo antitumor efficacy, we established spontaneous SMOA1 MB model, of which tumors contain mutation at the amino acid residue 539 of SMO (SMOW539L), and are resistant to current SMO inhibitor vismodegib (GDC-0449) in ND2:SMOA1 mice48. As shown in Fig. 5A, treatment with compound 25 at 10 mg/kg by oral administration (p.o., bid) realized 88.2% tumor growth inhibition (TGI), whereas the SMO inhibitor GDC-0449 failed to affect the tumor growth at 20 mg/kg (p.o., bid), a dosage resulting in complete tumor growth inhibition in SMO wild MB mice49. However, despite its higher potency of 35 in vitro than compound 25, treatment of this compound at the same dosage was not tolerated leading to significant casualty (data are not shown). However, a lower dose of 35 at 1 mg/kg was well tolerant with TGI of 68.7%. No obvious body weight loss was observed in both the 10 mg/kg group of 25 and the 1 mg/kg group of 35 during treatment (Fig. 5B). These results suggest that both compounds 25 and 35 are capable of combating SMO inhibitor-resistant MB tumors.
Figure 5.

Antitumor activity of compounds 25 and 35 in SMOA1 MB allograft model. Nude mice with subcutaneously transplanted SMOA1 MB allografts were treated with vehicle control (0.5% CMC-Na, p.o., bid), compounds 25 and 35 (p.o., bid) or GDC-0449 (p.o., bid). (A) Tumor volume and (B) body-weight change over time in treated SMOA1 MB allograft. Data represent group means ± SEM (control, n = 4; GDC-0449, n = 4; 35, n = 4; 25, n = 3).
Since compound 25 functioned as an efficacious and more tolerant GLI inhibitor than compound 35 to overcome the resistant MB tumors caused by SMOW539L mutation, we continued to evaluate this compound at lower dosage. As shown in Fig. 6A, compound 25, even at a lower dosage of 5 mg/kg (p.o., bid) still shows 83.3% tumor growth inhibition, similar to the effect of 10 mg/kg dose (TGI = 87.6%). Meanwhile, the new HH inhibitor 25 was well tolerant in both doses without significant overall toxicity (Fig. 6B).
Figure 6.

Antitumor activity of compound 25 in SMOA1 MB allograft model. Nude mice with subcutaneously transplanted SMOA1 MB allografts were treated with vehicle control (0.5% CMC-Na, p.o., bid), or compound 25 (p.o., bid). (A) Tumor volume and (B) body-weight change over time in treated SMOA1 MB allograft. Data represent group means ± SEM (control, n = 4; 25, 5 mg/kg, n = 4; 25, 10 mg/kg, n = 5).
3. Conclusions
In summary, by repurposing a series of the clinically investigational BRD4 inhibitors as the HH signaling pathway inhibitors, the AbbVie's phase I clinical pan-BET inhibitor 2 (ABBV-075) was selected for a systemic medicinal chemistry optimization. The established SAR indicates that the inhibitory activity against BRD4 does not parallel with the potency for the HH pathway. Notably, BRD4 is well tolerant to various substitution patterns, whereas the activity for the HH pathway is much different and highly dependent on the steric and electrostatic natures of the substituents. A number of compounds were identified showing IC50 values less than 1.0 nmol/L against the HH signaling pathway, and further investigation of their inhibition on hERG and PK parameters elected compounds 25 and 35 as the high potency HH inhibitors. Similar to compound 2, these new compounds are non-selective BET inhibitors (Supporting Information Fig. S2 and Table S2) that might be more effective to modulate GLI activity. Further mechanism profiling showed that the two new compounds suppressed HH signaling by interaction with the transcriptional factor GLI, and are equally potent against both the two clinically identified resistant SMO mutants (SMOD473H and SMOW535L) and SMO wild type, with IC50 values around 1 nmol/L or less. Although the PK parameters are not optimal, both compounds showed significant antitumor efficacy in vivo in the resistant MB allograft mice. Compared to the dose-limited toxicity of compound 35, compound 25 was more tolerant and significantly suppressed tumor growth at both 5 mg/kg (TGI = 83.3%) and 10 mg/kg (TGI = 87.6%) doses. Therefore, the new GLI inhibitor 25 represents not only a high potent HH inhibitor both in vitro and in vivo, but also a relatively safe and tolerant lead compound capable of suppressing HH-driven MB, that is resistant to current SMO antagonists.
4. Experimental
4.1. Chemistry
All reactions were performed in glassware containing a teflon-coated stir bar. All commercially purchased reagents and solvents were chemical pure and used without futher purification. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury 300, 400 or 500 NMR spectrometer (Bruker Biospin AG, Romanshorn, Switzerland) and referenced to deuterium dimethyl sulfoxide (DMSO-d6), deuterium chloroform (CDCl3), deuterium methanol (CD3OD) or deuterium dichloromethane (CD2Cl2). Chemical shifts (δ) were reported in ppm downfield from an internal TMS standard. High-resolution mass spectrometry (HRMS) analysis was recorded at anionizing voltage of 70 eV on a Finnigan/MAT95 spectrometer (Agilent, Palo Alto, CA, USA). Flash column chromatography on silica gel (200–300 mesh, Yucheng chemical, Shanghai, China) was used for the routine purification of reaction products. All reactions were monitored by TLC on silica gel plates (50 mm × 15 mm) and spots were visualized under UV light (Changbo, Shanghai, China). Melting points were determined using a SGW X-4 hot stage microscope (INESA, Shanghai, China) and are uncorrected. HPLC analysis was conducted for all bioassayed compounds on an Agilent Technologies 1260 series LC system (ZORBAX-C18 (150 mm × 4.6 mm, Daicel, Shanghai, China), 5 μmol/L, MeOH/H2O or MeCN/H2O, r.t.) with two ultraviolet wavelengths (UV 254 and 214 nm). The purities of these compounds were above 95%.
4.2. Synthetic procedures
Synthesis of target compounds and key intermediates was shown in Supporting Information. Compounds 46, 51, 60a, 66 and 75 were commericially purchased, and compounds 4735, 5349, 5549, 6042 and 6934 were prepared according to corresponding literature procedures.
4.3. Biological studies
4.3.1. Homogeneous time-resolved fluorescence
The binding of compounds to BRD4 was assessed using an homogeneous time-resolved fluorescence assay. Recombinant BRD4 proteins (Active Motif, Cat#31380 and 31446, Carlsbad, CA, USA), compounds and peptide were diluted to desired working concentrations with EPIgenerous binding buffer (Cisbio, Cat#62DLBDDF, Codolet, France). 4 μL of 5 × BRD4 protein at 165 ng/mL, 4 μL of 5 × biotin labeled acetylated H4K5/K8/K12/K16 at 200 ng/mL and 2 μL of serial diluted test compounds were added to the wells of a 384 ProxiPlate (PerkinElmer, Cat#6008289, Waltham, MA, USA). After 30 min incubation at 37 °C, a 10 μL mixture of IgG antibody labeled with Eu and streptavidin labeled with XL665 were prepared and added to each well for 3 h to reach equilibrium at room temperature. The signal of fluorescence was detected on Envison (PerkinElmer) and analyzed in GraphPad Prism (San Diego, CA, USA).
4.3.2. Dual luciferase reporter assay
Light2 cells (ATCC, Manassas, VA, USA) were treated with compounds as indicated for 36 h. Relative-luciferase assays were performed using dual-luciferase reporter assay kit (Promega, Madison, WI, USA) according to manufacturer's instructions. Relative luciferase values were normalized to TK-renilla value.
4.3.3. Cell growth inhibition assays
SKM-1 cells (JCRB, Osaka, Japan) were seeded into a 96-well plate at a suitable density in a volume of 100 μL medium. After incubation overnight, compounds dissolved in DMSO stock solutions were thawed at room temperature and diluted to the desired concentrations with saline. The compounds were added and cultured for 72 h, the IC50 (or GI50) value was measured with the Cell Counting Kit-8 (CCK-8) assay (Dojindo, Kumamoto, Japan).
4.3.4. GLI protein expression assays
Light2 cells were transfected with MYC-GLI2-FL or FLAG-GLI1 (Addgene, Cambridge, MA, USA) using Lipo2000 (Invitrogen, Carlsbad, CA, USA) as manufacturer's protocol. Cells were treated with indicated compounds 24 h after transfection.
4.3.5. Western blot analysis
Cells were lysed with LDS Sample Buffer (NP0008, Life, Waltham, MA, USA). The protein samples were analyzed with SDS-PAGE gel (Cell Signaling Technology, Boston, MA, USA) and immunoblotted with the following primary antibodies: anti-HA, anti-FLAG, anti-GLI2 (1:1000, Cell Signaling Technology) or GAPDH (1:5000, Santa Cruz Biotechnology, Santa Cruz, CA, USA).
4.3.6. Medulloblastoma allograft model
SMOA1 medulloblastoma model was derived from ND2:SMOA1 transgenic mice (Shanghai SLAC Laboratory Animal Co., Shanghai, China) as previously described48. Briefly, primary SMOA1 allografts were subcutaneously grafted into nude mice. Mice allografted with SMOA1 MB were administered compounds GDC-0449, 25 and 35 or vehicle twice a day by oral administration (p.o., n ≥ 3) once the tumor size reached 100–150 mm3. Tumor volumes were measured with calipers every 3 days and calculated as 0.5 × length × width2. All animal procedures were approved by Animal Care and Use Committee of Fudan University, Shanghai, China.
Acknowledgments
This work was supported by grants from National Natural Science Foundation (Grants Nos. 81773565, 81703327, 81430080, 81573452, and 81773767). Supporting grants from the Key Program of the Frontier Science (Grant No. 160621, China) of the Chinese Academy of Sciences, the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA12020374, China) are also appreciated. We also thank the support from the State Key Laboratory of Esophageal Cancer Prevention and Treatment, Ministry of Education of China, Zhengzhou University (Grant No. K2020-0012, China). In addition, we thank Prof. Zhaobing Gao and Dr. Xueqin Chen of Shanghai Institute of Materia Medica (SIMM, Shanghai, China), and Prof. Qingsong Liu and Dr. Aoli Wang of High Magnetic Field Laboratory, Chinese Academy of Sciences (Hefei, China) for the test of hERG inhibition and cellular antiproliferative activity, respectively.
Author contributions
Ao Zhang, Wenfu Tan and Hongchun Liu conceived and designed the study. Xiaohua Liu performed the synthesis of the target compounds and analyzed the data. Yu Zhang, Yalei Li and Juan Wang performed the biological experiments. Huaqian Ding and Wenjing Huang assisted with carrying out experiments. Ao Zhang and Xiaohua Liu wrote the manuscript. Chunyong Ding, Wenfu Tan, Hongchun Liu and Juan Wang revised the manuscript. All the authors approved the final version of the manuscript.
Conflicts of interest
The authors have no conflicts of interest to declare.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.07.007.
Contributor Information
Hongchun Liu, Email: hchliu@simm.ac.cn.
Wenfu Tan, Email: wftan@fudan.edu.cn.
Ao Zhang, Email: ao6919zhang@sjtu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Northcott P.A., Robinson G.W., Kratz C.P., Mabbott D.J., Pomeroy S.L., Clifford S.C. Medulloblastoma. Nat Rev Dis Primers. 2019;5:11. doi: 10.1038/s41572-019-0063-6. [DOI] [PubMed] [Google Scholar]
- 2.Pui C.H., Gajjar A.J., Kane J.R., Qaddoumi I.A., Pappo A.S. Challenging issues in pediatric oncology. Nat Rev Clin Oncol. 2011;8:540–549. doi: 10.1038/nrclinonc.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ostrom Q.T., Gittleman H., Truitt G., Boscia A., Kruchko C., Barnholtz-Sloan J.S. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011‒2015. Neuro Oncol. 2018;20:iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalli F.M.G., Remke M., Rampasek L., Peacock J., Shih D.J.H., Luu B. Intertumoral heterogeneity within medulloblastoma subgroups. Canc Cell. 2017;31:737–754. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capozza M.A., Trombatore G., Triarico S., Mastrangelo S., Attinà G., Maurizi P. Adult medulloblastoma: an overview on current and future strategies of treatment. Expert Opin Orphan Drugs. 2019;7:383–389. [Google Scholar]
- 6.Xin M., Ji X., de La Cruz L.K., Thareja S., Wang B. Strategies to target the hedgehog signaling pathway for cancer therapy. Med Res Rev. 2018;38:870–913. doi: 10.1002/med.21482. [DOI] [PubMed] [Google Scholar]
- 7.Miranda Kuzan-Fischer C., Juraschka K., Taylor M.D. Medulloblastoma in the molecular era. J Kor Neurosurg Soc. 2018;61:292–301. doi: 10.3340/jkns.2018.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rimkus T.K., Carpenter R.L., Qasem S., Chan M., Lo H.W. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancer. 2016;8:22. doi: 10.3390/cancers8020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gould S.E., Low J.A., Marsters J.C., Jr., Robarge K., Rubin L.L., de Sauvage F.J. Discovery and preclinical development of vismodegib. Expet Opin Drug Discov. 2014;9:969–984. doi: 10.1517/17460441.2014.920816. [DOI] [PubMed] [Google Scholar]
- 10.Dlugosz A., Agrawal S., Kirkpatrick P. Vismodegib. Nat Rev Drug Discov. 2012;11:437–438. doi: 10.1038/nrd3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan S., Wu X., Jiang J., Gao W., Wan Y., Cheng D. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med Chem Lett. 2010;1:130–134. doi: 10.1021/ml1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin V.T., Esmaeli B. Targeting the hedgehog pathway for locally advanced and metastatic basal cell carcinoma. Curr Pharmaceut Des. 2017;23:655–659. doi: 10.2174/1381612822666161208100325. [DOI] [PubMed] [Google Scholar]
- 13.Danial C., Sarin K.Y., Oro A.E., Chang A.L. An investigator-initiated open-label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin Cancer Res. 2016;22:1325–1329. doi: 10.1158/1078-0432.CCR-15-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gajjar A., Stewart C.F., Ellison D.W., Kaste S., Kun L.E., Packer R.J. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19:6305–6312. doi: 10.1158/1078-0432.CCR-13-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson G.W., Orr B.A., Wu G., Gururangan S., Lin T., Qaddoumi I. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog-subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol. 2015;33:2646–2654. doi: 10.1200/JCO.2014.60.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y., Song Q., Day B.W. Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: a systemic review and meta-analysis. Acta Neuropathol Commun. 2019;7:123. doi: 10.1186/s40478-019-0773-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Millard N.E., de Braganca K.C. Medulloblastoma. J Child Neurol. 2016;31:1341–1353. doi: 10.1177/0883073815600866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lannering B., Rutkowski S., Doz F., Pizer B., Gustafsson G., Navajas A. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol. 2012;30:3187–3193. doi: 10.1200/JCO.2011.39.8719. [DOI] [PubMed] [Google Scholar]
- 19.Esbenshade A.J., Kocak M., Hershon L., Rousseau P., Decarie J.C., Shaw S. A Phase II feasibility study of oral etoposide given concurrently with radiotherapy followed by dose intensive adjuvant chemotherapy for children with newly diagnosed high-risk medulloblastoma (protocol POG 9631): a report from the Children's Oncology Group. Pediatr Blood Cancer. 2017;64 doi: 10.1002/pbc.26373. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X., Ding C., Tan W., Zhang A. Medulloblastoma: molecular understanding, treatment evolution, and new developments. Pharmacol Ther. 2020;210:107516. doi: 10.1016/j.pharmthera.2020.107516. [DOI] [PubMed] [Google Scholar]
- 21.Liu G., Huang W., Wang J., Liu X., Yang J., Zhang Y. Discovery of novel macrocyclic hedgehog pathway inhibitors acting by suppressing the Gli-mediated transcription. J Med Chem. 2017;60:8218–8245. doi: 10.1021/acs.jmedchem.7b01185. [DOI] [PubMed] [Google Scholar]
- 22.Dong X., Wang C., Chen Z., Zhao W. Overcoming the resistance mechanisms of smoothened inhibitors. Drug Discov Today. 2018;23:704–710. doi: 10.1016/j.drudis.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Infante P., Alfonsi R., Botta B., Mori M., Di Marcotullio L. Targeting GLI factors to inhibit the hedgehog pathway. Trends Pharmacol Sci. 2015;36:547–558. doi: 10.1016/j.tips.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Villavicencio E.H., Walterhouse D.O., Iannaccone P.M. The Sonic hedgehog-Patched-Gli pathway in human development and disease. Am J Hum Genet. 2000;67:1047–1054. doi: 10.1016/s0002-9297(07)62934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lauth M., Bergström A., Shimokawa T., Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104:8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J., Huang S., Tian R., Chen J., Gao H., Xie C. The protective autophagy activated by GANT-61 in MYCN amplified neuroblastoma cells is mediated by PERK. Oncotarget. 2018;9:14413–14427. doi: 10.18632/oncotarget.24214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J., Aftab B.T., Tang J.Y., Kim D., Lee A.H., Rezaee M. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013;23:23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Y., Gholamin S., Schubert S., Willardson M.I., Lee A., Bandopadhayay P. Epigenetic targeting of hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med. 2014;20:732–740. doi: 10.1038/nm.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long J., Li B., Rodriguez-Blanco J., Pastori C., Volmar C.H., Wahlestedt C. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J Biol Chem. 2014;289:35494–35502. doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z., Wang P., Chen H., Wold E.A., Tian B. Drug discovery targeting bromodomain-containing protein 4. J Med Chem. 2017;60:4533–4558. doi: 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filippakopoulos P., Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 32.Burns C.J. Chromatin targeting: a BET inhibitor workaround. Nat Chem Biol. 2016;12:469–470. doi: 10.1038/nchembio.2107. [DOI] [PubMed] [Google Scholar]
- 33.Henssen A., Thor T., Odersky A., Heukamp L., EI-Hindy N., Beckers A. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget. 2013;4:2080–2095. doi: 10.18632/oncotarget.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDaniel K.F., Wang L., Soltwedel T., Fidanze S.D., Hasvold L.A., Liu D. Discovery of N-(4-(2,4-difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/mivebresib), a potent and orally available bromodomain and extraterminal domain (BET) family bromodomain inhibitor. J Med Chem. 2017;60:8369–8384. doi: 10.1021/acs.jmedchem.7b00746. [DOI] [PubMed] [Google Scholar]
- 35.Gautam B., Johannes W., Ibrahim A., Beibei H., Minh D., Alison T. First-in-human study of ABBV-075 (mivebresib), a pan-inhibitor of bromodomain and extra terminal (BET) proteins, in patients (Pts) with relapsed/refractory (RR) acute myeloid leukemia (AML): preliminary data. J Clin Oncol. 2018;36:7019. [Google Scholar]
- 36.Bui M.H., Lin X., Albert D.H., Li L., Lam L.T., Faivre E.J. Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. 2017;77:2976–2989. doi: 10.1158/0008-5472.CAN-16-1793. [DOI] [PubMed] [Google Scholar]
- 37.Piha-Pau S.A., Sachdev J.C., Barve M., LoRusso P., Szmulewitz R., Patel S.P. First-in-human study of mivebresib (ABBV-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with pelapsed/pefractory solid tumors. Clin Cancer Res. 2019;25:6309–6319. doi: 10.1158/1078-0432.CCR-19-0578. [DOI] [PubMed] [Google Scholar]
- 38.Faivre E.J., Wilcox D., Lin X., Hessler P., Torrent M., He W. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol Cancer Res. 2017;15:35–44. doi: 10.1158/1541-7786.MCR-16-0221. [DOI] [PubMed] [Google Scholar]
- 39.Matzuk M.M., McKeown M.R., Filippakopoulos P., Li Q., Ma L., Agno J.E. Small-molecule inhibition of BRDT for male contraception. Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohammad H.P., Barbash O., Creasy C.L. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019;25:403–418. doi: 10.1038/s41591-019-0376-8. [DOI] [PubMed] [Google Scholar]
- 41.Wang L., Pratt J.K., Soltwedel T., Sheppard G.S., Fidanze S.D., Liu D. Fragment-based, structure-enabled discovery of novel pyridones and pyridone macrocycles as potent bromodomain and extra-terminal domain (BET) family bromodomain inhibitors. J Med Chem. 2017;60:3828–3850. doi: 10.1021/acs.jmedchem.7b00017. [DOI] [PubMed] [Google Scholar]
- 42.Leroy J., Porhielb E., Bondonb A. Synthesis and characterization of partially β-fluorinated 5,10,15,20-tetraphenylporphyrins and some derivatives. Tetrahedron. 2002;58:6713–6722. [Google Scholar]
- 43.Liu G., Xue D., Yang J., Wang J., Liu X., Huang W. Design, synthesis, and pharmacological evaluation of 2-(2,5-dimethyl-5,6,7,8-tetrahydroquinolin-8-yl)-N-aryl propanamides as novel smoothened (Smo) antagonists. J Med Chem. 2016;59:11050–11068. doi: 10.1021/acs.jmedchem.6b01247. [DOI] [PubMed] [Google Scholar]
- 44.Bradbury R.H., Callis R., Carr G.R., Chen H., Clark E., Feron L. Optimization of a series of bivalent triazolopyridazine based bromodomain and extraterminal inhibitors: the discovery of (3R)-4-[2-[4-[1-(3-methoxy-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2-one (AZD5153) J Med Chem. 2016;59:7801–7807. doi: 10.1021/acs.jmedchem.6b00070. [DOI] [PubMed] [Google Scholar]
- 45.Wick J.Y. The history of benzodiazepines. Consult Pharm. 2013;28:538–548. doi: 10.4140/TCP.n.2013.538. [DOI] [PubMed] [Google Scholar]
- 46.AbbVie A study evaluating the safety and pharmacokinetics of ABBV-075 in subjects with cancer (ClinicalTrials.gov Identifier: NCT02391480) https://clinicaltrials.gov/ct2/show/NCT02391480 Available from: (March 18 2015)
- 47.Atwood S.X., Sarin K.Y., Whitson R.J., Li J.R., Kim G., Rezaee M. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hatton B.A., Villavicencio E.H., Tsuchiya K.D., Pritchard J.I., Ditzler S., Pullar B. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768–1776. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 49.Robarge K.D., Brunton S.A., Castanedo G.M., Cui Y., Dina M.S., Goldsmith R. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett. 2009;19:5576–5581. doi: 10.1016/j.bmcl.2009.08.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.