

Abstract
Little is known about the role of TBX1 in post-otocyst stages of inner ear development. Here, we report on mice with a missense mutation of Tbx1 that are viable with fully developed but abnormally formed inner ears. Mutant mice are deaf due to an undeveloped stria vascularis and show vestibular dysfunction associated with abnormal semicircular canal formation. We show that TBX1 is expressed in endolymph-producing strial marginal cells and vestibular dark cells of the inner ear and is an upstream regulator of Esrrb, which previously was shown to control the developmental fate of these cells. We also show that TBX1 is expressed in sensory cells of the crista ampullaris, which may relate to the semicircular canal abnormalities observed in mutant mice. Inner ears of mutant embryos have a non-resorbed fusion plate in the posterior semicircular canal and a single ampulla connecting anterior and lateral canals. We hypothesize that the TBX1 missense mutation prevents binding with specific co-regulatory proteins. These findings reveal previously unknown functions of TBX1 during later stages of inner ear development.
Keywords: TBX1, inner ear development, stria vascularis, semicircular canal, mouse mutation
Introduction
Morphogenesis of the complex structures of the inner ear depends on highly regulated expression patterns of signaling molecules and transcription factors. TBX1 belongs to the T-box protein family of transcription factors that play important roles in tissue and organ formation during embryonic development, including the inner ear. TBX1 haploinsufficiency is thought to be a major contributor to DiGeorge syndrome (DGS, OMIM #188400) and velocardiofacial syndrome (VCFS, OMIM #192430), which often include hearing loss as a clinical feature. Both of these syndromes are caused by a 1.5–3.0 Mb hemizygous deletion of chromosome 22q11.2 and are here collectively referred to as DGS/VCFS. Characteristics of DGS/VCFS include cardiac outflow tract abnormalities, hypoplasia or aplasia of the thymus and parathyroid gland, craniofacial defects, abnormal vertebrae, and cleft palate. Hearing loss occurs in roughly 30% of DGS/VCFS cases (Bassett et al., 2005; Ryan et al., 1997). The hearing loss in most patients is conductive in nature and often associated with chronic otitis media; however, sensorineural hearing loss related to the inner ear has been identified in some patients (Digilio et al., 1999). A hemizygous mouse model of the DGS/VCFS 22q11.2 deletion also showed a high incidence of hearing loss related to chronic otitis media (Fuchs et al., 2013).
TBX1 is considered a critical determinant in the pathogenesis of DGS/VCFS because many of the characteristic abnormalities of the syndromes are also displayed in Tbx1 knockout mice (Jerome and Papaioannou, 2001), in mice with altered Tbx1 dosage (Liao et al., 2004), and in humans with TBX1 missense mutations (Yagi et al., 2003; Zweier et al., 2007). TBX1 is required for the morphogenesis of many of the biological structures affected in DGS/VCFS, including cardiac outflow tract (Vitelli et al., 2002a; Xu et al., 2004), pharyngeal apparatus (Choe and Crump, 2014; Jackson et al., 2014), skeleton (Funato et al., 2015; Vitelli et al., 2006), cerebral cortex (Ogata et al., 2014; Paylor et al., 2006), outer and middle ear (Arnold et al., 2006; Moraes et al., 2005), and inner ear (Raft et al., 2004; Vitelli et al., 2003).
TBX1 has been postulated to contribute to inner ear morphogenesis through expansion of cellular subpopulations (Vitelli et al., 2003), suppression of neural cell fate (Raft et al., 2004), regulation of proliferation and cell fate determination (Xu et al., 2007b), and spatial pattern stabilization (Moraes et al., 2005). Tbx1 knockout mice exhibit perinatal lethality, and inner ear development in these mice is stopped at the otocyst stage around embryonic day 10 (E10), before formation of the sensory organs (Vitelli et al., 2003). Although Pax2-Cre Tbx1 conditional knockout mice, in which expression of the knockout allele is specific to the otic vesicle, are viable, inner ear development in these mice is also arrested at the otocyst stage (Arnold et al., 2006). A reporter gene study in non-mutant heterozygous knockout mice showed that Tbx1 is broadly expressed in sensory and non-sensory epithelial structures of the inner ear in E18.5 mouse embryos (Xu et al., 2007a). This late embryonic expression pattern suggests that Tbx1 has specific functions beyond the otocyst stage that are not accessible by studies of null and inner ear conditional knockout mice.
In this study, we report on a recessive ENU-induced mutation (nmf219) originally identified by the circling behavior and deafness of homozygous mutant mice. We show that nmf219 is a missense mutation of Tbx1. In contrast to previously reported Tbx1 knockout mice, Tbx1nmf219 mutant mice are viable with fully formed but abnormally developed inner ears. They thus provide a valuable model to study the roles of Tbx1 in inner ear development beyond the early otocyst stage. We show that the deafness and vestibular dysfunction of Tbx1nmf219 mutant mice are primarily due to defects in stria vascularis and semicircular canal development, demonstrating the importance of TBX1 function in the morphogenesis and function of these inner ear structures.
Materials and Methods
Mice
Experimental mice were bred and housed in the Research Animal Facility of the Jackson Laboratory (JAX) in Bar Harbor, Maine. Mice were fed ad libitum with a 6% fat mouse diet. Water bottles were changed weekly and filled with acidified (pH 2.5–3.0) water. The light cycle was 12 hours light and 12 hours dark. All procedures involving the use of experimental mice were approved by Institutional Animal Care and Use Committee at JAX. All methods used in the study were performed in accordance with the guidelines and regulations of the U.S. National Institutes of Health (NIH) Office of Laboratory Animal Welfare (OLAW) and the Public Health Service (PHS) Policy on the Humane Care and Use of Laboratory Animals.
The nmf219 mutation is an ENU-induced mutation generated by the discontinued Neuroscience Mutagenesis Facility at JAX. The strain carrying this mutation was originally designated C57BL/6J-nmf219/J and is available as JAX Stock #4831. Mutant mice (nmf219/nmf219) can be identified without genotyping by their overt circling and head tilting behaviors, and the colony is maintained by mating +/nmf219 (female) × nmf219/nmf219 (male) mice. In the present paper, we show that nmf219 is a mutation of Tbx1 (Tbx1nmf219).
Genetic mapping and whole-exome sequencing
To genetically map the nmf219 mutation, individual DNA samples from linkage cross mice were typed for multiple polymorphic DNA markers located throughout the mouse genome. Standard PCR methods were used to genotype the chromosomal markers, which were then analyzed for co-segregation with the mutant phenotype (circling behavior, presumed nmf219/nmf219 genotype). PCR primer pairs designed to amplify specific markers were purchased from Integrated DNA Technologies (Coralville, IA, USA).
Whole-exome sequencing was used to identify the nmf219 mutation in the Tbx1 gene, with particular reference to the genetically determined candidate gene region. Purified genomic DNA from C57BL/6J-nmf219/nmf219 mice was used to create a library for whole-exome sequence capture. Exon sequences from this library were compared with those of the C57BL/6J reference sequence. DNA purification, library construction, deep sequencing, and data quality control were performed by the Jackson Laboratory’s Next Generation Sequencing service, and data analysis and annotation were performed by the Computational Sciences-Biostatistics service.
DNA sequence confirmation and genotyping of the nmf219 mutation
PCR for comparative DNA analysis between nmf219 mutant and control mice was performed according to the same conditions as described above for genetic mapping. PCR primers used to genotype the nmf219 mutation of the Tbx1 gene were 5′- CGATGTTGCCCTAGGTATGC -3′ (forward) and 5′-GGCCTACAACAGGAGACAGC -3′ (reverse), which amplify a 232 bp product from genomic DNA. The nmf219 mutation destroys a TaqI restriction enzyme site within the amplified region (Fig. 1A). PCR product sizes after digestion with TaqI are 140 bp and 92 bp for the wild-type allele and 232 bp for the nmf219 allele, which are easily separated on a 3.5 % agarose gel. For DNA sequence confirmation of genotypes, PCR products were purified with the QIAquick PCR Purification Kit (Qiagen Inc., Valencia, CA), and sequencing was performed using the same primers as for DNA amplification, then run on an applied biosystems 3700 DNA Sequencer with an optimized Big Dye Terminator Cycle Sequencing method.
Figure 1. Molecular characterization and consequences of the Tbx1nmf219 mutation.

A. DNA sequence of Tbx1 exon 3 with encoded amino acids shown below in blue font. The adenine (A) nucleotide at coding position 653 of Tbx1 cDNA (shown in red) is changed to guanine (G) by the nmf219 mutation (c.635A>G), which changes the GAC codon (boldface, boxed) for aspartic acid (Asp) to glycine (Gly) at position 212 of the TBX1 protein (p.Asp212Gly). The mutation also destroys a TaqI restriction enzyme recognition site (TCGA, highlighted in green). B. Schematic diagram of the TBX1 protein with regions encoded by exons 1–7 depicted as alternating purple and orange bars. The nmf219 mutation occurs in exon 3, which encodes part of the T-box DNA binding domain, demarcated by the black bar below the protein diagram. Corresponding amino acid positions are shown at the bottom of the diagram. C. The 3D structure of the DNA-bound T-box domain of the human TBX1 protein. TBX1 binds DNA as a dimer; the individual monomers are shown in blue and magenta in the diagram. Red arrows point to the Asp212 residues of the monomers, which are mutated to Gly in nmf219 mutant mice. The two strands of the DNA helix are shown in gold and cyan. The 3D structure of the human protein was retrieved from the Molecular Modeling Database (MMDB) and provides an accurate proxy for the mouse protein because the 190 amino acid sequence of the T-box domain of the human TBX1 protein (aa 110–299, NP-542377) is identical to the 190 amino acid sequence of the T-box domain of the mouse TBX1 protein (aa 108–297, NP-035 662).
Auditory brainstem response
ABR thresholds were measured at 8, 16 and 32 kHz in a sound attenuating chamber using the SmartEP auditory evoked potential diagnostic system from Intelligent Hearing Systems (IHS, Miami, FL) as described previously (Zheng et al., 1999). Briefly, mice were anesthetized with tribromoethanol (0.2 ml of 20 mg/ml stock per 10 g of body weight, i.p.) and placed on a temperature controlled heating pad to maintain body temperature at 37°C. Three subdermal electrodes, placed at the vertex and behind each ear, were used to record brain stem responses to defined tone-bursts (3 ms duration. 1.5 ms cosine-gated rise/fall time). The responses were then amplified, filtered (100–3000 Hz) and averaged (25 kHz sampling rate, 10 ms analysis window). Stimulus intensity was initially decreased in 10 dB steps until the response began to disappear and then lowered in 5 dB steps; ABR threshold was defined as the lowest intensity at which an ABR response could be reliably obtained. With our testing system, average ABR thresholds for normal hearing mice are about 40, 20, and 45 dB SPL for 8, 16 and 32 kHz stimuli, respectively. Thresholds were assigned a value of 100 if no ABR response was obtained at the maximum test stimulus of 100 dB SPL.
Histology
Histological analyses of the middle and inner ears were performed following methods described previously (Johnson et al., 2003). Briefly, inner ears from Tbx1nmf219/nmf219 mice and wild-type mice were dissected and immersed in Bouin’s fixative and embedded in paraffin. Sections (7 μm) were cut and mounted onto Fisher Superfrost Plus slides (Fisher Scientific, Pittsburgh, PA) and counterstained in hematoxylin/eosin (H&E).
Immunofluorescence staining
Postnatal mouse inner ears were dissected and fixed in 4% formaldehyde/PBS overnight. Tissues were then washed with PBS 3× 5min, and then rinsed with 10%, 20%, and 30% glucose/PBS solution, and then frozen in Tissue-TekVR O.C.T. compound (Sakura Finetek USA Inc, Torrance, CA, USA) and sectioned at 10 mm. Sections were fixed in 4% formaldehyde/PBS for 10 min and permeabilized in 0.2% Triton X-100/PBS for 5 min. Inner ear sections were subsequently blocked in 3% goat or donkey serum plus 2% BSA in PBS for 1h, incubated with primary antibodies in the blocking solution at specific dilution ratio at 4C overnight, washed several times with PBS and incubated with secondary antibody in blocking solution for for 1h. After extensive washes with PBS, the stained inner ear sections were mounted on glass slides using ShandonTM Immu- MountTM aqueous non-fluorescing mounting medium (Thermo Electron Corporation, Pittsburgh, PA, USA) and imaged using an Olympus FV1000 confocal laser scanning microscope. Primary antibodies used were: TBX1 (Invitrogen, Cat # 34–9800, 1:500), KCNQ1 (Santa cruz, Cat # sc-10646, 1:200), KCNJ10 (Alomone labs, Cat # apc-035, 1:400), and GLUT1 (Santa cruz, Cat # sc-1605, 1:100). Secondary antibodies used were donkey anti-goat cy3 (Abcam, Cat # ab6949, 1:500) and goat anti-rabbit Alexa 488 (Invitrogen, Cat # A11008, 1:500).
Riboprobes and mRNA in situ hybridization
The following PCR primers (5’ – 3’) were used to amplify DNA templates for transcription of Esrrb, Aldh1a2, Dct, and Cldn11 riboprobes.
Esrrb forward: TTTGCCTGCTGTTTCTCCTT
Esrrb reverse: GCGATTTAGGTGACACTATAGTGTTTGTCCAACCCTTGCT
Aldh1a2 forward: TAACAATGAATGGCAGAACTCAG
Aldh1a2 reverse: GCGATTTAGGTGACACTATAGGCAATTTTCCAGGTGAAC
Dct forward: AATTCTTCAACCGGACATGC
Dct reverse: GCGATTTAGGTGACACTATAGGCATCTGTGGAAGGGTTGTT
Cldn11 forward: TGACCTGCAGCTACACCATC
Cldn11 reverse: GCGATTTAGGTGACACTATAGACCCAAGTCAGCAATGTTCC
The sequence for the SP6 promoter (underlined) was added as a 5′overhang to the reverse primers so that antisense probes could be generated directly from the PCR products. Digoxigenin-11-UTP (DIG) labeled antisense riboprobes were produced from the purified PCR products by in vitro transcription according to the manufacturer’s instructions (Roche Applied Science, Indianapolis, IN).
In situ hybridization on tissue cryosections was performed as described (Schaeren-Wiemers and Gerfin-Moser, 1993) with the following modifications. Tissue was fixed overnight at 4°C in 4% paraformaldehyde. Sections were mounted on Superfrost plus slides (Fisher Scientific, Suwanee, GA). Slides were air dried at room temperature for 1–2 hours, followed by fixation in 4% parafomaldehyde for 10 minutes. Digestion was carried out for 10 min at room temperature in buffer consisting of 50 mM Tris, pH 7.5, 5 mM EDTA, and 1 μg/ml proteinase K followed by fixation in 4% paraformaldehyde for 5 min. Hybridization was performed at 65° C overnight in buffer consisting of 50% deionized formamide, 0.3 M NaCl, 20 mM Tris, pH 8.0, 5 mM EDTA, pH 8.0, 10% dextran sulfate, 1× Denhardt’s solution, 0.5 mg/ml tRNA from baker’s yeast (Sigma, St. Louis, MO), and 500 ng/ml of the denatured DIG labeled probe. Washes were also carried out at 65° C. Following immunological detection, slides were mounted with VECTASHIELD mounting medium (Vector Laboratories, Burlingame, CA).
Inner ear paint fills
Paint fills of inner ears were performed using previously described procedures (Kiernan, 2006; Tian et al., 2017). E14.5 and E16.5 mouse embryos were decapitated and the heads bisected and fixed overnight in Bodian’s fixative. P0 mice were decapitated and half-heads were fixed in Bodian’s fixative after the brain was removed. Heads were then dehydrated in 75% ethanol (2× 2 hours), 95% ethanol (2× 2 hours), and 100% ethanol (2× 2 hours) and cleared overnight in methyl salicylate. Several microliters of 1% Wite-Out correction fluid in methyl salicylate were injected into the middle turn of the cochlea using a Hamilton syringe, with a pulled glass capillary needle broken to a tip diameter of 20–40 μm. For P0 inner ears, a second injection was made in the common cruss. The paint-filled inner ears were then dissected away from the rest of the head. Each of the ears of 3 mutants and 1 heterozygote were examined at E14.5, both ears of 2 mutants and 1 heterozygote were examined at E16.5, and both ears of 2 mutants and 1 heterozygote were examined at P0.
RNA-Seq
Five E16.5 mutant embryos and 5 littermate controls were used for the analysis. For each sample, RNA was isolated from 2 inner ears using the MagMAX mirVana Total RNA Isolation Kit (ThermoFisher) and the KingFisher Flex purification system (ThermoFisher). Tissues were lysed and homogenized in TRIzol Reagent (ThermoFisher). After the addition of chloroform, the RNA-containing aqueous layer was removed for RNA isolation according to the manufacturer’s protocol, beginning with the RNA bead binding step. RNA concentration and quality were assessed using the Nanodrop 2000 spectrophotometer (Thermo Scientific) and the RNA Total RNA Nano assay (Agilent Technologies). Libraries were prepared by the Genome Technologies core facility at The Jackson Laboratory using the KAPA mRNA HyperPrep Kit (KAPA Biosystems), according to the manufacturer’s instructions. Briefly, the protocol entails isolation of polyA containing mRNA using oligo-dT magnetic beads, RNA fragmentation, first and second strand cDNA synthesis, ligation of Illumina-specific adapters containing a unique barcode sequence for each library, and PCR amplification. Libraries were checked for quality and concentration using the DNA 1000 assay (Agilent Technologies) and quantitative PCR (KAPA Biosystems), according to the manufacturers’ instructions. Libraries were pooled and sequenced by the Genome Technologies core facility at The Jackson Laboratory, 75 bp single-end on the NextSeq 500 (Illumina) using NextSeq High Output Kit v2 reagents (Illumina).
Yeast two-hybrid screen
A yeast two-hybrid (Y2H) screen for TBX1 interacting proteins was performed by Hybrigenics Services, Paris, France (www.hybrigenics-services.com). RNA from inner ears of E16.5 C57BL/6J mouse embryos was used to synthesize random-primed cDNA fragments, which were then ligated into the prey expression vector pP6 and used to construct a yeast cDNA library containing about 10 million independent prey fragments. The translation products of 66.6 million prey fragments (about 6 times the number of independent fragments of the yeast-transformed cDNA library) were screened for interactions with Mus musculus TBX1 (amino acids 1–488) expressed in the bait expression vector pB27 (N-LexA-TBX1-C). Sixty-eight positive clones were processed by PCR and sequencing of their cDNA inserts to identify putative interacting domains of candidate TBX1-interacting proteins by reference to the GenBank database.
Co-immunoprecipitation assay
Co-immunoprecipitation (Co-IP) assays to validate TBX1 interactions with the candidate proteins from the Y2H screen in cultured mammalian cells were performed by Creative BioMart, Shirley, NY (http://www.creativebiomart.net), according to their standard procedures. The reciprocal Co-IP assays were carried out in mammalian HEK293 (human embryonic kidney) cells transfected with expression plasmids for a Flag-tagged TBX1 bait protein and four individual Myc-tagged prey proteins, followed by Western blot analysis.
Results
Origin and overt phenotype of nmf219 mutant mice
The ENU-induced nmf219 mutation was identified through the discontinued NIH-funded Neuroscience Mutagenesis Facility (NMF) at The Jackson Laboratory (JAX). The mutant mice exhibit hyperactive circling and head tossing behavior but otherwise appear to be normal. In preliminary analyses performed by the NMF, the nmf219 mutation was genetically mapped to a 16.4 Mb region of mouse Chr 16 (http://www.informatics.jax.org/marker), and auditory brainstem response (ABR) tests of two mice at six weeks of age showed that they were deaf (https://www.jax.org/strain/004831). We obtained mice carrying the nmf219 mutation to identify and molecularly characterize the mutated gene and to further analyze the mutant phenotype. We verified that homozygous mutant mice are deaf by ABR testing of additional mice (Table 1). None of the seven mutant mice (nmf219/nmf219) tested at 4–6 weeks of age showed a detectable ABR at the maximum stimulus amplitude of 100 dB SPL for any of the three auditory test frequencies (8, 16, and 32 kHz). The ABR thresholds of five age-matched heterozygotes (+/nmf219) were not statistically significantly different from those of wildtype (+/+) mice of the B6 parental strain, verifying the recessive nature of the mutation.
Table 1.
Hearing assessments of C57BL/6J-Tbx1nmf219 strain mice and controls.
| Genotype | Age days | Sex | ABR thresholds (dB SPL) | ||
|---|---|---|---|---|---|
| 8kHz | 16kHz | 32kHz | |||
| nmf219/nmf219 | 26 | f | 100 | 100 | 100 |
| nmf219/nmf219 | 26 | f | 100 | 100 | 100 |
| nmf219/nmf219 | 36 | f | 100 | 100 | 100 |
| nmf219/nmf219 | 36 | f | 100 | 100 | 100 |
| nmf219/nmf219 | 41 | f | 100 | 100 | 100 |
| nmf219/nmf219 | 41 | f | 100 | 100 | 100 |
| nmf219/nmf219 | 47 | f | 100 | 100 | 100 |
| +/nmf219 | 26 | m | 35 | 15 | 35 |
| +/nmf219 | 26 | m | 35 | 20 | 35 |
| +/nmf219 | 26 | f | 35 | 15 | 35 |
| +/nmf219 | 36 | f | 35 | 10 | 35 |
| +/nmf219 | 41 | f | 40 | 15 | 40 |
| mean | 36.0 | 15.0 | 36.0 | ||
| std dev | 2.2 | 3.5 | 2.2 | ||
| +/+ | 29 | m | 35 | 20 | 30 |
| +/+ | 37 | m | 30 | 20 | 40 |
| +/+ | 36 | f | 40 | 10 | 45 |
| +/+ | 36 | f | 40 | 25 | 35 |
| +/+ | 52 | f | 30 | 15 | 40 |
| +/+ | 52 | f | 30 | 15 | 40 |
| +/+ | 52 | f | 30 | 15 | 40 |
| mean | 33.6 | 17.1 | 38.6 | ||
| std dev | 4.8 | 4.9 | 4.8 | ||
| 2-tail Student’s t-test probability, +/nmf219 vs . +/+ | 0.318 | 0.424 | 0.292 | ||
Identification of nmf219 as a missense mutation of Tbx1
To refine the Chr 16 map position of the nmf219 mutation, an intercross of (B6-nmf219/nmf219 × CAST/EiJ) F1 hybrids (nmf219/+) was used to generate a total of 263 mutant progeny with nmf219/nmf219 genotypes assigned on the basis of their vestibular phenotype (head tilting/circling). The F2 mutant mice were then genotyped for multiple Chr 16 MIT microsatellite markers that differed in size between strains C57BL/6J (B6) and CAST/EiJ. Haplotype analysis of genotype-phenotype associations narrowed the candidate gene interval to a 2.7 Mb region of Chr 16, between D16Mit143 (18.1 Mb position, GRCm38) and D16Mit145 (20.8 Mb position), which contains more than 50 protein-coding genes.
Because of the large number of candidate genes, we used an exome sequencing approach to identify the underlying DNA lesion caused by the nmf219 nutation. A total of 15 putative exon sequence differences were identified between DNA samples from B6-nmf219/nmf219 and B6 +/+ control mice in the 2.7 Mb candidate gene interval. Only three of these putative variants had mismatch ratios greater than 0.6, and of these only the Tbx1 variant was predicted to have a negative effect on protein function. The Tbx1 variant equivalent to the nmf219 mutation is a single base pair change in exon 3 (c.665A>G) that changes a codon for aspartic acid (GAC) to a codon for glycine (GGC) at amino acid position 212 (p.Asp212Gly) of the mouse TBX1 protein (Fig. 1A). The altered aspartic acid is evolutionarily highly conserved, and both the PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) computer programs predict that the p.Asp212Gly substitution is likely damaging to protein function. The c.665A>G mutation destroys a TaqI restriction enzyme recognition site (TCGA), which provides an easy method for Tbx1nm219 genotyping by visualizing differently sized PCR products after gel separation. The predicted p.Asp212Gly substitution occurs within the highly conserved T-Box region of the TBX1 protein (Fig. 1B). TBX1 binds DNA as a dimer, and Asp212, the amino acid changed to guanine by the nmf219 mutation, is predicted to be located at the protein surface, as indicated in the 3D structure of the DNA-bound monomers (Fig. 1C).
Mutant mice have an undeveloped stria vascularis and malformed semicircular canals
To identify histological abnormalities that could underlie the deafness of Tbx1nmf219 mutant mice, we examined cross-sections of paraffin-embedded postnatal cochleae from age-matched mutant and control mice (Fig. 2). At postnatal day 7 (P7), cochleae of mutant mice show an undeveloped stria vascularis, a reduced scala media, and a displaced Reissner’s membrane. At 5 months of age, cochleae of mutant mice still exhibit an undeveloped stria vascularis, an extremely reduced scala media, and a completely collapsed Reissner’s membrane. Mutant mice at 5 months of age also display organ of Corti degeneration and loss of spiral ganglion neurons, which are likely secondary effects of the undeveloped stria vascularis. The stria vascularis of Tbx1nmf219 mutant mice is undeveloped because precursor epithelial cells fail to transform into mature marginal cells, as seen in the cochlear duct of P3 mutant mice (Fig. 3).
Figure 2. Cross-sections of cochleae from Tbx1nmf219/nmf219 (mutant) and Tbx1+/nmf219 (control) mice.

Mid-modiolar sections of cochleae from control (A, B) and mutant (C, D) mice are compared at 7 days (A, C) and 5 months (B, D) of age, stained with H&E. Cochleae of mutant mice show a progressive collapse of Reissner’s membrane (rm), a reduced endolymph volume in the scala media (SM), and an undeveloped stria vascularis (sv). At 5 months of age, mutant cochleae also show degeneration of the organ of Corti (oc) and loss of spiral ganglion cells (sg). All scale bars, 50 micrometers.
Figure 3. Failure of marginal cell maturation in Tbx1nmf219 mutant mice.
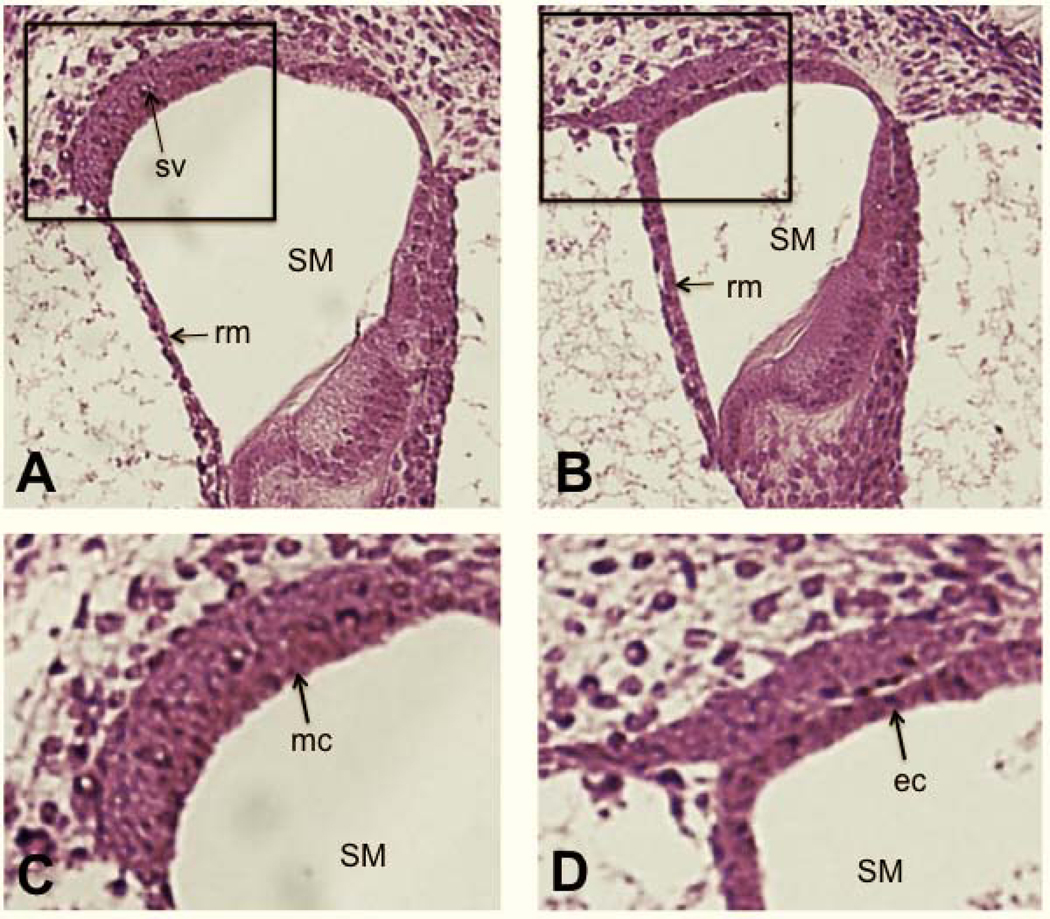
Transverse sections through the cochlear ducts of a +/+ control mouse (A) and a Tbx1nmf219 mutant mouse (B) at P3, stained with H&E. The boxed region of A encloses the stria vascularis and is enlarged in C, and the similarly positioned boxed region of B is enlarged in D. The +/+ control mouse (A) has a normally developed a stria vascularis (sv), but a mature stria vascularis does not form in Tbx1nmf219 mutant mice (B). The arrow in C points to the layer of mature marginal cells (mc) in the stria vascularis of the +/+ control mouse, which have interdigitated with the overlying intermediate cells. The arrow in D points to the layer of precursor epithelial cells (ec) lining the scala media (SM) in the Tbx1nmf219 mutant mouse, which have not matured into marginal cells and do not interdigitate with intermediate cells. For orientation Reissner’s membrane (rm) is marked in A and B.
We examined paintfills of inner ears to visualize in detail any morphological differences that may occur between the inner ears of B6-Tbx1nmf219 mutant and control embryos at embryonic days E14.5 and E16.5 and in newborns (P0). At all ages examined, the inner ears of mutant mice have normal appearing cochlea, saccule, utricle, and anterior and lateral semicircular canals; however, there are obvious malformations of the semicircular canals (Fig. 4). The posterior semicircular canal and common crus of Tbx1nmf219 mutant mice are fused into one plate, indicating failed resorption of the fusion plate, and the posterior canal ampulla fails to form. Fusion plates are formed at the beginning of semicircular canal formation through attachment of apposing lateral and medial epithelial extrusions of the dorsal otic vesicle (Hwang et al., 2019). Cells at the fusion plate intercalate to form a single sheet and normally disappear through a process of resorption. In addition to failed resorption of the posterior canal fusion plate, the anterior and lateral ampullae are fused in the mutant inner ear. The inner ear abnormalities first observed at E14.5 cannot be the result of delayed morphogenesis because they persist at P0.
Figure 4. Paint fills of inner ears reveal semicircular canal abnormalities of Tbx1nmf219 mutant mice.

Inner ears of a heterozygous Tbx1nmf219/+ control (HET) and two littermate Tbx1nmf219 mutants (MUT) are shown at each time point (E14.5, E16.5, and P0). The endolymphatic sac (es), endolymphatic duct (ed), utricle (ut), saccule (sa), cochlear duct (cd), common crus (cc), anterior semicircular canal (asc), posterior semicircular canal (psc), lateral semicircular canal (lsc), anterior ampulla (aa), posterior ampulla (pa), and lateral ampulla (la) are labeled in the inner ears of the heterozygous control mice (A, D, G). In mutant inner ears (B, C, E, F, H, I), the cells that form the fusion plate of the posterior semicircular canal have not resorbed (long arrows), and the posterior ampulla is not formed. A single fused ampulla connects to the anterior and lateral canals (short arrows). Scale bars, 0.5 mm.
Expression of TBX1 and strial markers in inner ears of mutant and control mice
Tbx1 expression patterns previously have been described in inner ears of E9.5-E18.5 embryos (Vitelli et al., 2003; Xu et al., 2007a); however, there are no previous reports of Tbx1 expression during early postnatal stages. We used TBX1 antibody to detect and localize TBX1 protein expression in the inner ear of Tbx1nmf219 mutant mice and littermate controls at P3 and P15 (Fig. 5). At P3, TBX1 expression is observed in marginal cells of the stria vascularis, in hair cells and supporting cells of the crista ampullaris, and in vestibular dark cells of both mutant and control inner ears. At P15, TBX1 is still expressed in the sensory cells of the crista, but is no longer expressed in the nonsensory endolymph-producing marginal cells and dark cells. TBX1 immunostaining intensity and localization in Tbx1nmf219 mutant mice was similar to that seen in the control mice, indicating that the p.D212G missense mutation has little effect on the stability or localization of the TBX1 protein.
Figure 5. TBX1 expression patterns in inner ears from Tbx1+/nmf219 (control) and Tbx1nmf219/nmf219 (mutant) mice.

Immunolabeling on transverse sections through the inner ear reveals TBX1 expression in marginal cells of the lateral wall (arrow) in both control (A) and mutant (B) mice at P3. TBX1 expression is no longer detected in marginal cells (arrow) of the mature stria vascularis at P15 (C). TBX1 is also expressed in cells of the crista ampullaris and adjacent dark cells (small arrows) in inner ears of both control (D) and mutant (E) mice at P3. TBX1 expression is retained in the sensory cells of the crista ampullaris at P15 (F) but can no longer be detected in the adjacent nonsensory dark cells (small arrows). All scale bars, 50 micrometers.
Because the stria vascularis is undeveloped in Tbx1nmf219 mutant mice (Fig. 2), we examined markers for marginal cells, intermediate cells, and basal cells to evaluate the effects of the mutation on these three primary strial cell types. Immunohistochemical results at P1 (Fig. 6) showed that the voltage-gated potassium channel protein KCNQ1, a marginal cell marker, is strongly expressed in control mice but absent or greatly reduced in Tbx1nmf219 mutant mice. Expression levels of the inwardly rectifying potassium channel protein KCNJ10, an intermediate cell marker, and the glucose transporter protein GLUT1, a marker for basal cells and intraepithelial capillaries, are high in control mice and reduced but not absent in mutants, with strongest expression in the strial region nearest Reissner’s membrane.
Figure 6. KCNQ1, KCNJ10, and GLUT1 protein expression in cochleae from Tbx1+/nmf219 (control) and Tbx1nmf219/nmf219 (mutant) mice.

The expression patterns of immunolabeled marker proteins for cells of the stria vascularis are shown on transverse sections through the cochlear duct of newborn (P1) mice. The white dashed lines in panels A and D demarcate the approximate location of the developing stria vascularis in the lateral wall of the cochlear duct. Only this region of the cochlear duct is shown in panels B, C, E, and F. KCNQ1 is expressed in marginal cells of the stria vascularis in control mice (A), but its expression was not detected in mutants (D). KCNJ10 is expressed in intermediate cells of the stria vascularis in control mice (B), but its expression is reduced in mutants (E). GLUT1 is expressed in basal cells and intraepithelial capillaries of the stria vascularis in control mice (C), but its expression is reduced in mutants (F). The expression of KCNJ10 (E) and GLUT1 (F) in the cochleae of mutant mice appears stronger in the strial region nearest Reissner’s membrane (rm). The images in B,C and E,F are from different inner ear sections. Scale bars, 50 micrometers.
We also examined strial gene expression by mRNA in situ hybridization analysis (Fig. 7). The estrogen related receptor beta gene (Esrrb) was strongly expressed in marginal cells of E16.5 and P3 control mice but was not detected in age-matched mutant mice. Likewise, the aldehyde dehydrogenase family 1, subfamily A2 gene (Aldh1a2) was expressed in marginal cells of P3 control mice but not detected in age-matched mutants. The dopachrome tautomerase gene (Dct), an intermediate cell marker, was very strongly expressed in the stria of control mice at P3 but its expression was much reduced in mutant mice. The claudin 11 gene (Cldn11), a basal cell marker, was expressed in the stria of control mice at P3, but its expression was reduced in mutant mice. These results demonstrate that the Tbx1nmf219 mutation primarily affects marginal cell gene expression, with secondary effects on gene expression in intermediate and basal cells.
Figure 7. Esrrb, Aldh1a2, Kcnq1, Dct, and Cldn11 mRNA expression in cochleae of Tbx1+/nmf219 (control) and Tbx1nmf219/nmf219 (mutant) mice.

In situ hybridizations of mRNAs were performed on transverse cryosections through the cochlear duct. Esrrb is expressed in marginal cells of the stria vascularis in control E16.5 embryos (A) and control P3 mice (C), but its expression was not detected in mutants at either age (B, D). Aldh1a2 is also expressed in marginal cells of the stria vascularis in P3 control mice (E) but not in mutants (F). Dct is highly expressed in intermediate cells of the stria vascularis of P3 control mice (G), but its expression is much reduced in mutants (H). Cldn11 expression appeared diffuse through intermediate and basal cells of the stria vascularis in P3 control mice (I) but is reduced in mutants (J). The expression of Dct (H) and Cldn11 (J) in the cochleae of mutant mice appears stronger in the strial region nearest Reissner’s membrane (rm). Scale bars, 50 micrometers.
Differential gene expression in inner ears of Tbx1nm219 mutant and control E16.5 embryos
Because TBX1 is a transcription factor, we sought to evaluate the effects of the Tbx1nmf219 mutation on transcription of downstream genes during inner ear development by comparing expression differences between Tbx1nmf219 mutant and Tbx1+/+ control mice. We used RNA-Seq to identify genes that showed differential expression in inner ears of E16.5 embryos, the age at which cells of the future stria vascularis start to mature. We identified 470 genes that had significant expression differences with adjusted probabilities of occurrence by chance less than 0.05 (corrected for multiple samples): 94 were downregulated and 376 upregulated in inner ears of mutant embryos. Genes that were most affected by the Tbx1nmf219 mutation (with greater than 2.5-fold changes in expression) are listed in Table 2, 12 were downregulated and 27 upregulated. Down regulation of Esrrb was consistent with our in situ hybridization results (Fig. 7) and provides evidence for the reliability of the RNA-Seq results. There was no difference in Tbx1 mRNA expression between inner ear samples from Tbx1nmf219 mutant and control embryos (Table 2), consistent with our immunohistochemical results, which also showed similar expression of the TBX1 protein in mutant and control mice (Fig. 5). A full list of genes detected by our RNA-Seq analysis is available in Gene Expression Omnibus under accession number GSE 134756.
Table 2. Differentially expressed genes in inner ears of E16.5 Txb1nmf219/nmf219 embryos compared with wild-type controls.
The top 12 downregulated and top 27 upregulated genes with greater than 2.5-fold differences in expression between Tbx1nmf219/nmf219 mutant and +/+ control mice are shown.
| Gene symbol | Fold-change | p-adj1 | logCPM2 | GO ID3 | Stria cells? |
|---|---|---|---|---|---|
| Tbxl | 1.04 | 1 | 4.63 | control | marginal |
| 12 most downregulated genes in Tbx1nmf219 mutant embryos: | |||||
| Kcnel | −13.63 | 2.70761E-93 | 1.34 | GO:0015075 | marginal |
| Kcne2 | −5.82 | 8.4506E-33 | 0.19 | GO:0015075 | marginal |
| Bsnd | −4.12 | 1.64628E-19 | −0.14 | GO:0015075 | marginal |
| Atp2c2 | −3.22 | 1.93355E-33 | 2.04 | GO:0015075 | |
| Slc4a5 | −3.20 | 4.96613E-22 | 0.83 | GO:0015075 | |
| Pcdh20 | −3.11 | 1.1674E-46 | 4.73 | ||
| Slc32a1 | −2.86 | 2.61157E-08 | −0.72 | GO:0015075 | |
| Taar5 | −2.83 | 2.85379E-17 | 0.74 | ||
| Tbx3os1 | −2.82 | 1.4394E-14 | 0.37 | ||
| Esrrb | −2.59 | 2.0551E-32 | 4.37 | marginal | |
| Atp13a4 | −2.52 | 3.55968E-12 | 0.42 | GO:0015075 | |
| Tfap2b | −2.51 | 7.17646E-12 | 0.28 | ||
| 27 most upregulated genes in Tbx1nmf219 mutant embryos: | |||||
| Myh2 | 4.22 | 9.66929E-24 | 0.35 | GO:0003012 | |
| Myoz1 | 4.22 | 4.58965E-48 | 2.22 | ||
| Smtnl1 | 3.63 | 1.58197E-16 | −0.13 | ||
| Mylk2 | 3.32 | 1.10131 E-16 | 0.27 | GO:0003012 | |
| Ckm | 3.26 | 1.48278E-51 | 5.64 | ||
| Myh8 | 3.12 | 6.39578E-50 | 7.80 | GO:0003012 | |
| Hspb3 | 3.06 | 1.28262E-10 | −0.38 | ||
| Col6a4 | 2.97 | 6.58941E-20 | 0.91 | ||
| Trim54 | 2.91 | 4.66823E-12 | 0.01 | ||
| Tnnt3 | 2.91 | 1.25745E-42 | 5.65 | GO:0003012 | |
| Tnni2 | 2.87 | 1.81765E-38 | 4.24 | GO:0003012 | |
| Cox8b | 2.85 | 1.10417E-07 | −0.68 | ||
| Ppp1r3a | 2.83 | 7.25681E-19 | 1.12 | ||
| Atp2a1 | 2.82 | 1.69714E-39 | 5.25 | GO:0003012 | |
| Tnnc2 | 2.80 | 1.16943E-38 | 5.07 | GO:0003012 | |
| Trdn | 2.79 | 1.67599E-24 | 1.94 | GO:0003012 | |
| Acta1 | 2.76 | 4.44014E-40 | 7.65 | ||
| Clec3b | 2.75 | 3.6703E-20 | 1.41 | ||
| Ckmt2 | 2.69 | 6.7443E-15 | 0.81 | ||
| Myh1 | 2.65 | 2.00181E-10 | 0.04 | GO:0003012 | |
| Casq2 | 2.61 | 3.47009E-33 | 4.78 | GO:0003012 | |
| Myl1 | 2.60 | 6.97107E-35 | 6.21 | GO:0003012 | |
| Lmod2 | 2.58 | 4.45897E-18 | 1.46 | GO:0003012 | |
| Mlip | 2.56 | 1.45137E-13 | 0.69 | GO:0003012 | |
| Myot | 2.56 | 5.5135E-24 | 2.57 | ||
| Myf6 | 2.54 | 5.68208E-10 | 0.19 | ||
| Dhrs7c | 2.52 | 1.3394E-11 | 0.51 | ||
p-adj, the adjusted probability of expression difference occurring by chance (Benjamini and Hochberg’s false discovery rate), which corrects for multiple testing.
logCPM, the log-2 transformed counts per million (CPM) averaged across all samples used in the pairwise comparison, which provides a measure of relative abundance. Values ranged from +12.5 to −0.1.08, average 2.76.
ID numbers of Gene Ontology terms that are most common to the listed genes (http://www.informatics.jax.org/gotools/MGI_Term_Finder.html)
GO:0015075, ion transmembrane transporter activity
7/12 downregulated genes, corrected p-value 9.627e-08
GO:0003012, muscle system process
13/27 upregulated genes, corrected p-value 1.738e-17
Identification of candidate TBX1 interacting proteins in inner ears of E16.5 embryos
The Tbx1nmf219 mutation is located in the highly conserved T-box DNA binding domain of the protein, which is not only important for DNA binding but is also known to mediate interactions with transcriptional co-regulators (Stoller et al., 2010). The conserved Asp212 residue of TBX1, which is altered in Tbx1nmf219 mutant mice, is located on the surface of the T-box domain of the protein (Fig. 1C), thus making it accessible for potential interactions with other proteins. Because the effects of the Tbx1nmf219 mutation appear to be primarily manifested in the inner ear, we reasoned that the mutation might prevent specific interactions of TBX1 with a co-regulatory protein or proteins expressed during inner ear development, rather than interfere with the more general DNA-binding property of the T-box, which would be expected to have more wide-ranging effects. We used yeast two-hybrid (Y2H) analysis as an unbiased method to identify candidate TBX1 interacting proteins expressed in a cDNA library prepared from inner ears of E16.5 mouse embryos. Y2H analysis, carried out by Hybrigenics Services (see Methods), identified 23 proteins that putatively interact with TBX1 with moderate to high confidence (Table 2). We chose four of the candidate TBX1 interacting proteins from the Y2H screen (MDF1, KDM2A, NOTCH3, and PIAS2) for reciprocal co-immunoprecipitation (Co-IP) experiments, performed by Creative BioMart (see Methods). The Co-IP results verified that all four proteins interact with TBX1 in cultured mammalian HEK293 cells (Fig. 8).
Figure 8. Co-IP validation of candidate TBX1 interacting proteins from the Y2H screen.

Western blot analysis of reciprocal Co-IP assays were used to validate the interaction between bait protein TBX1 and four prey proteins, MDFI (MyoD family inhibitor), KDM2A (lysine-specific demethylase 2A), NOTCH3 (notch 3) and PIAS2 (protein inhibitor of activated STAT 2). HEK293 cells were co-transfected transiently with expression vectors for Flag-tagged TBX1 (pcDNA3.1-Tbx1-Flag) and each of four Myc-tagged candidate interacting proteins (MDFI-Myc, KDM2A-Myc, Notch3-NICD-Myc, and PIAS-Myc). IP, antibody used for Co-IP; IB, antibody used for western blot. GAPDH expression is shown as a loading control. Red arrows indicate the TBX1-candidate protein interactions.
Discussion
The partially retained function of the nmf219 missense mutation can explain the viability and less severe inner ear phenotype of Tbx1nmf219 mutant mice compared with Tbx1 knockout mice. Other mouse ENU mutagenesis screens have identified two other Tbx1 missense mutations that cause hyperactive circling behavior suggestive of inner ear vestibular dysfunction (Chen et al., 2016; Liu et al., 2011); however, mice homozygous for these dominant-acting mutations have not been produced and the effects of these mutations on inner ear morphogenesis have not been examined.
In contrast to the undeveloped inner ears of Tbx1 knockout mice, the inner ears of B6-Tbx1nmf219 mutant mice are fully developed but with abnormalities similar to those of mice with Kcne1 (Letts et al., 2000; Nicolas et al., 2001), Kcnq1 (Casimiro et al., 2001; Lee et al., 2000), and Slc12a2 (Delpire et al., 1999; Dixon et al., 1999; Flagella et al., 1999) deficiencies, all of which exhibit a collapse of cochlear and vestibular endolymphatic spaces related to hearing and balance defects. The proteins encoded by these genes are all involved in the production of inner ear endolymph. The potassium channel proteins KCNE1 and KCNQ1 are coexpressed in the apical membrane of both cochlear marginal cells and vestibular dark cells, and release K+ to the endolymph (Jespersen et al., 2005; Nicolas et al., 2001). The Na-K-Cl cotransporter SLC12A2 is located in strial marginal cells and is important for K+ transport into the cochlear endolymph.
Our TBX1 protein localization results in P3 mice agree with results for Tbx1 mRNA expression deduced from reporter gene β-gal activity in Tbx1+/lacZ late stage E18.5 embryos, where expression also was detected in stria marginal cells and in cells of the crista ampullaris and adjacent dark cells (Xu et al., 2007a). Interestingly, we found that TBX1 expression in the nonsensory endolymph-producing cochlear marginal cells and vestibular dark cells disappears between P3 and P15, whereas expression remains in the sensory cells of the crista ampullaris (Fig. 5). The cochlear marginal cells and vestibular dark cells are responsible for K+ transport to the endolymph of the inner ear and possess many similarities. Consistent with the marginal cell localization of TBX1, there was a lack of expression of the marginal cell marker KCNQ1 in Tbx1nmf219 mutants (Fig. 6) and reduced expression of intermediate and basal cell markers (Figs. 6 and 7), which are likely secondary effects of the marginal cell defect.
ESRRB, an estrogen-related nuclear receptor, controls the differentiation and maturation of strial marginal cells and vestibular dark cells, and multiple genes expressed in the stria vascularis are down regulated in Esrrb−/− mutant mice (Chen and Nathans, 2007), similar to our results for Tbx1nmf219 mutant mice. Our mRNA in situ hybridization results (Fig. 7) show a severe reduction or absence of Esrrb expression in the developing stria vascularis of Tbx1nmf219 mutant mice as early as E16.5, indicating that TBX1 is a direct or indirect upstream regulator of Essrb expression. Previous to this discovery, Esrrb was the only gene known to control development of the endolymph-producing epithelial cells of the inner ear. Together, our results support an important role for TBX1 as an upstream regulator of Esrrb controlling marginal cell maturation, essential for stria vascularis development and function, and in the maturation of vestibular dark cells, essential for vestibular endolymph production. Thus, although TBX1 may not directly specify marginal cells or vestibular dark cells, its upstream regulation of Esrrb transcription indirectly controls the fate and maturation of these cells (Fig. 3).
Paintfills of E14.5 inner ears from B6-Tbx1nmf219/nmf219 mice revealed a single ampulla connecting to the anterior and lateral canals and non-resorption of the fusion plate of the posterior semicircular canal, a vestibular phenotype similar to that observed in Foxg1−/− mice (Hwang et al., 2009). TBX1 probably does not play a direct role in resorption of the posterior canal fusion plate because it does not appear to be expressed in the non-sensory portions of the canal plate. TBX1 is, however, expressed in the sensory cells of the cristae, which have been hypothesized to induce the formation of semicircular canal structures (Hwang et al., 2009). Dysfunction of TBX1 in sensory cells of the cristae (Fig. 5) in Tbx1nmf219 mutant mice, therefore, might possibly be related to the semicircular canal malformations of these mice. Mouse embryos exhibiting reduced Fgf10 expression also exhibit posterior canal defects (Urness et al., 2015) that are similar to those of Tbx1nmf219 mutant mice, suggesting that Tbx1 and Fgf10 interactions may be involved in semicircular canal development. Expression patterns of Fgf8 and Fgf10 overlap with that of Tbx1 in embryonic pharyngeal pouches and are altered in Tbx1−/− mutant mice (Vitelli et al., 2002b), and in vitro evidence suggests that TBX1 regulates Fgf10 expression in the second heart field (Watanabe et al., 2012).
Lineage analysis of mouse inner ears indicates that vestibular hair cells and supporting cells in the posterior crista are clonally related and raises the possibility that nonsensory epithelial cells bordering the crista may be clonally related to the vestibular sensory epithelium (Jiang et al., 2013). Our findings that TBX1 is expressed in sensory hair cells and supporting cells of the crista ampullaris and also in the adjacent nonsensory dark cells in P3 mice (Fig. 5) support the results and interpretations of the lineage analysis. TBX1 dysfunction in nonsensory dark cells, which help maintain ion concentrations in the vestibular endolymph, may also contribute to the balance dysfunction of Tbx1nmf219 mutant mice, in addition to semicircular canal malformations.
To elucidate the effects of the Tbx1nmf219 mutation on downstream transcription, we performed RNA-Seq analysis with inner ears from mutant and control E16.5 embryos, the age at which epithelial cells of the future stria vascularis begin to mature. A number of genes that are expressed in the stria vascularis and involved in stria development and function are down regulated in inner ear tissue from Tbx1nmf219 mutant embryos (Table 2), including the transcriptional regulator Esrrb, which our in situ RNA hybridization results also showed is down regulated (Fig. 7), thus supporting the reliability of the RNA-Seq data. We also observed drastic reductions in Kcne1 and Kcne2 expression, which is not surprising because KCNE1 and KCNE2 subunits form potassium channel complexes with KCNQ1 that are important for K+ secretion and production of inner ear endolymph. Bsnd, a marker for marginal cells, also was down regulated in Tbx1nmf219 mutant mice (Table 2), as was Aldh1a2 (Fig. 7), which encodes the RA-synthesizing enzyme retinaldehyde dehydrogenase. Expression of Aldh1a2 was previously reported in the stria vascularis, Reissner’s membrane, and dark cells of the inner ear, suggesting that RA is synthesized in these regions (Romand et al., 2001; Romand et al., 2006; Romand et al., 2004), and TBX1 previously was shown to regulate retinoic acid metabolic genes during cochlear morphogenesis (Braunstein et al., 2009). A disproportionate number of the most down regulated genes in inner ears of Tbx1nmf219 mutant embryos (7 out of 12) encode proteins with ion transmembrane transporter activity (Table 2), suggesting that the negative effects of the mutation on stria marginal cells and vestibular dark cells may identify additional genes that are involved in inner ear fluid homeostasis.
Surprisingly more genes were up regulated than down regulated by the Tbx1nmf219 mutation, suggesting that TBX1 inhibits expression of more genes than it enhances in the E16.5 inner ear. A disproportionate number of the most upregulated genes in mutant mice (13 out of 27) are involved in muscle system processes, including multiple myosin family members and troponins (Table 2), indicating that TBX1 normally functions to suppress the expression of these genes in the developing inner ear. Consistent with this possible role, TBX1 was shown to suppress the differentiation of cardiac progenitor cells (Chen et al., 2009) and act as a negative modulator of the myocyte enhancer factor gene Mef2c in E9.5 mouse embryos (Pane et al., 2012). TBX1 also is known to be a regulator of myogenic differentiation in other tissues (Buckingham, 2017; Dastjerdi et al., 2007).
We hypothesized that the Tbx1nmf219 mutation may alter the conformation and ability of TBX1 to interact with co-regulatory proteins, which may limit its effects to particular cell types or developmental stages. Our Y2H screen of a cDNA library from inner ears of E16.5 mouse embryos identified 23 putative TBX1 interacting proteins (Table 3). GRN (granulin), a secreted growth factor best known for its association with inflammation and neurodegeneration (Cenik et al., 2012), was classified as a TBX1 interacting protein with very high confidence primarily because five independent clones showed a positive interaction; however, we could not find any connections between the known functions of granulin and its possible roles in inner ear development. We selected four other candidate proteins (KDM2A, MDF1, NOTCH3, PIAS2) for secondary Co-IP analysis and confirmed their interactions with TBX1 in cultured mammalian cells (Fig. 8).
Table 3.
Candidate TBX1-interacting proteins identified from Y2H screen of an inner ear cDNA library from mouse E16.5 embryos.
| Protein symbol1 | Confidence level | Clones | Accession number | Protein size | Interaction domains (aa’s) |
|---|---|---|---|---|---|
| GRN | Very high | 5 | NP_032201 | 589 aa | 93–113, 215–364 |
| * MDFI | High | 3 | NP_034913 | 246 aa | 117–227 |
| MMADHC | High | 2 | NP_001335128 | 235 aa | 138–235 |
| THBS1 | High | 2 | NP_001300843 | 1170 aa | 478–713 |
| TLN1 | High | 2 | NP_035732 | 2541 aa | 1494–1657 |
| UBQLN1 | High | 3 | NP_081118 | 582 aa | 114–241 |
| ATRN | Good | 2 | NP_033860 | 1428 aa | 256–490 |
| AARS | Moderate | 1 | NP_666329 | 968 aa | 754–913 |
| ABTB2 | Moderate | 1 | NP_849221 | 1024 aa | 256–490 |
| COMP | Moderate | 1 | NP_057894 | 755 aa | 5–136 |
| CREBZF | Moderate | 1 | NP_660133 | 358 aa | 182–358 |
| FBN1 | Moderate | 1 | NP_032019 | 2873 aa | 267–512 |
| * KDM2A | Moderate | 1 | XP_006531786 | 721 aa | 1–204 |
| NID1 | Moderate | 1 | NP_035047 | 1245 aa | 676–928 |
| * NOTCH3 | Moderate | 1 | NP_032742 | 2318 aa | 1500–2318 |
| OTOG | Moderate | 1 | NP_038652 | 2911 aa | 421–540 |
| PBX2 | Moderate | 1 | NP_059491 | 430 aa | 110–385 |
| * PIAS2 | Moderate | 1 | NP_032628 | 621 aa | 1–258 |
| RNF138 | Moderate | 1 | NP_997506 | 245 aa | 1–188 |
| STAB1 | Moderate | 1 | NP_619613 | 2571 aa | 21–2571 |
| THBS4 | Moderate | 1 | NP_035712 | 963 aa | 245–418 |
| WIF1 | Moderate | 1 | NP-036045 | 379 aa | 109–282 |
| XPC | Moderate | 1 | NP_033557 | 930 aa | 556–771 |
Asterisks indicate proteins that underwent secondary Co-IP tests for TBX1 interactions in cultured mammalian cells.
KDM2A (lysine-specific demethylase 2A) is perhaps the most interesting of the TBX1-interacting proteins we detected. KDMA2 demethylates both mono- and di-methylated lysine-36 of histone H3 (H3K36) and was shown to play an essential role in embryonic development by regulating cell proliferation and survival (Kawakami et al., 2015). Many cell-type specific gene expression patterns in the inner ear are regulated by epigenetic modifications, including histone methylation and demethylation (Layman and Zuo, 2014). For example, the histone lysine demethylase KDM4B regulates otic vesicle invagination through epigenetic regulation of Dlx3 expression (Uribe et al., 2015). TBX1 has been shown to interact with and recruit several histone demethylases and methyltranasferases. ASH2L1, a core component of a multimeric histone methyltransferase complex, physically and functionally interacts with TBX1, with overlapping expression patterns in the developing mouse embryo (Stoller et al., 2010). Specific regions within the conserved T-box domain of T-box transcription factors were shown to interact with H3K27 demethylase to mediate activation of developmental gene expression (Miller et al., 2008), and TBX1 interaction with (and postulated recruitment of) histone methyltransferases positively regulates monomethylation of H3K4me1 (Fulcoli et al., 2016). Loss of TBX1-KDMA2 interaction in inner ears of Tbx1nmf219 mutant E16.5 embryos, therefore, may possibly disrupt a TBX1-localized histone modification that is important for particular aspects of inner ear development.
The other three candidate TBX1-interacting proteins we chose for secondary Co-IP analysis also have possible connections to processes that may be related to the gene expression and phenotypic effects we observed in inner ears of Tbx1nmf219 mutant mice. MDFI (MyoD family inhibitor) is a transcription factor that negatively regulates myogenic family proteins (Chen et al., 1996). MDFI interaction with TBX1 may suppress expression of these proteins in cells of the developing inner ear. This suppression may not occur if mutant TBX1 is not able to interact with MDFI, which could possibly explain our RNA-Seq results showing an overexpression of proteins involved in muscle system processes in Tbx1nmf219 mutant mice (Table 2). NOTCH3 functions as a receptor for membrane-bound ligands jagged1, jagged2 and delta1 to regulate cell-fate determination. Notch signaling plays an important role in inner ear development (Kiernan, 2013). The cleaved intracellular domain of NOTCH3 translocates to the nucleus (where it could interact with TBX1) to form a complex that recruits coactivators for transcription. PIAS2 (protein inhibitor of activated STAT 2) functions as a transcriptional co-regulator. PIAS proteins have the ability to differentially regulate signals from the growth factors activin, bone morphogenetic protein 4 (BMP4), and Wnt8 (Burn et al., 2011). BMP4 is essential for the formation of all three inner ear cristae and their associated canals (Chang et al., 2008), which suggests the possibility that BMP4 dysregulation via the loss of TBX1-PIAS2 interaction might contribute to the canal abnormalities of Tbx1nmf219 mice.
Further study is needed to determine whether TBX1 interacts with any of these proteins in vivo in the developing inner ear and if the mutant TBX1 protein fails to interact. The ability of wildtype but not mutant TBX1 to interact with one (or more) of these proteins with functional consequences would provide evidence that this molecular mechanism underlies the Tbx1nmf219 phenotype.
Highlights:
A missense mutation of Tbx1 causes inner ear dysfunction in mice
The non-lethal mutation enabled analysis of TBX1 during post-otocyst development
TBX1 is essential for normal stria vascularis and semicircular canal development
RNA-seq analysis revealed downstream transcriptional effects of the Tbx1 mutation
A yeast 2-hybrid screen identified candidate TBX1-interacting proteins
Acknowledgments
We thank Melissa Berry for helpful comments on the manuscript, Sandra Gray for general mouse husbandry, Leona Gagnon for linkage mapping, and Chantal Longo-Guess for in situ hybridization experiments. We also thank personnel of the Jackson Laboratory’s Scientific Research Services for whole exome sequencing and RNA-Seq analyses. This work was supported by the National Institutes of Health (NIH) grant R01 DC-0004301 (K.R.J.). The Jackson Laboratory shared services are supported by NIH grant P30 CA-034196.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold JS, Braunstein EM, Ohyama T, Groves AK, Adams JC, Brown MC, Morrow BE, 2006. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Human molecular genetics 15, 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA, 2005. Clinical features of 78 adults with 22q11 Deletion Syndrome. American journal of medical genetics. Part A 138, 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein EM, Monks DC, Aggarwal VS, Arnold JS, Morrow BE, 2009. Tbx1 and Brn4 regulate retinoic acid metabolic genes during cochlear morphogenesis. Bmc Dev Biol 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckingham M, 2017. Gene regulatory networks and cell lineages that underlie the formation of skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America 114, 5830–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burn B, Brown S, Chang C, 2011. Regulation of early Xenopus development by the PIAS genes. Developmental dynamics : an official publication of the American Association of Anatomists 240, 2120–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casimiro MC, Knollmann BC, Ebert SN, Vary JC, Greene AE, Franz MR, Grinberg A, Huang SP, Pfeifer K, 2001. Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange- Nielsen Syndrome. Proceedings of the National Academy of Sciences of the United States of America 98, 2526–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G, 2012. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. The Journal of biological chemistry 287, 32298–32306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Lin Z, Kulessa H, Hebert J, Hogan BL, Wu DK, 2008. Bmp4 is essential for the formation of the vestibular apparatus that detects angular head movements. PLoS genetics 4, e1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, Kraut N, Groudine M, Weintraub H, 1996. I-mf, a novel myogenic repressor, interacts with members of the MyoD family. Cell 86, 731–741. [DOI] [PubMed] [Google Scholar]
- Chen J, Nathans J, 2007. Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell 13, 325–337. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang X, Li J, Song C, Jia Y, Xiong W, 2016. Identification of a Novel ENU-Induced Mutation in Mouse Tbx1 Linked to Human DiGeorge Syndrome. Neural plasticity 2016, 5836143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fulcoli FG, Tang S, Baldini A, 2009. Tbx1 regulates proliferation and differentiation of multipotent heart progenitors. Circulation research 105, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe CP, Crump JG, 2014. Tbx1 controls the morphogenesis of pharyngeal pouch epithelia through mesodermal Wnt11r and Fgf8a. Development 141, 3583–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastjerdi A, Robson L, Walker R, Hadley J, Zhang Z, Rodriguez-Niedenfuhr M, Ataliotis P, Baldini A, Scambler P, Francis-West P, 2007. Tbx1 regulation of myogenic differentiation in the limb and cranial mesoderm. Developmental dynamics : an official publication of the American Association of Anatomists 236, 353–363. [DOI] [PubMed] [Google Scholar]
- Delpire E, Lu J, England R, Dull C, Thorne T, 1999. Deafness and imbalance associated with inactivation of the secretory Na- K-2Cl co-transporter. Nature genetics 22, 192–195. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Pacifico C, Tieri L, Marino B, Giannotti A, Dallapiccola B, 1999. Audiological findings in patients with microdeletion 22q11 (di George/velocardiofacial syndrome). British journal of audiology 33, 329–333. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP, 1999. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Human molecular genetics 8, 1579–1584. [DOI] [PubMed] [Google Scholar]
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE, 1999. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. The Journal of biological chemistry 274, 26946–26955. [DOI] [PubMed] [Google Scholar]
- Fuchs JC, Zinnamon FA, Taylor RR, Ivins S, Scambler PJ, Forge A, Tucker AS, Linden JF, 2013. Hearing loss in a mouse model of 22q11.2 Deletion Syndrome. PloS one 8, e80104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcoli FG, Franzese M, Liu X, Zhang Z, Angelini C, Baldini A, 2016. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nature communications 7, 11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H, 2015. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Human molecular genetics 24, 424–435. [DOI] [PubMed] [Google Scholar]
- Hwang CH, Keller J, Renner C, Ohta S, Wu DK, 2019. Genetic interactions support an inhibitory relationship between bone morphogenetic protein 2 and netrin 1 during semicircular canal formation. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CH, Simeone A, Lai E, Wu DK, 2009. Foxg1 is required for proper separation and formation of sensory cristae during inner ear development. Developmental dynamics : an official publication of the American Association of Anatomists 238, 2725–2734. [DOI] [PubMed] [Google Scholar]
- Jackson A, Kasah S, Mansour SL, Morrow B, Basson MA, 2014. Endoderm-Specific Deletion of Tbx1 Reveals an FGF-Independent Role for Tbx1 in Pharyngeal Apparatus Morphogenesis. Dev Dynam 243, 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE, 2001. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature genetics 27, 286–291. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, Olesen SP, 2005. The KCNQ1 potassium channel: from gene to physiological function. Physiology 20, 408–416. [DOI] [PubMed] [Google Scholar]
- Jiang H, Wang L, Beier KT, Cepko CL, Fekete DM, Brigande JV, 2013. Lineage analysis of the late otocyst stage mouse inner ear by transuterine microinjection of a retroviral vector encoding alkaline phosphatase and an oligonucleotide library. PloS one 8, e69314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY, 2003. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Human molecular genetics 12, 3075–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami E, Tokunaga A, Ozawa M, Sakamoto R, Yoshida N, 2015. The histone demethylase Fbxl11/Kdm2a plays an essential role in embryonic development by repressing cell-cycle regulators. Mechanisms of development 135, 31–42. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, 2006. The paintfill method as a tool for analyzing the three-dimensional structure of the inner ear. Brain research 1091, 270–276. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, 2013. Notch signaling during cell fate determination in the inner ear. Seminars in cell & developmental biology 24, 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layman WS, Zuo J, 2014. Epigenetic regulation in the inner ear and its potential roles in development, protection, and regeneration. Frontiers in cellular neuroscience 8, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP, 2000. Targeted disruption of the kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest 106, 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts VA, Valenzuela A, Dunbar C, Zheng QY, Johnson KR, Frankel WN, 2000. A new spontaneous mouse mutation in the Kcne1 gene. Mammalian genome : official journal of the International Mammalian Genome Society 11, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Kochilas L, Nowotschin S, Arnold JS, Aggarwal VS, Epstein JA, Brown MC, Adams J, Morrow BE, 2004. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Human molecular genetics 13, 1577–1585. [DOI] [PubMed] [Google Scholar]
- Liu X, Dobbie M, Tunningley R, Whittle B, Zhang Y, Ittner LM, Gotz J, 2011. ENU mutagenesis screen to establish motor phenotypes in wild-type mice and modifiers of a pre-existing motor phenotype in tau mutant mice. Journal of biomedicine & biotechnology 2011, 130947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS, 2008. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes & development 22, 2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes F, Novoa A, Jerome-Majewska LA, Papaioannou VE, Mallo M, 2005. Tbx1 is required for proper neural crest migration and to stabilize spatial patterns during middle and inner ear development. Mechanisms of development 122, 199–212. [DOI] [PubMed] [Google Scholar]
- Nicolas M, Dememes D, Martin A, Kupershmidt S, Barhanin J, 2001. KCNQ1/KCNE1 potassium channels in mammalian vestibular dark cells. Hearing research 153, 132–145. [DOI] [PubMed] [Google Scholar]
- Ogata T, Niihori T, Tanaka N, Kawai M, Nagashima T, Funayama R, Nakayama K, Nakashima S, Kato F, Fukami M, Aoki Y, Matsubara Y, 2014. TBX1 Mutation Identified by Exome Sequencing in a Japanese Family with 22q11.2 Deletion Syndrome-Like Craniofacial Features and Hypocalcemia. PloS one 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pane LS, Zhang Z, Ferrentino R, Huynh T, Cutillo L, Baldini A, 2012. Tbx1 is a negative modulator of Mef2c. Human molecular genetics 21, 2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, Murphy KC, Monks S, Williams N, O’Donovan MC, Owen MJ, Scambler PJ, Lindsay E, 2006. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. P Natl Acad Sci USA 103, 7729–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raft S, Nowotschin S, Liao J, Morrow BE, 2004. Suppression of neural fate and control of inner ear morphogenesis by Tbx1. Development 131, 1801–1812. [DOI] [PubMed] [Google Scholar]
- Romand R, Albuisson E, Niederreither K, Fraulob V, Chambon P, Dolle P, 2001. Specific expression of the retinoic acid-synthesizing enzyme RALDH2 during mouse inner ear development. Mech Dev 106, 185–189. [DOI] [PubMed] [Google Scholar]
- Romand R, Kondo T, Fraulob V, Petkovich M, Dolle P, Hashino E, 2006. Dynamic expression of retinoic acid-synthesizing and -metabolizing enzymes in the developing mouse inner ear. J Comp Neurol 496, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romand R, Niederreither K, Abu-Abed S, Petkovich M, Fraulob V, Hashino E, Dolle P, 2004. Complementary expression patterns of retinoid acid-synthesizing and -metabolizing enzymes in pre-natal mouse inner ear structures. Gene expression patterns : GEP 4, 123–133. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brondum-Nielsen K, Scambler PJ, et al. , 1997. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. Journal of medical genetics 34, 798–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A, 1993. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100, 431–440. [DOI] [PubMed] [Google Scholar]
- Stoller JZ, Huang L, Tan CC, Huang F, Zhou DD, Yang J, Gelb BD, Epstein JA, 2010. Ash2l interacts with Tbx1 and is required during early embryogenesis. Exp Biol Med (Maywood) 235, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Gagnon LH, Longo-Guess C, Korstanje R, Sheehan SM, Ohlemiller KK, Schrader AD, Lett JM, Johnson KR, 2017. Hearing loss without overt metabolic acidosis in ATP6V1B1 deficient MRL mice, a new genetic model for non-syndromic deafness with enlarged vestibular aqueducts. Human molecular genetics 26, 3722–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe RA, Buzzi AL, Bronner ME, Strobl-Mazzulla PH, 2015. Histone demethylase KDM4B regulates otic vesicle invagination via epigenetic control of Dlx3 expression. The Journal of cell biology 211, 815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness LD, Wang X, Shibata S, Ohyama T, Mansour SL, 2015. Fgf10 is required for specification of non-sensory regions of the cochlear epithelium. Developmental biology 400, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A, 2002a. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Human molecular genetics 11, 915–922. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Taddei I, Morishima M, Meyers EN, Lindsay EA, Baldini A, 2002b. A genetic link between Tbx1 and fibroblast growth factor signaling. Development 129, 4605–4611. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E, 2003. TBX1 is required for inner ear morphogenesis. Human molecular genetics 12, 2041–2048. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Zhang Z, Huynh T, Sobotka A, Mupo A, Baldini A, 2006. Fgf8 expression in the Tbx1 domain causes skeletal abnormalities and modifies the aortic arch but not the outflow tract phenotype of Tbx1 mutants. Dev Biol 295, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Zaffran S, Kuroiwa A, Higuchi H, Ogura T, Harvey RP, Kelly RG, Buckingham M, 2012. Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2–5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proceedings of the National Academy of Sciences of the United States of America 109, 18273–18280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Chen L, Baldini A, 2007a. In vivo genetic ablation of the periotic mesoderm affects cell proliferation survival and differentiation in the cochlea. Developmental biology 310, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A, 2004. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 131, 3217–3227. [DOI] [PubMed] [Google Scholar]
- Xu H, Viola A, Zhang Z, Gerken CP, Lindsay-Illingworth EA, Baldini A, 2007b. Tbx1 regulates population, proliferation and cell fate determination of otic epithelial cells. Developmental biology 302, 670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R, 2003. Role of TBX1 in human del22q11.2 syndrome. Lancet 362, 1366–1373. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC, 1999. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hearing research 130, 94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A, 2007. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. American journal of human genetics 80, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]