

Abstract
NIPA1 (nonimprinted in Prader-Willi/Angelman syndrome 1) mutations are known to cause hereditary spastic paraplegia type 6, a neurodegenerative disease that phenotypically overlaps to some extent with amyotrophic lateral sclerosis (ALS). Previously, a genomewide screen for copy number variants found an association with rare deletions in NIPA1 and ALS, and subsequent genetic analyses revealed that long (or expanded) polyalanine repeats in NIPA1 convey increased ALS susceptibility. We set out to perform a large-scale replication study to further investigate the role of NIPA1 polyalanine expansions with ALS, in which we characterized NIPA1 repeat size in an independent international cohort of 3955 patients with ALS and 2276 unaffected controls and combined our results with previous reports. Meta-analysis on a total of 6245 patients with ALS and 5051 controls showed an overall increased risk of ALS in those with expanded (>8) GCG repeat length (odds ratio = 1.50, p = 3.8×10−5). Together with previous reports, these findings provide evidence for an association of an expanded polyalanine repeat in NIPA1 and ALS.
Keywords: Amyotrophic lateral sclerosis, NIPA1, Repeat expansion
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder characterized by the loss of both upper and lower motor neurons leading to progressive weakness, spasticity, and ultimately respiratory failure (Hardiman et al., 2011; van Es et al., 2017). The complex genetic architecture of ALS is characterized by 5%‒15% of patients with a positive family history, where it is assumed that there is a single causal mutation (Andersen and Al-Chalabi, 2011). However, even in most seemingly sporadic patients, a large genetic contribution is expected and causal mutations have been reported despite a negative family history (Al-Chalabi et al., 2017; McLaughlin et al., 2015). To date, mutations in more than 20 different genes have been implicated in ALS, one of the most prominent being an intronic repeat expansion in C9orf72 (Al-Chalabi et al., 2017).
In addition to C9orf72, repeat expansions in other genes have been reported in ALS, including ATXN2 and NIPA1 (Blauw et al., 2012; Elden et al., 2010). NIPA1 (nonimprinted in Prader-Willi/Angelman syndrome 1) mutations are known to cause hereditary spastic paraplegia (HSP) type 6, a neurodegenerative disease characterized by slowly progressive upper motor neuron signs (pre-dominantly in the lower limbs) and is a condition that to some extent has phenotypic overlap with ALS (Rainier et al., 2003). Interestingly, a genomewide screen for copy number variants found an association with rare deletions in NIPA1 and ALS and subsequent genetic analyses revealed that long (or expanded) polyalanine repeats in NIPA1 confer increased disease susceptibility (Blauw et al., 2010, 2012). In most people (98%), the 5′-end of NIPA1 (NCBI: NM_144599.4) encodes for a stretch of 12 or 13 alanine residues of which 7 or 8 are encoded by a (GCG)n trinucleotide repeat (TNR), although both shorter and longer GCG stretches have been reported in nonaffected individuals (Chai et al., 2003). In the previous study, an analysis of an international cohort of 2292 patients with ALS and 2777 controls showed that “long” repeats (>8) in NIPA1 were enriched in ALS cases compared with controls (5.5% versus 3.6%; OR 1.71; p = 1.6 × 10−4) (Blauw et al., 2012).
Although interesting and potentially relevant, only a small fraction of initially positive results from candidate gene studies (such as that performed on NIPA1) replicated consistently (Hirschhorn et al., 2002). Therefore, additional steps, such as replication of the findings and imposing a proper significance threshold (such as exome or genomewide significance), are required to make any claims of causality (MacArthur et al., 2014).
We therefore set out to perform a large-scale replication study to further investigate the role of NIPA1 polyalanine expansions with ALS, in which we characterized NIPA1 repeat size in a large international cohort of patients with ALS and unaffected controls and then meta-analyze our results with previous reports.
2. Materials and methods
2.1. Subjects
All participants gave written informed consent and approval was obtained from the local, relevant ethical committees for medical research. Genotyping experiments were performed on 6231 samples comprising 3955 patients with ALS and 2276 healthy controls from 6 populations. All patients were diagnosed according to the revised El Escorial criteria. Control subjects were from ongoing population-based studies on risk factors in ALS. All related individuals were excluded from further analysis. The baseline characteristics for available samples are provided in Supplementary Table 1.
2.2. PCR, sequencing, and genotyping
Dutch samples obtained from 753 patients with ALS and 603 unaffected individuals were analyzed using PCR according to protocols described previously, and results were analyzed in a blinded and automated fashion with a call rate of 96.6% (Blauw et al., 2012). Samples that failed genotyping were additionally analyzed with Sanger sequencing to assess possible bias. An additional cohort of 767 unaffected controls and 764 ALS samples were genotyped using Sanger sequencing and automatically genotyped with a call rate of 99.1%. Primers: 5′ -GCCCCTCTTCCTGCTCCT-3′ (forward) and 5′-CGATGCCCTTCTTCTGTAGC -3′ (reverse). A total of 847 samples were analyzed using both methods (PCR and Sanger), with manual review of discordant genotypes (n = 35, 4.1%).
We analyzed NIPA1 repeat size in whole-genome sequencing (WGS) data of 3344 samples (2438 cases and 906 controls) from the HiSeq X Sequencing platform, available to us through Project MinE (Project MinE ALS Sequencing Consortium, 2018), using the Illumina ExpansionHunter tool (Dolzhenko et al., 2017). There was a 691 sample overlap genotyped using both ExpansionHunter and Sanger sequencing, showing a 99% concordance (n = 684). Considering this 99% concordance between ExpansionHunter and Sanger results in the Dutch data set, we did not perform additional validation experiments on the WGS samples and proceeded with the ExpansionHunter calls. C9orf72 status had been determined for 3907 ALS samples from the PCR, Sanger, and ExpansionHunter cohorts. In addition, the presence of rare nonsynonymous and loss-of-function variants in the established ALS-associated genes SOD1, FUS, and TARDBP was known for 5030 cases and controls from all cohorts as described previously (Dekker et al., 2016; Project MinE ALS Sequencing Consortium, 2018).
2.3. Statistical analysis
All statistical procedures were carried out in R 3.3.0 (http://www.r-project.org). For association analyses, we applied a logistic regression analysis to all subgroups, the effect of the expanded (>8) versus nonexpanded polyalanine repeat length on the disease status, adjusting for sex at birth, method of genotyping, and country of origin. Samples with missing sex at birth status (n = 108, 1%) were imputed using multivariate multiple imputation with the “mice” 2.46.0 package.
Subgroup effects were meta-analyzed using both fixed and random effects modeling using the “metafor” 2.0 package. For the joint analysis on individual data, we used a generalized linear model with fixed-effects covariates: sex, method of genotyping and country of origin. We additionally applied generalized linear mixed model on nonimputed data to account for possible random effects.
The survival after onset and age at onset analyses were performed using multivariate Cox regression with sex at birth, site of onset, age at onset (for survival only), and C9orf72 status as covariates.
To assess whether the observed frequency of co-occurring genetic risk variants for ALS was in excess of what would be expected on the basis of chance, we used a method described previously by Dekker et al., 2016. The expected frequency of co-occurring variants was calculated using the following formula: (the observed number of patients carrying a variant/the total number of patients) × (the observed number of controls carrying a variant/the total number of controls). This formula was used to take into account the higher frequency of just one variant in patients with ALS (= frequency of variants in patients), multiplied by the chance probability of a second variant (= frequency of variants in controls). Then, a binomial test was performed to compare the observed frequency of co-occurring variants in patients with ALS with the calculated expected frequency.
We specified a formal null model for an increase in repeat expansion with consideration of repeat confounding variables such as the genomic frequency and repeat size. Previous studies have shown that there are a total of 878 genes in the genome that contain a coding TNR with a repeat size of 6 repeats or greater, 90 of which contain a polyalanine tract (Kozlowski et al., 2010). We therefore set 2 thresholds for significance in this study; 1) a relatively loose threshold, in which we correct for the number of genes that contain a polyalanine tract of 6 or larger resulting in p = 0.05/90 = 5.6×10−4 and 2) a more conservative threshold, in which we correct for the total number of genes in the genome that contain a coding TNR with a size of 6 or larger which gives p = 0.05/878 = 5.7×10−5.
3. Results
3.1. Replication
We first tried to replicate the initial findings in an independent Dutch cohort comprising 1517 ALS cases and 1370 unaffected controls by genotyping the GCG repeat length in NIPA1 using repeat PCR and/or Sanger sequencing. As was reported previously, we found the most frequent alleles to consist of either 7 or 8 (GCG)n repeats (25% and 72%, respectively) (Fig. 1). Our analysis showed a similar allele frequency difference of expanded or “long” alleles (repeat length of 9 or longer) between patients with ALS (n = 85, 2.80%) and controls (n = 51, 1.86%). Both ALS and control subgroups had only one single case with a homozygous expansion, indicating a dominant model for further analysis. This resulted in 84 individuals with ALS (5.54%) and 50 unaffected individuals (3.65%) as carriers of an expanded NIPA1 polyalanine repeat length. Logistic regression analysis, corrected for sex at birth and method of genotyping (PCR or Sanger), revealed an effect of expanded NIPA1 repeat length on disease susceptibility (OR = 1.54, p = 0.018).
Fig. 1.
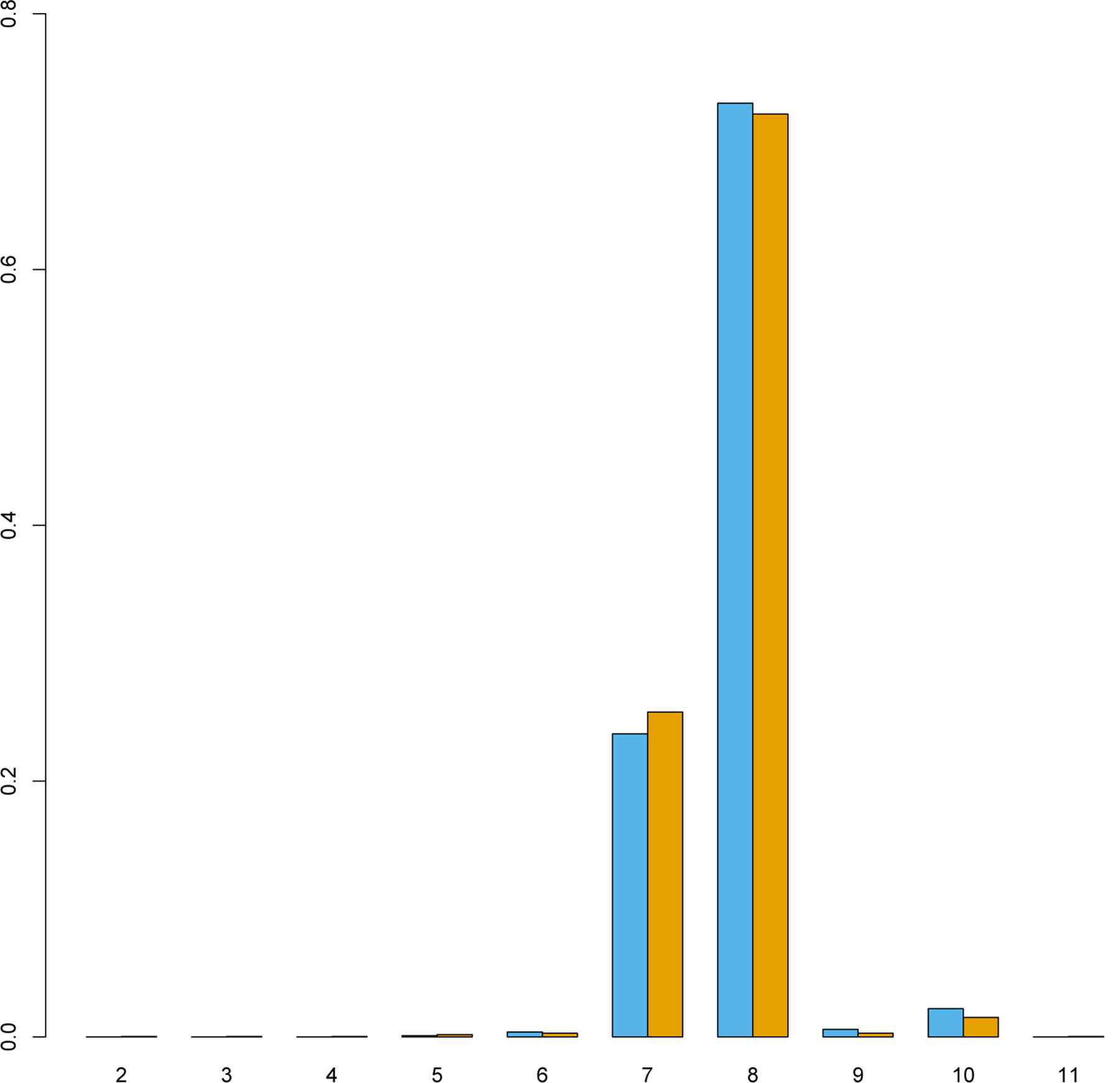
NIPA1 polyalanine repeat length distribution. Proportion of total alleles grouped per NIPA1 polyalanine repeat size. Alleles displayed were observed multiple times in the Dutch replication cohort of 1517 individuals affected with amyotrophic lateral sclerosis (blue) and 1370 unaffected controls (orange).
3.2. Project MinE
To further increase sample size and investigate cohorts other than the Dutch population, we then analyzed NIPA1 repeat expansion genotypes that were called using the Illumina ExpansionHunter tool in 2438 independent ALS cases and 906 controls whole-genome sequenced as part of the Project MinE ALS Sequencing Consortium (Project MinE ALS Sequencing Consortium, 2018). This multicohort WGS data showed a more equal distribution of expanded NIPA1 carriers in patients with ALS (114/2,438, 4.67%) and controls (40/906, 4.42%). A logistic regression analysis, corrected for country of origin and sex, showed no significant difference.
3.3. Meta-analysis
Finally, we sought to perform an analysis of all available NIPA1 polyalanine expansion data, combining our data with the original data published previously (Blauw et al., 2012). After exclusion of duplicate samples; individual level data were available for a total of 5056 samples (2290 cases and 2775 controls) in the discovery data set published by Blauw et al. (2012). Our replication cohort (including results from PCR, Sanger, and ExpansionHunter) comprised 3955 cases and 2276 controls. The final data set included 6245 patients with ALS and 5051 controls, reaching a final number of 11,296 unique individuals. We combined these data in a fixed-effects meta-analysis and found an overall risk of expanded NIPA1 repeat length on ALS (odds ratio (OR) = 1.50, p = 3.8×10−5) (Fig. 2). Because individual level data were available, we additionally performed a multivariate logistic regression analysis, using sex at birth, method of genotyping, and country of origin as covariates in the pooled data, resulting in an equal effect and significance (OR = 1.48, p = 6.2×10−5). Other association models that account for random effects, such as random effect meta-analysis and a generalized linear mixed model gave similar results (data not shown). Repeating the analysis excluding the 322 C9orf72 repeat expansion carriers yielded a p value of 7.7×10−5 for the fixed-effects meta-analysis (OR = 1.49, 95% confidence interval [CI] = 1.22‒1.81) and a p value of 1.0×10−4 for the multivariate logistic regression analysis (OR = 1.47, 95% CI = 1.21‒1.78). Exclusion of an additional 171 samples (133 cases and 38 controls) carrying a nonsynonymous or loss-of-function mutation in SOD1, FUS, or TARDBP did not alter the results (fixed-effects meta-analysis p value = 7.5 ×10−5, OR = 1.49, 95% CI = 1.22‒1.81) (Supplementary Fig. 1).
Fig. 2.
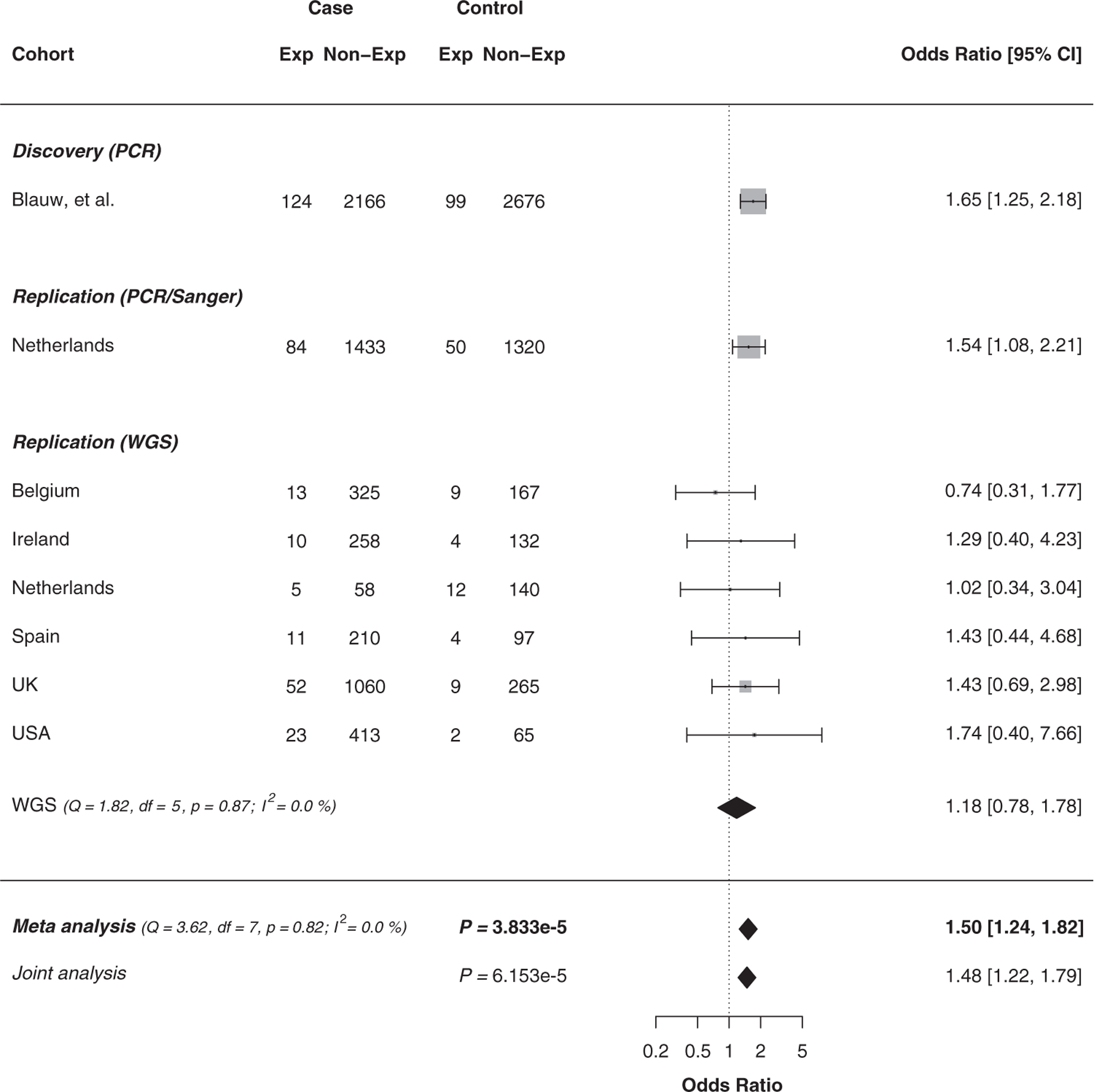
NIPA1 polyalanine repeat expansion meta-analysis. Forest plot for the fixed-effect meta-analysis and joint analysis on individual level data of the effect of expanded NIPA1 polyalanine (>8 GCG repeats) on amyotrophic lateral sclerosis risk with the initial discovery reports (Blauw et al., 2012) and current replication using PCR, Sanger, or whole-genome sequencing (WGS) grouped per cohort/country of origin. Weights depending on number of participants. CI, confidence interval.
3.4. Survival
Clinical data and survival data were available for 1954 of 3955 patients with ALS from the combined replication cohorts (Supplementary Table 2). After correction for sex, age at onset, bulbar site of onset, and C9orf72 status, we used a Cox regression model in this mixed population to test if NIPA1 conferred any risk for shorter survival time; we found no evidence for such an effect (hazard ratio = 1.16; 95% CI = 0.94‒1.45; p = 0.16) (Supplementary Fig. 2). In addition, there was no significant association between NIPA1 repeat length and age at onset in this replication cohort with correction for sex, site of onset, and the presence of a C9orf72 expansion (Supplementary Fig. 3).
3.5. Co-occurrence with C9orf72 repeat expansion
Because a significant number of NIPA1 expansion carriers was reported in a subgroup of patients with ALS that also carried a C9orf72 repeat expansion (Dekker et al., 2016), we evaluated this co-occurrence in 4619 participants genotyped for both loci in all cohorts (n = 712 for the discovery cohort; n = 3907 for the combined replication cohorts).
Although we did observe a higher than expected frequency of co-occurrence of the repeat expansions, our data did not robustly replicate the previously published finding (0.37% observed versus 0.26% expected; p = 0.06) (Supplementary Table 3).
4. Discussion
In this study, we included a large international cohort and additionally meta-analyzed the NIPA1 expansion genotypes in a total of 6245 patients with ALS and 5051 controls. Given that we were able to replicate our previous results in an independent cohort and observed an increase in significance in the overall meta-analysis, our data add to the evidence that expanded NIPA1 repeats are a risk factor for sporadic ALS. Mutations in NIPA1 were already known to cause HSP type 6, a neurodegenerative disease with motor neuron involvement, whereas the 15q11.2 micro-deletions are better known for low penetrant neurodevelopmental phenotypes, further adding to the complexity of the NIPA1 locus (Butler, 2017; Rainier et al., 2003). Interestingly, genetic pleiotropy between HSP and ALS appears to be more widespread, as recently it has been shown that mutations in different domains in KIF5A either cause HSP or ALS (Brenner et al., 2018; Nicolas et al., 2018).
After C9orf72 and ATXN2, NIPA1 is the third reported expanded genomic repeat motif associated with an increased risk for ALS. Its initial discovery in ALS by identification of copy number variants in the chromosome 15q11.2 locus containing NIPA1, was followed by further genetic screening in a large international cohort consisting of Belgian, Dutch, and German subjects (Blauw et al., 2010, 2012). This subsequent study in 2292 patients with ALS and 2777 controls revealed that, although NIPA1 deletions and missense mutations were identified in patients with ALS, it actually was an increase of the (GCG)n repeat motif in the 5′-end of NIPA1 that seemed to associate with ALS (OR = 1.71 with p = 1.6×10−4). Knowing that positive results derived from candidate gene studies often fail to replicate, we sought to replicate the NIPA1 finding in ALS, particularly given the complex genotypic and phenotypic architecture of the NIPA1 locus (Hirschhorn et al., 2002).
Our results showed a very similar effect of increased NIPA1 polyalanine expansions on ALS susceptibility in a new Dutch cohort of 1517 ALS cases and 1370 unaffected controls tested via PCR or Sanger sequencing. Given the high concordance between Sanger/PCR results and the calls from the bioinformatic tool ExpansionHunter on WGS data, we were able to further increase the sample size of our study by including data from Project MinE (Dolzhenko et al., 2017; Project MinE ALS Sequencing Consortium, 2018). This allowed us to additionally evaluate the role of NIPA1 repeat sizes in non-Dutch cohorts. The size of this cohort was similar in the number of cases compared with the original discovery cohort, but smaller in number of controls compared with that original cohort. In addition, the Project MinE data set is more heterogeneous compared with the original discovery cohort. This is a possible explanation as to why the overall NIPA1 signal was not replicated in the WGS data. However, we did find a similar direction and effect size in 4 of the 6 WGS cohorts (Ireland, Spain, the United States of America and the United Kingdom).
While empirical thresholds for genomewide and exomewide significance have been derived for studies assessing associations between phenotypes and single nucleotide variants, these thresholds are likely to be too stringent in the context of screening for coding repeat expansions, as the genome contains only ~900 genes with a coding TNR tract with a length of 6 or more, 90 of which code for a polyalanine tract (Kozlowski et al., 2010). We therefore set the significance threshold for associations with TNRs to be approximately p = 5.6×10−4, correcting for polyalanine only, or (more conservative) p = 5.7×10−5, correcting for all TNRs with a length of 6 repeats or more. The meta-analysis results are significant regardless of the threshold applied. Furthermore, exclusion of samples carrying a mutation in established ALS genes (C9orf72, SOD1, TARDBP, and FUS) yielded somewhat lower p values (due to loss of power corresponding to lower number of included samples) with similar magnitude of effect, further supporting the role of NIPA1 as independent risk factor for developing ALS.
Although we did see a higher than expected number of ALS cases carrying both NIPA1 and C9orf72 repeat expansions in this study (n = 17, p = 0.06), we did not robustly reproduce the co-occurrence of C9orf72 expansion carriers in the NIPA1 expanded cases described by Dekker et al. (2016). This might be attributed to the relatively small sample size in the original study (755 patients with ALS), resulting in broad confidence intervals that overlap with our results (frequency = 0.004 [0.002‒0.006] in the present study; frequency = 0.009 [0.004‒0.019] in Dekker et al. (2016)). Alternatively, the co-occurrence might be relevant in some, but not all included populations. In addition, we were unable to replicate the effect of NIPA1 expansions on ALS survival and age at onset (Blauw et al., 2012). These findings again re-emphasize the necessity for replication and the importance of tracking clinical characteristics in large genetic databases. Currently, we were able to perform a survival analysis on just 50% of our replication set and further evaluation in a larger and complete data set is therefore recommended.
Interestingly, the increase in the NIPA1 repeat size seems to be limited to the addition of mostly 2 GCG repeats. However, this seemingly small addition might well have protein conformational effects as has been shown in vitro; polyalanine stretches between 7 and 15 alanines transition from a monomeric alpha helix to a predominant macromolecular beta sheet, which in turn may lead to stronger protein‒protein interactions and aggregation (Shinchuk et al.,2005). In addition, a patient with a mutation in NIPA1 suffering from a progressive motor neuron phenotype was shown to have TDP-43 inclusions, very similar to effects seen in ALS and ALS-FTD cases (Martinez-Lage et al., 2012). These findings might explain how alterations in NIPA1 could increase ALS risk.
In conclusion, our data add to the evidence for an association of NIPA1 expansions and ALS. Future investigations may provide further insights in the role of NIPA1 and polyalanine stretches in the development and possibly treatment of motor neuron disease.
Supplementary Material
Acknowledgements
This study was supported by the ALS Foundation Netherlands, the Belgian ALS Liga and National Lottery, and Agency for Innovation by Science and Technology (IWT), and the MND Association (UK) (Project MinE, www.projectmine.com). Research leading to these results has received funding from the European Community’s Health Seventh Framework Program (FP7/2007–2013). This study was supported by ZonMW under the frame of E-Rare-2, the ERA Net for Research on Rare Diseases (PYRAMID). This is an EU Joint Programme‒Neurodegenerative Disease Research (JPND) project (STRENGTH, MEND, SOPHIA, ALS-CarE). The project is supported through the following funding organizations under the aegis of JPND: UK, Medical Research Council (MR/L501529/1; MR/R024804/1) and Economic and Social Research Council (ES/L008238/1); Ireland, Health Research Board; the Netherlands, ZonMw; Belgium, FWO-Vlaanderen. Samples used in this research were in part obtained from the UK National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. This project was supported by the MND Association of England, Wales and Northern Ireland and the Netherlands Organisation for Health Research and Development (Vici scheme to L.H. van den Berg and veni scheme to M.A. van Es). NDAL cordially thanks Suna and Inan Kirac Foundation for their generous support. M.A. van Es is supported by the Thierry Latran Foundation, the Dutch ALS foundation and the Rudolf Magnus Brain Center Talent Fellowship. C.E. Shaw and A Al-Chalabi receive salary support from the National Institute for Health Research (NIHR) Dementia Biomedical Research Unit and Biomedical Research Center in Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London. O. Hardiman is funded by the Health Research Board Clinician Scientist Program and Science Foundation Ireland. J.E. Landers is supported by the US National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (R01NS073873) and the American ALS Association. R.L. McLaughlin is supported by the Thierry Latran Foundation and the ALS Association (2284). P. Van Damme holds a senior clinical investigatorship from FWO-Vlaanderen.
Footnotes
Members and affiliations of the Project MinE ALS Sequencing Consortium are listed in Supplementary Information.
This article contains supplementary material available from the authors by request or via the Internet at https://doi.org/10.1016/j.neurobiolaging.2018.09.012.
Disclosure statement
L.H. van den Berg serves on scientific advisory boards for the Prinses Beatrix Spierfonds, Thierry Latran Foundation, Biogen, and Cytokinetics; and serves on the editorial board of Amyotrophic Lateral Sclerosis And Frontotemporal Degeneration and the Journal of Neurology, Neurosurgery, and Psychiatry. O. Hardiman has received speaking honoraria from Novarits, Biogen Idec, Sanofi Aventis, and Merck-Serono; has been a member of advisory panels for Biogen Idec, Allergen, Ono Pharmaceuticals, Novartis, Cytokinetics, and Sanofi Aventis; and serves as the editor-in-chief of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. A. Al-Chalabi has consulted for OrionPharma, Biogen Idec, Cytokinetics Inc, Treeway Inc, and Chronos Therapeutics. J.H. Veldink reports that his institute received consultancy fees from Vertex Pharmaceuticals outside the submitted work. M.A. van Es received grants from the Netherlands Organization for Health Research and Development (Veni scheme), the Thierry Latran foundation, the Netherlands ALS foundation (Stichting ALS Nederland), and the Joint Program Neurodegeneration (JPND). He has received travel grants from Baxalta and serves on the biomedical research advisory panel of the motor neurone disease association (MNDA). Other authors have no reported conflicts of interest.
References
- Al-Chalabi A, van den Berg PLH, Veldink J, 2017. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat. Rev. Neurol 13, 96–104. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Al-Chalabi A, 2011. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol 7, 603–615. [DOI] [PubMed] [Google Scholar]
- Blauw HM, Al-Chalabi A, Andersen PM, van Vught PWJ, Diekstra FP, van Es MA, Saris CGJ, Groen EJN, van Rheenen W, Koppers M, Van’t Slot R, Strengman E, Estrada K, Rivadeneira F, Hofman A, Uitterlinden AG, Kiemeney LA, Vermeulen SHM, Birve A, Waibel S, Meyer T, Cronin S, McLaughlin RL, Hardiman O, Sapp PC, Tobin MD, Wain LV, Tomik B, Slowik A, Lemmens R, Rujescu D, Schulte C, Gasser T, Brown RH, Landers JE, Robberecht W, Ludolph AC, Ophoff RA, Veldink JH, van den Berg PLH, 2010. A large genome scan for rare CNVs in amyotrophic lateral sclerosis. Hum. Mol. Genet 19, 4091–4099. [DOI] [PubMed] [Google Scholar]
- Blauw HM, van Rheenen W, Koppers M, Van Damme P, Waibel S, Lemmens R, van Vught PWJ, Meyer T, Schulte C, Gasser T, Cuppen E, Pasterkamp RJ, Robberecht W, Ludolph AC, Veldink JH, van den Berg PLH, 2012. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet 21, 2497–2502. [DOI] [PubMed] [Google Scholar]
- Brenner D, Yilmaz R, Müller K, Grehl T, Petri S, Meyer T, Grosskreutz J, Weydt P, Ruf W, Neuwirth C, Weber M, Pinto S, Claeys KG, Schrank B, Jordan B, Knehr A, Günther K, Hübers A, Zeller D, Kubisch C, Jablonka S, Sendtner M, Klopstock T, de Carvalho M, Sperfeld A, Borck G, Volk AE, Dorst J, Weis J, Otto M, Schuster J, Del Tredici K, Braak H, Danzer KM, Freischmidt A, Meitinger T, Strom TM, Ludolph AC, Andersen PM, Weishaupt JH, The German ALS Network MND-NET, Weyen U, Hermann A, Hagenacker T, Koch JC, Lingor P, Göricke B, Zierz S, Baum P, Wolf J, Winkler A, Young P, Bogdahn U, Prudlo J, Kassubek J, 2018. Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, 2017. Clinical and genetic aspects of the 15q11.2 BP1-BP2 microdeletion disorder. J. Intellect. Disabil. Res 61, 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J-H, Locke DP, Greally JM, Knoll JHM, Ohta T, Dunai J, Yavor A, Eichler EE, Nicholls RD, 2003. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am. J. Hum. Genet 73, 898–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker AM, Seelen M, van Doormaal PTC, van Rheenen W, Bothof RJP, van Riessen T, Brands WJ, van der Kooi AJ, de Visser M, Voermans NC, Pasterkamp RJ, Veldink JH, van den Berg PLH, van Es MA, 2016. Large-scale screening in sporadic amyotrophic lateral sclerosis identifies genetic modifiers in C9orf72 repeat carriers. Neurobiol. Aging 39, 220.e9–220.e15. [DOI] [PubMed] [Google Scholar]
- Dolzhenko E, Van Vugt Joke JFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, Ajay SS, Rajan V, Lajoie BR, Johnson NH, Kingsbury Z, Humphray SJ, Schellevis RD, Brands WJ, Baker M, Rademakers R, Kooyman M, Tazelaar GHP, van Es MA, McLaughlin R, Sproviero W, Shatunov A, Jones A, Al Khleifat A, Pittman A, Morgan S, Hardiman O, Al-Chalabi A, Shaw C, Smith B, Neo EJ, Morrison K, Shaw PJ, Reeves C, Winterkorn L, Wexler NS, The USeVenezuela Collaborative Research Group, Housman DE, Ng CW, Li AL, Taft RJ, van den Berg PLH, Bentley DR, Veldink JH, Eberle MA, 2017. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 27, 1895–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elden AC, Kim H-J, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, Padmanabhan A, Clay-Falcone D, McCluskey L, Elman L, Juhr D, Gruber PJ, Rüb U, Auburger G, Trojanowski JQ, Lee VMY, Van Deerlin VM, Bonini NM, Gitler AD, 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardiman O, van den Berg PLH, Kiernan MC, 2011. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol 7, 639–649. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K, 2002. A comprehensive review of genetic association studies. Genet. Med 4, 45–61. [DOI] [PubMed] [Google Scholar]
- Kozlowski P, de Mezer M, Krzyzosiak WJ, 2010. Trinucleotide repeats in human genome and exome. Nucleic Acids Res 38, 4027–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C, 2014. Guidelines for investigating causality of sequence variants in human disease. Nature 508, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lage M, Molina-Porcel L, Falcone D, McCluskey L, Lee VMY, Van Deerlin VM, Trojanowski JQ, 2012. TDP-43 pathology in a case of hereditary spastic paraplegia with a NIPA1/SPG6 mutation. Acta Neuropathol 124, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin RL, Vajda A, Hardiman O, 2015. Heritability of amyotrophic lateral sclerosis. JAMA Neurol 72, 857–862. [DOI] [PubMed] [Google Scholar]
- Nicolas A, Kenna KP, Renton AE, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, Rivera AM, van Rheenen W, Murphy NA, Van Vugt Joke JFA, Geiger JT, Van Der Spek Rick A, Pliner HA, Shankaracharya Smith BN, Topp SD, Abramzon Y, Gkazi A-S, Eicher JD, Kenna A, Consortium I, Simone I, Logroscino G, Bartolomei I, Borghero G, Murru MR, Costantino E, Pani C, Puddu R, Caredda C, Piras V, Cuccu S, Corongiu D, Melis M, Milia A, Marrosu F, Marrosu MG, Floris G, Cannas A, Tranquilli S, Capasso M, Mancardi G, Origone P, Mandich P, Conforti FL, Cavallaro S, Mora G, Marinou K, Sideri R, Penco S, Mosca L, Lunetta C, Pinter GL, Corbo M, Riva N, Carrera P, Volanti P, Mandrioli J, Fini N, Fasano A, Tremolizzo L, Arosio A, Ferrarese C, Tedeschi G, Monsurrò MR, Piccirillo G, Femiano C, Ticca A, Ortu E, Spataro R, Colletti T, Zollino M, Conte A, Luigetti M, Lattante S, Marangi G, Santarelli M, Petrucci A, Pugliatti M, Pirisi A, Parish LD, Occhineri P, Giannini F, Ricci C, Benigni M, Cau TB, Loi D, Moglia C, Brunetti M, Barberis M, Restagno G, Casale F, Marrali G, Fuda G, Ossola I, Cammarosano S, Canosa A, Ilardi A, Manera U, Grassano M, Tanel R, Pisano F, Calvo A, Caponnetto C, Battistini S, La Bella V, Messina S, Simone IL, Trojsi F, Salvi F, Logullo FO, Ferrucci L Consortium GTFACG, Goldstein DB, Goutman S, Simmons Z, Chandran S, Pal S, Manousakis G, Appel SH, Simpson E, Wang L, Gibson S, Bedlack R, Lacomis D, Sareen D, Sherman A, Bruijn L, Penny M, de Araujo Martins Moreno C, Kamalakaran S Consortium AS, Allen AS, Appel S, Bedlack RS, Boone BE, Brown R, Carulli JP, Chesi A, Chung WK, Cirulli ET, Cooper GM, Couthouis J, Day-Williams AG, Dion PA, Han Y, Harris T, Hayes SD, Jones AL, Keebler J, Krueger BJ, Lasseigne BN, Levy SE, Lu YF, Maniatis T, McKenna-Yasek D, Miller TM, Petrovski S, Raphael AR, Ravits JM, Ren Z, Rouleau GA, Sapp PC, Sims KB, Staropoli JF, Waite LL, Wang Q, Wimbish JR, Xin WW, Gitler AD, Myers RM Consortium NA, Phatnani H, Kwan J, Sareen D, Broach JR, Simmons Z, Arcila-Londono X, Lee EB, Shneider NA, Fraenkel E, Ostrow LW, Zaitlen N, Berry JD, Malaspina A, Cox GA, Thompson LM, Finkbeiner S, Dardiotis E, Hornstein E, MacGowan DJ, Heiman-Patterson T, Hammell MG, Patsopoulos NA, Dubnau J, Nath A, Phatnani H, Musunuri RL, Evani US, Abhyankar A, Zody MC Answer ALS Foundation, Kaye J, Finkbeiner S, Wyman S, LeNail A, Lima L, Fraenkel E, Rothstein JD, Svendsen CN, Thompson LM, Van Eyk J, Maragakis NJ, Berry JD, Cudkowicz M, Baxi E, Kaye J, Finkbeiner S, Wyman SK, LeNail A, Lima L, Fraenkel E, Svendsen CN, Thompson LM, Van Eyk JE, Berry JD, Kolb SJ, Cudkowicz M, Baxi E Consortium CRIAARDFTDC, Benatar M, Wu G, Rampersaud E, Wuu J, Rademakers R, Züchner S, Schule R, McCauley J, Hussain S, Cooley A, Wallace M, Clayman C, Barohn R, Statland J, Ravits J, Swenson A, Jackson C, Trivedi J, Khan S, Katz J, Jenkins L, Burns T, Gwathmey K, Caress J, McMillan C, Elman L, Pioro E, Heckmann J, So Y, Walk D, Maiser S, Zhang J, Taylor JP, Rampersaud E, Wu G, Consortium S, Gellera C, Taroni F, Lauria G, Fogh I, Comi GP, Sorarù G, Cereda C, D’Alfonso S, Corrado L, De Marchi F, Corti S, Ceroni M, Mazzini L, Siciliano G, Filosto M, Inghilleri M, Peverelli S, Colombrita C, Poletti B, Maderna L, Del Bo R, Gagliardi S, Querin G, Bertolin C, Pensato V, Castellotti B, Verde F, Tiloca C, Consortium FA, Mouzat K, Lumbroso S, Meininger V, Besson G, Lagrange E, Clavelou P, Guy N, Couratier P, Vourc’h P, Danel V, Bernard E, Lemasson G, Corcia P, Laaksovirta H, Myllykangas L, Jansson L, Valori M, Ealing J, Hamdalla H, Rollinson S, Pickering-Brown S, Orrell RW, Sidle KC, Hardy J, Singleton AB, Johnson JO, Arepalli S, Polak M, Asress S, Al-Sarraj S, King A, Troakes C, Vance C, de Belleroche J, Baas F, ten Asbroek ALMA, Muñoz-Blanco JL, Hernandez DG, Ding J, Gibbs JR, Scholz SW, Floeter MK, Campbell RH, Landi F, Bowser R, Pulst SM, MacGowan DJL, Kirby J, Pioro EP, Pamphlett R, Broach J, Gerhard G, Dunckley TL, Brady CB, Kowall NW, Troncoso JC, Le Ber I, Mouzat K, Lumbroso S, Heiman-Patterson TD, Kamel F, Van Den Bosch L, Baloh RH, Strom TM, Meitinger T, van Eijk KR, de Carvalho M, Kooyman M, Middelkoop B, Moisse M, McLaughlin RL, van Es MA, Boylan KB, van Blitterswijk M, Morrison KE, Basak AN, Mora JS, Drory VE, Shaw PJ, Turner MR, Talbot K, Hardiman O, Williams KL, Fifita JA, Nicholson GA, Blair IP, Esteban-Pérez J, García-Redondo A, Al-Chalabi A, Consortium PMAS, Al Kheifat A, Andersen P, Chiò A, Cooper-Knock J, Dekker A, Drory V, Redondo AG, Gotkine M, Hide W, Iacoangeli A, Glass J, Kenna K, Kiernan M, Kooyman M, Landers J, McLaughlin R, Middelkoop B, Mill J, Neto MM, Moisse M, Pardina JM, Morrison K, Newhouse S, Pinto S, Pulit S, Robberecht W, Shatunov A, Shaw P, Shaw C, Silani V, Sproviero W, Tazelaar G, Ticozzi N, Van Damme P, van der Spek R, van Eijk K, van Es M, van Vugt J, Veldink J, Weber M, Zatz M, Bauer DC, Twine NA, Rogaeva E, Zinman L, Ostrow LW, Maragakis NJ, Simmons Z, Cooper-Knock J, Brice A, Goutman SA, Feldman EL, Gibson SB, Ratti A, Fratta P, Sabatelli M, Ludolph AC, Andersen PM, Weishaupt JH, Camu W, Trojanowski JQ, Van Deerlin VM, Brown RH Jr., van den Berg PLH, Veldink JH, Harms MB, Glass JD, Stone DJ, Tienari P, Shaw CE, Traynor BJ, Landers JE, 2018. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1282.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Project MinE ALS Sequencing Consortium, 2018. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur. J. Hum. Genet 26, 1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainier S, Chai J-H, Tokarz D, Nicholls RD, Fink JK, 2003. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am. J. Hum. Genet 73, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinchuk LM, Sharma D, Blondelle SE, Reixach N, Inouye H, Kirschner DA, 2005. Poly-(L-alanine) expansions form core β-sheets that nucleate amyloid assembly. Proteins 61, 579–589. [DOI] [PubMed] [Google Scholar]
- van Es MA, Hardiman O, Chiò A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH, 2017. Amyotrophic lateral sclerosis. Lancet 390, 2084–2098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.