

Abstract
Non-obstructive azoospermia (NOA), the lack of spermatozoa in semen due to impaired spermatogenesis affects nearly 1% of men. In about half of cases, an underlying cause for NOA cannot be identified. This study aimed to identify novel variants associated with idiopathic NOA. We identified a nonconsanguineous family in which multiple sons displayed the NOA phenotype. We performed whole exome sequencing in three affected brothers with NOA, their two unaffected brothers and their father, and identified compound heterozygous frameshift variants (one novel and one extremely rare) in Telomere Repeat Binding Bouquet Formation Protein 2 (TERB2) that segregated perfectly with NOA. TERB2 interacts with TERB1 and Membrane Anchored Junction Protein (MAJIN) to form the tripartite meiotic telomere complex (MTC), which has been shown in mouse models to be necessary for completion of meiosis and both male and female fertility. Given our novel findings of TERB2 variants in NOA men, along with the integral role of the three MTC proteins in spermatogenesis, we subsequently explored exome sequence data from 1,499 NOA men to investigate the role of MTC gene variants in spermatogenic impairment. Remarkably, we identified two NOA patients with likely damaging rare homozygous stop and missense variants in TERB1 and one NOA patient with a rare homozygous missense variant in MAJIN. Available testis histology data from three of the NOA patients indicate germ cell maturation arrest, consistent with mouse phenotypes. These findings suggest that variants in MTC genes may be an important cause of NOA in both consanguineous and outbred populations.
Keywords: TERB1, TERB2, MAJIN, meiotic telomere complex, non-obstructive azoospermia, male infertility, exome sequencing, maturation arrest
INTRODUCTION
Infertility, clinically defined as failure to conceive after 12 months of unprotected sexual intercourse, is common, affecting an estimated 15% of couples worldwide (Boivin et al. 2007). Male infertility is implicated in about half of infertile couples and takes many forms, from the complete lack of sperm production to normal numbers of sperm with some functional defect. Unfortunately, an underlying cause for infertility cannot be identified in the majority of cases (Krausz and Riera-Escamilla 2018; Tüttelmann et al. 2018).
Spermatogenic failure (SPGF) affects nearly 1% of all men, accounting for approximately 20% of all cases of male infertility. In its most severe form, SPGF manifests as non-obstructive azoospermia (NOA), the lack of detectable spermatozoa in semen due to impaired spermatogenesis. Currently, physicians are severely limited in their ability to offer useful direction to NOA patients regarding the chances for successful sperm retrieval from testicular biopsies (testicular sperm extraction, TESE) or prognosis for treatment using assisted reproductive techniques (ART) if sperm are identified on TESE. Moreover, in cases of idiopathic NOA, physicians are unable to counsel patients about the chances of transmission of infertility or health risks to ART-derived offspring.
The identification of clinically relevant causes of NOA is critical to providing appropriate counselling and care to patients, including genetic counselling. Importantly, men with severe spermatogenic defects are more likely to develop some cancers as well as a number of comorbidities (Ventimiglia et al. 2016; Glazer et al. 2017; Nagirnaja et al. 2018). Understanding the genetic underpinnings of NOA is an important first step in identifying risk factors associated with these associated conditions.
Approximately 14% of men with NOA will show a numerical or structural chromosomal abnormality on routine karyotype, with 47,XXY (Klinefelter syndrome) being the most common (Ferlin et al. 2007). An additional 8–12% will have chromosome Yq microdeletions of the so-called Azoospermia Factor loci (AZFa, AZFb, or AZFc) (Krausz et al. 2014). AZF deletions are Mb-scale deletions of the euchromatic region of the q arm of the Y chromosome that encompass multiple genes required for spermatogenesis. As important as these genetic factors are in the diagnosis of male factor infertility, progress in identifying new genetic variants responsible for spermatogenic failure since the discovery of Yq microdeletions nearly four decades ago has been extremely limited (Tiepolo and Zuffardi 1976). Nevertheless, discoveries have accelerated in recent years, largely due to increased application of whole exome sequencing (WES) in infertile men (Krausz and Riera-Escamilla 2018; Mbango et al. 2019; Vockel et al. 2019; Oud et al. 2019; Kasak and Laan 2020; Xavier et al. 2020).
In the current study we performed WES in six individuals (five brothers and their father) from a non-consanguineous family and identified compound heterozygous frameshift variants in TERB2 specifically in the three brothers with NOA. Given that TERB2 forms a molecular complex with two other proteins necessary for meiosis (TERB1 and MAJIN), we searched for variants in all three genes in two exome datasets representing nearly 1,500 men with NOA and identified three additional patients with homozygous, likely pathogenic missense variants in TERB1 or MAJIN. The genomes of the three additional patients displayed increased regions of homozygosity, indicative of recent consanguinity. Consistent with animal models, which display male infertility with no other apparent phenotype, these results strongly suggest that disruption of any of the three genes that encode the meiotic telomere complex (MTC) can result in NOA in men.
MATERIALS AND METHODS
Ethics Statement
The study was approved by the Ethics Committee of all collaborative centers (Utah: IRB_00063950, Münster: 2010-578-f-S and Porto: PTDC/SAU-GMG/101229/2008). Written, informed consent was obtained for all participants prior to recruitment, and the study was carried out in compliance with the Helsinki Declaration.
Study design
Members of a large non-consanguineous family from Utah, United States were initially recruited due to the finding of idiopathic NOA in 3 brothers. The subsequent search for related variants in other NOA men was performed in two pre-existing infertility cohorts: first, the NIH-funded Genetics of Male Infertility Initiative (GEMINI) that includes 926 patients with NOA (https://gemini.conradlab.org/) and second, the large-scale Male Reproductive Genomics (MERGE) study, that currently comprises >1000 men including 569 NOA cases with a clinically standardized characterization (Kliesch 2014).
Eligible participants for both the GEMINI and MERGE cohorts included men between 18–55 years old with idiopathic NOA. Exclusion criteria included azoospermia due to acquired or congenital obstruction, absence of vas deferens, radical pelvic surgery, ejaculatory disorders, spinal cord injury, radiation or chemotherapy treatments, exposure to environmental risk factors associated with male infertility, Y chromosome microdeletions or karyotypic abnormalities.
Histological profiling of testicular samples
When possible, testicular histology of the affected patients was evaluated. Testis biopsies were fixed in Bouin’s solution and subsequently washed in 70% ethanol, embedded in paraffin and sectioned and stained for histological analysis (Nieschlag et al. 2010). In one patient, more specific histological investigation was performed. In this case, immunohistochemical detection of cAMP response element modulator (CREM) was performed following a published protocol (Schlatt et al. 2019). CREM is expressed in haploid spermatids, mostly round spermatids (Weinbauer et al. 1998). Histologic evaluation was performed by trained histopathology experts.
Whole exome sequencing
We conducted WES of genomic DNA from peripheral blood leukocytes of one father (I:1) and five of his sons (II:1–II:5) in a non-consanguineous family including three infertile sons diagnosed with NOA (Family 1; Fig. 1a). Exome capture was performed using the Agilent SureSelect XT2 Human All Exon v7 kit, and libraries were sequenced at the Huntsman Cancer Institute to >160X mean depth across coding regions using the NovaSeq sequencer (Illumina). We subsequently queried existing data from GEMINI and MERGE cohorts to search for additional individuals with variants in TERB2, TERB1 and MAJIN.
Fig. 1.
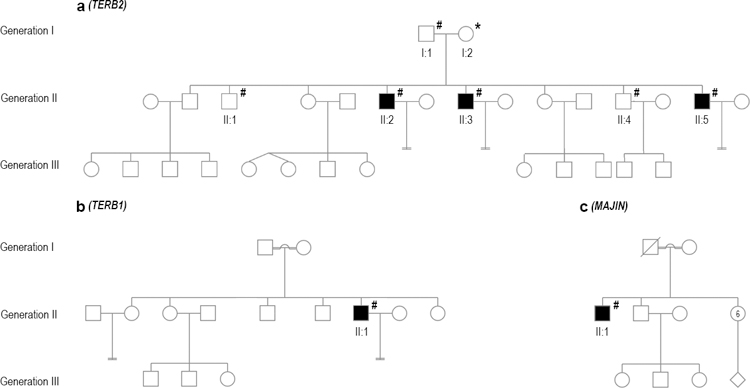
Pedigree structure of three families. a) Non-consanguineous Family 1. b) Consanguineous Family (Individual 3; M2073). c) Consanguineous Family (Individual 4; M1646). Filled symbols indicate members affected with NOA, and unfilled symbols indicate unaffected members. WES with subsequent confirmation by Sanger sequencing was performed on the individuals indicated with #, and * indicates a case in which only Sanger sequencing was performed. Pedigree information was unavailable for Individual 2.
Briefly, for GEMINI samples, genomic DNA sequencing libraries were prepared at McDonnell Genome Institute of Washington University in St. Louis, MO, USA using an in-house exome targeting reagent which captures 39.1 Mb of exome, and libraries were sequenced on Illumina HiSeq 4000 to an average coverage of 80X across coding regions. The exomes of individuals M1646 and M2073 were sequenced as part of the MERGE cohort. For M1646, target enrichment was performed by SureSelect QXT Target Enrichment Kit according to the manufacturer’s protocol using the capture libraries Agilent SureSelect Human All Exon V6. For M2073, target enrichment was performed by Human Core Exome Enrichment Kit according to the manufacturer’s protocol using the capture libraries TwistHumanCoreExome 1.3 plusRefSeq. Sequencing for both subjects was performed on the Illumina NextSeq®500 system, achieving a mean sequence coverage of more than 200x, with more than 99% of the target bases having at least 10x coverage.
Data analysis and variant prioritization
For all individuals, sequence reads were aligned against the human reference genome using BWA-MEM (Li and Durbin 2010) and duplicate reads were marked for exclusion from variant calling. Family 1 and MERGE exomes were aligned to GRCh37 while GEMINI exomes were aligned to GRCh38 and then lifted over to GRCh37 (for variant prioritization). Estimation of homozygosity was performed using the R package mclust (Scrucca et al. 2016), and prediction of nonsense mediated decay (NMD) was performed using the R package mansomd (Hu et al. 2017).
For Family 1, analysis of the exome data was performed by the Utah Center for Genetic Discovery (UCGD). Variants were called using the Sentieon suite, a computationally efficient replacement for GATK, according to a pipeline adapted from the GATK best practices. Alignment metrics were generated using alignstats, and sample gender, ancestry, and relatedness were verified using Peddy (Pedersen and Quinlan 2017). Variants were annotated with Annovar (Wang et al. 2010) and filtered to select those that met the following criteria: were non-synonymous or within 6 bp of a splice site, or a start-gain in the 5’ untranslated region; had less than 1% alt allele frequency in European populations from the gnomAD and 1000 Genomes variant databases; and co-segregated with the NOA phenotype according to a recessive inheritance model. Read level support for putative candidate variants was examined using the Integrative Genomics Viewer to identify likely false positives arising from alignment artifacts or inadequate coverage.
Exome data from Individual 2 was genotyped jointly with all GEMINI study subjects using GATK v3.6.0 as per the GATK best practices. Genotypes that are unusual in the context of the human population variation and likely deleterious, were subsequently prioritized using a modified version of an n-of-one framework called Population Sampling Probability (PSAP) (Wilfert et al. 2016). Only variations that had PSAP P<10−3, minor allele frequency <0.01 across populations in gnomAD and were rare in the GEMINI cohort were considered. Additionally, only genes either with enhanced expression in testis, known to cause male infertility in mice or humans or genes with loss-of-function variation were included.
In individual 3 (M2073) and individual 4 (M1646) from MERGE, both GATK v3.8 with HaplotypeCaller and freebayes v1.2.0 (Garrison and Marth 2012) were used to identify small insertions/deletions (indels) and single nucleotide substitutions, in accordance with best practice recommendations (McKenna et al. 2010). Called variants were annotated with Ensembl Variant Effect Predictor (McLaren et al. 2016). The Münster in-house pipeline Sciobase© was used to further annotate each variant with transcript, categories, functional consequences, population frequencies, and in-silico predicted relevance. Likely causative variants were identified by filtering the data according to the population frequency in the Genome Aggregation Database (Karczewski et al. 2019) (gnomAD, minor allele frequency [MAF] < 0.01) and the functional impact of the variant.
As a standardized analysis for all three patient exomes, PSAP was used to prioritize variants (Wilfert et al. 2016). Following generation of the PSAP output, variants were filtered according to the following criteria: 1) minor allele frequency [MAF] <0.01 in gnomAD, 1000 Genomes, NHLBI Exome Sequencing Project and ExAC databases, 2) CADD-score ≥20) and 3) PSAP p-value ≤ 0.001. The filtered list was additionally screened for variants in 170 candidate genes that were previously reported to be associated with impaired spermatogenesis according to Oud et al. 2019 (see Table S1 for gene list) as well as in eight recently published genes ADAD2, M1AP, MSH4, RAD21L1, RNF212, SHOC1, STAG3, SYCP2 associated with non-obstructive azoospermia (Krausz et al. 2020).
Experimental validation by Sanger sequencing
Variants in TERB1, TERB2, and MAJIN were confirmed using Sanger sequencing in all available individuals. Sanger sequencing was performed at the University of Utah DNA Sequencing Core Facility (Family 1), at the Institute of Human Genetics, University of Münster (Individual 3-M2073 and Individual 4-M1646), and at the Institute of Molecular Pathology and Immunology of the University of Porto (Individual 2) according to standard procedures. While not available for WES, the mother’s DNA from Family 1 was subsequently obtained and Sanger sequenced. Primers were designed with Primer3 and tested by UCSC in-silico PCR (primer sequences are indicated in Fig. S1, S4 and S6).
RESULTS
Characterization of the families
The non-consanguineous family of European ancestry from Utah (Family 1) includes the father and mother, two fertile daughters and six sons; three of the sons are infertile and diagnosed with NOA, and three have normal sperm count (including two known fertile) (Fig. 1a). The NOA patients were diagnosed at the Andrology and IVF laboratory at the University of Utah. WES was performed in the father (I:1) and five sons (II:1, II:2, II:3, II:4 and II:5). NOA-affected patients were ages 34 (II:2), 33 (II:3) and 31 (II:5) years at phenotyping (Table 1). DNA was subsequently obtained from the mother and subjected to Sanger sequencing, targeting the two variants identified in TERB2. While testicular histology records were not available for any individuals from this family, patient II:2 did undergo testicular biopsy more than a decade ago with no sperm identified and was informed that the histology indicated maturation arrest (no images collected).
Table 1.
Clinical characteristics of the study subjects at the clinical assessment.
| Family 1 (TERB2) | Individual 2 (TERB1) | Individual 3 (TERB1) | Individual 4 (MAJIN) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pedigree code | I:1 | II:1 | II:2 | II:3 | II:4 | II:5 | ND | II:1 | II:1 | Reference values |
| NOA affected? | No | No | Yes | Yes | No | Yes | Yes | Yes | Yes | - |
| Demographic and lifestyle parameters | ||||||||||
| Age (years) | 70 | 38 | 34 | 33 | 29 | 31 | 28 | 31 | 37 | - |
| Smoking | No | No | No | Yes | No | No | ND | Yes | No | - |
| Alcohol consumption | No | No | No | Yes | No | No | ND | No | No | - |
| Illegal drugs consumption | No | No | No | No | No | No | ND | No | No | - |
| Fertility parameters | ||||||||||
| Karyotype | 46,XY | 46,XY | 46,XY | 46,XY | 46,XY | 46,XY | 46,XY | 46,XY | 46,XY | - |
| Y microdeletion | ND | ND | Negative | Negative | ND | Negative | Negative | Negative | Negative | - |
| Testicular volume: left (ml) | 21 | 30 | 25 | 17 | 30 | 25 | ND | 17 | 8 | - |
| Testicular volume: right (ml) | 20 | 30 | 26 | 16 | 30 | 26 | ND | 19 | 7 | - |
| Varicocele | Grade II | Grade II | Grade II | Negative | Grade I | Negative | ND | Grade I | Negative | - |
| Semen volume (ml) | ND | 3.3 | 3.5 | 4.2 | 1.8 | 2 | ND | 2.9 | 1.2 | >1.5 |
| Total motility (%) | ND | 51 | - | - | 79 | - | - | - | - | >40 |
| Progressive motility (%) | ND | 46 | - | - | 74 | - | - | - | - | >32 |
| Vitality (%) | ND | 54 | - | - | 63 | - | - | - | - | >58 |
| Normal heads (%) | ND | 21 | - | - | 35 | - | - | - | - | >30 |
| Normal tails (%) | ND | 78 | - | - | 68 | - | - | - | - | >65 |
| Sperm concentration (x106) | ND | 33.4 | 0 | 0 | 324 | 0 | 0 | 0 | 0 | >15 |
| Total sperm count (x106) | ND | 110.1 | 0 | 0 | 583.2 | 0 | 0 | 0 | 0 | >39 |
| Total progressive motile sperm count (x106) | ND | 50.7 | 0 | 0 | 431.5 | 0 | 0 | 0 | 0 | >30 |
| E2 (pg/ml) | 24.2 | 19.0 | 13.5 | 23.8 | <5.0 | 6.0 | ND | <5.0 | 20.7 | 10–40.0 |
| FSH (mIU/ml) | 5.3 | 4.4 | 6.4 | 10.5 | 2.7 | 3.5 | ND | 2.3 | 12.2 | 1.5–12.4 |
| LH (mIU/ml) | 3.5 | 2.6 | 4.5 | 6.9 | 2.5 | 2.1 | ND | 1.6 | 2.9 | 1.8–12.0 |
| SHBG (nmol/l) | 85.1 | 48.6 | 55.3 | 38.8 | 42.7 | 51.0 | ND | 10.0 | 19.0 | 10–57.0 |
| T (ng/dl) | 549.2 | 380.3 | 483.0 | 424.3 | 388.2 | 363.5 | ND | 167.3 | 317.3 | 300–1000 |
Abbreviations: E2, Estradiol; FSH, Follicle-stimulating hormone; LH, Luteinizing hormone; ND, No Data; NOA, non-obstructive azoospermia; SHBG, Sex hormone binding globulin; T, Testosterone.
Individual 2 is an infertile man from the GEMINI cohort with NOA and was diagnosed at the Departamento de Genética Humana, Instituto Nacional de Saúde Dr Ricardo Jorge from Lisbon (Portugal). Other family members were not available for genetic analysis, and pedigree information was unavailable. The patient was age 28 years at phenotyping (Table 1) and is suspected to have third-degree consanguinity with 6.6% of autosomal sequence being homozygous as determined based on mclust analysis of the WES data. Testicular histology was not available for this patient.
Individual 3 (M2073) is from a consanguineous family from Lebanon with 6.3% of autosomal sequence in a homozygous state (parents were first cousins; Fig. 1b). He attended the Centre of Reproductive Medicine and Andrology (CeRA), Münster, at 31 years of age (Table 1) for a second opinion on couple infertility. He was diagnosed with NOA, and a biopsy had been performed previously. The biopsy showed germ cell maturation arrest with the furthest progressed cell type being spermatocytes (quantification not available). No postmeiotic germ cells were observed, and TESE was unsuccessful.
Individual 4 (M1646) is from a consanguineous family of Middle Eastern ancestry from Syria comprising the father and mother, two sons, and six daughters, with only one son (II:1) diagnosed as infertile with NOA (Fig. 1c). Other family members were not available for genetic analysis. Approximately 2.6% of this patient’s autosomal sequence is homozygous, suggesting relatively distant (4th or 5th degree) parental consanguinity. The NOA patient was diagnosed at age 37 at the CeRA, University Hospital Münster. He underwent bilateral TESE with no sperm identified. Histological analysis indicated germ cell maturation arrest with occasional postmeiotic round spermatids present in 2–4% of tubules containing apparently post meiotic cells (Fig. 2).
Fig. 2.
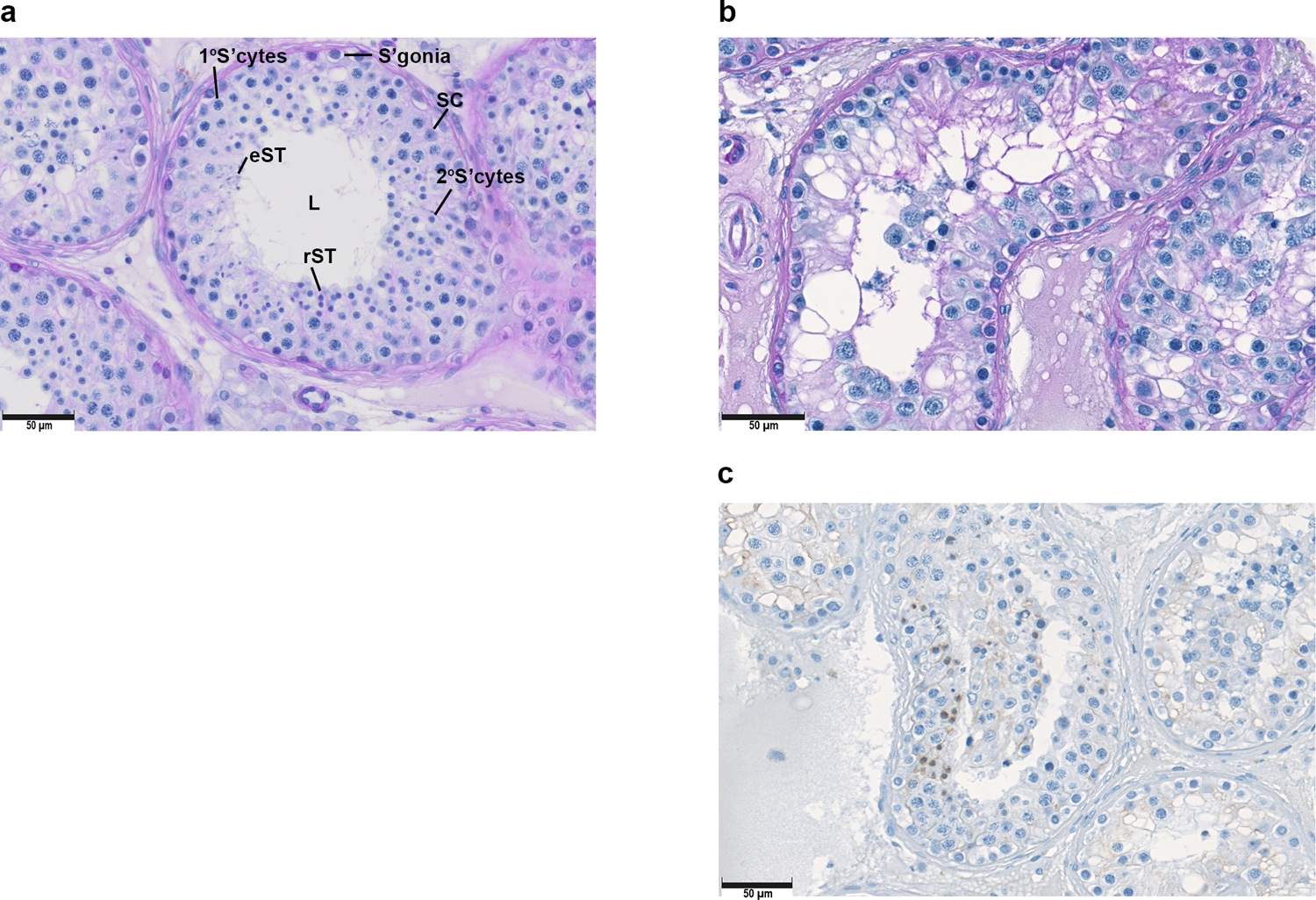
a) Periodic acid-Schiff (PAS) staining of a control individual with normal spermatogenesis (M2211). b) PAS staining, and c) immunohistochemistry (CREM staining) of testis from individual 4 (M1646; MAJIN), indicating an arrest at round spermatid stage. Abbreviations: L=Lumen, S’gonia=Spermatogonia, SC=Sertoli cell, 1°S’cyte=Primary spermatocyte, 2°S’cyte =Secondary spermatocyte, rST= Round spermatids, eST=Elongated spermatids.
Variants detected
Family 1 exomes were sequenced to a mean depth across protein-coding sequence of at least 160X, and all samples had 98% of coding bases covered to a depth of at least 10X (Table S2). All sample genders and relationships were confirmed from the exome data using Peddy.
In Family 1 we detected two rare coding variants in the TERB2 gene that were both shared among all three affected brothers and not among the two unaffected brothers. TERB2 was the only gene for which variants met our filtering criteria for rare, high-impact, recessively inherited variants. One variant is a novel 2 bp deletion [NM_152448.3(TERB2):c.457_458del] and the other is a very rare (2.8E-5) complex variant (c.[544dup;547_551del]) resulting in a net 4 bp deletion (Table 2). Both variants are predicted to cause frameshifts in the coding sequence (p.Thr153fs*17 and p.Met182fs*31, respectively) that result in premature stop codons and both variants were confirmed by Sanger sequencing in all family members (Fig. S1). The father (I:1) as well as unaffected sons were heterozygous for p.Met182fs*31 but did not carry p.Thr153fs*17. Subsequent Sanger sequencing of the mother’s DNA confirmed that the p.Thr153fs*17 variant, present in the affected brothers, was inherited from her. Based on the positions of the frameshifts, they were not predicted by masonmd to trigger NMD; however both variants result in truncated proteins and changes in highly conserved amino acids. The p.Thr153fs*17 (CADD score=34) variant results in a change in 16 highly conserved amino acids followed by a premature stop codon that truncates nearly a quarter of the protein (51 of the 220 residues in the normal protein; Fig. S2). The functional consequence of the p.Met182fs*31 (CADD score=34) variant is less certain based on its position in the last exon, however the frameshift results in missense changes of amino acids 182–211 and a loss of the final 9 amino acids. Importantly, of the final 39 amino acids that are changed or truncated, 12 are perfectly conserved in 60/60 mammals, and another 8 are conserved in 59/60 mammals (Fig. S3), suggesting that these residues may be functionally important, and the mutated protein may exhibit impaired function.
Table 2.
Summary of the predicted pathogenic variants detected in the analyzed subjects.
| Position (hg19) (Chromosome:Start-End) |
dbSNP ID | Gene | Region | cDNA change | Protein change | Frequency gnomAD v2.1.1 | Frequency gnomAD v3 | gnomAD homozygotes | CADD score |
|---|---|---|---|---|---|---|---|---|---|
| Family 1 | |||||||||
| chr15:45266087–45266088 | NA | TERB2 | Exon 6 | c.457_458del | p.Thr153fs*17 | 0 | 0 | 0 | 34 |
| chr15:45270706–45270706 | rs1218912028 | TERB2 | Exon 7 | c.[544dup;547_551del] | p.Met182fs*31 | 0 | 2.80E-05 | 0 | 34 |
| chr15:45270710–45270714 | rs1266809459 | TERB2 | Exon 7 | 0 | 2.79E-05 | 0 | 33 | ||
| Individual 2 | |||||||||
| chr16:66811114–66811114 | rs780206976 | TERB1 | Exon 11 | c.977A>G | p.Glu326Gly | 8.27E-05 | 7.68E-05 | 0 | 28.4 |
| Individual 3 (M2073) | |||||||||
| chr16:66801395–66801395 | NA | TERB1 | Exon 16 | c.1703C>G | p.Ser568Ter | 0 | 0 | 0 | 41 |
| Individual 4 (M1646) | |||||||||
| chr11:64717892–64717892 | rs375342082 | MAJIN | Exon 5 | c.158G>A | p.Arg53His | 6.76E-05 | 7.70E-05 | 0 | 27 |
Abbreviations: Arg, Arginine; CADD, Combined Annotation Dependent Depletion; chr, chromosome; del, deletion; dup, duplication; fs, frameshift; Glu, Glutamate; Gly, Glycine; His, Histidine; MAJIN, Membrane Anchored Junction Protein; Met, Methionine; NA, not applicable; Ser, Serine; Ter, Termination (codon); TERB1, Telomere Repeat Binding Bouquet Formation Protein 1; TERB2, Telomere Repeat Binding Bouquet Formation Protein 2; Thr, Threonine.
The complex variant in TERB2 exon 7 is described as a single variant for cDNA and protein nomenclature, but as two variants for database annotation. Frequencies in gnomAD v2.1.1 are reported for exomes only because gnomAD v2.1.1 genomes are included in gnomAD v3. CADD score is Phred-scaled.
Following PSAP filtering in GEMINI and MERGE exomes, 15 variants remained for Individual 2, 64 for Individual 3 and 26 for Individual 4 (Table S3), of which, the top ranked variants for Individuals 2 and 3 were in TERB1 and for individual 4 were in MAJIN, as described below.
In the GEMINI cohort, we identified a single third-degree suspected consanguineous NOA patient (Individual 2) with a rare homozygous missense variant [NM_001136505.2(TERB1):c.977A>G] in the TERB1 gene (or CCDC79), which was confirmed by Sanger sequencing (Table 2, Fig. S4). This substitution p.Glu326Gly has never been observed in a homozygous state in the gnomAD database and is predicted to be deleterious (CADD score=28.4). The affected residue is conserved in 74 of 76 available vertebrate species, suggesting low tolerance for mutation at this position (Fig. S5). Further, the variant is located in a long run of homozygosity (23.5 Mb) resulting in the homozygous state of this variant. There were no other variants in this individual that passed filtering criteria for rare variants in either testis-expressed or known infertility-associated genes that had PSAP P values <10−3.
Individual 3 (M2073) from the MERGE cohort carries a novel homozygous nonsense variant [NM_001136505.2(TERB1):c.1703C>G] in the TERB1 gene The variant is located within a 11.1 Mb run of homozygosity and was confirmed by Sanger sequencing. It results in a premature stop codon p.Ser568Ter (CADD score=41) and is predicted to result in NMD based on analysis with masonmd R package.
In individual 4 (M1646) from the MERGE cohort, we identified a 9.5 Mb region of homozygosity that included a rare homozygous missense variant [NM_001037225.3(MAJIN):c.158G>A] in the MAJIN gene (Table 2, Fig. S6). This variant results in a substitution p.Arg53His at a residue that is perfectly conserved in 70 of 70 vertebrate species for which orthologous protein sequence is available, and it is predicted to be deleterious (CADD score=27) (Fig. S7). This patient is of Syrian ancestry, and the c.158G>A variant is present in the Greater Middle Eastern (GME) Variome at a frequency of 0.00151(Scott et al. 2016); there are 3 heterozygotes and 0 homozygotes among 993 GME individuals with sequence coverage at that site. Based on the observed variant frequency in this population, the predicted homozygote frequency in the GME population would be 0.001512 or 2.28E-6, assuming Hardy-Weinberg equilibrium.
DISCUSSION
The search for genetic variants responsible for spermatogenic failure presents unique challenges given the complexity of the process of spermatogenesis (Djureinovic et al. 2014; Kasak and Laan 2020), with more than 1,000 genes displaying testis-specific or testis-enriched expression. Consequently, there are several thousand genes that when disrupted could result in spermatogenic failure. Given this complexity, it is not surprising that the majority of NOA-associated variants identified to date have been found in a very small percentage of men with the disease (Xavier et al. 2020). The assembly of large cohorts of infertile men, thorough phenotyping, and the development of genome analysis tools with deep annotation of reproductive data and efficient use of animal models are critical for continued gene discovery (Houston et al. 2020; Xavier et al. 2020).
Here we report novel or rare, biallelic coding variants with high CADD scores in all three genes that comprise the MTC (TERB1, TERB2 and MAJIN) and whose expression is highly enriched in the testis relative to other tissues (Fig. S8). To our knowledge, genetic variants in TERB2 and MAJIN have not previously been implicated in male infertility, while TERB1 variants were very recently reported in three men with NOA (Krausz et al. 2020) as well as in two distantly related NOA men (Alhathal et al. 2020). Subsequent to submission of the study by Krausz et al., a new batch of sequence data from the MERGE cohort was completed, and a patient (M2073) was found to likewise carry homozygous variants in TERB1. Importantly, no homozygous loss of function variants were identified in any of the three genes in 125,748 exomes and 71,702 genomes in gnomAD. In all cases, the variants were identified in men diagnosed with NOA, and in the three cases for which testicular biopsies were performed, histology analysis indicated meiotic arrest of spermatogenesis. These findings are consistent with phenotypes observed in mice with any of the three genes disrupted. Disruption of Terb1 in the mouse results in meiotic arrest and impairment of homologous pairing and synapsis, ultimately resulting in infertility in both males and females (Shibuya et al. 2014). Likewise, mice disrupted for either Majin or Terb2 display impaired synapsis, zygotene arrest, a lack of postmeiotic cells and infertility (Shibuya et al. 2015; Zhang et al. 2017). Notably, while disruption of any of the three genes in mice resulted in complete infertility, animals were otherwise healthy with no overt somatic phenotype (Shibuya et al. 2014, 2015; Zhang et al. 2017).
The MTC plays an essential role in meiotic prophase I; telomeres attach to the nuclear envelope via the complex, regulating homologous chromosome pairing and movement to ensure telomere tethering for correct chromosome recombination (Wang et al. 2019). The complex also interacts with other essential proteins; in meiotic prophase I, TERB1 and TERB2 are sequestered to the inner nuclear membrane by the transmembrane DNA-binding protein MAJIN. Telomere attachment is facilitated by another protein, the telomeric repeat-binding factor 1 (TERF1). Following telomere binding to the nuclear envelope, another protein complex, SUN1-CCDC155 (or KASH5) triggers chromosome movement by linking telomeres to the microtubules of the outer nuclear membrane and its associated dynein-dynactin motors (Fig. 3). It has been demonstrated previously that disruption of any of the MTC genes impairs telomere association with the inner nuclear membrane, which is requisite for chromosomal synapsis and chromosome movement during meiosis. Failure of these events triggers meiotic checkpoints leading to meiotic arrest, and consequently to male infertility/NOA (Dunce et al. 2018).
Fig. 3.
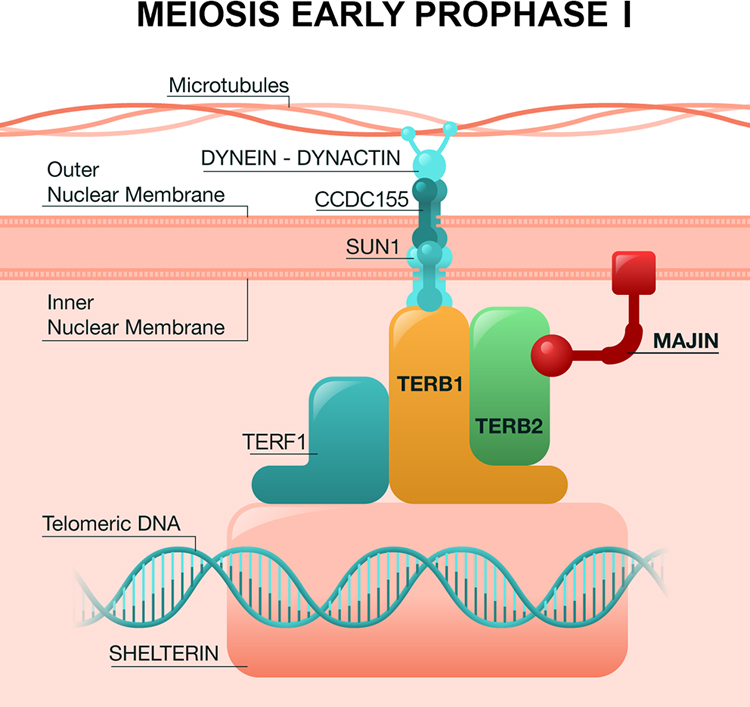
Schematic model of the connection between telomeres and the nuclear membrane in prophase I of meiosis through the MTC (TERB1-TERB2-MAJIN) in mammals. Figure adapted from the information of (Shibuya et al. 2015; Zhang et al. 2017; Wang et al. 2019).
The genesis of this study was the identification of an unusual family in which three brothers displayed idiopathic NOA and share the same TERB2 genotype, c.[457_458del]; [544dup;547_551del]. Genomic analysis of families has proven to be an efficient and cost-effective approach for discovering high-confidence causal variants in a variety of rare diseases including male infertility (Okutman et al. 2015; Kasak et al. 2018; Martinez et al. 2018). The discovery of TERB2 variations in this family motivated us to search for variants in all three MTC genes in two large cohorts of NOA men (GEMINI and MERGE cohorts) for whom exome sequencing had recently been completed. This approach allowed the identification of three additional individuals that carried biallelic variants in related genes [TERB1 (n = 2) and MAJIN (n = 1)].
Robust knockout studies in the mouse, and conservation of the MTC coding sequence across species lend strong support for the critical role of these genes in spermatogenesis, and it is expected that biallelic knockout of these genes would result in meiotic arrest of spermatogenesis and NOA in men. It is important to note that while it’s intriguing that we have identified rare homozygous or compound heterozygous variants in all three genes that comprise the MTC, the evidence for a causal role of these variants varies by gene. Testicular histology information was only available for three individuals, each harboring a variant in one of the three MTC genes, however in each case available information indicated concordance with the testicular phenotype of the respective knockout mouse, specifically meiotic arrest.
The compound heterozygous frameshift variants in TERB2 identified in three NOA brothers are highly likely to result in complete ablation of gene activity; both frameshifts result in a premature stop codon, and each has a CADD score of 34. This observation, along with the perfect segregation of the variants by phenotype in a large family, the evolutionary conservation, phenotypic similarity with the mouse model and extreme rarity of the variants in the general population, provides additional evidence for pathogenicity of these variants.
Evidence for causality of the TERB1 variant is also compelling, with the identification of two unrelated NOA men from consanguineous families carrying homozygous, premature stop and predicted damaging missense variants with CADD scores of 41 and 28.4 respectively. Moreover, likely pathogenic variants in TERB1 were recently reported in two NOA brothers and a single unrelated NOA patient (Krausz et al. 2020) as well as in two distantly related NOA men (Alhathal et al. 2020), significantly strengthening the evidence for the role of TERB1 disruption in male infertility.
Finally, even though the deleterious MAJIN variant was found in a single homozygous patient the available evidence supports a role for this gene in spermatogenic failure and represents the third possible candidate responsible for NOA due to disruptions to the MTC function. The affected residue in MAJIN is perfectly conserved across 70 vertebrates and the variant is observed at extremely low frequency, while homozygosity of the variant has not been reported previously.
We report here evidence suggesting that, consistent with mouse models, disruption of any one of the three MTC genes in humans may underlie non-obstructive azoospermia. Additional studies are encouraged to further elucidate the functional consequences of the MTC gene variants reported here and in other recent studies (Alhathal et al. 2020; Krausz et al. 2020). Although we know that the described variants are rare in the general population, we do not know the sum contribution of variants in these three genes to idiopathic/sporadic NOA. We therefore encourage additional well-designed cohort-based studies to assess the prevalence of these and other pathogenic MTC gene variants in men with severe spermatogenic impairment. In addition, given the fundamental role of the MTC in meiotic prophase I and the observation of female infertility in MTC mouse models, we propose that MTC gene variants should be considered as a candidate for female infertility as well.
These findings along with recently published studies suggest that biallelic, pathogenic mutations in any one of the three MTC genes (TERB1, TERB2 or MAJIN) may be a recurrent cause of NOA with maturation arrest. Further studies may help to identify additional variants in these and additional related genes underlying the NOA phenotype and will aid in genetic screening and diagnosis of male and potentially female infertility.
Supplementary Material
Table S2. Sequencing quality and coverage metrics for Family 1.
Table S3. Filtered PSAP output for three patients with TERB1 and MAJIN variants based on the following criteria: 1) minor allele frequency [MAF] <0.01 in gnomAD, 1000 Genomes, NHLBI Exome Sequencing Project and ExAC databases, 2) CADD-score ≥20) and 3) PSAP p-value ≤ 0.001. Tabs 1–3 represent the highest ranked variants for Individual 2, M2073 and M1646, respectively, and tab 4 includes the legend to define column headers in PSAP tables.
Fig. S1. Protein, genetic context, validation and alignment of the TERB2 variants detected in Family 1. a) Position of TERB2 on chromosome 15. b) Sequence length and exon structure for TERB2. c) Corresponding protein domain structure. d) Primers for region of interest in exon 6, wild type sequence and validation of exon 6 TERB2 variant using Sanger sequencing in all samples analyzed. e) Primers for region of interest in exon 7, wild type sequence and validation of exon 7 TERB2 variant using Sanger sequencing in all the analyzed samples.
Fig. S2. Protein sequence conservation of the exon 6 TERB2 variant (p.Thr153fs*17) detected in Family 1 was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser. * Coelacanth was excluded from the species count because its sequence is truncated.
Fig. S3. Protein sequence conservation of the exon 7 TERB2 variant (p.Met182fs*31) detected in Family 1 was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser. * Coelacanth was excluded from the species count because its sequence is truncated.
Fig. S4. Protein, genetic context, validation and alignment of the TERB1 variants detected in Individuals 2 and 3. a) Position of TERB1 on chromosome 16. b) Sequence length and exon structure for TERB1. c) Corresponding protein domain structure. d) Primers for the region of interest and validation of exon 11 TERB1 variant using Sanger sequencing in individual 2. e) Primers for the region of interest and validation of exon 18 TERB1 variant using Sanger sequencing in individual 3 (M2073).
Fig. S5. Protein sequence conservation of the TERB1 variant (c.977A>G, p.Glu326Gly) detected in Individual 2 was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser.
Fig. S6. Protein, genetic context, validation and alignment of the MAJIN variant detected in Individual 4 (M1646). a) Position of MAJIN on chromosome 11. b) Sequence length and exon structure for MAJIN gene. c) Corresponding protein domain structure. d) Primers for the region of interest and validation of exon 5 MAJIN variant using Sanger sequencing in the analyzed sample.
Fig. S7. Protein sequence conservation of the MAJIN variant (c.158G>A, p.Arg53His) detected in Individual 4 (M1646) was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser.
Fig. S8. RNA expression of TERB2, TERB1 and MAJIN genes with NX data (Consensus Normalized eXpression) levels for 55 tissue types and 6 blood cell types, showing strong enrichment in the testis, created by combining the data from the three transcriptomic datasets (HPA, GTEx and FANTOM5).
Table S1. List of 170 candidate genes that were previously reported to be associated with impaired spermatogenesis according to a systematic review by Oud et al. 2019 and eight recently published genes associated with non-obstructive azoospermia (Krausz et al. 2020).
Acknowledgments
Funding/Acknowledgments
We thank all the participants and their families for their enthusiastic collaboration. Variant calling and interpretation for Family 1 were performed at the Utah Center for Genetic Discovery Core, part of the Health Sciences Center Cores at University of Utah. We are grateful to Jochen Wistuba for help with the CREM staining of case M1646, and we thank Nadja Rotte for her help with testicular histology in controls. This work was supported in part by funding from the National Institutes of Health (R01HD078641) and the German Research Foundation (Clinical Research Unit ‘Male Germ Cells: from Genes to Function’, DFG CRU326).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflicts of interest
The authors declare no competing interests.
Consortia information
GEMINI Consortium members:
Donald F. Conrad, Kenneth I. Aston, Douglas T. Carrell, James M. Hotaling, Liina Nagirnaja, Timothy G. Jenkins, Moira K. O’Bryan, Rob McLachlan, Peter N. Schlegel, Michael L. Eisenberg, Jay I. Sandlow, James F. Smith, Puneet Kamal, Carole Ober, Mark Sigman, Kathleen Hwang, Emily S. Jungheim, Kenan R. Omurtag, Alexandra M. Lopes, Filipa Carvalho, Susana Fernandes, Alberto Barros, João Gonçalves, Maris Laan, Margus Punab, Ewa Rajpert-De Meyts, Niels Jørgensen, Kristian Almstrup, Csilla G. Krausz, Keith A. Jarvi, Davor Jezek
Data and code availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Web resources
Gene Cards, https://www.genecards.org/
UCSC Genome Browser, http://genome.ucsc.edu/
Genome Aggregation Database, https://gnomad.broadinstitute.org/
Greater Middle East Variome, http://igm.ucsd.edu/gme/
OMIM, http://www.omim.org/
Protein Atlas, https://www.proteinatlas.org/
PubMed, http://www.ncbi.nlm.nih.gov/PubMed/
UCSC In-Silico PCR, https://genome.ucsc.edu/cgi-bin/hgPcr
UniProt, https://www.uniprot.org/
REFERENCES
- Alhathal N, Maddirevula S, Coskun S, et al. (2020) A genomics approach to male infertility. Genet Med In Press. 10.1038/s41436-020-0916-0 [DOI] [PubMed]
- Boivin J, Bunting L, Collins JA, Nygren KG (2007) International estimates of infertility prevalence and treatment-seeking: potential need and demand for infertility medical care. Hum Reprod 22:1506–1512. 10.1093/humrep/dem046 [DOI] [PubMed] [Google Scholar]
- Djureinovic D, Fagerberg L, Hallström B, et al. (2014) The human testis-specific proteome defined by transcriptomics and antibody-based profiling. Mol Hum Reprod 20:476–488. 10.1093/molehr/gau018 [DOI] [PubMed] [Google Scholar]
- Dunce JM, Milburn AE, Gurusaran M, et al. (2018) Structural basis of meiotic telomere attachment to the nuclear envelope by MAJIN-TERB2-TERB1. Nat Commun 9:1–18. 10.1038/s41467-018-07794-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlin A, Raicu F, Gatta V, et al. (2007) Male infertility: role of genetic background. Reprod Biomed Online 14:734–745. 10.1016/S1472-6483(10)60677-3 [DOI] [PubMed] [Google Scholar]
- Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing. arXiv 1207.3907
- Glazer CH, Bonde JP, Giwercman A, et al. (2017) Risk of diabetes according to male factor infertility: A register-based cohort study. Hum Reprod 32:1474–1481. 10.1093/humrep/dex097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston BJ, Conrad DF, O’Bryan MK (2020) A framework for high-resolution phenotyping of candidate male infertility mutants: from human to mouse. Hum Genet In press. 10.1007/s00439-020-02159-x [DOI] [PMC free article] [PubMed]
- Hu Z, Yau C, Ahmed AA (2017) A pan-cancer genome-wide analysis reveals tumour dependencies by induction of nonsense-mediated decay. Nat Commun 8:1–9. 10.1038/ncomms15943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, et al. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210. 10.1101/531210 [DOI]
- Kasak L, Laan M (2020) Monogenic causes of non-obstructive azoospermia: challenges, established knowledge, limitations and perspectives. Hum Genet In press. 10.1007/s00439-020-02112-y [DOI] [PubMed]
- Kasak L, Punab M, Nagirnaja L, et al. (2018) Bi-allelic Recessive Loss-of-Function Variants in FANCM Cause Non-obstructive Azoospermia. Am J Hum Genet 103:200–212. 10.1016/j.ajhg.2018.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliesch S (2014) Diagnosis of male infertility: Diagnostic work-up of the infertile man. Eur Urol Suppl 13:73–82. 10.1016/j.eursup.2014.08.002 [DOI] [Google Scholar]
- Krausz C, Hoefsloot L, Simoni M, Tüttelmann F (2014) EAA/EMQN best practice guidelines for molecular diagnosis of Y-chromosomal microdeletions: State-of-the-art 2013. Andrology 2:5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz C, Riera-Escamilla A (2018) Genetics of male infertility. Nat Rev Urol 15:369–384. 10.1038/s41585-018-0003-3 [DOI] [PubMed] [Google Scholar]
- Krausz C, Riera-Escamilla A, Moreno-Mendoza D, et al. (2020) Genetic dissection of spermatogenic arrest through exome analysis: clinical implications for the management of azoospermic men. Genet Med In Press. 10.1038/s41436-020-0907-1 [DOI] [PMC free article] [PubMed]
- Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G, Kherraf Z-E, Zouari R, et al. (2018) Whole-exome sequencing identifies mutations in FSIP2 as a recurrent cause of multiple morphological abnormalities of the sperm flagella. Hum Reprod 33:1973–1984. 10.1093/humrep/dey264 [DOI] [PubMed] [Google Scholar]
- Mbango JN, Coutton C, Arnoult C, et al. (2019) Genetic causes of male infertility: snapshot on morphological abnormalities of the sperm flagellum. Basic Clin Androl 9:2 10.1186/s12610-019-0083-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, et al. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. 10.1101/gr.107524.110.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, et al. (2016) The Ensembl Variant Effect Predictor. Genome Biol 17:1–14. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagirnaja L, Aston KI, Conrad DF (2018) Genetic intersection of male infertility and cancer. Fertil Steril 109:20–26. 10.1016/j.fertnstert.2017.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieschlag E, Behre HM, Nieschlag S (2010) Andrology-Male Reproductive Health and Dysfunction. Springer [Google Scholar]
- Okutman O, Muller J, Baert Y, et al. (2015) Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum Mol Genet 24:5581–5588. 10.1093/hmg/ddv290 [DOI] [PubMed] [Google Scholar]
- Oud MS, Volozonoka L, Smits RM, et al. (2019) A systematic review and standardized clinical validity assessment of male infertility genes. Hum Reprod 34:932–941. 10.1093/humrep/dez022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BS, Quinlan AR (2017) Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am J Hum Genet 100:406–413. 10.1016/j.ajhg.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatt S, Pohl E, H V, et al. (2019) Ageing in men with normal spermatogenesis alters spermatogonial dynamics and nuclear morphology in Sertoli cells. Andrology 7:827–839. 10.1111/andr.12665 [DOI] [PubMed] [Google Scholar]
- Scott EM, Halees A, Itan Y, et al. (2016) Characterization of greater middle eastern genetic variation for enhanced disease gene discovery. Nat Genet 48:1071–1079. 10.1038/ng.3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrucca L, Fop M, Murphy T, Raftery A (2016) mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J 8:289–317. 10.1016/j.physbeh.2017.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya H, Hernández-Hernández A, Morimoto A, et al. (2015) MAJIN Links Telomeric DNA to the Nuclear Membrane by Exchanging Telomere Cap. Cell 163:1252–1266. 10.1016/j.cell.2015.10.030 [DOI] [PubMed] [Google Scholar]
- Shibuya H, Ishiguro KI, Watanabe Y (2014) The TRF1-binding protein TERB1 promotes chromosome movement and telomere rigidity in meiosis. Nat Cell Biol 16:145–156. 10.1038/ncb2896 [DOI] [PubMed] [Google Scholar]
- Tiepolo L, Zuffardi O (1976) Localization of Factors Controlling Spermatogenesis in the Nonfluorescent Portion oi the Human Y Chromosome Long Arm. Hum Genet 124:119–124. 10.1007/bf00278879 [DOI] [PubMed] [Google Scholar]
- Tüttelmann F, Ruckert C, Röpke A (2018) Disorders of spermatogenesis: Perspectives for novel genetic diagnostics after 20 years of unchanged routine. Medizinische Genet 30:12–20. 10.1007/s11825-018-0181-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventimiglia E, Montorsi F, Salonia A (2016) Comorbidities and male infertility: A worrisome picture. Curr Opin Urol 26:146–151. 10.1097/MOU.0000000000000259 [DOI] [PubMed] [Google Scholar]
- Vockel M, Riera-Escamilla A, Tüttelmann F, Krausz C (2019) The X chromosome and male infertility. Hum Genet In press. 10.1007/s00439-019-02101-w [DOI] [PMC free article] [PubMed]
- Wang K, Li M, Hakonarson H (2010) ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:1–7. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Chen Y, Chen J, et al. (2019) The meiotic TERB1-TERB2-MAJIN complex tethers telomeres to the nuclear envelope. Nat Commun 10:1–19. 10.1038/s41467-019-08437-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbauer GF, Behr R, Bergmann M, Nieschlag E (1998) Testicular cAMP responsive element modulator (CREM) protein is expressed in round spermatids but is absent or reduced in men with round spermatid maturation arrest. Mol Hum Reprod 4:9–15. 10.1093/molehr/4.1.9 [DOI] [PubMed] [Google Scholar]
- Wilfert AB, Chao KR, Kaushal M, et al. (2016) Genome-wide significance testing of variation from single case exomes. Nat Genet 48:1455–1461. 10.1038/ng.3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier M, Salas-Huetos A, Oud M, et al. (2020) Disease gene discovery in male infertility: past, present and future. Hum Genet In press. 10.1007/s00439-020-02202-x [DOI] [PMC free article] [PubMed]
- Zhang J, Tu Z, Watanabe Y, Shibuya H (2017) Distinct TERB1 Domains Regulate Different Protein Interactions in Meiotic Telomere Movement. Cell Rep 21:1715–1726. 10.1016/j.celrep.2017.10.061 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Sequencing quality and coverage metrics for Family 1.
Table S3. Filtered PSAP output for three patients with TERB1 and MAJIN variants based on the following criteria: 1) minor allele frequency [MAF] <0.01 in gnomAD, 1000 Genomes, NHLBI Exome Sequencing Project and ExAC databases, 2) CADD-score ≥20) and 3) PSAP p-value ≤ 0.001. Tabs 1–3 represent the highest ranked variants for Individual 2, M2073 and M1646, respectively, and tab 4 includes the legend to define column headers in PSAP tables.
Fig. S1. Protein, genetic context, validation and alignment of the TERB2 variants detected in Family 1. a) Position of TERB2 on chromosome 15. b) Sequence length and exon structure for TERB2. c) Corresponding protein domain structure. d) Primers for region of interest in exon 6, wild type sequence and validation of exon 6 TERB2 variant using Sanger sequencing in all samples analyzed. e) Primers for region of interest in exon 7, wild type sequence and validation of exon 7 TERB2 variant using Sanger sequencing in all the analyzed samples.
Fig. S2. Protein sequence conservation of the exon 6 TERB2 variant (p.Thr153fs*17) detected in Family 1 was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser. * Coelacanth was excluded from the species count because its sequence is truncated.
Fig. S3. Protein sequence conservation of the exon 7 TERB2 variant (p.Met182fs*31) detected in Family 1 was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser. * Coelacanth was excluded from the species count because its sequence is truncated.
Fig. S4. Protein, genetic context, validation and alignment of the TERB1 variants detected in Individuals 2 and 3. a) Position of TERB1 on chromosome 16. b) Sequence length and exon structure for TERB1. c) Corresponding protein domain structure. d) Primers for the region of interest and validation of exon 11 TERB1 variant using Sanger sequencing in individual 2. e) Primers for the region of interest and validation of exon 18 TERB1 variant using Sanger sequencing in individual 3 (M2073).
Fig. S5. Protein sequence conservation of the TERB1 variant (c.977A>G, p.Glu326Gly) detected in Individual 2 was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser.
Fig. S6. Protein, genetic context, validation and alignment of the MAJIN variant detected in Individual 4 (M1646). a) Position of MAJIN on chromosome 11. b) Sequence length and exon structure for MAJIN gene. c) Corresponding protein domain structure. d) Primers for the region of interest and validation of exon 5 MAJIN variant using Sanger sequencing in the analyzed sample.
Fig. S7. Protein sequence conservation of the MAJIN variant (c.158G>A, p.Arg53His) detected in Individual 4 (M1646) was assessed across 100 vertebrate species using Multiz alignment in the UCSC Genome Browser.
Fig. S8. RNA expression of TERB2, TERB1 and MAJIN genes with NX data (Consensus Normalized eXpression) levels for 55 tissue types and 6 blood cell types, showing strong enrichment in the testis, created by combining the data from the three transcriptomic datasets (HPA, GTEx and FANTOM5).
Table S1. List of 170 candidate genes that were previously reported to be associated with impaired spermatogenesis according to a systematic review by Oud et al. 2019 and eight recently published genes associated with non-obstructive azoospermia (Krausz et al. 2020).