

Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive fatal neurodegenerative disease affecting one in 350 people. The aim of Project MinE is to elucidate the pathophysiology of ALS through whole-genome sequencing at least 15,000 ALS patients and 7500 controls at 30× coverage. Here, we present the Project MinE data browser (databrowser.projectmine.com), a unique and intuitive one-stop, open-access server that provides detailed information on genetic variation analyzed in a new and still growing set of 4366 ALS cases and 1832 matched controls. Through its visual components and interactive design, the browser specifically aims to be a resource to those without a biostatistics background and allow clinicians and preclinical researchers to integrate Project MinE data into their own research. The browser allows users to query a transcript and immediately access a unique combination of detailed (meta)data, annotations and association statistics that would otherwise require analytic expertise and visits to scattered resources.
Keywords: Databrowser, amyotrophic lateral sclerosis, whole-genome sequencing, open-access
Introduction
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive fatal neurodegenerative disease affecting one in 350 people. While research over the past years has revealed an increasing number of genetic variants contributing to ALS risk, the bulk of heritability in ALS remains to be elucidated. In addition to known rare variants, there is evidence for a central role of low-frequency and rare genetic variation in ALS susceptibility (1). Well-powered genetic studies enabled through large-scale collaboration are crucial for identifying these variants and improving our understanding of ALS pathophysiology (2,3).
Project MinE, an international collaboration, was initiated precisely with the challenge of sample aggregation in mind. The Project MinE ALS sequencing Consortium has set out to collect whole-genome sequencing (WGS) of 15,000 ALS patients and 7,500 controls (4). Currently, the Project MinE initiative has sequenced 4,366 ALS patients and 1,832 age- and sex-matched controls. Project MinE is a largely crowd-funded initiative. As such, we are committed to sharing data and results with the scientific and healthcare communities, as well as the public more broadly. Data sharing within the genetics community facilitated large-scale genome-wide association studies and ignited initiatives such as the Gene Atlas, LDhub GWAShare Center, and MRbase, places where people can share, explore and analyze data with few restrictions (5,6). In this same spirit, we aim to share raw sequence data, provide results from our analyses, and facilitate interpretation through integration with existing datasets to serve researchers and the public across disciplines.
We, therefore, created the Project MinE databrowser (databrowser.projectmine.com). We integrated multi-level association statistics, metadata, and public resources including gnomAD, GTEx, and ClinVar in an intuitive and flexible framework (Figure 1) (7–9). These data are freely available through the browser for any research initiative. We aim for the data to serve several purposes, including providing a backbone for new gene discovery, serving as a costless replication dataset, and aiding clinical interpretation of individual ALS patient genomes or specific genetic variants.
Figure 1.
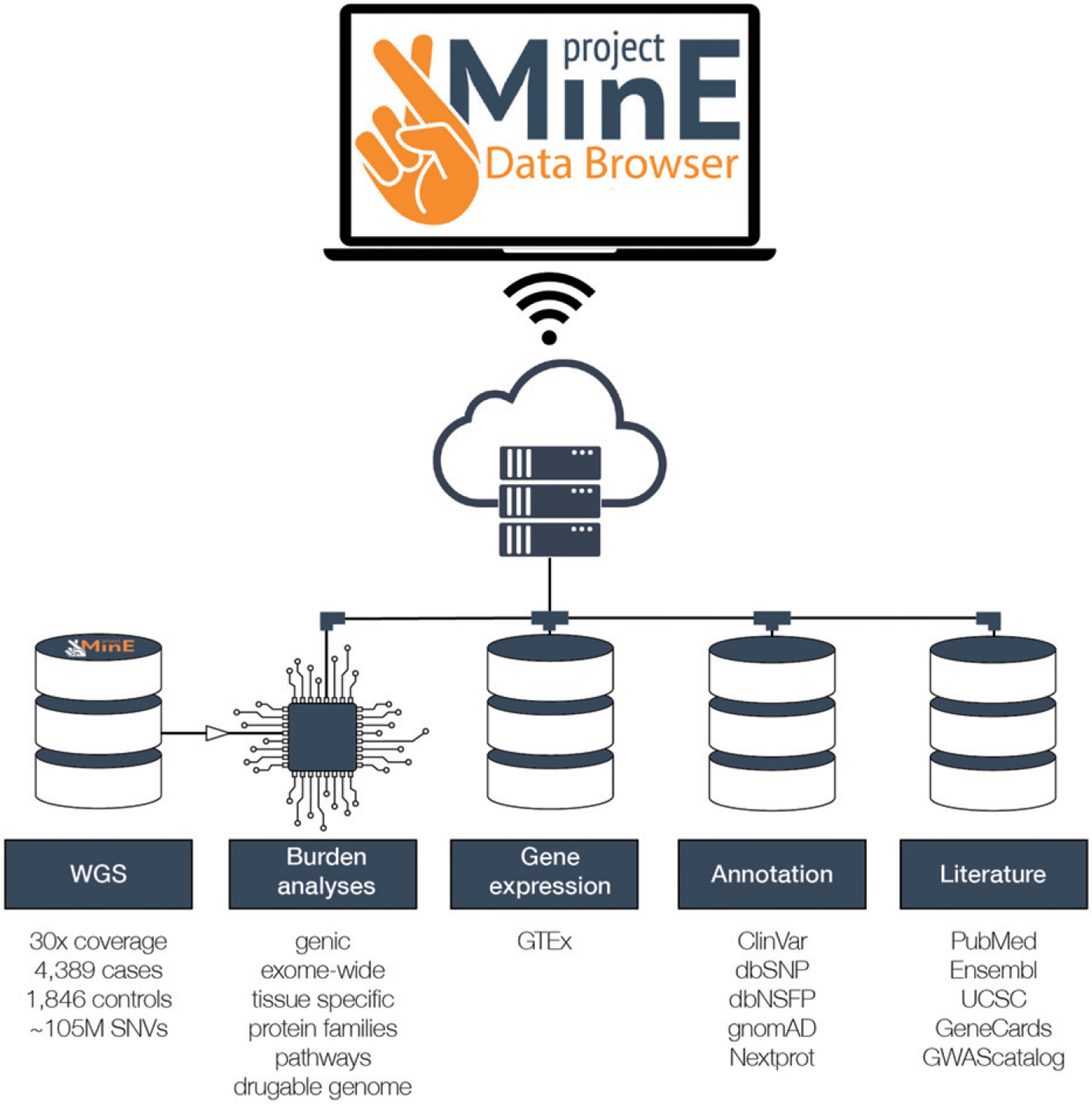
Schematic representation of the databrowser. Whole genomes generated by Project MinE are openly available for research and the public. The databrowser does not have a login requirement. It integrates multiple public resources and provides a wide range of robust statistical analyses.
Materials and methods
Additional information regarding sample selection, data merging, sample- and variant level quality control and source-code can be found in the Supplementary Material.
Association analyses
The main association analysis consists of several rare-variant burden analyses for an association with ALS risk. For quality control, we have performed single variant association analysis using a mixed linear model, including a genetic relationship matrix and the first 20 PCs, as implemented in GCTA (10). We set genome-wide significance in single variant association analyses at p < 5 × 10−9 (11), to account for the increased number of independent SNVs tested in sequence data.
We performed rare-variant burden tests using firth logistic regression in R, adjusting for the first 10 PCs, sex and platform (12,13). Variants for the rare-variant burden tests have been aggregated on multiple levels; gene, protein superfamilies, pathways, druggable categories, and exome-wide. Genic regions were defined as all transcripts in the GRCh37.p13 version of Ensembl Biomart (14). Higher level aggregation for burden analysis was performed by creating genesets. These genesets are based on: (a) protein superfamilies (15); (b) druggable categories as defined by the drug–gene interaction database (16); and (c) pathways downloaded from GSEA, using curated genesets v6.1 from KEGG, BioCarta or Reactome (17,18).
We tested genes or genesets when we could identify ≥5 individuals with ≥1 variant. We used three definitions for “rare”: minor allele frequency cutoffs 1% and 0.5% and variants not observed in ExAC (7). We classified variants based on their functional annotation (disruptive, damaging, missense-non-damaging, and synonymous, and described previously (19)). Briefly, frame-shift, splice site, exon loss, stop gained, stoploss, startloss, and transcription ablation variants were regarded as disruptive variants. We defined damaging variants as missense variants (resulting in an amino-acid change) predicted as damaging by all of seven methods: SIFT, Polyphen-2, LRT, Mutation Taster, Mutations Assessor, and PROVEAN (19). Missense-non-damaging variants are missense variants that are not classified as damaging. Synonymous variants do not result in an amino-acid change. From these annotations, we created three variant sets for burden testing: (1) disruptive variants, (2) disruptive + damaging variants, (3) disruptive + damaging + missense-non-damaging variants. The synonymous category functions as a null category to check for biases when testing for association. We set the threshold for exome-wide significance in genic rare-variant burden analyses at p < 1.7 × 10-6. We acknowledge that this threshold does not fully account for the multiple testing burden introduced by the different variant sets, allele-frequency cutoffs, and various burden testing approaches.
Data integration and annotation
After quality control, we performed functional annotation of all variants using snpEff V4.3T and SnpSift using the GRCh37.75 database (including Nextprot and Motif), dbSNFP v2.9, dbSNP b150 GRCh37p13, and ClinVar GRCh37 v2.0 (9,20–24). We obtained population frequency estimates from gnomAD (7). To visualize the variant-level coverage from Project MinE and external sources, we included coverage information from Project MinE samples, gnomAD database (123,136 exome sequences plus 15,496 genome sequences). We further integrated tissue-specific gene expression profiles for 53 tissues from the GTEx resource (https://gtexportal.org/home/datasets) (25). Finally, the available literature on each gene is presented through an iframe linking to either PubMed, UCSC, GeneCards, Ensembl, WikiGenes, GTEx or the GWAScatalog.
Existing ALS datasets.
The browser also includes freely available summary-level data for the 2016 ALS GWAS for download. Additionally, downloadable SKAT and SKAT-O burden testing results from 610 ALS cases and 460 controls with Chinese ancestry (3) are available.
Results
Dataset
The databrowser currently comprises 4,366 ALS cases and 1,832 age- and sex-matched controls whole-genome sequenced and quality control processed as part of the broader Project MinE effort.
Quality control and association analysis
The quality controlled dataset includes 6198 individuals and describes more than 105 million SNVs and indels. In this sample, we have limited power to detect genome-wide significant association in a single variant framework and as a result we did not find any variants reaching genome-wide significance. In our rare-variant burden framework, we find that the excess of disruptive and damaging variants at MAF < 1% in the canonical transcript of NEK1 in ALS patients compared to controls reaches exome-wide significance (p = 2.31 × 10−7, odds ratio = 3.55 [95% confidence interval = 2.02–6.26], Figure 2). We also noticed that some genes might contain a transcript specific burden, most notably in TARDBP (Supplementary Table 3 and Supplementary Fig. 12).
Figure 2.
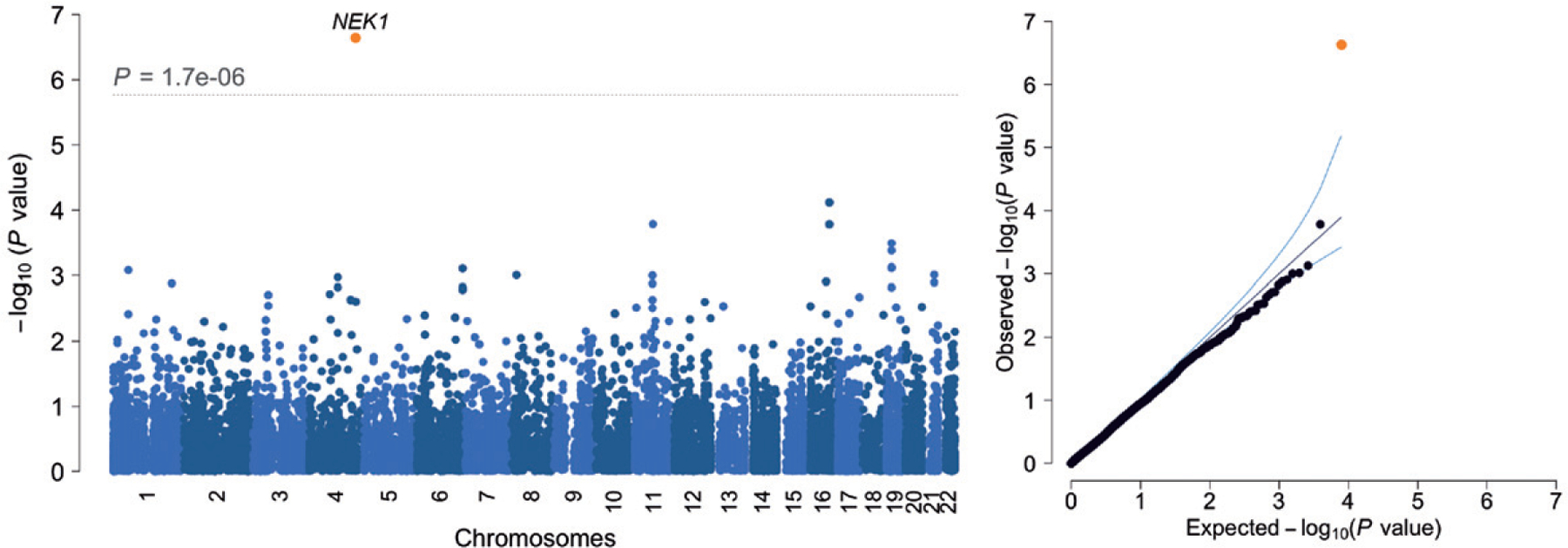
Results are shown for genic (canonical transcripts only) firth logistic regression including variants with a MAF < 1% and categorized as disruptive and damaging. λGC = 0.907, λ1000 = 0.964.
Next, we aggregated all variants across the exome. We observed no difference in the exome-wide burden of synonymous variants between cases and controls, which provides no indication for systemic confounding of burden analyses using higher-order variant aggregation strategies. Therefore, we proceeded to test a genome-wide excess of rare non-synonymous variants among ALS patients. In contrast to similar analyses in schizophrenia (19) and educational attainment (26), we found no evidence for such excess in any variant set combining all allele frequency cutoffs and functional classification. Furthermore, we do not find any protein families, druggable categories significantly enriched for rare variants after collapsing allele-frequency cutoffs and variant classification. All association analysis results are available for download at the browser website.
Databrowser
By entering a gene or transcript in the databrowser you will be shown a visualization of the rare-variant burden tests, as well as several other components (Figure 3).
Figure 3.
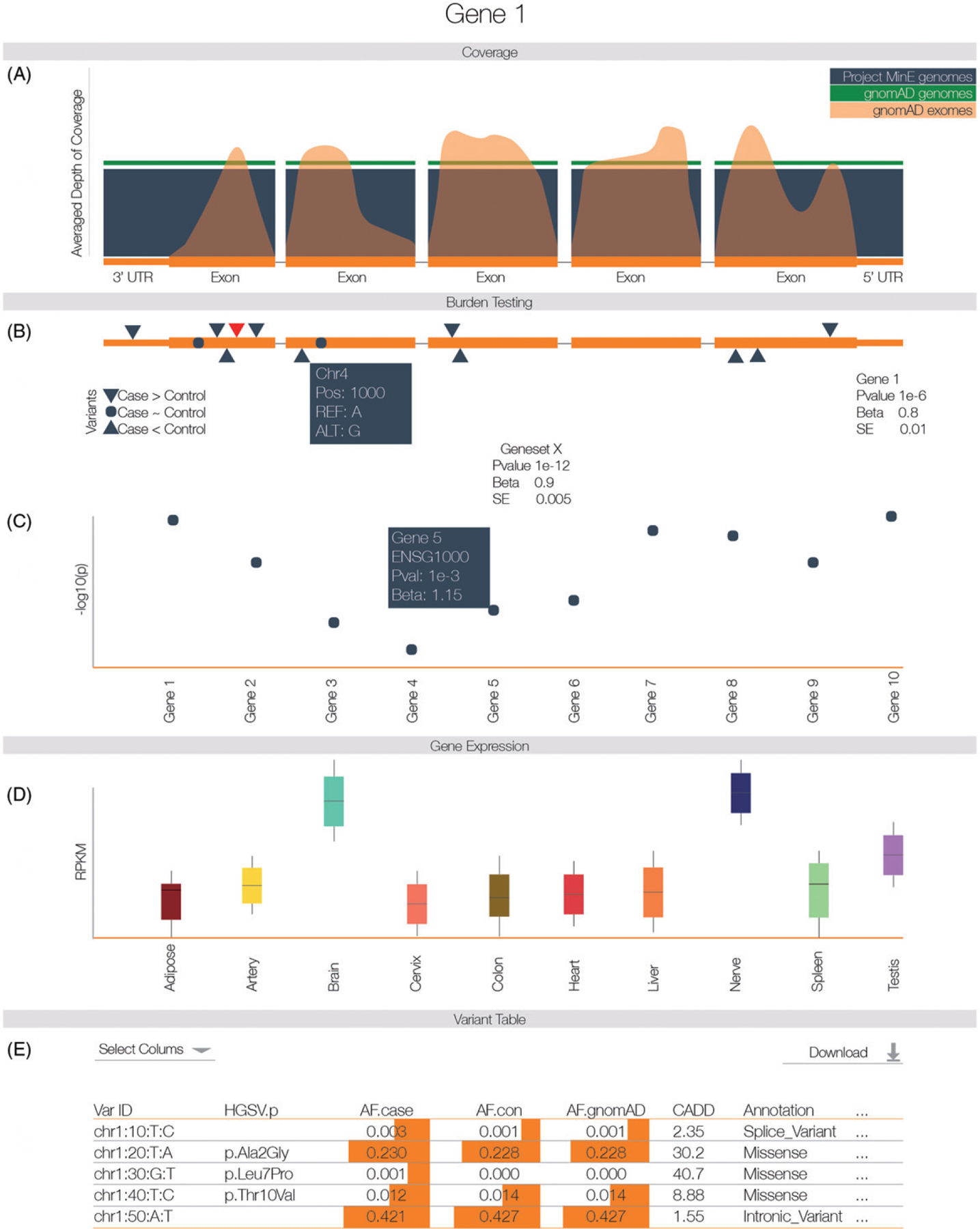
After entering the gene name (HGNC, Ensembl gene (ENSG), or transcript (ENST) identifier) in the search box on the homepage, you will be directed to the gene-specific page. (A) Averaged depth of coverage in the Project MinE dataset, compared to public data and indicating quality of coverage in the region. (B) Firth logistic regression-based genic burden tests. Triangles indicate variant locations. Red triangles reach nominal significance in the single variants association test. Hovering over the triangles to obtain more information about that variant. (C) Firth logistic regression-based geneset burden test. Tests are based on pathways, gene families or druggable gene categories. To elucidate the gene or genes driving a signal in the geneset, a Manhattan plot indicates the genic burden results for each of the genes included in the geneset. Hovering over individual genes will reveal more information about that gene. (D) Gene expression profiles extracted from GTEx. (E) Variant table. By default, a subset of variant information is shown; columns of interest can be selected from the dropdown menu. Minor allele frequency is based on all unrelated and QC passing samples in the Project MinE dataset (6198 genomes). Frequency information is also stratified by phenotypic status and compared to public exome and whole genome data. For comparison, we have indicated the allele frequency on a log scale with orange bars; the longer the bar, the higher the allele frequency. Variant filtering can be customized using the search boxes below the header of each column. All data, including case/control frequencies, are available for download in a tab-delimited file. For a more detailed view of the databrowser, see Supplementary Fig. 11.
Transcript details.
Here, we describe the elementary transcript details for the gene of interest. This includes the Ensembl transcript ID, Ensembl Gene ID, number of exons and genomic coordinates as described in the GRCh37 build.
Coverage information (Figure 3(a)).
To illustrate whether a particular gene/transcript or exon has been adequately covered to detect variation, we have included a graphical representation of average depth of coverage. This graph also includes the coverage information from the ExAC database to illustrate the difference in coverage between genome- and exome-sequencing. Optionally, the coverage across introns can be visualized.
Genic burden results (Figure 3(b)).
Burden testing, by definition, aggregates many variants. This approach can increase statistical power to find an association, but can obscure which variant(s) are driving a potential association. Therefore, we have included an interactive graphical representation of the gene indicating where variants are located and whether these variants are case or control-specific. Hovering over a specific variant will reveal the position, alleles, heterozygous and homozygous allele counts in ALS cases and controls, and functional annotation of the variant. We additionally provide the burden test statistics. To further facilitate interpretation, we describe the burden test properties and relevant references in a dropdown menu “Burdentest Info”. We have performed genic burden results for all transcripts.
Geneset burden results (Figure 3(c)).
Here, we show burden test results for genesets such as protein families and druggable targets to which the selected gene belongs. This includes a mini-Manhattan plot generated to indicate which genes might be driving an association signal in the geneset by plotting their individual genic burden results.
Tissue-specific gene expression (Figure 3(d)).
This panel shows gene expression levels across all general tissues included in GTEx.
Variant annotation table (Figure 3(e)).
Each variant has been extensively annotated and aggregated in a customizable table. By default, only allele frequency in cases and controls, comparison to gnomAD genomes and exomes, and amino acid change, impact and functional consequence are shown. All information can be downloaded in tabular form. We have not stratified by country of origin due to the small sample sizes per country. This small sample size makes proving the ethnic specificity of an SNP unreliable while raising serious privacy concerns through attribute disclosure attacks via DNA (ADAD) (27).
Gene-specific literature.
To provide background information on the gene’s function and disease association from literature, we have included an iframe linking to PubMed, UCSC, GeneCards, Ensembl, WikiGenes, GTEx, and the GWAScatalog. This allows a user to rapidly extract information from various resources while staying on the same page.
Group and individual level data sharing
The summary statistics for the latest GWAS, WGS single variant association and all WGS burden analyses can be downloaded directly. Access to individual-level data can be requested by providing a digital form with a brief research proposal (https://www.projectmine.com/research/data-sharing/).
Duplicate and relatedness checks
We have created sumchecks for each individual in our dataset. Sumchecks are hashes which have been created, based on a small subset of SNPs, which allow for the identification of duplicates without sharing the genetic data itself. If researchers wish to check duplicates with our dataset, they can simply request the sumchecks, create hashes for their own data and compare the hashes. Due to the fact that these hashes are strong identifiers of individuals, they are not shared publicly. The code to generate the hashes and the list of SNPs used is available on the Project MinE Bitbucket. These hashes only identify duplicate samples, and in some instances relatedness information can be valuable, e.g. extending pedigrees or meta-analyses. Relatedness checks can be requested through info@projectmine.com, we will need a statement that this information will be used for academic purposes only and will not be used to re-identify individuals without consent perform the relatedness checks. These checks do not require a data-access request nor approval.
Technical details
The whole website, including data storage, runs on a dual core server with 4 Gb RAM and needs <50 Gb of storage. As of July 2018, we have had over 6200 sessions from over 1500 users.
Discussion
Both research and clinical work increasingly rely on open-access databases to find newly associated variants and interpret genetic findings when counseling patients (28). Therefore, sharing de-identified data is instrumental to ensuring scientific and clinical progress, and patient-derived data should not be regarded as intellectual property nor as trade secret (29,30). Also, most genetic browsers are based on healthy individuals, or unselected individuals who might carry specific rare genetic variants which hampers adequate comparison to a sample of patients from another geographical region. With exactly this in mind, we developed a unique, publicly available, disease-specific databrowser which serves as a transparent framework for sharing data and results in ALS genetics. The Project MinE Databrowser contains an unprecedented amount of WGS data from ALS patients, more than doubling the currently-available exome based databases, and provides (meta)data in far greater detail. The intuitive design facilitates interpretation of robust statistical association analyses by presenting detailed metadata and through integration with population-based observations, biological/functional context and literature. As a result, we make our data and results accessible to a broad public of diverse backgrounds and for any research initiative. The databrowser provides an easy framework for other consortia who are generating similar genetic data and results in ALS and other diseases.
The data have already provided a backbone for new gene discovery and variant interpretation in ALS. For example, subsets of the current dataset have been incorporated in previous publications which identified C21orf2, NEK1, and KIF5A (1,31,32). The resource will continue to grow as the Project MinE consortium does, and will thus increasingly allow for more reliable identification of true positives (33,34). The growth in both sample size and ancestral diversity will increasingly reflect the ALS mutation spectrum and yield increasingly accurate estimations of effect sizes in the general population. The browser can also offer researchers quick, easy to access to a reliable dataset for significant improvement in statistical power without financial burden. The next data freeze will include summary results from data where large amounts of external controls are included to boost statistical power and will be made available through the browser.
One of the major goals of the databrowser is to allow cross-disciplinary interrogation and interpretation of the data with minimal effort. We enable this through the intuitive display of individual variant level data, statistical results and through the integration with databases including GTEx and gnomAD. The databrowser ensures transparency and continued reevaluation of established associations, vitally important for clinical laboratories to make appropriate variant classifications (34). Furthermore, we aim to facilitate the design of functional experiments by showing which variants, might be driving a genic burden signal and if these are located in specific exons and therefore specific protein domains.
Project MinE is largely crowd-funded and the ALS-community is highly engaged in the scientific progress in our field. Consequently, we feel an obligation to give something back to the community and promote data sharing in general. We hope that our databrowser will inspire similar efforts in other fields. The Project MinE databrowser is a light-weight and open-source R script that can easily be adapted to serve other consortia and thus share similarly important data. Further, we aim to improve data sharing by encouraging fellow researchers to gain access to individual-level data by submitting an analysis proposal to the consortium. This procedure includes a swift review by the data access committee (all Project MinE PIs) without the need for sending in local proof of IRB approval or yearly renewals of the request, to encourage the process of datasharing. After access is granted, (g)vcf data can be obtained. Analyses on read level data can be performed on the compute facilities of SURFsara, a supercomputer based in Amsterdam, The Netherlands. Researchers will only need to pay a minimal fee to compensate costs for their core hours and data storage requirements.
Project MinE continues to work forward to its ultimate goal of WGS 15,000 cases and 7500 matched controls, as well as combining the data with publicly available control data. Current efforts also focus on single SNV and aggregated SNV analyses of autosomal chromosomes. Future efforts will aim to include sex chromosomes, indels, structural variation (in particular, repeat expansions (35)) and non-coding burden analyses. Additionally, Project MinE is collecting methylation data on all samples using the Infinium Human Methylation 450K and EPIC BeadChip. These data and analyses will also be shared expeditiously through our databrowser prior to publication. As the project proceeds and data generation continues apace, we intend for the browser to pave the way for more accurate diagnosis and prognosis, aid in the identification of novel disease-associated genes, and elucidate potential novel therapeutic targets.
Supplementary Material
Acknowledgements
This work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.
Funding
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program [grant agreement no. 772376 – EScORIAL].
Appendix A. Project MinE ALS Sequencing Consortium
Rick A.A. van der Spek1,‡, Wouter Van Rheenen1,‡, Sara L. Pulit2, Kevin P. Kenna1, Russell L. McLaughlin3, Matthieu Moisse4,5,6, Annelot M. Dekker1, Gijs H.P. Tazelaar1, Brendan Kenna1, Kristel R. Van Eijk1, Joke J.F.A. Van Vugt1, Perry T.C. Van Doormaal1, Bas Middelkoop1, Raymond D. Schellevis1, William J. Brands1, Ross Byrne3, Johnathan Cooper-Knock7, Ahmad Al Khleifat8, Yolanda Campos9, Atay Vural10, Jonathan D. Glass11,12, Alfredo Iacoangeli13, Aleksey Shatunov8, William Sproviero8, Ersen Kavak14, Tuncay Seker10, Fulya Akçimen10, Cemile Kocoglu10, Ceren Tunca10, Nicola Ticozzi15,16, Maarten Kooyman17, Alberto G. Redondo18, Ian Blair19, Naomi R. Wray20, Matthew C. Kiernan21, Mamede de Carvalho22, Vivian Drory23, Marc Gotkine24, Peter M. Andersen25,26, Philippe Corcia27,28, Philippe Couratier27,28, Vera Fominyh29, Mayana Zatz30, Miguel Mitne-Neto30, Adriano Chio31,32, Vincenzo Silani15,16, Boris Rogelj33,34, Blaž Koritnik35, Janez Zidar35, Markus Weber36, Guy Rouleau37, Nicolas Dupre38,39,40, Ian Mackenzie41, Ekaterina Rogaeva42,43, Gabriel Miltenberger-Miltenyi44, Lev Brylev29, Ervina Bilić45, Ivana Munitic46, Victoria López Alonso47, Karen E. Morrison48, Stephen Newhouse49,50, Johnathan Mill51,52, Pamela J. Shaw53, Christopher E. Shaw8, Monica P. Panades54, Jesus S. Mora55, Wim Robberecht4,5,6, Philip Van Damme4,5,6, A. Nazli Basak10, Orla Hardiman56,57, Michael A. Van Es1, Ammar Al-Chalabi8, John E. Landers58, Leonard H. Van den Berg1,§ & Jan H. Veldink1,§,*
1. Department of Neurology, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, The Netherlands; 2. Department of Medical Genetics, Center for Molecular Medicine, University Medical Center Utrecht, Utrecht, The Netherlands; 3. Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Republic of Ireland; 4. KU Leuven - University of Leuven, Department of Neurosciences, Experimental Neurology and Leuven Research Institute for Neuroscience and Disease (LIND), B-3000 Leuven, Belgium; 5. VIB, Vesalius Research Center, Laboratory of Neurobiology, Leuven, Belgium; 6. University Hospitals Leuven, Department of Neurology, Leuven, Belgium; 7. Sheffield Institute for Translational Neuroscience (SITraN), University of Sheffield, Sheffield, UK; 8. Maurice Wohl Clinical Neuroscience Institute, King’s College London, Department of Basic and Clinical Neuroscience, London, UK; 9. Mitochondrial pathology Unit, Instituto de Salud Carlos III, Madrid, Spain; 10. Neurodegeneration Research Laboratory, Bogazici University, Istanbul, Turkey; 11. Department Neurology, Emory University School of Medicine, Atlanta, GA, USA; 12. Emory ALS Center, Emory University School of Medicine, Atlanta, GA, USA; 13. Department of Biostatistics, IoPPN, King’s College London, London, US; 14. Genomize Inc. Bogazici University, Technology Transfer Region, ETAB, Istanbul, Turkey; 15. Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milano, Italy; 16. Department of Pathophysiology and Transplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy; 17. SURFsara, Amsterdam, the Netherlands; 18. Hospital Carlos III, Madrid, Spain; 19. Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Macquarie University, Sydney, New South Wales, Australia; 20. Queensland Brain Institute, The University of Queensland, Brisbane, Queensland, Australia; 21. Brain and Mind Centre, The University of Sydney, New South Wales 2050, Australia; 22. Physiology Institute, Faculty of Medicine, Instituto de Medicina Molecular, University of Lisbon, Lisbon, Portugal; 23. Department of Neurology Tel-Aviv Sourasky Medical Centre, Israel; 24. Hadassah University Hospital, Jerusalem, Israel; 25. Department of Neurology, Ulm University, Ulm, Germany; 26. Department of Pharmacology and Clinical Neuroscience, Umea University, Umea, Sweden; 27. Centre SLA, CHRU de Tours, Tours, France; 28. Federation des Centres SLA Tours and Limoges, LITORALS, Tours, France; 29. Neurology Department, Bujanov Moscow City Clinical Hospital; 30. Human Genome and stem-cell center, Biosciences Institute, Universidade de São Paulo, Brazil; 31. Rita Levi Montalcini, Department of Neuroscience, ALS Centre, University of Torino, Turin, Italy; 32. Azienda Ospedaliera Citta della Salute e della Scienza, Torino, Italy; 33. Department of Biotechnology, Jozef Stefan Institute, Ljubljana, Slovenia; 34. Biomedical Research Institute BRIS, Ljubljana, Slovenia; 35. Ljubljana ALS Centre, Institute of Clinical Neurophysiology, University Medical Centre Ljubljana, SI-1000 Ljubljana, Slovenia; 36. Neuromuscular Diseases Unit/ALS Clinic, Kantonsspital St. Gallen, 9007, St. Gallen, Switzerland; 37. Montreal Neurological Institute, McGill University, Montreal, Quebec, Canada; 38. Department of Human Genetics, McGill University, Montreal, Quebec, Canada; 39. Montreal Neurological Institute, McGill University, Montreal, Quebec, Canada; 40. Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada; 41. Department of Pathology and Laboratory Medicine, University of British Columbia, Canada; 42. Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto, Toronto, Ontario, Canada; 43. Department of Medicine, Division of Neurology, University of Toronto, Toronto, Ontario, Canada; 44. Physiology Institute, Faculty of Medicine, Instituto de Medicina Molecular, University of Lisbon, Lisbon, Portugal; 45. Department of Neurology Clinical Hospital Center Zagreb, University of Zagreb School of Medicine; 46. Department of Biotechnology, University of Rijeka; 47. Computational Biology Unit, Instituto de Salud Carlos III, Madrid, Spain; 48. Faculty of Medicine, University of Southampton, Southampton, UK; 49. Department of Biostatistics, IoPPN, King’s College London, London, US; 50. Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, UK; 51. Maurice Wohl Clinical Neuroscience Institute, King’s College London, Department of Basic and Clinical Neuroscience, London, UK; 52. University of Exeter Medical School, Exeter University, St Luke's Campus, Magdalen Street, Exeter EX1 2LU, UK; 53. Sheffield Institute for Translational Neuroscience (SITraN), University of Sheffield, Sheffield, UK; 54. Neurology Department, Hospital Universitari de Bellvitge, Barcelona, Spain; 55. Hospital San Rafael, Madrid, Spain; 56. Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Republic of Ireland; 57. Department of Neurology, Beaumont Hospital, Dublin, Republic of Ireland; 58. Department of Neurology, University of Massachusetts Medical School, Worcester, MA, USA
Footnotes
A full list of Project MinE ALS Sequencing Consortium members is given in Appendix A.
Supplemental data for this article can be accessed here.
Code availability
Source-code for the databrowser is available at https://bitbucket.org/ProjectMinE/databrowser
Declaration of interest
The authors report no conflict of interest.
References
- 1.van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 2016;48:1043–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schijven D, van Rheenen W, Van Eijk KR, Brien MOR, Kahn RES, Ophoff RA, et al. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat Commun 2017;8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benyamin B, He J, Zhao Q, Gratten J, Garton F, Leo PJ, et al. Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat Commun 2017;8:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Rheenen W, Pulit SL, Dekker AM, Khleifat Al A, Brands WJ, Iacoangeli A, et al. Project MinE: study design and pilot analyses of a large-scale whole-genome sequencing study in amyotrophic lateral sclerosis. Eur J Hum Genet 2018;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canela-Xandri O, Rawlik K, Tenesa A. An atlas of genetic associations in UK Biobank. Nature Genetics 2018;50: 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 2017;33:272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Consortium GTEx. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Lee SH, Goddard ME, Visscher PM. REPOR T GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pulit SL, de With SAJ, de Bakker P. Resetting the bar: statistical significance in whole-genome sequencing-based association studies of global populations. Genet Epidemiol 2017;41:145–51. [DOI] [PubMed] [Google Scholar]
- 12.Firth D Bias reduction of maximum likelihood estimates. Biometrika 1993;80:27. [Google Scholar]
- 13.Heinze G, Ploner M. SAS and SPLUS programs to perform Cox regression without convergence problems. Comput Methods Prog Biomed 2002;67:217–23. [DOI] [PubMed] [Google Scholar]
- 14.Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 2015;43:W589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson D, Madera M, Vogel C, Chothia C, Gough J. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Res 2007;35:D308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, et al. DGIdb 3.0: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Res 2018;46:D1068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34: 267–73. [DOI] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 2016;19:1433–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruden DM. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Frontiers in Genetics 2012;3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaudet P, Michel P-A, Zahn-Zabal M, Britan A, Cusin I, Domagalski M, et al. The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res 2017; 45:D177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 2016;37:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consortium GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganna A, Genovese G, Howrigan DP, Byrnes A, Kurki MI, Zekavat SM, et al. Ultra-rare disruptive and damaging mutations influence educational attainment in the general population. Nat Neurosci 2016;19:1563–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erlich Y, Narayanan A. Routes for breaching and protecting genetic privacy. Nat Rev Genet 2014;15: 409–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonàs-Guarch S, Guindo-Martínez M, Miguel-Escalada I, Grarup N, Sebastian D, Rodriguez-Fos E, et al. Reanalysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat Commun 2018;2018:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Directors ABO. Laboratory and clinical genomic data sharing is crucial to improving genetic health care: a position statement of the American College of Medical Genetics and Genomics. Genet Med 2017;19:721–2. [DOI] [PubMed] [Google Scholar]
- 30.Data sharing and the future of science. Nat Commun 2018;9:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 2018;97:1268–82.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenna KP, van Doormaal PTC, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet 2016;48:1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van der Spek RA, van Rheenen W, Pulit SL, Kenna KP, Ticozzi N, Kooyman M, et al. Reconsidering the causality of TIA1 mutations in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2018;19:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Project MinE ALS Sequencing Consortium. CHCHD10variants in amyotrophic lateral sclerosis: where is the evidence? Ann Neurol 2018;84:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolzhenko E, van Vugt J, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 2017;27:1895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.