

Abstract
Endophytic fungi are promising producers of bioactive small molecules. Bioinformatic analysis of the genome of an endophytic fungus Penicillium dangeardii revealed 43 biosynthetic gene clusters, exhibited its strong ability to produce numbers of secondary metabolites. However, this strain mainly produce rubratoxins alone with high yield in varied culture conditions, suggested most gene clusters are silent. Efforts for mining the cryptic gene clusters in P. dangeardii, including epigenetic regulation and one-strain-many-compounds (OSMAC) approach were failed probably due to the high yield of rubratoxins. A metabolic shunting strategy by deleting the key gene for rubratoxins biosynthesis combining with optimization of culture condition successfully activated multiple silent genes encoding for other polyketide synthases (PKSs), and led to the trace compounds detectable. As a result, a total of 23 new compounds including azaphilone monomers, dimers, trimers with unprecedented polycyclic bridged heterocycle and spiral structures, as well as siderophores were identified. Some compounds showed significant cytotoxicities, anti-inflammatory or antioxidant activities. The attractive dual PKSs gene clusters for azaphilones biosynthesis were mined by bioinformatic analysis and overexpression of a pathway specific transcription factor. Our work therefor provides an efficient approach to mine the chemical diversity of endophytic fungi.
KEY WORDS: Endophytic fungi, Penicillium dangeardii, Genome mining, Silent gene cluster, Azaphilones, Trimers, Metabolic shunting
Graphical abstract
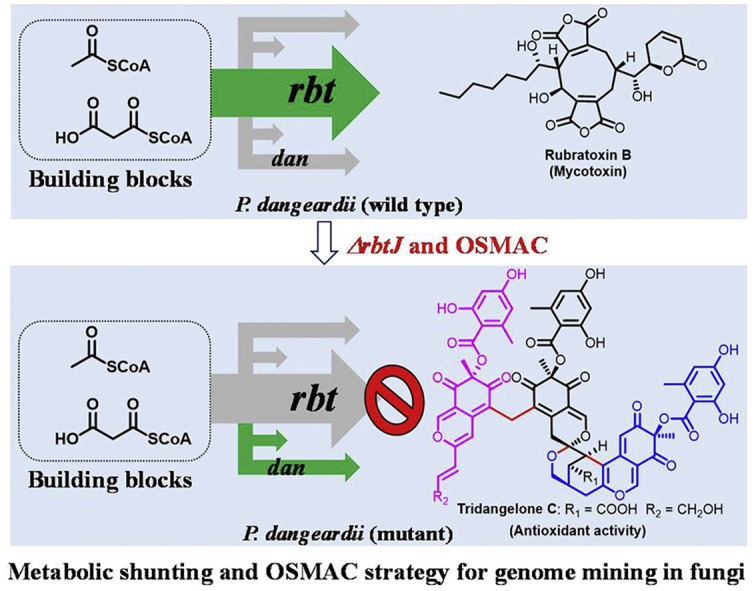
A combinational genome mining strategy based on metabolic shunting and OSMAC by deleting the key gene (rbtJ) for the biosynthesis of main products rubratoxins in fungus Penicillium dangeardii successfully aroused abundant silent gene clusters and led to the production of numerous novel metabolites with unprecedented structures and diverse bioactivities.
1. Introduction
Endophytic fungi, which live inside plants interacting with the host and promoting plant growth, have been proved to be promising producer of secondary metabolites with novel structures and a remarkable range of activities such as antibacterial, insecticidal, anticancer, plant hormone and antioxidant1,2. For instance, Taxomyces andreanae, isolated from the phloem of Taxus brevifolia, could produce taxol as an anti-tumor agent in breast and ovarian cancers3. However, advances in genome sequencing and bioinformatics revealed that fungi with many biosynthetic gene clusters have far greater ability to encode secondary metabolites than which were produced under standard laboratory culture conditions. So various genome mining strategies have been applied to activate silent gene clusters for the biosynthesis of natural products4,5, including one-strain-many-compounds (OSMAC)6,7, co-cultivation8, chemical epigenetics9, ribosome engineering10, epigenetic modification11,12, overexpression of transcription factor13 and heterologous expression14, and achieved amazing results.
Penicillium dangeardii is a filamentous fungus isolated from a toxic plant Lysidice rhodostegia (Leguminosae sp.)15. Investigation on its chemical compositions mainly led to the isolation of large anhydrides such as rubratoxins A−C (1–3, Fig. 1A)16,17 and penicillactones A−C15. Due to the highly oxidized complex structure and bioactivity, the biosynthesis pathway of rubratoxins A and B has been elucidated18. However, analysis of genomic information of P. dangeardii revealed 43 gene clusters responsible for natural products biosynthesis, which are significantly more than the number of above known anhydrides metabolites, suggesting that most gene clusters are cryptic.
Figure 1.

Analyses of metabolites from P. dangeardii. (A) The structures of anhydrides. (B) LC‒MS analyses of crude extracts from wild type (WT) and ΔrbtJ mutant strain fermented in PDB and rice culture media. The number on the peaks corresponds to the products shown in Figure 1, Figure 2. The anhydrides are partially present as diacids (labeled 1′ and 2′) in aqueous solvent.
To activate the cryptic gene clusters in P. dangeardii, OSMAC strategy and epigenetic regulation11 were firstly applied, respectively. Unfortunately, it failed probably due to the super-high yield of rubratoxins (0.65 g/L). Alternatively, a metabolic shunting strategy based on deletion of the key gene for rubratoxins biosynthesis and optimization of culture condition successfully activated multiple cryptic gene clusters involved in the biosynthesis of polyketides, which resulted in the isolation of a large number of new metabolites, including azaphilone monomers, dimers, and trimers featuring unprecedented polycyclic bridged heterocycle and spiral structures, as well as siderophores. All isolated compounds were evaluated for their cytotoxicities, anti-inflammatory and antioxidant activities, and the gene clusters for azaphilones biosynthesis were characterized.
2. Results and discussion
2.1. Bioinformatic analysis of genome of P. dangeardii
Bioinformatic analysis of the genome sequence from P. dangeardii showed 43 gene clusters that encode biosynthesis of natural products, including 22 polyketide synthases (PKSs), 8 nonribosomal peptide synthases (NRPSs), 8 PKSs/NRPSs hybrids, and 5 terpene synthases (TSs; Supporting Information Tables S1–S3), which far exceeds the number of the predominant anhydrides rubratoxins A−C (Fig. 1A). This indicated that the title strain possesses potential to produce more diverse natural compounds.
2.2. OSMAC strategy and epigenetic modification applied on P. dangeardii
To develop its potential for biosynthesis of pleiotropic second metabolites, an OSMAC strategy was applied, where P. dangeardii was cultured in different media, including potato dextrose agar (PDA), potato dextrose broth (PDB), Czapek concentrate yeast extract (CY), yeast extract glucose (YG), yeast extract malt extract glucose (YMEG), and rice (Secition 4.1. Strains and cultivation conditions). However, no additional metabolites except rubratoxins were produced, indicated by LC‒MS analysis (Supporting Information Fig. S1). Moreover, deletion of the four epigenetic regulation genes separately did not lead to the generation of any new products (data not show). With the high yield of rubratoxins in wild type or mutant strains, we believe that acetyl CoA, the starting unit for polyketide synthase was largely consumed by rubratoxins biosynthesis, which inhibits the biosynthesis of other polyketides and makes efforts to activate cryptic gene cluster unsuccessful. In addition, the presence of high yield of rubratoxins in P. dangeardii also leads to the trace secondary metabolites undetectable. Therefore, a strategy based on inhibition of rubratoxins production was believed to be efficient for mining the chemical diversity in P. dangeardii.
2.3. Combinational genome mining leads to the production of abundant metabolites
To inhibit the production of rubratoxins, the gene rbtJ encoding PKS for rubratoxins biosynthesis was deleted, to give a mutant strain ΔrbtJ, which was screened for appropriate culture media. Among the screening rice, PDA, PDB, CY, YG, and YMEG media, rice medium was found to be a perfect medium for strain ΔrbtJ, where abundant metabolites were detectable through LC‒MS analysis (Fig. 1B and Supporting Information Fig. S2). Comprehensive reverse transcription-PCR (RT-PCR) analysis of all 43 core biosynthetic genes exhibited that most core genes in the wild type showed weak to no expression except for the PKS gene for biosynthesis of rubratoxins in contig 20, while inhibiting the production of rubratoxins resulted in significantly increased expression of four additional PKS genes in contigs 4 and 60 (Supporting Information Fig. S3). Therefore, the combinational strategy applied here successfully up-regulated the expression of multiple cryptic genes encoding for PKSs and led to the pleiotropic production of secondary metabolism.
To identify the newly produced compounds in the ΔrbtJ strain, a large-scale fermentation (13 kg) in rice was carried out, following by various chromatography, which resulted in the isolation of a total of 36 compounds from part fractions (Fig. 2), of which 23 compounds possess new structure, including 9 azaphilone monomers (4–9, 18–19 and 34), 8 azaphilone dimers (20–25, 27 and 28), 5 azaphilone trimers (29–33), and 1 unclassified polyketide (35), along with 13 known ones including polyketides Sch 725680 (10)19, pinophilin B (11)20, pinazaphilone B (12)21, Sch 1385568 (13)22, talaraculone F (14)23, (+)-mitorubrinic acid B (15)24, (−)-mitorubrin (16)25,26, (−)-mitorubrinol (17)25, diazaphilonic acid (26)27,28, (±)-asperlones A (36)29 and orsellinic acid (37)30, alkaloid terreusinone (38)31, and a siderophore desferritriacetylfusigen (39)32,33. Remarkably, three types of novel azaphilones polymers were discovered for the first time, including 5 unusual methylene-bridged azaphilone dimers (20–24), 2 rare spiro-polycyclic azaphilone dimers (27 and 28), and 5 unprecedented azaphilone trimers (29–33) featuring both methylene and polycyclic bridged heterocycle and spiral structures, tetrahydro-3-oxaspiro[bicyclo[3.3.1]nonane-2,2′-pyran] core.
Figure 2.

Compounds purified from P. dangeardii ΔrbtJ mutant strain.
Compound 4 was isolated as a yellow amorphous powder. The molecular formula C21H22O7 was determined according to its high resolution ESI MS (HR-ESI-MS) spectrum ([M+H]+, m/z 387.1438; Calcd. for C21H23O7, 387.1438) combined with the 1H NMR and 13C NMR data (Supporting Information Tables S4 and S5). IR spectrum showed the presence of hydroxy (3355 cm−1), conjugated carbonyl (1645 cm−1), and phenyl (1583 and 1447 cm−1) groups. The 1H NMR and 13C NMR spectra of 4 showed two aromatic proton signals at δH 6.11 (s, 2H), a methyl protons at δH 2.12 (s, 3H), two phenolic hydroxy protons at δH 9.96 (s), 10.63 (s), a conjugated carbonyl carbon at δC 169.0, and six aromatic carbon signals in the region of δC 100.3–161.1, indicating the existence of 2,4-dihydroxy-6-methyl benzoic acid (orsellinic acid) moiety19,21. In addition, the NMR data also displayed resonances attributable to a conjugated ketene at δC 198.5 (C-6), two olefinic protons at δH 5.72 (d, J = 2.1 Hz, H-5), 5.77 (s, H-4), two E-configured olefinic protons at δH 6.33 (dq, J = 15.5, 6.9 Hz, H-10), 6.01 (dd, J = 15.5, 1.6 Hz, H-9), two methyl groups at δH 1.81 (d, J = 6.9, 1.6 Hz, H3-11), 1.33 (s, H3-12), an oxygenated methylene at δH 3.75 (dd, J = 12.9, 10.6 Hz, H-1a), 4.51 (dd, J = 10.6, 5.2 Hz, H-1b), δC 66.8 (C-1), an oxygen-bearing methine at δH 5.60 (d, J = 2.8 Hz, H-8), δC 77.2 (C-8), a methine at δH 3.43 (m, H-8a), δC 35.7 (C-8a), an alcoholic hydroxy proton at δH 5.36 (s, 7-OH), and three quaternary carbons at δC 158.5 (C-3), 149.1 (C-4a), 74.8 (C-7).
Analysis of the 1H–1H COSY NMR data of 4 identified the C-1/C-8a/C-8 and C-9/C-10/C-11 moieties (Fig. 3). HMBC correlations from olefin proton H-5 to C-7/C-8a, from oxygenated methine proton H-8 to ketone carbonyl C-6/C-4a/C-7, and from Me-12 to C-6/C-7/C-8 established the 7-methyl cyclohexenone ring. HMBC correlations from olefinic proton H-4 to C-3/C-5/C-8a, from oxygenated methylene proton H-1 to C-3/C-4a/C-8a displayed the presence of a dihydropyran ring. The side chain C-9/C-10/C-11 was adjacent to C-3 supported by HMBC correlations from H-9 to C-3 and from H-4 to C-9. Thus, the azaphilone skeleton was established. HMBC correlations from oxygen-bearing methine proton H-8 to carbonyl carbon C-13 suggested that the orsellinic acid moiety attached to C-8 (Fig. 3). Therefore, the planar structure of 4 was determined, which was same as that of a known azaphilone analogue Sch 725680 (10)19, but with different relative configuration.
Figure 3.

1H–1H COSY and key HMBC correlations of compounds 4, 27, and 35.
The relative configuration of 4 was determined on the basis of 1H–1H coupling constants and NOESY experiments. The coupling constant of J8,8a = 2.8 Hz indicated the syn relationship of H-8/H-8a. In the NOESY spectrum, NOE correlations of H-8 with Me-12 and H-8a suggested that these protons were placed on the same side of the molecule (Fig. 4). The absolute configuration of 4 was determined by the circular dichroism (CD) exciton chirality method34,35. The CD spectrum of 4 showed Cotton effects involved in the conjugated trienone chromophores interacting with benzoate at 360 and 306 nm (Δε −1.61 and 0.57), suggesting the 8S configuration. Therefore, the absolute configuration of 4 was established as 7S, 8S, 8aR and it was named dangelone A.
Figure 4.

Key NOESY correlations of compounds 4, 27, and 28.
Analysis of NMR data (Tables S4 and S5) of compounds 5 and 6 suggested that they had the similar azaphilone plane frame as 4 and the distinction between them lay the respective side chains at C-3. In the side chain of 5, a hydroxymethyl group at C-11 [δH 4.09 (d, J = 13.6 Hz, 2H), δC 60.6, and δH 4.98 (t, J = 5.1 Hz, OH-11)] was present instead of C-11 methyl group (δC 18.1) in 4. The COSY correlations of H-9/H-10/H-11/OH-11 and HMBC correlations from H-11 to C-9/C-10 confirmed the side chain assignment of 5. In the side chain of 6, a free C-11 carboxyl group (δC 166.8) replaced the C-11 methyl in 4, which was verified by HMBC correlation from H-9 to C-11. The relative configurations of 5 and 6 were in accordance with that of 4 based on NOESY spectra. By comparing the CD spectra and optical rotation with those of 4, the absolute configurations of 5 and 6 were both determined as 7S, 8S, 8aR and they were named dangelones B and C, respectively.
Compound 7 possessed the molecular formula C23H24O9 on the basis of HRESIMS ([M+Na]+, m/z 467.1328; Calcd. for C23H24O9Na, 467.1313). The 1H and 13C NMR data (Tables S4 and S5) of 7 showed an additional acetyl group signals at δH 2.02 (3H, s, H-22) and δC 20.7 (C-22), 170.6 (C-21) by comparison to those of pinophilin B (11)20. HMBC correlations from oxygenated methylene protons H2-11 (δH 4.70) to carbonyl carbon C-21 (δC 170.6) and from methyl protons H3-22 (δH 2.02) to C-21 confirmed that the acetyl group was attached to the terminal C-11 hydroxy in 11. Thus, 7 was an acetylated derivative of 11 and named dangelone D.
Compound 8 was established the molecular formula C21H22O9 by positive HRESIMS at m/z 419.1352 [M+H]+ (Calcd. for C21H23O9, 419.1337), two more hydrogens than that of pinazaphilone B (12)21. Comparing the 1H and 13C NMR data (Tables S4 and S5) of 8 with those of 12 indicated that a trans-olefin double bond (δC 137.5, 125.5) at C-9 (C-10) in 12 was replaced by two methylene [δH 2.55 (2H, overlap, H-9), 2.56 (2H, overlap, H-10) and δC 31.1 (C-9), 30.5 (C-10)] in 8, suggesting that 8 was a hydrogenated derivative of 12 at C-9 and C-10, which was confirmed by HMBC correlations from methylene protons H2-9 (δH 2.55) to C-3/C-4/C-10/C-11. The relative configurations of 7 and 8 were established to be same as that of 12 by the similar J8,8a coupling constants and NOESY spectra, respectively. Since the CD spectra of 7 and 8 agree well with those of 1019 and 1221, respectively (Supporting Information Figs. S283 and S284), the absolute configurations of 7 and 8 were both assigned as 7S, 8S, 8aS. Then, 8 was named dangelone E.
Comparing the NMR data (Tables S4 and S5) of compound 9 with those of 1221, an oxygenated methine was observed at δH 4.00 (d, J = 4.4 Hz, H-6) and δC 74.3 (C-6) in 9, with the disappearance of ketone carbonyl carbon at δC 197.3 in 12. HMBC correlations from the oxygenated methine proton H-6 to C-4a/C-5/C-7/C-12, from H-12 to C-6/C-7/C-8, and from H-5 to C-4/C-7/C-8a indicated that ketone carbonyl group at C-6 in 12 was reduced to a hydroxy in 9. The trans-diaxial relationship of H-8/H-8a was deduced from the coupling constant (J8,8a = 10.1 Hz). The NOESY correlations between H-6/H-12/H-8 suggested that they were placed on the same side of the molecule. To determine the absolute configuration of 9, the CD spectrum was measured in methanol and compared with its calculated electronic circular dichroism (ECD) of 6R,7R,8S,8aS-9 and 6S,7S,8R,8aR-9 using the quantum chemical method36. The predicted ECD spectrum of 6R,7R,8S,8aS-9 exhibited an excellent fit with the experimental one (Supporting Information Fig. S285). Thus, the absolute configuration of 9 was assigned to be 6R, 7R, 8S, 8aS and it was named dangelone F.
Compound 18 was obtained as a yellow amorphous powder and had a molecular formula of C26H28O13 on the base of HRESIMS [M+Na]+ m/z 571.1429 (Calcd. for C26H28O13Na, 571.1422). The 1H and 13C NMR data (Supporting Information Table S6) exhibited additional five oxygenated carbons (δC 101.1, 73.0, 70.7, 88.1, 62.9), an anomeric proton at δH 5.67 (d, J = 4.5 Hz), and six hydrogens between δH 3.66−4.21 by comparing their NMR data with those of 6, indicating the presence of a α-ribose37, which was confirmed by 1H–1H COSY and HMBC correlations. The α-ribose was connected to C-17 in orsellinic acid moiety supported by HMBC correlation from anomeric proton H-1′ to C-17. The sugar moiety in 18 was determined as d-ribose by comparing the retention time in LC‒MS of the thiocarbamoyl-thiazolidine derivative of the acid hydrolysate of 18 with those of authentic d-ribose and l-ribose (Supporting Information Fig. S7). Due to the same relative configuration and similar CD spectrum by comparing with those of 6, the absolute configuration of the aglycone in 18 was established as 7S, 8S, 8aR, which was further confirmed by ECD calculations36. The calculated weighted ECD spectrum of 7S,8S,8aR-18 agrees well with the experimental spectrum of 18 (Supporting Information Fig. S286). Thus, 18 was determined to be 17-O-α-d-ribosyl-dangelone C and named dangeloside A.
The NMR spectroscopic data (Table S6) of compound 19 closely resembled those of Sch 725680 (10)19, except for the presence of an additional ribose moiety, the 13C NMR data of which was in good accordance with those of α-ribose moiety in 18. HMBC correlation from anomeric proton H-1′ to C-17 suggested that ribose was linked to C-17. Due to the limitation of the trace amount of 19, the configuration of d-ribose in 19 was determined according to the same biosynthetic origin of suger moiety in 18. The relative configuration of the aglycone in 19 was same as that of 10 based on NOESY spectrum. The absolute configuration of the aglycone in 19 was identified as 7S, 8S, 8aS by comparison of the CD spectrum with those of 10 (Supporting Information Fig. S287). Thus, 19 was determined to be 17-O-α-d-ribosyl-Sch 725680 and named dangeloside B.
Compound 20 was isolated as a light yellow powder, HRESIMS analysis of 20 gave an ion [M+H]+ at m/z 777.2205 consistent with a molecular formula of C43H36O14 (Calcd. for C43H37O14, 777.2178). Acquisition of 1H, 13C, HSQC NMR spectra of 20 showed only 21 carbons, the presence of an orsellinic acid moiety, and signals of azaphilone skeleton, including a downfield shifted olefinic proton at δH 8.08 (s), two ketone carbonyl at δC 193.8, 193.6, a propenyl group at δH 5.78 (dd, J = 15.6, 1.3 Hz), 6.51 (dq, J = 15.6, 6.9 Hz), 1.67 (d, J = 6.9 Hz), δC 124.1, 137.0, 18.7 (Supporting Information Tables S7 and S9). These signals were very similar to those of 16, a known compound (−)-mitorubrin25, except for the presence of an additional methylene (δH 3.67, δC 19.6) in 20. According to its molecular formula, 20 was presumed to be composed of two same azaphilone monomer moieties similar to 16. Furthermore, HMBC correlations from the additional methylene protons H2-21 (δH 3.67) to C-4a/C-5/C-6, from H-1 to C-3/C-4a/C-8/C-8a, from H-4 to C-3/C-4a/C-8a/C-9, and from H-9 to C-3/C-4/C-10/C-11 confirmed that 20 was a symmetric azaphilone dimer connected by a C-21 methylene bridge to C-5 and C-5′ in two same azaphilone monomer moieties, respectively. Then, 20 was named didangelone A.
Compound 21 was isolated as yellow powder; its molecular formula was determined as C43H36O15 by HRESIMS ([M+H]+, m/z 793.2121; Calcd. for C43H37O15, 793.2127), one more oxygen than that of 20. The 13C NMR data (Table S9) of 21 were closely related to those of 20 except that the signal for C-11 at δC 18.7 in 20 was shifted downfield to δC 62.9 in 21, which indicated that 21 was the 11-hydroxy derivative of 20. HMBC correlations from H-11 to C-9/C-10 confirmed the location of hydroxy group at C-11. Then, 21 was named didangelone B.
The molecular formula C43H36O16 of compound 22 was established by HRESIMS ([M+Na]+, m/z 831.1918; Calcd. for C43H36O16Na, 831.1896). Analysis of the NMR spectra indicated that 22 also was a completely symmetrical azaphilone dimer. The NMR data (Tables S7 and S9) of 22 showed very similar signals to those of 20. The differences were that a methyl group (δH 1.67, δC 18.7) at C-11/C-11′ in 20 was replaced by a hydroxymethyl group (δH 4.07, δC 60.6) in 22, respectively, which was elucidated by 1H–1H COSY correlations of H-9/H-10/H-11 and HMBC correlations from hydroxymethyl proton H-11 to C-9/C-10. Then, 22 was named didangelone C.
Compound 23 was isolated as a red amorphous powder. The molecular formula of 23, C43H37NO13, was established by its HRESIMS spectrum ([M+H]+, m/z 776.2358; Calcd. for C43H38NO13, 776.2338), which showed that a nitrogen atom replaced an oxygen atom comparing with that of 20. The 1H and 13C NMR spectra (Tables S7 and S9) showed the typical signals of methylene-bridged azaphilone dimer. Detailed analysis of its NMR spectrum revealed that comparing with C-1′ and C-3′, the resonances for C-1 and C-3 were shifted up-field by 15.6 and 11.3 ppm, respectively, which suggested that an oxygen atom from one of pyran ring in 20 was replaced by a nitrogen atom to become a pyridine ring. Then, 23 was named didangelone D.
Compound 24 was also a red amorphous powder, its molecular formula was determined as C43H37NO15 by HRESIMS ([M+H]+, m/z 808.2291; Calcd. for C43H38NO15, 808.2295). The NMR data (Tables S7 and S9) of 24 were similar to those of 23, indicating that 24 was also a nitrogenous azaphilone dimer with a pyridine ring. The differences from 23 were that two methyl group (δH 1.51 and 1.55, δC 18.74 and 18.65) at C-11 and C-11′ in 23 was replaced by two hydroxymethyl group (δH 3.85 and 3.81, δC 60.7 and 60.9) in 24, respectively, which was confirmed by HMBC correlations from H-11 (δH 3.85) to C-9/C-10 and from H-11′ (δH 3.81) to C-9′/C-10′. Then, 24 was named didangelone E.
The absolute configuration of 20 was determined by ECD calculations36. It was found that the calculated weighted ECD spectra of 7R,7′R-20 were in good accordance with the experimental CD spectra of 20 (Supporting Information Fig. S288). Consequently, the absolute configuration of 20 was identified as 7R, 7′R. Likewise, the CD curve similarities between 21, 22, 23, 24 and 20 indicated that they bear the same configurations.
Compound 25 was isolated as a light yellow amorphous powder. Its molecular formula C42H34O17 was established by HRESIMS ([M+H]+, m/z 811.1860; Calcd. for C42H35O17, 811.1869). The 1D NMR (Supporting Information Tables S8 and S9) and HSQC data showed four ketone carbonyl carbons C-6/C-8/C-6′/C-8′ (δC 194.8/194.0/195.0/193.9), two oxygenated quaternary carbon C-7/C-7′ (δC 86.6/86.2), two downfield shifted olefin protons H-1/H-1′ (δH 8.15/8.23), and two orsellinic acid moieties. Based on these features, the NMR data of 25 were closely related to those of a known azaphilone dimer diazaphilonic acid (26)27,28 except that the carboxyl group at C-11 (δC 174.7) in 26 was replaced by a hydroxymethyl group [δH 3.65 (d, J = 7.2 Hz, 2H), δC 63.7] in 25, which was confirmed by HMBC correlations from H-11 (δH 3.65) to C-9/C-10 and 1H–1H COSY correlations of H-11/H-10(H-10′)/H-9. Thus, the planar structure of 25 was determined. The relative configuration of 25 was established on the basis of NOESY experiment. The NOE correlations of H-11 with H-9/H-10′/H-9′a suggested that these protons were placed on the same side of the molecule. According to its biosynthesis pathway same as that of azaphilone monomer, the absolute configuration of C-7 and C-7′ was assigned as R. ECD calculations were conducted to clarify the absolute configuration of 2536. The calculated weighted ECD spectra of 9R,10R,10′R,7R,7′R-25 and 9S,10S,10′S,7R,7′R-25 have obvious differences, revealing that the calculated ECD spectrum of 9R,10R,10′R,7R,7′R-25 agrees well with the experimental one (Supporting Information Fig. S289). Therefore, the absolute configuration of 25 was assigned as 9R, 10R, 10′R, 7R, 7′R and it was named didangelone H.
Compound 27 was isolated as a light yellow amorphous powder. HRESIMS spectrum established its molecular formula as C42H34O17 ([M+H]+, m/z 811.1829; Calcd. for C42H35O17, 811.1869), showing 26 degrees of unsaturation. 1D NMR (Tables S8 and S9) and HSQC spectra displayed typical signals for azaphilone dimer, including four ketone carbonyl carbons C-6/C-8/C-6′/C-8′ (δC 195.4/193.4/194.7/194.3), two downfield shifted olefin protons H-1/H-1′ (δH 7.90/8.13), and two orsellinic acid moieties. The 13C NMR data and HMBC interactions from H-1 to C-3/C-4a/C-8/C-8a and from H-1′ to C-3′/C-4a′/C-8′/C-8a′ established two orsellinic acyl substituted azaphilone pyranoquinone bicyclic cores similar to that of 16. In one of azaphilone cores, a double bond at C-3 and C-4 was reduced, which was supported by HMBC correlations of H-1 to C-3 (δC 104.3) and of H2-4 (δH 3.19/2.82) to C-3/C-4a/C-5/C-8a. In addition to above mentioned two orsellinic acyl substituted azaphilone cores, the remaining 13C NMR data showed only one unsaturated carboxyl carbon signal at δC 176.3 (C-11), indicating two unassigned degrees of unsaturation. Therefore, 27 was presumed to be dimerized though two rings by two orsellinic acyl substituted azaphilone bicyclic cores. 1H–1H COSY cross-peaks of H-9/H-10/H-10′(H-9′)/H-11′ and HMBC interactions from methine proton H-9 to C-3, and from oxygenated methylene protons H-11′ to C-3/C-9′ established a tetrahydropyran ring connecting with a dihydropyran ring of azaphilone core though spiral carbon C-3 (Fig. 3). HMBC correlations from H-9 to C-3′/C-4′/C-4a′, and from methylene proton H-9′ to C-3′/C-10′/C-11′ indicated a cyclohexene ring fused with a pyran ring of another azaphilone core though double bond at C-3′(C-4′), then establishing the polycyclic bridged heterocycle and spiral structures, tetrahydro-3-oxaspiro [bicyclo [3.3.1]nonane-2,2′-pyran] core. HMBC correlation from H-10 to C-11 suggested a carboxyl group attached to C-10. Thus, the planar structure of 27 was established. The signals for two active conjugated olefin protons H-5 and H-5′ did not show in the 1H NMR spectrum due to deuterium exchange in CD3OD.
The relative configuration of 27 was assigned by NOESY experiment. NOESY correlations of axial H-11′a with H-1/H-10 and of H-10 with H-9/H-10′/H-11′a indicated that these protons were in the same side of the tetrahydropyran ring, whereas that of H-9′a with H-11′b suggested the other side orientation (Fig. 4). According to the same biosynthetic pathway for azaphilone monomer, the absolute configuration of C-7 and C-7′ was both assigned as R. ECD calculations were performed to clarify the absolute configuration of 2738. The calculated weighted ECD spectra of 3R,9R,10S,10′R,7R,7′R-27 and 3S,9S,10R,10′S,7R,7′R-27 have obvious differences from 210 to 300 nm, revealing that the calculated ECD spectrum of 3R,9R,10S,10′R,7R,7′R-27 agrees well with the experimental one of 27 (Supporting Information Fig. S290). Therefore, the absolute configuration of 27 was determined as 3R, 9R, 10S, 10′R, 7R, 7′R and it was named didangelone F.
Compound 28 possessed the molecular formula C42H34O17 deduced from its HRESIMS ([M+Na]+, m/z 833.1708; Calcd. for C42H35O17Na, 833.1688) and 13C NMR. The planar structure of 28 was found to be same as that of 27 by detailed interpretation of its 1D and 2D NMR data (Tables S8 and S9). 28 differs from 27 only in the stereochemistry at C-3/C-9/C-10/C-10′, of which the relative configuration for C-3/C-9/C-10/C-10′ was same as that of 27 according to analysis of NOESY spectrum (Fig. 4). Using the same computational methods as that of 27, the absolute configuration of 28 was finally determined as 3S, 9S, 10R, 10′S, 7R, 7′R based on its CD curve similarities with ECD spectra of 3S,9S,10R,10′S,7R,7′R-28 (Supporting Information Fig. S290)38, which is actually a diastereoisomer of 27 and it was named didangelone G.
Compound 29 was isolated as a yellow amorphous powder. Its molecular formula was identified as C64H52O24 according to HRESIMS ([M+H]+, m/z 1205.2882; Calcd. for C64H53O24, 1205.2921). 1D NMR (Supporting Information Tables S10 and S11) and HSQC data showed three sets of typical azaphilone monomer signals, including six ketone carbonyl carbons, three oxygenated olefin protons, and three orsellinic acid moieties, indicating that 29 was probably an azaphilone trimer. A detailed comparison of 1H and 13C NMR data between 29, 27 and 20 disclosed that except for the presence of an azaphilone dimer moiety similar to 27, an additional methylene (δH 3.25/3.12, δC 20.9) and an orsellinic acyl substituted azaphilone monomer moiety, resemble to the half fragment of 20, were present in 29. HMBC correlations from the additional methylene protons H2-21 (δH 3.25/3.12) to C-5/C-6/C-4a″/C-5″ revealed that a C-21 methylene bridge connected C-5″ in an azaphilone monomer to C-5 in an azaphilone dimer moiety similar to 27. Thus, the planar structure of 29 was established. According to NOESY correlations, the relative configuration for C-3/C-9/C-10/C-10′ of 29 was same as that of 27. As the CD spectra of 29 with 27 highly coincidence, the absolute configurations of 29 was assigned as 3R, 9R, 10S, 10′R, 7R, 7′R, 7″R. Then, 29 was named tridangelone A.
Compound 30 has the same molecular formula as 29, C64H52O24, deduced from HRESIMS ([M+Na]+, m/z 1227.2653; Calcd. for C64H52O24Na, 1227.2682). Interpretation of 1D and 2D NMR data (Tables S10 and S11) disclose that 30 and 29 possess the same planar structure. 30 differs from 29 only in the stereochemistry at C-3/C-9/C-10/C-10′. According to NOESY correlations, the relative configuration for C-3/C-9/C-10/C-10′ of 30 was same as that of 28. The CD curve similarities between 30 and 28 indicated that the absolute configuration of 30 was established as 3S, 9S, 10R, 10′S, 7R, 7′R, 7″R, which is actually a diastereoisomer of 29 and it was named tridangelone B.
Compounds 31 and 32 were assigned to have the same planar structure by analysis of their HRESIMS and NMR data. Comparison of the 1H and 13C NMR data (Tables S10 and S11) between 31 and 29 suggested that 31 was a 11″-hydroxy derivative of 29, which was supported by HMBC correlations from hydroxymethyl proton H-11″ (δH 4.14) to C-9″/C-10″. According to NOESY correlations, the relative configurations for C-3/C-9/C-10/C-10′ of 31 and 32 were same as those of 29 and 30, respectively. The absolute configurations of 31 and 32 were respectively determined as 3R, 9R, 10S, 10′R, 7R, 7′R, 7″R and 3S, 9S, 10R, 10′S, 7R, 7′R, 7″R by comparing their CD spectra with those of 29 and 30. Then, 31 and 32 were named tridangelones C and D, respectively.
Compound 33 was isolated as a yellow amorphous powder. The molecular formula of 33 was determined to be C64H54O24 on the basis of HRESIMS data ([M+Na]+, m/z 1229.2950, Calcd. for C64H54O24Na, 1229.2897), one fewer oxygen and two more hydrogens than 31. The 1H and 13C NMR spectra (Tables S10 and S11) of 33 and 31 showed very similar signals, with the exception that the carboxyl carbon signal at δC 172.6 (C-11) in 31 was replaced by the signals assigned to hydroxymethyl group at δH 3.27/3.19 and δC 60.9 in 33. These data indicated that the carboxyl group at C-11 in 31 was substituted by a hydroxymethyl group in 33, which was supported by 1H–1H COSY correlation of H-11/H-10 and HMBC correlation from H-11 to C-9/C-10. According to NOESY correlations, the relative configuration for C-3/C-9/C-10/C-10′ of 33 was same as that of 31. Likewise, the CD curve similarities between 33 and 31 suggest that they bear the same absolute configurations and 33 was named tridangelone E.
Compound 34 was obtained as a yellow powder. HRESIMS analysis gave an [M+Na]+ ion at 585.1382 (Calcd. for C30H26O11Na, 585.1367), consistent with a molecular formula of C30H26O11. Comparison of the NMR data (Supporting Information Table S12) of 34 with those of (−)-mitorubrin (16)25 indicated the presence of three fragments, an orsellinic acyl substituted azaphilone monomer moiety similar to 16 (fragment A), an additional methylene (δH 3.40/3.76, δC 17.8), and a 3-substituted 2,4-dihydroxy-6-methylbenzoic acid moiety (fragment B). HMBC correlations from the methylene protons H-21 (δH 3.40/3.76) to C-5/C-4a/C-6/C-22/C-23/C-27 suggested that the methylene bridge at C-21 respectively linked to C-5 in fragment A and C-22 in fragment B. Thus, the planar structure of 34 was established. The absolute configuration of 34 was determined as 7R by comparing its CD spectra with those of (−)-mitorubrin (16)25,39 and it was named dangelone G.
Compound 35 was obtained as a yellow amorphous powder. Its molecular formula of C20H18O6 was determined on the basis of HRESIMS ([M−H]−, m/z 353.1032; Calcd. for C20H17O6, 353.1030). The 1H, 13C, and HSQC NMR (Table S12) spectra showed the presence of an aldehyde group at δH 9.81(s), δC 195.0, two methyl groups at δH 1.86/2.00 and δC 21.2/7.2, six aromatic protons at δH 6.07/6.11/6.21/5.89/6.34/7.46, and a methine at δH 6.42 and δC 39.3. HMBC correlations (Fig. 3) from methyl proton H-13 (δH 2.00) to C-9/C-10/C-11, from aldehyde proton H-14 (δH 9.81) to C-8/C-9, and from aromatic proton H-12 (δH 6.07) to C-7/C-8/C-10/C-11 established a 6-substituted 2,4-dihydroxy-3-methylbenzaldehyde (fragment C). 1H–1H COSY correlations of H-4/H-5/H-6 and HMBC correlations from H-4 to C-2/C-6 and from H-6 to C-2/C-4/C-5 confirmed a 2-substituted furan ring (fragment D). Additionally, the 1H NMR spectrum showed the presence of two meta-position protons of benzene ring at δH 6.21 (d, J = 2.3 Hz, H-17), 6.11 (d, J = 2.3 Hz, H-19). HMBC correlations from methyl protons H-21 (δH 1.86) to C-15/C-19/C-20, from H-17 to C-15/C-18/C-19, and from H-19 to C-15/C-17/C-18 established a 4-substituted 1,3-dihydroxy-5-methylbenzene (fragment E). The key HMBC correlations from methine proton H-1 (δH 6.42) to C-2/C-6/C-7/C-8/C-12/C-15/C-16/C-20 indicated that the C-1 methine bridge connected to C-7 in fragment C, C-2 in fragment D, and C-15 in fragments E, respectively (Fig. 3). Thus, the planar structure of 35 was established. Finally, ECD computational methods were used to clarify the absolute configuration of 35. The conformers of 1R-35 and 1S-35 were calculated by using the Gaussian 0936 software (Revision C.01, Gaussian Inc., Wallingford, CT, USA, 2010). It was found that the calculated weighted ECD spectra of 1R-35 are in good accordance with the experimental CD spectrum of 35 (Supporting Information Fig. S291). Consequently, the 1R configuration of 35 was assigned and it was named dangelone H.
2.4. Bioactivities evaluation
Cytotoxic activities of all isolated compounds were evaluated against two human cancer lines HepG-2 and MCF-7 in the MTT assay. Taxol was used as a positive control, with IC50 values of 1.28 (HepG2) and 2.31 (MCF-7) μmol/L, respectively. Compounds 14, 5, 10, and 11 exhibited cytotoxicities against HepG2 cells with IC50 values of 2.83, 6.82, 7.78, and 11.4 μmol/L, respectively. Compounds 14, 10, and 5 showed potent inhibition against MCF-7 cells with IC50 values of 5.53, 14.63, and 14.98 μmol/L, respectively, while the other compounds were inactive (IC50 > 20 μmol/L, Supporting Information Table S13). Comparing the structure of compounds 14, 5, and 11 with that of the other azaphilone monomers, orsellinic acid esterification at C-8 (14 vs. 17), double bond between C-8a and C-1 (14 vs. 5 and 11), and hydroxy at C-11 (14, 5, 11 vs. the others) significantly increase the activity, indicating the important contribution effects on cytotoxicity.
In addition, compounds 4–39 were also assessed for their anti-inflammatory and antioxidant inhibitory activities, with curcumin as a positive control (inhibitory rates 93.7 ± 2.5% at 10−5 mol/L and 91.2 ± 3.2% at 10−4 mol/L, respectively). At a concentration of 10−5 mol/L, compound 13 was found to be significant inhibitory effect on NO production induced by lipopolysaccharide (LPS) in murine microglial BV2 cell with inhibitory rate of 96.0 ± 4.6% (Supporting Information Table S14). Four compounds 21, 31, 34, and 36 were found to possess potent antioxidant activities against lipid peroxidation induced by Fe2+-cystine in rat liver microsomal with inhibitory rates of 68.7 ± 4.2%, 64.8 ± 2.6%, 66.7 ± 4.5%, and 63.5 ± 5.2%, respectively, at a concentration of 10−4 mol/L (Supporting Information Table S15). Interestingly, although the azaphilone monomers 16 and 17 showed no antioxidant activities and cytotoxicities, the azaphilone dimer 21 and trimer 31 exhibited potent antioxidant activities.
The antioxidant activities of compounds 4–39 were evaluated by measuring the inhibitory rates of malondialdehyde (MDA) in rat liver microsomal lipid peroxidation induced by Fe2+-cystine in vitro according to the reported anti-inflammatory activities of compounds 4–39 were assessed by measuring the inhibitory effects on NO production induced by lipopolysaccharide (LPS) in murine microglial BV2 cell line according to the reported procedures.
2.5. Characterization of the gene cluster for azaphilones biosynthesis
To understand the biosynthesis of these azaphilones, we analyzed the PKSs in the genome of P. dangeardii. BLAST search against the non-reducing PKS (NR-PKS) AzaA in the aza cluster40 and MrpigA in the MonAzPs cluster41, which were responsible for the biosynthesis of azanigerones and Monascus azaphilone pigments (MonAzPs), respectively, revealed two NR-PKSs (DanE and DanE′) with high protein identities (>55%) on Contig 60. A detailed bioinformatic analysis of adjacent genes of these two NR-PKSs identified two attractive neighbouring dual PKSs gene clusters, which were designated as dan1 and dan2 (Supporting Information Fig. S292A). Since multiple PKS genes located in a same gene cluster are common for fungal genomes42 and it has been reported that two or three PKSs synthesize a polyketide product either organized in sequence43,44 or in convergence45, the combination of dan1 and dan2 clusters responsible for the biosynthesis of isolated azaphilones is reasonable. In the dan1 and dan2 clusters, the predicted domains of NR-PKSs DanE and DanE′ were same as starter unit: ACP transacylase (SAT), ketosynthase (KS), acyltransferase (AT), product template (PT), C-methyltransferase (MT), acyl carrier protein (ACP) and reductase (R; Supporting Information Tables S16 and S17). Another NR-PKS (DanV) with SAT-KS-AT-PT-ACP-ACP domains in the dan2 cluster was proposed to involve in the formation of orsellinic acid. However, the fatty acid synthase DanG with α/β subunits in the dan1 cluster was supposed to inactive since the absence of ACPS domain. Most of the genes in the dan1 and dan2 clusters have one or more corresponding homolog in the aza gene cluster40 (Table S16). However, no key AzaH-like flavin-dependent monooxygenase, which is involved in C-7 hydroxylation and pyran-ring formation to afford the characteristic pyrano-quinone bicyclic core in azaphilones40, was presented in the dan2 cluster. These features suggest that the pair of gene clusters might redundantly or collaboratively biosynthesize the novel azaphilone derivatives with structure diversity.
To verify the gene clusters responsible for azaphilones biosynthesis, a pathway specific transcriptional factor danS was overexpressed through a gpdA gene as a strong promoter based on the homologous recombination method. The metabolites from the overexpressed mutant gpdA::danS were extracted and analyzed by LC‒MS (Supporting Information Fig. S4). Comparing to the metabolites from wild type P. dangeardii which did not produce any azaphilones, various azaphilones were detected in the mutant gpdA::danS, suggested that the dan gene clusters were responsible for azaphilones biosynthesis.
2.6. Proposed biosynthetic pathway for azaphilones from the dan1 and dan2 clusters
A putative biosynthetic pathway of azaphilones was proposed (Supporting Information Fig. S292B). Started from the NR-PKS DanE or DanE′ assembling the hexaketide precursor, an aldehyde intermediate was released. Then, the terminal ketone is reduced to alcohol i by the ketoreductase DanU (61% identity to AzaE). The FAD-dependent monooxygenase DanB, a homology of AzaH (59% identity) which is the only enzyme responsible for benzaldehyde hydroxylation and the formation of pyran ring during azanigerone biosynthesis40, is proposed to catalyze the hydroxylation of C-7 in i, which triggers the pyran ring formation to yield ii. Then, O-acetyltransferase DanN/DanN′ catalyzes the acetylation of ii at 10-OH, followed by the acetic acid elimination catalyzed by the dehydratase DanW to afford iii. In parallel, NR-PKS DanV is responsible for the generation of orsellinic acyl chain and O-acetyltransferase DanC/DanC′ directly transferred the orsellinic acyl-ACP to the C-7 hydroxy of iii. Previous studies demonstrated that cytochrome P450 oxygenases are able to catalyze the terminal methyl into alcohol, aldehyde, or acid through one to three consecutive oxidation steps, examples including P450-1 and P450-4 for gibberellin biosynthesis, CYP71AV1 for artemisinic acid biosynthesis, and AsCYP51H10 for the biosynthesis of antimicrobial triterpenes46. The terminal C-11 methyl in 16, 13, 4, and 10 could be hydroxylated by cytochrome P450 DanM to afford 17, 14, 5, and 11, respectively, and further oxidized to yield 15, 6, and 12 catalyzed by DanM.
For the biosynthesis of azaphilone polymers, we hypothesize that the C-5(4a) double bond of the azaphilone monomer I is reduced by aldoketoreductase DanK (Fig. S292B), an orthologue of NAD(P)H-dependent oxidoreductase MrPigE41 (69% identity), to yield intermediate II. The aldolase DanY is proposed to catalyze the enolization of C-6 ketone in II and introduce a hydroxymethyl at C-5 from formaldehyde, followed by dehydration to generate the reactive enone intermediate III. The enzyme with similar function has been reported, such as the α-methylserine aldolase from Variovorax paradoxus, which is responsible for conversion from l-alanine to α-methyl-l-serine accompanied by hydroxymethyl transfer from formaldehyde47. Michael addition between I and III yields the methylene-bridged azaphilone dimers (20–24). While non-enzymatic intermolecular Diels–Alder cycloaddition between two molecular of I results in the production of azaphilone dimers. Finally, azaphilone trimers (29–33) could be generated from combination of Michael addition and Diels–Alder cycloaddition between different azaphilone dimers with a third azaphilone monomer.
2.7. Discussion
Due to the potential of endophytic fungi to produce structurally diverse and biologically active secondary metabolites, various strategies for inducing the expression of silent biosynthetic gene clusters (BGCs) have been developed. However, no any strategies could be universal for all strains. The metabolic shunting strategy applied here is basically adapted from “Robin Hood” who robbed the wealthy to relieve the indigent. Inhibiting the biosynthesis of main metabolites in wild type strain would save the common building blocks (such as acetyl-CoA and malonyl-CoA for PKSs) to be readily competitively acquired by other BGCs, and induce the expression of cryptic gene clusters responsible for the biosynthesis of natural products required the saved building blocks. Results from RT-PCR (Fig. S3) also supported the up-regulated expression of four additional genes encoding PKS in the mutant strain. Since polyketide precursors such as acetyl-CoA and malonyl-CoA were reserved by elimination of rubratoxins biosynthesis, three genes encoding PKS in contig 60 responsible for azaphilones biosynthesis were activated and showed high expression, which consequently led to the predominant azaphilones production in the mutant strain. In addition, trace metabolites could become detectable without the presence of main metabolites. Consequently, a siderophore peptide desferritriacetylfusigen (39) was found only from the mutant strain ΔrbtJ although the gene encoding NRPS in contig 23 was expressed in both wild type and mutant strain.
Previously, an approach of genetic dereplication has been reported48, 49, 50 to discover novel natural products (especially trace metabolites) from fungi, which eliminated the production of major metabolites and made minor compounds detectable. Our strategy based on metabolic shunting and OSMAC is different in principle from genetic dereplication, and leads to the activation of cryptic gene clusters, besides letting trace metabolites detectable. The combinational application of metabolic shunting and OSMAC strategy is helpful to predict the biosynthesis and accumulation of specific types of natural products according to the deleted core gene type.
Although azaphilone monomers have been isolated from many other Penicillium strains21, such as Penicillium commune51 and Penicillium pinophilum20,52, inducing the expression of silent BGCs in P. dangeardii led to the production of many structurally complex azaphilone dimers and trimers with methylene and polycyclic bridged heterocycle and spiral structures.
Therefore, our established combinational strategy based on metabolic shunting and OSMAC is efficient and readily handled, and would be a perfect complement to other approaches of globally activating silent BGCs. This strategy has large potential to be applied for genome mining of other fungi or bacteria for discovering numerous novel secondary metabolites with a range of attractive bioactivities.
3. Conclusions
An approach combined metabolic shunting and OSMAC strategy was applied to successfully activate cryptic BGCs in the endophytic fungus P. dangeardii, which led to the production of numerous new compounds with diverse structure and biological activities. Remarkably, a group of unprecedented methylene-bridged and polycyclic azaphilone dimers and trimers were found for the first time. The biosynthetic gene cluster of these azaphilones was identified through overexpression of a pathway transcription factor in azaphilones cluster. Bioinformatic analysis of the interesting pair of dan1 and dan2 gene clusters helps to uncover some intriguing aspects of azaphilones biosynthetic pathway. Consequently, our work successfully develops a new strategy to rapidly and globally activate cryptic gene clusters and discover the amazing novel bioactive natural products in endophytic fungi.
4. Experimental
4.1. Strains and cultivation conditions
The endophytic fungus P. dangeardii was grown on PDA (BD Difco, Sparks, NV, USA) plate at 28 °C for 6 days for sporulation. P. dangeardii wild type and its mutant strains (ΔrbtJ and gpdA::danS) were stationarily cultured in PDA, PDB (BD Difco), CY (0.01% (v/v) Czapek concentrate, 0.5% yeast extract, 3% sucrose, 0.1% K2HPO4), YG (0.5% yeast extract, 2% glucose, 0.04% (v/v) trace element), YMEG (0.4% yeast extract, 1% malt extract, 0.4% glucose, 0.2% CaCO3) and rice at 28 °C for 6 days for production of secondary metabolites. Escherichia coli XL1-Blue was used for standard gene cloning. Saccharomyces cerevisiae YEH02 was used for yeast homologous recombination in vitro.
4.2. Gene knockout in P. dangeardii
The deletion of the target gene rbtJ (PKS) in P. dangeardii was carried out by homologous recombination using the S. cerevisiae YEH02. The genomic DNA of P. dangeardii was prepared using the plant genomic DNA extraction kit (Tiangen, Beijing, China). Primers for gene knockout were listed in Supporting Information Table S18. Pairs of primers, KC78uFor/KC78uRev, KC78dFor/KC78dRev and hphFor/hphRev were used for amplification of the upstream (2.5 kb) and downstream (2.5 kb) homologous regions of rbtJ and hph (hygromycin resistance). These PCR products were ligated into linearized vector backbone derived from plasmid pRS423, introduced to YEH02 using PEG/LiAc mediated transformation, and selected on uracildropout semisynthetic media, then constructed the deletion cassette. The plasmid in the correct transformant screened by colony PCR was rescued using Zymoprep Yeast Plasmid Miniprep Kit (Zymo Research, Orange County, CA, USA) and transferred to E. coli for gene cloning. The correct plasmids were used as template to amplify the knockout cassette. The PCR products of knockout cassette were recovered by gel recycling kit (Axygen, Union, CA, USA) and dissolved in STC buffer (1.2 mol/L sorbitol, 10 mmol/L calcium chloride and 10 mmol/L Tris-HCl, pH 7.5).
P. dangeardii spores were induced to young germ tubes in 50 mL PDB with 1% yeast extract at 28 °C for 17 h with 180 rpm agitation (Zhicheng ZWY-103 B, Shanghai, China). Cells were collected by centrifuging at 4 °C (5000 rpm) for 5 min (Sigma 3-18 K, Gettingen, Germany), washed twice with osmotic buffer (1.2 mol/L MgCl2, 10 mmol/L sodium phosphate, pH 5.8), and resuspended in the 12 mL enzyme cocktail solution (36 mg lysing enzyme and 24 mg yatalase in 1.2 mol/L MgSO4, 10 mmol/L sodium phosphate buffer) in 150 mL flask at 30 °C for 6 h with 80 rpm (Zhicheng ZWY-103B). After the digestion, the enzyme solution was transferred to a new tube and an equal volume of trapping buffer (0.6 mol/L sorbitol, 10 mmol/L Tris-HCl, pH 7.0) was added before centrifuging at 4 °C (5500 rpm, Sigma 3-18 K) for 10 min. The protoplast layer was collected and washed twice with two volumes of STC buffer and centrifuged (5000 rpm, Sigma 3-18 K) for 5 min at 4 °C. Protoplast precipitated in the lower layer suspension with 100 μL STC buffer and gently mixed with 10 μg of knockout cassette DNA fragment and incubated for 1 h on the ice. 1.2 mL of PEG 4000 solution (60% PEG 4000, 50 mmol/L CaC12, 50 mmol/L Tris-HCl, pH 7.5) was added to reaction mixture, incubated at 28 °C for 20 min and plated on the regeneration selection medium (PDA, 1.2 mol/L sorbitol, 800 mg/L hygromycin B). After culturing at 28 °C for 4–5 days, the transformants were stationarily inoculated on PDB for 3 days and then extracted the genomic DNA and confirming the genotype by PCR.
4.3. RNA preparation of P. dangeardii and reverse transcription-PCR (RT-PCR)
P. dangeardii wild type and the ΔrbtJ mutant were grown on rice at 25 °C for 6 days. The mycelia were collected and used for RNA extraction by TRIZOL® Reagent (Invitrogen, Carlsbad, CA, USA). Residual genomic DNA in the extracts was digested by DNase I (2 U/μL, NEB) at 37 °C for 45 min. The cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Mannheim, Germany) with Oligo-dT primers following directions from the user manual. PCR was performed with 2 × Hieff® PCR Master Mix (YEASEN, Shanghai, China) with cDNA. Primers for RT-PCR were listed in Supporting Information Table S19.
4.4. Overexpression of transcription factor danS in P. dangeardii
The overexpression cassette was constructed in plasmid pRS423 including a gpdA gene which as a strong promoter replaced the native promoter in transcription factor danS, an upstream (2.5 kb) and a downstream (2.5 kb) homologous regions of the native promoter, and a selection marker hph. The genes of gpdA and hph, upstream and downstream homologous regions were amplified with the primers of OETF52uFor/OETF52uRev, hphFor/hphRev, gpdaFor/gpdaRev and OETF52dFor/OETF52dRev. Primers for overexpression of transcription factor danS were listed in Supporting Information Table S20. The yeast recombinant in vitro, preparation of the protoplasts and transformation and screening were performed as above described in gene knockout.
4.5. General experimental procedures
Optical rotations were measured in MeOH using a JASCO P-2000 spectropolarimeter (Tokyo, Japan). IR spectra were recorded using a Thermo Nicolet 5700 FT-IR spectrophotometer (Waltham, MA, USA). UV spectra were determined with JASCO V-650 instrument. High-resolution electrospray ionization mass spectra (HRESIMS) were carried out using an Agilent Technologies 6520 Accurate Mass Q-TOF LC/MS spectrometer (Santa Clara, CA, USA). The CD spectra were obtained on a JASCO J-815 spectrometer. Medium pressure liquid chromatography (MPLC) was performed using ISCO-CombiFlash®Rf 200 (Teledyne Isco, Lincoln, NE, USA) with different types C18 column. Semipreparative HPLC was carried out on a SSI instrument (Scientific Systems Inc., Philadelphia, PA, USA) with a Series 1500 PDA detector and using a YMC-Pack ODS-A column (YMC, 5 μm, 250 mm × 10 mm) or a phenyl-hexyl column (phenomenex, 5 μm, 250 mm × 10 mm). Column chromatography (CC) was performed using middle chromatogram isolated (MCI) gel CHP 20P/P120 (Mistubishi Chemical Co., Tokyo, Japan) and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Thin layer chromatography (TLC) was performed on GF254 (Qingdao Marine Chemical Factory, Qingdao, China). All LC‒MS analyses were carried out on a Waters ACQUITY H-Class UPLC‒MS (Milford, MA, USA) with QDA mass detector (ACQUITY UPLC® BEH, 1.7 μm, 50 mm × 2.1 mm, C18 column) using positive and negative mode electrospray ionization with a linear gradient of 5%–99% MeCN/H2O (v/v, 0.02% formic acid) in 8 min followed by 99% MeCN/H2O (v/v, 0.02% formic acid) for 4 min with a flow rate of 0.4 mL/min 1D and 2D NMR spectra were determined with Bruker AVIII-600 spectrometer (Rheinstetten, Germany) with a 5 mm dual cryoprobe or a Bruker AVIII-500 spectrometer or Bruker AVANCE III 800 MHz with a 5 mm broadband probe. All solvents and chemicals used in this study are of analytical grade (for extraction) or LC‒MS grade (for LC‒MS analysis).
4.6. Analysis, extract, and isolation of metabolites
For metabolites analysis, spores of wild type and mutant strains were inoculated in different culture media and stationarily cultured for 7 days at 28 °C. The mycelia and medium were extracted with ethyl acetate (EtOAc) and analyzed by UPLC‒MS.
The strain was incubated on PDA at 28 °C for 6 days to afford spores and the spores were inoculated into the PDB at 28 °C for 4 days with 180 rpm (Zhicheng ZWY-103 B) shake to prepare the seed culture. 5 mL seed culture was inoculated into 130 Erlenmeyer flasks (500 mL), previously sterilized by autoclaving, each containing 100 g rice and 100 mL distilled water. All flasks were incubated at 28 °C for 30 days. The culture was extracted three times with EtOAc at room temperature and the EtOAc was removed under reduced pressure to yield a crude extract. The crude extract was dissolved with petroleum ether (PE) to remove the fatty oil (27 g) and the residue which is insolvable in PE was evaporated to dryness and separated by MCI CC with a gradient of H2O, 30%, 70% and 100% MeOH, acetone to obtain five fractions (Frs. A–E).
Fr. B (5 g) was further separated with MCI CC eluted with a gradient of 10%, 20%, 50% and 100% MeOH to obtain four fractions (B1–B4). Fr. B4 (1.1 g) was applied to a Sephadex LH-20 CC eluted with MeOH to obtain 10 fractions (Frs. B4a–j), based on TLC analysis.
Fr. B4e was purified by semi-preparative HPLC on phenyl-hexyl column eluted with 29% MeCN/H2O (v/v, 0.02% formic acid) at a flow rate of 2.0 mL/min to yield 6 (5.0 mg, tR = 42.0 min), 8 (2.5 mg, tR = 50.0 min), 9 (2.4 mg, tR = 26.0 min) and 15 (4.0 mg, tR = 34.0 min).
Fr. B4f was purified by semi-preparative HPLC on phenyl-hexyl column eluted with 34% MeCN/H2O (v/v, 0.02% formic acid) at a flow rate of 2.0 mL/min to afford 18 (5.0 mg, tR = 23.0 min).
Fr. C (12 g) was divided into two parts and separated by MPLC eluted with a linear gradient of 5%–95% MeOH/H2O (v/v, 0.02% formic acid) in 130 min and 100% MeOH for 30 min at a flow rate of 50 mL/min on C18 column (SepaFlash C18, 20–45 μm, SW330, Changzhou, China) to give twenty fractions (Frs. C1–20).
Fr. C7 (340 mg) was separated on Sephadex LH-20 CC (50% MeOH/H2O) to obtain 16 additional fractions (Frs. C7a–p). Then Fr. C7m (70 mg) purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 29% MeCN, v/v 0.02% formic acid) to yield 5 (9 mg, tR = 32 min), 14 (9 mg, tR = 27 min) and Fr. C7o (37 mg) purified by semi-preparative HPLC on C18 column (2.0 mL/min, MeCN/H2O, 36%, v/v 0.02% formic acid) to yield 17 (4.4 mg, tR = 26 min).
Fr. C9 (888 mg) was separated on Sephadex LH-20 CC (50% MeOH/H2O) to obtain 11 additional fractions (Frs. C9a–k). Fr. C9b (590 mg) was further purified by semi-preparative HPLC on C18 column eluted with 44% MeOH/H2O (v/v, 0.02% formic acid) at a flow rate of 5.0 mL/min to yield 39 (150 mg, tR = 73.0 min).
Fr. C10 (440 mg) was subjected to a Sephadex LH-20 CC eluted with MeOH to yield 6 subfractions (Frs. C10a–f), based on TLC analysis. Fr. C10a (43 mg) was purified by semi-preparative HPLC on C18 column eluted with 37% MeCN/H2O (v/v, 0.02% formic acid) at a flow rate of 2.0 mL/min to yield 38 (7.7 mg, tR = 17.0 min).
Fr. C11 (67 mg) was further purified by semi-preparative HPLC on C18 column eluting with 35% MeCN/H2O (v/v, 0.02% formic acid) at a flow rate of 2.0 mL/min to yield 11 (2.2 mg, tR = 23.0 min), 12 (15.5 mg, tR = 26.0 min), 37 (20 mg, tR = 11.0 min), respectively.
Fr. C12 (1.24 g) was separated on a Sephadex LH-20 CC (MeOH) to obtain 15 additional fractions (Frs. C12a–o) and then Fr. C12j (154 mg) purified by semi-preparative HPLC on C18 column (5.0 mL/min, 54% MeOH/H2O, v/v 0.02% formic acid) to yield 25 (48.2 mg, tR = 54.0 min), 35 (2.2 mg, tR = 95.0 min). Fr. C12k (88 mg) purified by semi-preparative HPLC on C18 column (2.0 mL/min, 38% MeCN/H2O, v/v 0.02% formic acid) to yield 26 (36 mg, tR = 36.5 min). Fr. C12l (33 mg) purified by semi-preparative HPLC on C18 column (2.0 mL/min, 41% MeCN/H2O, v/v 0.02% formic acid) to yield 28 (9.8 mg, tR = 41 min). Fr. C12n (33 mg) purified by semi-preparative HPLC on C18 column (2.0 mL/min, 42% MeCN/H2O, v/v 0.02% formic acid) to yield 27 (4.1 mg, tR = 44 min).
Fr. C13 (1.15 g) was applied to a Sephadex LH-20 column eluted with MeOH to give 11 subfractions (Frs. C13a–k), based on TLC analysis. Then Subfraction Fr. C13f (45 mg) was purified by semi-preparative HPLC on C18 column eluted with 42% MeCN/H2O (v/v, 0.02% formic acid) at a flow rate of 2.0 mL/min to yield 19 (1.0 mg, tR = 19.0 min). Fr. C13g (247 mg) was purified by semi-preparative HPLC on phenyl-hexyl column eluted with MeCN/H2O (2.0 mL/min, 47% MeCN/H2O, v/v 0.02% formic acid) to yield 10 (10.2 mg, tR = 27 min), 16 (11.7 mg, tR = 35 min).
Fr. C14 (1.0 g) was subjected to a Sephadex LH-20 column eluted with MeOH to give 14 subfractions (Frs. C14a–n), based on TLC analysis. Fr. C14l (120 mg) was further separated by Sephadex LH-20 CC eluted with MeOH to give 6 subfractions (Frs. C14l1–6). Fr. C14l4 (29 mg) was purified by semi-preparative HPLC on C18 column (2.0 mL/min, 45%–48% MeCN/H2O 40 min, 48%–60% MeCN/H2O 10 min, v/v 0.02% formic acid) to yield 24 (3.5 mg, tR = 15 min). Fr. C14l6 (12 mg) was purified by semi-preparative HPLC on C18 column (2.0 mL/min, 46% MeCN/H2O, v/v 0.02% formic acid) to yield 22 (8.0 mg, tR = 34 min).
Fr. C15 (483 mg) was applied to Sephadex LH-20 CC eluted with MeOH to obtain eight subfractions (Frs. C15a–h). Fr. C15f (75 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 46% MeCN/H2O, v/v 0.02% formic acid) to yield 31 (3.5 mg, tR = 33 min), 33 (3.0 mg, tR = 39 min), and 32 (16.4 mg, tR = 43 min).
Fr. C17 (293 mg) was subjected to Sephadex LH-20 CC eluted with MeOH to get 6 subfractions (Frs. C17a–f). Fr. C17d (41 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 50% MeCN/H2O, v/v 0.02% formic acid) to yield 29 (3.9 mg, tR = 41 min) and 30 (6.2 mg, tR = 52 min).
Fr. C18 (481 mg) was further separated by Sephadex LH-20 CC eluted with MeOH to obtain eight subfractions (Frs. C18a–h). Fr. C18f (42 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 53% MeCN/H2O, v/v 0.02% formic acid) to yield 21 (11.2 mg, tR = 43 min) and 23 (4.4 mg, tR = 33 min). Fr. C18g (22 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 55% MeCN/H2O, v/v 0.02% formic acid) to yield 34 (1.5 mg, tR = 27 min).
Fr. C20 (275 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 60% MeCN/H2O, v/v 0.02% formic acid) to yield 20 (13 mg, tR = 47 min).
Fr. D (2.0 g) was applied to Sephadex LH-20 CC eluted with MeOH to obtain 14 subfractions (Frs. D1–14). Fr. D7 (80 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 47% MeCN/H2O, v/v 0.02% formic acid) to yield 4 (4.0 mg, tR = 31 min), 7 (0.9 mg, tR = 24 min), and 13 (0.9 mg, tR = 26 min). Fr. D12 (80 mg) was purified by semi-preparative HPLC on phenyl-hexyl column (2.0 mL/min, 48% MeCN/H2O, v/v 0.02% formic acid) to yield 36 (2.0 mg, tR = 31 min).
4.6.1. Dangelone A (4)
Yellow powder –181.3 (c 0.8, MeOH); UV (MeOH) λmax (logε) 215 (4.28), 265 (4.03), 346 (4.21) nm. IR (KBr) νmax 3355, 2918, 1645, 1583, 1447, 1255, 1204, 1169, 1104, 962, 797 cm−1; CD (MeOH) λmax (ε): 227 (−3.29), 259 (+0.49), 275 (−0.31), 306 (+0.57), 360 (−1.61) nm. 1H NMR (800 MHz, in DMSO-d6) and 13C NMR (200 MHz, in DMSO-d6), Tables S4 and S5. ESI-MS m/z 387.16 [M+H]+ and 385.34 [M–H]−. HRESIMS: 387.1438 [M+H]+, Calcd. for C21H23O7, 387.1438.
4.6.2. Dangelone B (5)
Yellow powder –132.6 (c 0.4, MeOH); UV (MeOH) λmax 211, 262, 346 nm. IR (KBr) νmax 3305, 2978, 1649, 1585, 1456, 1257 cm−1; CD (MeOH) λmax (ε): 226 (−7.4), 249 (+0.98), 272 (−1.45), 307 (+0.71), 359 (−3.76) nm. 1H NMR (800 MHz, in DMSO-d6) and 13C NMR (200 MHz, in DMSO-d6), Tables S4 and S5. ESI-MS m/z 403.38 [M+H]+ and 401.32 [M–H]−. HRESIMS: 403.1387 [M+H]+, Calcd. for C21H23O8, 403.1387.
4.6.3. Dangelone C (6)
Yellow powder –173.7 (c 0.8, MeOH); UV (MeOH) λmax (logε) 215 (4.14), 265 (3.96), 347 (4.10) nm. IR (KBr) νmax 3295, 2917, 1649, 1591, 1257 cm−1; CD (MeOH) λmax (ε): 230 (−2.0), 255 (+0.91), 275 (−0.27), 358 (−1.63) nm. 1H NMR (800 MHz, in DMSO-d6) and 13C NMR (200 MHz, in DMSO-d6), Tables S4 and S5. ESI-MS m/z 417.17 [M+H]+ and 415.23 [M–H]−. HRESIMS: 417.1190 [M+H]+ Calcd. for C21H21O9, 417.1180.
4.6.4. Dangelone D (7)
Yellow powder +52.9 (c 0.3, MeOH); UV (MeOH) λmax 215, 263, 349 nm. IR (KBr) νmax 3386, 2934, 1716, 1647, 1619, 1585, 1447, 1381, 1255, 1202, 1171, 1054 cm−1; CD (MeOH) λmax (ε): 210 (+2.27), 225 (−0.35), 258 (+1.92), 301 (−3.95), 331 (+1.63), 361 (+1.51) nm. 1H NMR (600 MHz, in acetone-d6) and 13C NMR (150 MHz, in acetone-d6), Tables S4 and S5. ESI-MS m/z 445.22 [M+H]+ and 443.38 [M–H]−. HRESIMS: 467.1328 [M+Na]+, Calcd. for C23H24O9Na, 467.1313.
4.6.5. Dangelone E (8)
Yellow powder +43.1 (c 1.4, MeOH); UV (MeOH) λmax (logε) 214 (4.03), 268 (3.77), 314 (3.87) nm. IR (KBr) νmax 3325, 2918, 1712, 1644, 1588, 1449, 1255, 1168 cm−1; CD (MeOH) λmax (ε): 230 (−1.52), 266 (+2.67), 296 (−1.11), 320 (+1.72), 352 (+1.38) nm. 1H NMR (600 MHz, in acetone-d6) and 13C NMR (150 MHz, in acetone-d6), Tables S4 and S5. ESI-MS m/z 419.17 [M+H]+, 417.21 [M–H]−. HRESIMS: 419.1352 [M+H]+, Calcd. for C21H23O9, 419.1337.
4.6.6. Dangelone F (9)
Yellow powder +60.7 (c 1.9, MeOH); UV (MeOH) λmax (logε) 215 (4.13), 266 (3.92), 308 (3.85) nm. IR (KBr) νmax 3363, 2975, 1696, 1645, 1450, 1259, 1204 cm−1; CD (MeOH) λmax (ε): 212 (+1.87), 254 (+1.95), 288 (−2.11), 320 (+2.21), 374 (−0.18) nm. 1H NMR (600 MHz, in acetone-d6) and 13C NMR (150 MHz, in acetone-d6), Tables S4 and S5. ESI-MS m/z 441.16 [M+Na]+ and 417.26 [M–H]−. HRESIMS: 441.1138 [M+Na]+, Calcd. for C21H22O9Na, 441.1156.
4.6.7. Dangeloside A (18)
Light yellow powder –52.9 (c 0.2, MeOH); UV (MeOH) λmax (logε) 206 (4.04), 264 (3.51), 346 (3.02) nm. IR (KBr) νmax 3383, 2920, 1647, 1614, 1419, 1257, 1203 cm−1; CD (MeOH) λmax (ε): 209 (−3.65), 232 (−0.46), 255 (+1.07), 276 (−0.45), 305 (+0.64), 358 (−2.58) nm. 1H NMR (600 MHz, in acetone-d6) and 13C NMR (150 MHz, in acetone-d6), Table S6. ESI-MS m/z 549.17 [M+H]+ and 547.23 [M–H]−. HRESIMS: 571.1429 [M+Na]+, Calcd. for C26H28O13Na, 571.1422.
4.6.8. Dangeloside B (19)
Light yellow powder +134.5 (c 0.6, MeOH); UV (MeOH) λmax (logε) 214 (3.73), 262 (3.42), 348 (3.55) nm. IR (KBr) νmax 3409, 2924, 1722, 1650, 1582, 1453, 1257, 1051 cm−1; CD (MeOH) λmax (ε): 214 (+5.25), 262 (+5.37), 305 (−2.29), 372 (+4.04) nm. 1H NMR (500 MHz, in CD3OD) and 13C NMR (125 MHz, in CD3OD), Table S6. ESI-MS m/z 519.17 [M+H]+ and 517.23 [M–H]−. HRESIMS: 519.1868 [M+H]+, Calcd. for C26H31O11, 519.1861.
4.6.9. Didangelone A (20)
Yellow powder –299.4 (c 0.2, MeOH); UV (MeOH) λmax (logε) 216 (4.53), 267 (4.43), 300 (4.22), 361 (4.25) nm. IR (KBr) νmax 3367, 2918, 1716, 1620, 1587, 1449, 1262, 1169 cm−1; CD (MeOH) λmax (ε): 216 (−5.56), 262 (+9.64), 295 (+11.63), 352 (+8.52), 402 (−8.35) nm. 1H NMR (500 MHz, in CD3OD) and 13C NMR (125 MHz, in CD3OD), Tables S7 and S9. ESI-MS m/z 777.44 [M+H]+ 775.41 [M–H]−. HRESIMS: 777.2205 [M+H]+, Calcd. for C43H37O14, 777.2178.
4.6.10. Didangelone B (21)
Yellow powder –226.2 (c 0.2, MeOH); UV (MeOH) λmax (logε) 215 (4.56), 267 (4.43), 299 (4.22), 360 (4.24) nm. IR (KBr) νmax 3349, 3249, 2928, 2854, 1716, 1613, 1442, 1262, 1165 cm−1; CD (MeOH) λmax (ε): 215 (−2.08), 262 (+6.57), 295 (+10.50), 328 (−3.76), 356 (+3.99), 404 (−5.42) nm. 1H NMR (500 MHz, in CD3OD) and 13C NMR (125 MHz, in CD3OD), Tables S7 and S9. ESI-MS m/z 793.29 [M+H]+ and 791.74 [M–H]−. HRESIMS: 793.2121 [M+H]+, Calcd. for C43H37O15, 793.2127.
4.6.11. Didangelone C (22)
Yellow powder –261.8 (c 0.9, MeOH); UV (MeOH) λmax (logε) 211 (4.43), 265 (4.25), 295 (4.02), 358 (3.98) nm. IR (KBr) νmax 3358, 2919, 1715, 1619, 1449, 1262, 1170 cm−1; CD (MeOH) λmax (ε): 221 (+2.73), 244 (−0.53), 295 (+5.30), 331 (−7.43), 389 (−4.55) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S7 and S9. ESI-MS m/z 809.36 [M+H]+ and 807.53 [M–H]−. HRESIMS: 831.1918 [M+Na]+, Calcd. for C43H36O16Na, 831.1896.
4.6.12. Didangelone D (23)
Red powder –333.7 (c 1.6, MeOH); UV (MeOH) λmax (logε) 208 (4.28), 266 (4.05), 301 (3.89), 364 (3.71) nm. IR (KBr) νmax 3281, 2921, 1713, 1620, 1448, 1323, 1262, 1168 cm−1; CD (MeOH) λmax (ε): 214 (−6.24), 234 (+3.45), 295 (+16.97), 350 (+6.50), 391 (−12.70) nm. 1H NMR (600 MHz, in CD3OD) and 13C NMR (150 MHz, in CD3OD), Tables S7 and S9. ESI-MS m/z 776.34 [M+H]+ and 774.45 [M–H]−. HRESIMS: 776.2358 [M+H]+, Calcd. for C43H38NO13, 776.2338.
4.6.13. Didangelone E (24)
Red powder –248.6 (c 0.2, MeOH); UV (MeOH) λmax (logε) 215 (4.53), 267 (4.33), 300 (4.16), 363 (3.96) nm. IR (KBr) νmax 3275, 2921, 1710, 1618, 1595, 1452, 1322, 1264 cm−1; CD (MeOH) λmax (ε): 212 (−2.33), 232 (+1.31), 296 (+6.31), 387 (−5.60) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S7 and S9. ESI-MS m/z 808.30 [M+H]+ and 806.49 [M–H]−. HRESIMS: 808.2291 [M+H]+, Calcd. for C43H38NO15, 808.2295.
4.6.14. Didangelone H (25)
Yellow powder –365.4 (c 0.5, MeOH); UV (MeOH) λmax (logε) 216 (4.75), 267 (4.44), 333 (4.43) nm. IR (KBr) νmax 3258, 2920, 1719, 1621, 1545, 1451, 1323, 1262 cm−1; CD (MeOH) λmax (ε): 232 (+14.07), 303 (+48.21), 347 (−64.22) nm. 1H NMR (500 MHz, in CD3OD) and 13C NMR (125 MHz, in CD3OD), Tables S8 and S9. ESI-MS m/z 811.40 [M+H]+ and 809.38 [M–H]−. HRESIMS: 811.1860 [M+H]+, Calcd. for C42H35O17, 811.1869.
4.6.15. Didangelone F (27)
Light yellow powder –259.5 (c 0.3, MeOH); UV (MeOH) λmax (logε) 214 (4.34), 269 (4.04), 307 (3.97) nm. IR (KBr) νmax 3286, 2919, 2852, 1717, 1620, 1452, 1261, 1110 cm−1; CD (MeOH) λmax (ε): 223 (+10.76), 295 (+21.75), 335 (−25.94) nm. 1H NMR (600 MHz, in CD3OD) and 13C NMR (150 MHz, in CD3OD), Tables S8 and S9. ESI-MS m/z 811.41 [M+H]+ and 809.34 [M–H]−. HRESIMS: 811.1829 [M+H]+, Calcd. for C42H35O17, 811.1869.
4.6.16. Didangelone G (28)
Light yellow powder –182.5 (c 0.8, MeOH); UV (MeOH) λmax (logε) 215 (4.49), 268 (4.19), 307 (4.10) nm. IR (KBr) νmax 3363, 2920, 2851, 1715, 1620, 1452, 1324, 1263, 1171 cm−1; CD (MeOH) λmax (ε): 211 (+7.91), 268 (+12.63), 302 (+19.28), 336 (−45.34) nm. 1H NMR (600 MHz, in CD3OD) and 13C NMR (150 MHz, in CD3OD), Tables S8 and S9. ESI-MS m/z 811.38 [M+H]+ and 809.37 [M–H]−. HRESIMS: 833.1708 [M+Na]+, Calcd. for C42H34O17Na, 833.1688.
4.6.17. Tridangelone A (29)
Yellow powder –102.1 (c 0.4, MeOH); UV (MeOH) λmax (logε) 214 (4.54), 268 (4.29), 305 (4.16), 346 (4.01) nm. IR (KBr) νmax 3357, 3288, 2917, 1715, 1626, 1449, 1323, 1262, 1169 cm−1; CD (MeOH) λmax (ε): 227 (+16.86), 255 (−0.33), 295 (+26.64), 342 (−31.29) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S10 and S11. ESI-MS m/z 1205.38 [M+H]+ and 1203.54 [M–H]−. HRESIMS: 1205.2882 [M+H]+, Calcd. for C64H53O24, 1205.2921.
4.6.18. Tridangelone B (30)
Yellow powder –167.4 (c 1.8, MeOH); UV (MeOH) λmax (logε) 214 (4.66), 268 (4.44), 305 (4.32), 347 (4.17) nm. IR (KBr) νmax 3362, 2920, 2852, 1715, 1621, 1449, 1324, 1262, 1168, 1105 cm−1; CD (MeOH) λmax (ε): 209 (+11.07), 245 (−2.09), 306 (+27.55), 345 (−27.86) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S10 and S11. ESI-MS m/z 1205.43 [M+H]+ and 1203.54 [M–H]−. HRESIMS: 1227.2653 [M+Na]+, Calcd. for C64H52O24Na, 1227.2682.
4.6.19. Tridangelone C (31)
Yellow powder –143.2 (c 0.4, MeOH); UV (MeOH) λmax (logε) 205 (4.45), 269 (4.15), 308 (4.02), 349 (3.87) nm. IR (KBr) νmax 3394, 2920, 2851, 1734, 1645, 1466, 1325, 1262, 1103, 801 cm−1; CD (MeOH) λmax (ε): 227 (+25.79), 294 (+33.52), 340 (−39.96) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S10 and S11. ESI-MS m/z 1221.33 [M+H]+ and 1219.40 [M–H]−. HRESIMS: 1221.2883 [M+H]+, Calcd. for C64H53O25, 1221.2870.
4.6.20. Tridangelone D (32)
Yellow powder –203.8 (c 1.3, MeOH); UV (MeOH) λmax (logε) 215 (4.59), 268 (4.37), 307 (4.24), 348 (4.10) nm. IR (KBr) νmax 3247, 2920, 2851, 1619, 1449, 1324, 1262, 1169 cm−1; CD (MeOH) λmax (ε): 211 (+8.40), 246 (−1.68), 306 (+22.93), 344 (−24.60) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S10 and S11. ESI-MS m/z 1221.26 [M+H]+ and 1219.39 [M–H]−. HRESIMS: 1243.2625 [M+Na]+, Calcd. for C64H52O25Na, 1243.2690.
4.6.21. Tridangelone E (33)
Yellow powder –191.3 (c 1.2, MeOH); UV (MeOH) λmax (logε) 214 (4.43), 268 (4.21), 305 (4.06), 346 (3.90) nm. IR (KBr) νmax 3328, 2269, 2917, 1714, 1619, 1450, 1323, 1261, 1104 cm−1; CD (MeOH) λmax (ε): 226 (+16.04), 258 (−0.79), 296 (+20.80), 339 (−29.02) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Tables S10 and S11. ESI-MS m/z 1207.42 [M+H]+ and 1205.57 [M–H]−. HRESIMS: 1229.2950 [M+Na]+, Calcd. for C64H54O24Na, 1229.2897.
4.6.22. Dangelone G (34)
Yellow powder –186.3 (c 0.9, MeOH); UV (MeOH) λmax (logε) 216 (4.15), 262 (3.96), 301 (3.70), 355 (3.63) nm. IR (KBr) νmax 3199, 2924, 2851, 1715, 1642, 1619, 1449, 1325, 1261 cm−1; CD (MeOH) λmax (ε): 228 (+3.62), 255 (−2.77), 300 (+4.78), 352 (−4.69) nm. 1H NMR (600 MHz, in DMSO-d6) and 13C NMR (150 MHz, in DMSO-d6), Table S12. ESI-MS m/z 563.12 [M+H]+ and 561.23 [M–H]−. HRESIMS: 585.1382 [M+Na]+, Calcd. for C30H26O11Na, 585.1367.
4.6.23. Dangelone H (35)
Yellow powder –54.1 (c 0.9, MeOH); UV (MeOH) λmax (logε) 205 (4.15), 270 (3.60), 300 (3.56) nm. IR (KBr) νmax 3331, 2918, 1730, 1618, 1455, 1261, 1102 cm−1; CD (MeOH) λmax (ε): 217 (−2.00), 247 (+0.26), 301 (+1.30), 342 (−2.05) nm. 1H NMR (600 MHz, in CD3OD) and 13C NMR (150 MHz, in CD3OD), Table S12. ESI-MS m/z 353.25 [M–H]−. HRESIMS: 353.1032 [M–H]–, Calcd. for C20H17O6, 353.1030.
4.7. ECD calculations of compounds 9, 18, 20, 25, 27, 28, and 35
Conformational analyses of compounds 9, 18, 20, 25, and 35 were performed via Monte Carlo searching with the MMFF94 force field using the Spartan 10 software (Wavefunction, Inc., Irvine, CA, USA). All conformers with relative energies within 9 kcal/mol were optimized for density functional theory (DFT) calculations at the B3LYP/6-31G(d) level in vacuum using the Gaussian 09 program36. Subsequently, the B3LYP/6-31G(d)-optimized conformers were re-optimized at the WB97XD/DGDZVP level in methanol. The WB97XD/DGDZVP harmonic vibrational frequency analyses confirmed the stability of the conformers. ECD computations for the WB97XD/DGDZVP-optimized conformers were performed using time-dependent DFT (TD-DFT) at the CAM-B3LYP/DGDZVP level in methanol. The predicted ECD spectra were simulated by the Gaussum 2.25 program53. The final ECD spectra were acquired on the base of the Boltzmann distribution theory and the relative Gibbs free energy (ΔG).
Conformational analyses of compounds 27 and 28 were performed in Yinfo Cloud Platform (https://cloud.yinfotek.com/) using Confab54 with systematic algorithm at MMFF94 force field with RMSD threshold of 0.5 Å and energy window of 7 kcal/mol. The theoretical calculations were carried out using Gaussian 0938 (Revision D.01, Gaussian Inc., Wallingford, CT, USA, 2009). At first, conformers were optimized at PM6 and HF/6-31G(d) theory levels, consecutively. Room-temperature equilibrium populations were calculated on the basis of Boltzmann distribution law and dominative conformers with the value greater than 1% were saved. The chosen conformers were finally optimized at B3LYP/6-31G(d) in gas phase. Vibrational frequency analysis confirmed the stable structures. ECD calculations were conducted in methanol with IEFPCM model using TD-DFT. Rotatory strengths for 100 excited states were calculated. The ECD spectrum was simulated using the ECD/UV analysis tool in Yinfo Cloud Platform by overlapping Gaussian functions for each transition.
4.8. Determination of the absolute configuration of ribose moieties in compound 18
To verify the absolute configurations of ribose moiety in 18, a modified method based on UPLC‒MS analysis was adopted, where the parent compounds were hydrolyzed to afford the sugar samples, the retention times of which were compared with those of standard sugars (d/l)55.
Acid hydrolysis reaction: dissloving 18 (1.4 mg, each) in 1 mL HCl (1 mol/L) treated with 3 mL MeOH/H2O (v/v, 2:1) under 70 °C for 4 h. The reaction mixture was evaporated to dry extract, and extracted three times with EtOAc after extract dissloving in 2 mL H2O. The water layer was evaporated to afford a hydrolytic monosaccharide.
Preparation of monosaccharide derivatives: solutions of l-ribose (2.0 mg), d-ribose (2.0 mg) and hydrolytic monosaccharides (0.4 mg, each) in 1 mL pyridine were individually treated with l-cysteine methyl ester hydrochloride (2.0 mg) under 60 °C for 2 h, and then added O-tolyl isothiocyanate (2.0 mg) in each reaction system under 60 °C for 1 h. A blank control group was preapared at the same time.
Each reaction mixture was analyzed by UPLC‒MS. As shown in Fig. S7, the retention time of hydrolyzed monosaccharide derivative was in accordance with that of d-ribose derivative. Thus, the ribose in 18 was determined as d-configuration.
4.9. Cytotoxicity assays
The cytotoxicities of compounds 4–39 against human cancer cell lines HepG-2 and MCF-7 (NCI-60, Bethesda, USA) as well as the murine microglial BV2 cell line were measured using the MTT assay according to the reported procedures56. Taxol was used as the positive control.
4.10. Anti-inflammatory assays
The anti-inflammatory activities of compounds 4–39 were assessed by measuring the inhibitory effects on NO production induced by lipopolysaccharide (LPS) in murine microglial BV2 cell line according to the reported procedures56. Curcumin was selected as the positive control (inhibitory rate 93.7 ± 2.5% at 10−5 mol/L). The murine microglia BV2 cell line was purchased from the Cell Culture Center at the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
4.11. Antioxidant assays
The antioxidant activities of compounds 4–39 were evaluated by measuring the inhibitory rates of malondialdehyde (MDA) in rat liver microsomal lipid peroxidation induced by Fe2+-cystine in vitro according to the reported procedures57. Curcumin was selected as the positive control (inhibitory rate 91.0 ± 3.2% at 10−4 mol/L). Rat liver microsomes are prepared from male Sprague–Dawley rats (Hua Fu Kang Bioscience, Beijing, China).
Acknowledgments
This work was supported financially by the National Key Research and Development Program of China (2018YFA0901900), the CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-I2M-1-010, 2017-I2M-4-004), and Fundamental Research Funds for the Central Universities (2017PT35001). Jian Bai was supported by the Drug Innovation Major Project (2018ZX09711001-008-001). We are grateful to Jie Zhu from National Institute of Biological Sciences for part of cytotoxicity assays.
Author contributions
Qian Wei conducted most of the experiment and wrote the manuscript; Jian Bai analyzed data, elucidated the chemical structures, and wrote the manuscript; Daojiang Yan analyzed the BGCs of anzaphilones; Xiuqi Bao, Dan Zhang, Wenting Li and Xiangbing Qi performed anti-inflammatory, antioxidant assays and cytotoxicity assays; Bingyu Liu involved in NMR data analysis; Dequan Yu involved in discussion and design; Youcai Hu designed and guided the entire project and edited the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Appendix A. Supporting information
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2020.07.020.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Zhang H.W., Song Y.C., Tan R.X. Biology and chemistry of endophytes. Nat Prod Rep. 2006;23:753–771. doi: 10.1039/b609472b. [DOI] [PubMed] [Google Scholar]
- 2.Nisa H., Kamili A.N., Nawchoo I.A., Shafi S., Shameem N., Bandh S.A. Fungal endophytes as prolific source of phytochemicals and other bioactive natural products: a review. Microb Pathog. 2015;82:50–59. doi: 10.1016/j.micpath.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Stierle A., Strobel G., Stierle D. Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science. 1993;260:214–216. doi: 10.1126/science.8097061. [DOI] [PubMed] [Google Scholar]
- 4.Rutledge P.J., Challis G.L. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol. 2015;13:509–523. doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 5.Gross H. Strategies to unravel the function of orphan biosynthesis pathways: recent examples and future prospects. Appl Microbiol Biotechnol. 2007;75:267–277. doi: 10.1007/s00253-007-0900-5. [DOI] [PubMed] [Google Scholar]
- 6.Scherlach K., Hertweck C. Discovery of aspoquinolones A–D, prenylated quinoline-2-one alkaloids from Aspergillus nidulans, motivated by genome mining. Org Biomol Chem. 2006;4:3517–3520. doi: 10.1039/b607011f. [DOI] [PubMed] [Google Scholar]
- 7.Scherlach K., Schuemann J., Dahse H.M., Hertweck C. Aspernidine A and B, prenylated isoindolinone alkaloids from the model fungus Aspergillus nidulans. J Antibiot. 2010;63:375–377. doi: 10.1038/ja.2010.46. [DOI] [PubMed] [Google Scholar]
- 8.Schroeckh V., Scherlach K., Nutzmann H.W., Shelest E., Schmidt-Heck W., Schuemann J. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci. 2009;106:14558–14563. doi: 10.1073/pnas.0901870106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asai T., Yamamoto T., Shirata N., Taniguchi T., Monde K., Fujii I. Structurally diverse chaetophenol productions induced by chemically mediated epigenetic manipulation of fungal gene expression. Org Lett. 2013;15:3346–3349. doi: 10.1021/ol401386w. [DOI] [PubMed] [Google Scholar]
- 10.Hosaka T., Ohnishi-Kameyama M., Muramatsu H., Murakami K., Tsurumi Y., Kodani S. Antibacterial discovery in actinomycetes strains with mutations in RNA polymerase or ribosomal protein S12. Nat Biotechnol. 2009;27:462–464. doi: 10.1038/nbt.1538. [DOI] [PubMed] [Google Scholar]
- 11.Mao X.M., Xu W., Li D., Yin W.B., Chooi Y.H., Li Y.Q. Epigenetic genome mining of an endophytic fungus leads to the pleiotropic biosynthesis of natural products. Angew Chem Int Ed. 2015;54:7592–7596. doi: 10.1002/anie.201502452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai J., Mu R., Dou M., Yan D., Liu B., Wei Q. Epigenetic modification in histone deacetylase deletion strain of Calcarisporium arbuscula leads to diverse diterpenoids. Acta pharma Sin B. 2018;8:687–697. doi: 10.1016/j.apsb.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang T., Wan J., Zhan Z., Bai J., Liu B., Hu Y. Activation of an unconventional meroterpenoid gene cluster in Neosartorya glabra leads to the production of new berkeleyacetals. Acta pharma Sin B. 2018;8:478–487. doi: 10.1016/j.apsb.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biggins J.B., Liu X., Feng Z., Brady S.F. Metabolites from the induced expression of cryptic single operons found in the genome of Burkholderia pseudomallei. J Am Chem Soc. 2011;133:1638–1641. doi: 10.1021/ja1087369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Y., Ding G., Li Y., Qu J., Ma S., Lv H. Structures and absolute configurations of penicillactones A‒C from an endophytic microorganism, Penicillium dangeardii Pitt. Org Lett. 2013;15:5206–5209. doi: 10.1021/ol4023485. [DOI] [PubMed] [Google Scholar]
- 16.Buchi G., Snader K.M., White J.D., Gougoutas J.Z., Singh S. Structures of rubratoxins A and B. J Am Chem Soc. 1970;92:6638–6641. doi: 10.1021/ja00725a045. [DOI] [PubMed] [Google Scholar]
- 17.Chen R.D., Yan Z., Zou J.H., Wang N., Dai J.G., Rubratoxin C. A new nonadride derivative from an endophytic fungus Penicillium sp. F-14. Chin Chem Lett. 2014;25:1308–1310. [Google Scholar]
- 18.Bai J., Yan D., Zhang T., Guo Y., Liu Y., Zou Y. A cascade of redox reactions generates complexity in the biosynthesis of the protein phosphatase-2 inhibitor rubratoxin A. Angew Chem Int Ed. 2017;56:4782–4786. doi: 10.1002/anie.201701547. [DOI] [PubMed] [Google Scholar]
- 19.Yang S.W., Chan T.M., Terracciano J., Patel R., Patel M., Gullo V. A new hydrogenated azaphilone Sch 725680 from Aspergillus sp. J Antibiot. 2006;59:720–723. doi: 10.1038/ja.2006.96. [DOI] [PubMed] [Google Scholar]
- 20.Myobatake Y., Takeuchi T., Kuramochi K., Kuriyama I., Ishido T., Hirano K. Pinophilins A and B, inhibitors of mammalian A-, B-, and Y-family DNA polymerases and human cancer cell proliferation. J Nat Prod. 2012;75:135–141. doi: 10.1021/np200523b. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y., Yang Q., Xia G., Huang H., Li H., Ma L. Polyketides with alpha-glucosidase inhibitory activity from a mangrove endophytic fungus, Penicillium sp. HN29-3B1. J Nat Prod. 2015;78:1816–1822. doi: 10.1021/np500885f. [DOI] [PubMed] [Google Scholar]
- 22.Yang S.W., Chan T.M., Terracciano J., Loebenberg D., Patel M., Gullo V. Sch 1385568, a new azaphilone from Aspergillus sp. J Antibiot. 2009;62:401–403. doi: 10.1038/ja.2009.51. [DOI] [PubMed] [Google Scholar]
- 23.Ren J., Ding S.S., Zhu A., Cao F., Zhu H.J. Bioactive azaphilone derivatives from the fungus Talaromyces aculeatus. J Nat Prod. 2017;80:2199–2203. doi: 10.1021/acs.jnatprod.7b00032. [DOI] [PubMed] [Google Scholar]
- 24.Natsume M., Takahashi Y., Marumo S. (−)-Mitorubrinic acid, a morphogenic substance inducing chlamydospore-like cells, and its related new metabolite, (+)-mitorubrinic acid B, isolated from Penicillium funiculosum. Agric Biol Chem. 1985;49:2517–2519. [Google Scholar]
- 25.Buechi G., White J.D., Wogan G.N. The structures of mitorubrin and mitorubrinol. J Am Chem Soc. 1965;87:3484–3489. doi: 10.1021/ja01093a036. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J., Porco J.A.J. Asymmetric syntheses of (‒)-mitorubrin and related azaphilone natural products. Org Lett. 2006;8:5169–5171. doi: 10.1021/ol062233m. [DOI] [PubMed] [Google Scholar]
- 27.Tabata Y., Ikegami S., Yaguchi T., Sasaki T., Hoshiko S., Sakuma S. Diazaphilonic acid, a new azaphilone with telomerase inhibitory activity. J Antibiot. 1999;52:412–414. doi: 10.7164/antibiotics.52.412. [DOI] [PubMed] [Google Scholar]
- 28.Hoshiko S., Sasaki T., Yaguchi T., Tabata Y., Sakuma S., Ikegami S., inventors. Meiji Seika Kaisha Ltd. and Meiji Milk Prod Co. Ltd., assignees . 1999 Oct 05. New chemical compound PF1195 and its manufacturing method. Japan patent JP 11269164 A. [Google Scholar]
- 29.Xiao Z., Lin S., Tan C., Lu Y., He L., Huang X. Asperlones A and B, dinaphthalenone derivatives from a mangrove endophytic fungus Aspergillus sp. 16-5C. Mar Drugs. 2015;13:366–378. doi: 10.3390/md13010366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopes T.I., Coelho R.G., Yoshida N.C., Honda N.K. Radical-scavenging activity of orsellinates. Chem Pharm Bull. 2008;56:1551–1554. doi: 10.1248/cpb.56.1551. [DOI] [PubMed] [Google Scholar]
- 31.Lee S.M., Li X.F., Jiang H., Cheng J.G., Seong S., Choi H.D. Terreusinone, a novel UV-A protecting dipyrroloquinone from the marine algicolous fungus Aspergillus terreus. Tetrahedron Lett. 2003;44:7707–7710. [Google Scholar]
- 32.Moore R.E., Emery T. Nalpha-acetylfusarinines: isolation, characterization, and properties. Biochemistry. 1976;15:2719–2723. doi: 10.1021/bi00658a001. [DOI] [PubMed] [Google Scholar]
- 33.Middleton A.J., Cole D.S., Macdonald K.D. A hydroxamic acid from Aspergillus nidulans with antibiotic activity against Proteus species. J Antibiot. 1978;31:1110–1115. doi: 10.7164/antibiotics.31.1110. [DOI] [PubMed] [Google Scholar]
- 34.Koreeda M., Harada N., Nakanishi K. Exciton chirality methods as applied to conjugated enones, esters, and lactones. J Am Chem Soc. 1974;96:266–268. [Google Scholar]
- 35.Itabashi T., Ogasawara N., Nozawa K., Kawai K.I. Isolation and structures of new azaphilone derivatives, falconensins E–G, from Emericella falconensis and absolute configurations of falconensins A-G. Chem Pharm Bull. 1996;44:2213–2217. [Google Scholar]
- 36.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R. Gaussian Inc.; Wallingford, CT, USA: 2010. Gaussian 09, revision C.01. [Google Scholar]
- 37.Gorin P.A.J., Mazurek M. Further studies on the assignment of signals in 13C magnetic resonance spectra of aldoses and derived methyl glycosides. Can J Chem. 1975;53:1212–1223. [Google Scholar]
- 38.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R. Gaussian Inc.; Wallingford, CT, USA: 2009. Gaussian 09, revision D.01. [Google Scholar]
- 39.Steyn P.S., Vleggaar R. The structure of dihydrodeoxy-8-epi-austdiol and the absolute configuration of the azaphilones. J Chem Soc Perkin Trans. 1976;1:204–206. [PubMed] [Google Scholar]
- 40.Zabala A.O., Xu W., Chooi Y.H., Tang Y. Characterization of a silent azaphilone gene cluster from Aspergillus niger ATCC 1015 reveals a hydroxylation-mediated pyran-ring formation. Chem Biol. 2012;19:1049–1059. doi: 10.1016/j.chembiol.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W., Chen R., Liu Q., He Y., He K., Ding X. Orange, red, yellow: biosynthesis of azaphilone pigments in Monascus fungi. Chem Sci. 2017;8:4917–4925. doi: 10.1039/c7sc00475c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez J.F., Somoza A.D., Keller N.P., Wang C.C.C. Advances in Aspergillus secondary metabolite research in the post-genomic era. Nat Prod Rep. 2012;29:351–371. doi: 10.1039/c2np00084a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang Y.M., Szewczyk E., Davidson A.D., Keller N., Oakley B.R., Wang C.C.C. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, Asperfuranone, in Aspergillus nidulans. J Am Chem Soc. 2009;131:2965–2970. doi: 10.1021/ja8088185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui Z., Qiao K., Gao Z., Meehan M.J., Li J.W.H., Zhao X. Enzymatic synthesis of resorcylic acid lactones by cooperation of fungal iterative polyketide synthases involved in hypothemycin biosynthesis. J Am Chem Soc. 2010;132:4530–4531. doi: 10.1021/ja100060k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie X., Meehan M.J., Xu W., Dorrestein P.C., Tang Y. Acyltransferase mediated polyketide release from a fungal megasynthase. J Am Chem Soc. 2009;131:8388–8389. doi: 10.1021/ja903203g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X., Li S. Expansion of chemical space for natural products by uncommon P450 reactions. Nat Prod Rep. 2017;34:1061–1089. doi: 10.1039/c7np00028f. [DOI] [PubMed] [Google Scholar]
- 47.Nozaki H., Kuroda S., Watanabe K., Yokozeki K. Gene cloning of α-methylserine aldolase from Variovorax paradoxus and purification and characterization of the recombinant enzyme. Biosci Biotechnol Biochem. 2008;72:2580–2588. doi: 10.1271/bbb.80274. [DOI] [PubMed] [Google Scholar]
- 48.Chiang Y.M., Ahuja M., Oakley C.E., Entwistle R., Asokan A., Zutz C. Development of genetic dereplication strains in Aspergillus nidulans results in the discovery of aspercryptin. Angew Chem Int Ed. 2016;55:1662–1665. doi: 10.1002/anie.201507097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L., Wu H., Liu H., Li E., Ren J., Wang W. Genetic dereplication of trichoderma hypoxylon reveals two novel polycyclic lactones. Bioorg Chem. 2019;91:103185. doi: 10.1016/j.bioorg.2019.103185. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez J.F., Entwistle R., Hung J.H., Yaegashi J., Jain S., Chiang Y.M. Genome-based deletion analysis reveals the prenyl xanthone biosynthesis pathway in Aspergillus nidulans. J Am Chem Soc. 2011;133:4010–4017. doi: 10.1021/ja1096682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao S.S., Li X.M., Zhang Y., Li C.S., Cui C.M., Wang B.G. Comazaphilones A‒F, azaphilone derivatives from the marine sediment-derived fungus Penicillium commune QSD-17. J Nat Prod. 2011;74:256–261. doi: 10.1021/np100788h. [DOI] [PubMed] [Google Scholar]
- 52.Zhao D.L., Shao C.L., Zhang Q., Wang K.L., Guan F.F., Shi T. Azaphilone and diphenyl ether derivatives from a gorgonian-derived strain of the fungus Penicillium pinophilum. J Nat Prod. 2015;78:2310–2314. doi: 10.1021/acs.jnatprod.5b00575. [DOI] [PubMed] [Google Scholar]
- 53.O'Boyle N.M., Tenderholt A.L., Langner K.M. Cclib: a library for package-independent computational chemistry algorithms. J Comput Chem. 2008;29:839–845. doi: 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- 54.O'Boyle N.M., Vandermeersch T., Flynn C.J., Maguire A.R., Hutchison G.R. Confab-systematic generation of diverse low-energy conformers. J Cheminf. 2011;3:8. doi: 10.1186/1758-2946-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka T., Nakashima T., Ueda T., Tomii K., Kouno I. Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem Pharm Bull. 2007;55:899–901. doi: 10.1248/cpb.55.899. [DOI] [PubMed] [Google Scholar]
- 56.Qu J., Fang L., Ren X.D., Liu Y., Yu S.S., Li L. Bisindole alkaloids with neural anti-inflammatory activity from Gelsemium elegans. J Nat Prod. 2013;76:2203–2209. doi: 10.1021/np4005536. [DOI] [PubMed] [Google Scholar]
- 57.Hu Y., Ma S., Li J., Yu S., Qu J., Liu J. Targeted isolation and structure elucidation of stilbene glycosides from the bark of Lysidice brevicalyx Wei guided by biological and chemical screening. J Nat Prod. 2008;71:1800–1805. doi: 10.1021/np800083x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.