

Abstract
BACKGROUND
Human-induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) created from patients with catecholaminergic polymorphic ventricular tachycardia 1 (CPVT1) have been used to study CPVT1 arrhythmia.
OBJECTIVE
The purpose of this study was to evaluate the Ca2+ signaling aberrancies and pharmacological sensitivities of 3 CRISPR/Cas9-introduced CPVT1 mutations located in different molecular domains of ryanodine receptor 2 (RyR2).
METHODS
CRISPR/Cas9-engineered hiPSC-CMs carrying RyR2 mutations—R420Q, Q4201R, and F2483I—were voltage clamped, and their electrophysiology, pharmacology, and Ca2+ signaling phenotypes measured using total internal reflection fluorescence microscopy.
RESULTS
R420Q and Q4201R mutant hiPSC-CMs exhibit irregular, long-lasting, spatially wandering Ca2+ sparks and aberrant Ca2+ releases similar to F2483I unlike the wild-type myocytes. Large sarcoplasmic reticulum (SR) Ca2+ leaks and smaller SR Ca2+ contents were detected in cells expressing Q4201R and F2483I, but not R420Q. Fractional Ca2+ release and calcium-induced calcium release gain were higher in Q4201R than in R420Q and F2483I hiPSC-CMs. JTV519 was equally effective in suppressing Ca2+ sparks, waves, and SR Ca2+ leaks in hiPSC-CMs derived from all 3 mutant lines. Flecainide and dantrolene similarly suppressed SR Ca2+ leaks, but were less effective in decreasing spark frequency and durations.
CONCLUSION
CRISPR/Cas9 gene editing of hiPSCs provides a novel approach in studying CPVT1-associated RyR2 mutations and suggests that Ca2+-signaling aberrancies and drug sensitivities may vary depending on the mutation site.
Keywords: Ca2+ sparks, CPVT1, CRISPR/Cas9, hiPSC-CMs, JTV519, RyR2
Graphical Abstract
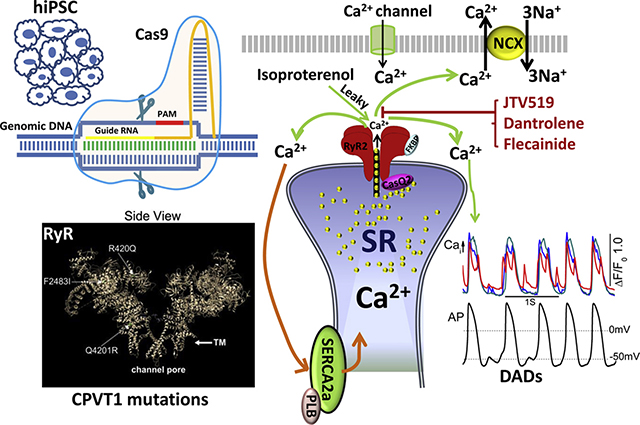
Introduction
Cardiac excitation-contraction coupling is mediated by activation of the calcium-induced calcium release (CICR) mechanism that triggers Ca2+ release from sarcoplasmic reticulum (SR) during each action potential.1 Dysfunctions in Ca2+ signaling may result in cardiac hypertrophy, failure, and arrhythmias.2–4 Missense mutations in human ryanodine receptor 2 (RYR2) have been shown to associate with catecholaminergic polymorphic ventricular tachycardia 1 (CPVT1), a life-threatening arrhythmia.5 Thus far, >150 missense mutations, spread all over the ~5000-amino acid RyR2 polypeptide, are identified to associate with CPVT1.6,7 Functional consequences of CPVT1 mutations have been studied by heterologous expression of the recombinant mutants of RyR2 and knock-in mutation in mouse models.8,9 These studies have led to proposals that CPVT1 pathology results either from phosphorylation-induced leakiness of the RyR2 channel10,11 or from the Ca2+ overload of SR.12 Such aberrant Ca2+ releases activate the Na/Ca exchanger to depolarize the myocytes, triggering early afterdepolarizations or delayed afterdepolarizations and arrhythmias.13,14 Despite the insights gained from such studies, a systematic mutation-dependent comparison of Ca2+-signaling aberrancies of CPVT1 pathology in human cardiomyocytes has not been possible and is desirable.
The advent of hiPSCs15 and CRISPR/Cas9 genome editing make it possible to carry out such comparisons using fibroblast-derived cardiomyocytes,16 where point mutations associated with CPVT1 are introduced in wild-type (WT) human cardiomyocytes to quantify the Ca2+ signaling aberrancies of different mutations and determine whether disease-associated mutations are causative or associative. We have already reported that CRISPR/Cas9 gene–edited human-induced pluripotent stem cell–derived cardiomyocytes (hiPSC-CMs) carrying F2483I-RyR2 mutation exhibit aberrant Ca2+ signaling properties indistinguishable from those recorded in patient-derived cells carrying F2483I mutation.17 Therefore, we undertook detailed comparisons of Ca2+ signaling and pharmacological profiles of 3 CPVT1 mutations, located in N-terminal R420Q, C-terminal Q4201R, and central-domain F2483I, to determine whether the molecular mechanism of CPVT1 pathology and its pharmacology depended on the RyR2 mutation site.
The data suggest that all 3 mutations express prolonged, wandering, and reigniting Ca2+ sparks, but differ in their CICR gain, the Ca2+ content of SR, its leakiness, and the sensitivity to antiarrhythmic drugs.
Methods
Maintenance of hiPSC lines
The hiPSC-K3 line was maintained in StemFlex Medium (Gibco, Thermo Fisher Scientific, Waltham, MA, US) and passaged with Versene as described previously.17,18
CRISPR/Cas9 genome editing in hiPSCs
Two CPVT1 associated RyR2 mutations—R420Q and Q4201R—were introduced into the RYR2 gene of WT hiPSCs by CRISPR/Cas9 gene editing, as described previously17 (see Online Supplemental Materials).
Differentiation of hiPSCs into functional cardiomyocytes
hiPSCs were differentiated into cardiomyocytes using protocols described previously.17,18 Three to four weeks after spontaneous beating started, beating clusters were enzymatically (TrypLE Select Enzyme [10×], Gibco) dissociated into single cardiomyocytes for Ca2+ signaling experiments. The same batch of differentiated cells was used for up to 3 months.
Quantitative reverse transcription polymerase chain reaction(RT-PCR) and Western blot
Total RNAs and proteins of hiPSC-CMs were collected at day 45 after differentiation for analysis. For method details and primer selections, see Online Supplemental Materials.
Measurements of Ca2+ transients and sparks
Fluo-4AM (Invitrogen, Thermo Fisher Scientific, Waltham, MA, US)–incubated hiPSC-CMs were imaged using multi-color total internal reflection fluorescence (TIRF) microscopy. Ca2+ sparks were recorded at 60–80 Hz with a depth of penetration of 110–150 nm. Sparks data were exported and analyzed with Leica LAS X (Leica Microsystems Inc, Wetzlar, Germany) and Con2i (custom developed by our lab) software as described previously.17
Electrophysiology
All electrophysiological and Ca2+ transient measurements were carried out simultaneously in whole-cell patch-clamped hiPSC-CMs at 32°C–35°C (see Online Supplemental Materials).
SR Ca2+ leak and load measurement
The SR Ca2+ content and diastolic Ca2+ leak of WT and mutant hiPSC-CMs were measured as described previously17 (see Online Supplemental Materials).
Statistical analysis
The results are presented as mean ± SEM. Comparative analysis was performed using 1-way analysis of variance followed by the Tukey test or Student t test. Significant differences are labeled with 1 (*P < .05) or 2 (**P < .01) asterisks.
Results
Creation of hiPSC lines harboring R420Q and Q4201R RyR2 mutations
We used CRISPR/Cas9 gene editing to introduce R420Q and Q4201R mutations into the RYR2 gene of control hiPSCs, as described for F2483I,17 where it was shown that heterozygous and homozygous F2483I mutants had similar arrhythmogenic profiles.17 The molecular sites of these mutations are shown in the 4.2 Å resolution cryoelectron microgram image of the purified pig RyR2 (see Graphical Abstract). We created 2 new homozygous—Q4201R and R420Q—and 1 heterozygous—Q4201R—hiPSC lines and verified the mutations by restriction enzyme digest (Figure 1C) and sequencing of the polymerase chain reaction (PCR) product amplified from the target loci (Figures 1A and 1B). Quantitative RT-PCR analyses showed that the expression level of RyR2 messenger RNA in R420Q mutant hiPSC-CMs were essentially similar to those of WT cells, but Q4201R mutant hiPSC-CMs had significantly lower levels of RyR2 (Figure 1D). RyR2 protein expression levels of all 3 homozygous mutants were essentially the same as those of WT cells (Western blots in Figures 1E and 1F).
Figure 1.

Introduction of R420Q and Q4201R mutations into the ryanodine receptor 2 (RYR2) gene of hiPSCs. A and B: Sequencing of polymerase chain reaction (PCR) products show R420Q (CGG to CAG) and Q4201R (CAG to CGG) mutations. C: PCR analysis of the mutations. PCR products from homozygous mutants were completely digested into 2 fragments: 323 and 126 bp for R420Q and 340 and 141 bp for Q4201R. D: Quantification of RyR2 transcription levels in wild-type (WT) and mutant hiPSC-CMs. **P < .01 vs WT. E and F: Western blot image and quantification of RyR2 protein levels in WT and 3 homozygous mutants. The F2483I homozygous clone was established in the previous study.17
Ca2+ sparks in WT and mutant hiPSC-CMs
Ca2+ signaling aberrancies of RyR2-CPVT1 mutations and their domain dependence were measured in 3 homozygous and 1 heterozygous mutations and compared with WT hiPSC-CMs. Figure 2 shows that spontaneously occurring Ca2+ sparks in WT hiPSC-CMs were brief, igniting, and decaying rapidly, whereas sparks of the 3 homozygous mutant hiPSC-CMs were longer lasting with variable durations, often reigniting and setting off localized releases of Ca2+ (Figures 2A–2D). Although these aberrancies were common to all 3 mutations, there were noticeable differences in spark durations and frequency among the 3 mutant lines. Figure 2E shows that the histogram of spark duration, measured at half-maximum amplitude, averaged 58.2 ± 2.1 ms in WT as compared with 113.0 ± 4.5 ms in R420Q-homozygous, 95.5 ± 3.4 ms in Q4201R-homozygous, 91.4 ± 3.7 ms in Q4201R-heterozygous, and 126.1 ± 4.9 ms in F2483I-homozygous mutants, consistent with the longer mean open time of mutant RyR2s. Despite the significantly increased spark durations, the frequency of the occurence of spontaneously triggered sparks did not vary significantly among the mutant lines compared with WT (Figure 2F). The mean spark frequency of the R420Q mutant was consistently lower, especially when compared with the F2483I mutant (Figure 2F).
Figure 2.

Ca2+ sparks recorded from wild-type (WT) and mutant hiPSC-CMs. A–D: Representative time courses of normalized Ca2+ sparks from the selected regions (different colors) in WT and 3 homozygous mutants. E: Histogram and distribution curves show the spark duration measured at half-maximum amplitude. **P < .01 vs WT. Spark durations in F2483I and R420Q cells are significantly longer than those in Q4201R hiPSC-CMs (P < .01). F: Spark frequency of WT and mutant cells. *P < .05, R420Q vs F2483I by 1-way analysis of variance. Part of the WT and F2483I-hom data are published previously.17
Figure 3 shows total internal reflection fluorescence images of spark evolution (indicated by an asterisk in Figure 2) in WT and the 3 homozygous mutants. The igniting and decaying times of the sparks from F2483I and R420Q hiPSC-CMs were much longer than those from WT or Q4201R cells. Taken together, our results indicated that all 3 mutants exhibited prolonged Ca2+ release times.
Figure 3.

Total internal reflection fluorescence images show the evolution of the asterisk labeled sparks (Figure 2) from wilde-type (A) and the 3 homozygous mutants (B–D). Note that R420Q and F2483I sparks are much longer.
SR Ca2+ leak and content in mutant hiPSC-CMs
To check whether the gene-edited RyR2 mutations had altered SR function, we quantified their caffeine-triggered Ca2+ store and their diastolic Ca2+ leak by application of a cocktail containing 1 mM tetracaine in zero Ca2+ and Na+ solutions.17 Figures 4A–4D show representative Ca2+ signals comparing the responses of R420Q, Q4201R, and F2483I homozygous myocytes with those of WT myocytes to application of the “tetracaine cocktail” followed by caffeine. The SR Ca2+ leak was significantly larger in F2483I and Q4201R homozygous hiPSC-CMs than in WT cells or a small leak of R420Q cells (Figure 4E). Consistent with the SR leak levels, the SR Ca2+ content of Q4201R and F2483I hiPSC-CMs were smaller than that of WT cells (Figure 4F), confirming that the larger diastolic leak results in smaller SR Ca2+ content.
Figure 4.

SR Ca2+ leak and load levels. A–D: Representative time courses of normalized Ca2+ fluorescence changes after rapid exposure to 1 mM tetracaine followed by 5 mM caffeine in wild-type (WT) and 3 homozygous mutants. E: Scatter dot graph shows quantified sarcoplasmic reticulum (SR) leak levels, normalized to the SR store size. F: SR Ca2+ content levels were determined by caffeine. *P < .05 and **P < .01 by 1-way analysis of variance. Part of the WT and F2483I-homozygous data are published previously.17
L-type Ca2+ current(ICa), fractional Ca2+ release, and CICR gain
To determine the fractional Ca2+ release and CICR gain, ICa and its triggered Ca2+ transient were measured in cells dialyzed with 0.1 mM Fluo-4 pentapotassium salt while the SR content was determined by applying 5 mM caffeine (see Methods). Figures 5A–5D show representative traces of ICa and ICa-induced Ca2+ release, ICa tail currents (activated by repolarization from +80 to −50 mV) and their triggered Ca2+ transients, caffeine-triggered Ca2+ release, and Na+/Ca2+ exchange current in WT and mutant myocytes. ICa densities from WT and the 3 mutant hiPSC-CMs averaged between 6.4 and 9.2 pA/pF, and the accompanying Ca2+ transients averaged from 0.96 to 1.8 pA/pF. The differences in ICa densities were not statistically significant between WT and mutant hiPSC-CMs (Figure 5E), but ICa-induced Ca2+ release in Q4201R heterozygote was significantly higher than that in F2483I and R420Q mutants, resulting in higher fractional Ca2+ release and CICR gains in Q4201R than in F2483I and R420Q myocytes (Figure 5F).
Figure 5.

L-type Ca2+ current(ICa), fractional Ca2+ release, and calcium-induced calcium release (CICR) gain. A–D: Representative traces of ICa and ICa-induced Ca2+ release (left panels), ICa tail currents and their induced Ca2+ release (middle panels), and caffeine-induced Ca2+ release and INCX (right panels) from WT and 3 homozygous mutant hiPSC-CMs. E: Quantified ICa density (bottom panel) and ICa-induced Ca2+ release (top panel) in WT and mutant cells. F: Fractional Ca2+ release (top panel) and CICR gain (bottom panel) in each group. *P < .05, **P < .01 by 1-way analysis of variance.
Pharmacological sensitivity of the mutant lines
Adrenergic enhancement of Ca2+ release
Figure 6 shows the effectiveness of 500 nM isoprenaline in enhancing ICa and aberrant Ca2+ releases in WT and mutant hiPSC-CMs. As expected, adrenergic stimulation enhanced aberrant Ca2+ releases in >30% of the mutant cells.
Figure 6.

Effect of adrenergic stimulation on L-type Ca2+ current(ICa) and aberrant Ca2+ releases in wild-type(WT) and mutant hiPSC-CMs. Cells were stimulated by depolarization at 0.2 Hz (pulse train). A–D: The time course of cellular Ca2+ transients (colored) and 7 ICa stimulation traces (black) before and after exposure to 500 nM isoprenaline in amphotericin B perforated patch-clamped WT and 3 homozygous mutant hiPSC-CMs. Enlarged ICa shows the first ICa trace from the pulse train.
Ca2+ sparks
Since all 3 homozygous mutants had irregular long-lasting and reigniting sparks as compared with WT cells, we examined the effectiveness of JTV519, flecainide, and dantrolene (drugs reported to inhibit RyR219–21) in altering the morphology and frequency of spontaneously occurring Ca2+ sparks. Figure 7 shows that 2 μM JTV519, a 1,4-benzothiazepine derivative,20 significantly shortened the spark duration and decreased the spark frequency in all 3 homozygous mutants. JTV519 also consistently suppressed the frequency and duration of sparks in the Q4201R heterozygous mutant (Online Supplemental Figure 1). Flecainide decreased the spark frequency in all 3 mutants but had little effect on spark duration even in the Q4201R heterozygous mutant (Figures 7B, 7E, and 7H and Online Supplemental Figure 1). Dantrolene, is an inhibitor of RyR1 and used in treating malignant hyperthermia, also decreased the spark frequency in R420Q and F2483I-RyR2 mutants, consistent with its suppression of CPVT1 in a mouse model and CPVT1 patient–derived hiPSC-CMs,21,22 but had little effect on spark characteristics of the Q4201R mutant. Dantrolene was less effective than JTV519 in shortening the spark duration (Figures 7C and 7I).
Figure 7.

Effects of JTV519, flecainide, and dantrolene on the Ca2+ sparks of 3 mutants. Effects of 2 μM JTV519, 5 μM flecainide, and 10 μM dantrolene on the spark duration and frequency of R420Q (A–C), Q4201R (D–F), and F2483I (G–I) homozygous hiPSC-CMs. *P < .05 and **P < .01 vs control.
Spontaneously beating mutant hiPSC-CMs often showed aberrant local Ca2+ transients, occurring in between the regular beats especially in Q4201R cells (Online Supplemental Figure 2), that were suppressed by 2 μM JTV519 in 88% of R420Q (22 of 25), 87% of Q4201R (13 of 15), and 79% of F2483I (11 of 14) mutant cells (Figure 8). JTV519 also abolished such aberrant Ca2+ releases in 91% of heterozygous Q4201R mutant cells (10 of 11) (Online Supplemental Figure 1). Ten micromolar dantrolene was less effective than JTV519, suppressing only 27% (4 of 15) of aberrant focal Ca2+ releases in R420Q, 40% (4 of 10) in Q4201R, and 22% (2 of 9) in F2483I cells. Flecainide (5 μM) was also effective in suppressing focal Ca2+ releases but only in 44% of R420Q (4 of 9), 55% of Q4201R (6 of 11), and 38% of F2483I (5 of 13) cells. The effects of all 3 drugs were reversible, allowing serial comparison of the drugs on the same cell (Figure 8). Our results suggest that JTV519 is equally effective in suppressing Ca2+ releases in all 3 mutants. The antiarrhythmic effectiveness of the 3 drugs was as follows: JTV519>flecainide≈dantrolene.
Figure 8.

Effects of JTV519, dantrolene, and flecainide on the aberrant Ca2+ releases of mutant hiPSC-CMs. Representative traces of spontaneous Ca2+ transients with aberrant Ca2+ releases before and after exposure to and washout of 2 μM JTV519, 10 μM dantrolene, and 5 μM flecainide on R420Q (A–C), Q4201R (D–F), and F2483I (G–I) homozygous hiPSC-CMs.
SR Ca2+ leak and load
Since hiPSC-CMs of WT and the 3 homozygous mutants showed different levels of SR Ca2+ leaks (Figure 4), we examined whether the 3 drugs preferentially suppressed the SR Ca2+ leak and restored the SR Ca2+ content. Online Supplemental Figure 3 shows that JTV519, dantrolene, and flecainide significantly, but to different extent, suppressed the Ca2+ leak of all 3 mutants. JTV519 was especially effective on the F2483I mutant, suppressing the leak by ~65% (Online Supplemental Figure 3A) and restoring the SR Ca2+ content (Online Supplemental Figure 3B). Similarly, the JTV519 suppression of the SR leak in R420Q or Q4201R cells was accompanied by recovery of the Ca2+ content of SR. Flecainide also suppressed the Ca2+ leak in all 3 lines, but its effect was somewhat weaker. In contrast, dantrolene was more effective in suppressing the SR Ca2+ leak and the recovery of SR Ca2+ load more effectively in Q4201R than in R420Q and F2483I hiPSC-CMs. These findings suggest that different mutant lines show different drug sensitivities, but more extensive dose-response relation is required to determine the specificity and effectiveness of the drugs.
Discussion
This is the first comparative report of creating 3 CRISPR/Cas9 gene–edited CPVT1-RyR2 mutations in hiPSC-CMs, where electrophysiological and Ca2+ imaging consequences of 3 point mutations in N, central, and C terminals of RyR2 are examined and their pharmacological sensitivities determined. We found that hiPSC-CMs from all 3 mutants exhibited long-lasting Ca2+ sparks that often reignited and spatially wandered, causing variable degrees of the SR Ca2+ leak. Only moderate differences in the drug sensitivities of the 3 mutants were found. JTV519 was effective in suppressing aberrant Ca2+ sparks and releases, but showed no site-dependent mutation specificity, while dantrolene and flecainide effects, though mutation dependent, were variable and moderate compared with JTV519.
Creation and selection of CRISPR/Cas9 gene–edited mutations
The 3 CPVT1 mutation lines were created from the same hiPSC-K3 line,23 not only to have lines with the same genetic background but also to represent the 3 domains of RyR2 protein. To confirm the reliability of the CRISPR approach, we chose point mutations that had been either introduced in mouse models or CPVT1 patient–derived hiPSC-CMs that expressed them.16,24,25 We used protospacer adjacent motif (PAM) mutations in single-stranded oligo donor nucleotide (ssODNs) to improve homology-directed repair (HDR) accuracy26 and introduce other mutations that created new restriction enzyme sites for restriction fragment length polymorphisms (RFLP) analysis.
Although CPVT1 is expressed as a heterozygous mutation in patients, we were more successful in creating the homozygous mutation. We felt that the homozygous mutation clones may better represent the pathology of CPVT1 in in vitro cell models, because the heterotetrameric structure of RyR2 may minimize the expression of Ca2+ signaling aberrancies. Nevertheless, we succeeded in obtaining heterozygous clones of Q4201R (Figure 1) as well as F2483I17 and found no obvious differences in their Ca2+ signaling phenotype regardless of whether only 1 allele or both alleles were mutated.
Electrophysiological and Ca2+ signaling consequences of the site-specific RyR2 mutations
hiPSC-CMs from all 3 mutants showed prolonged and wandering sparks interspersed with aberrant focal Ca2+ releases.18,19 Although there were significant differences in the spark durations between WT and mutants, there were only some differences in spark occurrence frequencies between F2483I and R420Q (Figure 2). There were, however, significant differences in the magnitude of the Ca2+ leak or SR Ca2+ content among the mutants. The SR leak was larger and the SR content was smaller in F2483I and Q4201R mutants than in control or the R420Q mutant. The higher fractional Ca2+ releases and CICR gain of Q4201R mutation may be caused by the higher occurrence of Ca2+ waves and larger SR Ca2+ leaks. In contrast, the lower spark frequency may counteract the prolonged spark duration in R420Q mutation and result in only moderate SR leaks. The higher spark frequency, longer spark duration, and large leaks of the F2783I mutant may be caused by the proximity of this site to the FK506-binding protein (FKBP) binding site, causing the dissociation of FKBP from RyR2 and producing significantly larger leaks.
Pharmacological and pathophysiological implications
hiPSC-CMs as a platform for Ca2+ signaling pathology
Although hiPSC-CMs have been used as in vitro models of human inherited cardiac pathologies, considerable controversy still exists as to their appropriateness for pathophysiological studies. Clearly, advantages include human cell sources of the same genetic background that are preferable to genetically engineered mice or recombinant human embryonic kidney (HEK) cell models. Nevertheless, there are developmental aspects of hiPSC-CM maturation, such as lack of T-tubular network, the polygonal-flat cellular shapes, disorganized sarcomeres, rhythmic automaticity, and heterogeneity of cell cultures, expressing atrial, ventricular, and nodal action potentials, suggesting that hiPSC-CMs may represent fetal rather than adult cardiomyocytes. Our recent review systematically compares the functional aspects of Ca2+ signaling of hiPSC-CMs with those of native mammalian cardiomyocytes and concludes that despite quantitative differences, the qualitative aspects of Ca2+ signaling are extremely similar to those of adult cardiomyocytes.27
In the present study, the development of adult-type sarcomere structure and T-tubular network do not appear to play a major functional role in RyR2 Ca2+ signaling, making it possible to use hiPSC-CMs as an appropriate platform for aberrancies associated with missense mutations of RyR2. We have identified significant differences between WT and mutant Ca2+ sparks characteristics, SR Ca2+ leaks and loads, and the rate of occurrence of aberrant focal Ca2+ releases. Nevertheless, we have found that 5%–10% WT hiPSC-CMs also show age and culture-dependent arrhythmogenic phenotypes and hope that efforts to mature hiPSC-CMs will further improve their reliability as pathophysiological models.
Pharmacological specificity
Three drugs, JTV519, dantrolene, and flecainide, known for targeting RyR2 showed preferential RyR2 mutation site sensitivity. Dantrolene, reported to bind to RyR2 N-terminus amino acids 601–62028,29 that rescues the arrhythmogenic phenotype of S406L RyR2 in patient-derived hiPSC-CMs21 and being antiarrhythmic in human CPVT1-associated RyR2 R2474S/+ knock-in mouse model, 22 was less effective than JTV519 in suppressing Ca2+ sparks and aberrant Ca2+ releases in all 3 mutants. Nevertheless, dantrolene was more effective in suppressing the SR leak (Online Supplemental Figure 3) and aberrant Ca2+ releases (Figure 8) in the Q4201R mutant than in the other 2 mutants, even though its effects on the Ca2+ spark of Q4201R cells were less clear. Although this finding appears inconsistent with the idea that spontaneous Ca2+ sparks play a dominant role in the SR Ca2+ leak, we note that Ca2+ sparks are not the only pathway that mediates the SR leak. Spark-independent RyR openings as well as RyR-independent pathway have been reported to contribute to SR leaks.30 Our finding that dantrolene was more effective in suppressing the SR Ca2+ leak and recovery of the Ca2+ load of SR without affecting the Ca2+ sparks of Q4201R mutant myocytes suggests that dantrolene may be suppressing the non-Ca2+ sparks–mediated SR leak of the Q4201R mutant. The possibility that different mechanisms may contribute to SR Ca2+ leaks among the various mutants has not been fully tested, but should be considered in future studies. Flecainide has been reported to abolish Ca2+ abnormalities in 33% of L4115F, 30% of V4653F, and 52% of exon 3 deletion RyR2 mutations in hiPSC-CMs from patients with CPVT.31 In our study, flecainide consistently suppressed the spark frequency in all 3 mutants without affecting the spark duration and moderately suppressed the aberrant Ca2+ releases in 44% of R420Q, 55% of Q4201R, and 38% in F2483I (Figure 8). JTV519, though effective in suppressing the spark frequency and aberrant focal releases in all 3 mutants, showed little mutation site specificity (Figure 7). The higher efficacy of JTV519, as compared with flecainide or dantrolene, may be related to its proposed interaction with the FKBP binding site of RyR2. The specificity of these drugs in suppressing the aberrant Ca2+ releases is weakened by the finding that the drugs also suppressed the regularly occurring beats.
Study limitations
The structural and biophysical maturity of hiPSC-CMs as models for pathophysiological studies must be carefully considered. A small population of WT cells showed arrhythmogenic behavior, likely related to the immaturity of hiPSC-CMs that may be alleviated by the availability of more mature hiPSC-CMs. Detailed drug dose-response studies may also provide better insights into the mutation site specificity of drugs and determine their sensitivities for control vs diseased tissues.
Conclusion
We have created CPVT1-associated mutations in 3 domains of RyR2 using CRISPR/Cas9 gene editing in hiPSC-CMs. Our results demonstrated that F2483I, R420Q, and Q4201R CPVT1 mutants express variable Ca2+ signaling phenotype and pharmacological sensitivity. It is likely that as the pharmacological profiles of more mutants are studied, the therapy of CPVT1 pathology will be moving into the realm of personalized medicine with mutation-specific pharmacotherapy.
Supplementary Material
Acknowledgments
We thank N. Yamaguchi, PhD for supervising the CRISPR/Cas9 experiments. Part of the wild-type and F2483I-homozygous sparks data were previously published in Cell Calcium.
Sources of funding: The study was supported by the National Institutes of Health grant 1R56HL147054-01, and Jennifer Friedman Hillis Family Foundation Inc. Disclosures: The authors have no conflicts of interest to disclose.
Footnotes
Appendix
Supplementary data
Supplementary data associated with this article can be found in the online version at https://doi.org/10.1016/j.hrthm.2020.09.007.
References
- 1.Nabauer M, Callewaert G, Cleemann L, Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science 1989; 244:800–803. [DOI] [PubMed] [Google Scholar]
- 2.Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol 2000;525:483–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang XH, Morad M. Calcium signaling in human stem cell-derived cardiomyocytes: evidence from normal subjects and CPVT afflicted patients. Cell Calcium 2016;59:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol 2012; 9:561–575. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001;103:196–200. [DOI] [PubMed] [Google Scholar]
- 6.Xiao Z, Guo W, Sun B, et al. Enhanced cytosolic Ca2+ activation underlies a common defect of central domain cardiac ryanodine receptor mutations linked to arrhythmias. J Biol Chem 2016;291:24528–24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimlicka L, Tung CC, Carlsson AC, Lobo PA, Yuchi Z, Van Petegem F. The cardiac ryanodine receptor N-terminal region contains an anion binding site that is targeted by disease mutations. Structure 2013;21:1440–1449. [DOI] [PubMed] [Google Scholar]
- 8.Jiang D, Xiao B, Yang D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci USA 2004;101:13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu N, Colombi B, Memmi M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 2006;99:292–298. [DOI] [PubMed] [Google Scholar]
- 10.Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol 2002;190:1–6. [DOI] [PubMed] [Google Scholar]
- 11.Wehrens XH, Lehnart SE, Huang F, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003;113:829–840. [DOI] [PubMed] [Google Scholar]
- 12.Jiang D, Wang R, Xiao B, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res 2005;97:1173–1181. [DOI] [PubMed] [Google Scholar]
- 13.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization: underlying mechanism and threshold for triggered action potentials. Circ Res 2000;87:774–780. [DOI] [PubMed] [Google Scholar]
- 14.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T. Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circ Res 2008;103:509–518. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 16.Zhang XH, Haviland S, Wei H, et al. Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium 2013;54:57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei H, Zhang XH, Clift C, Yamaguchi N, Morad M. CRISPR/Cas9 gene editing of RyR2 in human stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca2+ signaling. Cell Calcium 2018;73:104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang XH, Saric T, Mehrjardi NZ, Hamad S, Morad M. Acid sensitive ion channels are expressed in human induced pluripotent stem cell-derived cardiomyocytes. Stem Cells Dev 2019;28:920–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilliard FA, Steele DS, Laver D, et al. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol 2010;48:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lehnart SE, Terrenoire C, Reiken S, et al. Stabilization of cardiac ryanodine receptor prevents intracellular calcium leak and arrhythmias. Proc Natl Acad Sci U S A 2006;103:7906–7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung CB, Moretti A, Mederos y Schnitzler M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med 2012;4:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi S, Yano M, Uchinoumi H, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circulation 2010; 74:2579–2584. [DOI] [PubMed] [Google Scholar]
- 23.Mallanna SK, Cayo MA, Twaroski K, Gundry RL, Duncan SA. Mapping the cell-surface N-glycoproteome of human hepatocytes reveals markers for selecting a homogeneous population of iPSC-derived hepatocytes. Stem Cell Rep 2016; 7:543–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novak A, Barad L, Lorber A, et al. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J Cell Mol Med 2015;19:2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Penttinen K, Swan H, Vanninen S, et al. Antiarrhythmic effects of dantrolene in patients with catecholaminergic polymorphic ventricular tachycardia and replication of the responses using iPSC models. PLoS One 2015; 10:e0125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang XH, Morad M. Ca2+ signaling of human pluripotent stem cells-derived cardiomyocytes as compared to adult mammalian cardiomyocytes. Cell Calcium 2020;90:102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paul-Pletzer K, Yamamoto T, Ikemoto N, et al. Probing a putative dantrolene-binding site on the cardiac ryanodine receptor. Biochem J 2005;387:905–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi S, Yano M, Suetomi T, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol 2009;53:1993–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and –independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol 2010;588:4743–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polonen RP, Penttinen K, Swan H, Aalto-Setala K. Antiarrhythmic effects of carvedilol and flecainide in cardiomyocytes derived from catecholaminergic polymorphic ventricular tachycardia patients. Stem Cells Int 2018;2018:9109503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.