

Abstract
Spartalizumab is a humanized IgG4/κ mAb directed against human programmed cell death‐1 (PD‐1). In this phase I study, we investigated safety, pharmacokinetics, preliminary antitumor activity, and toxicity of spartalizumab in patients with advanced malignancies. Patients (n = 18) with a range of tumor types received spartalizumab i.v. at doses of 1, 3, and 10 mg/kg every 2 weeks until disease progression, unacceptable toxicity, or discontinuation at the discretion of the investigator or patient. Most patients (61%) had received five or more prior lines of therapy. No dose‐limiting toxicities were reported and, hence, the maximum tolerated dose was 10 mg/kg or more. Pharmacokinetics in Japanese patients aligned with those reported in a global dose‐escalation study. The safety profile was consistent with other approved anti‐PD‐1 mAbs; the most common drug‐related adverse events were maculopapular rash (22%), followed by malaise and increased blood alkaline phosphatase (11% each). Partial responses were reported in two patients (11%), one with transitional cell carcinoma and the other with hepatocellular carcinoma. In conclusion, this study confirmed the safety of spartalizumab given at a dose of up to 10 mg/kg every 2 weeks in Japanese patients with cancers.
Keywords: clinical trial, humanized monoclonal antibody, immunotherapy, Japan, maximum tolerated dose
This phase I study confirmed the safety of spartalizumab (PDR001), an anti‐programmed cell death‐1 (PD‐1) mAb, given at a dose of up to 10 mg/kg every 2 weeks in Japanese patients with advanced malignancies. No dose‐limiting toxicities were reported and the safety profile was consistent with other approved anti‐PD‐1 mAbs. The overall response rate was 11% in this heavily pretreated population.

1. INTRODUCTION
The programmed cell death‐1 (PD‐1) immune checkpoint receptor is expressed on activated T cells, regulatory T cells, and B cells. 1 , 2 Binding of PD‐1 to its ligands PD‐L1 and PD‐L2 results in the exhaustion or dysfunction of T cells, thereby increasing immune tolerance. 3 , 4 Blockade of the interaction between PD‐1 and PD‐L1 enhances antitumor immune responses by restoring effector T cell function. 2 Anti‐PD‐1 mAbs have shown clinical benefit in patients with advanced cancers, leading to their approval in a number of indications. 5 , 6 , 7 , 8
Spartalizumab (PDR001) is a humanized IgG4/κ mAb directed against human PD‐1; it binds to PD‐1 with high affinity, and blocks the binding of PD‐L1 and PD‐L2 to PD‐1. 9 Clinical data from a parallel global phase I/II dose‐escalation and ‐expansion study (NCT02404441) of spartalizumab in patients with advanced malignancies showed a well‐tolerated safety profile and dose‐proportional pharmacokinetics (PK) consistent with those observed with approved anti‐PD‐1 mAbs. 7 , 8 , 9 There was also some preliminary evidence of antitumor activity in diverse tumor types. 9 Spartalizumab has since shown antitumor activity in diseases known to be sensitive to PD‐1 inhibitors. 10 , 11 The potential of spartalizumab as the backbone for various combinations with immuno‐oncology or targeted agents, including drugs under clinical development, is being explored in multiple clinical trials with the aim of providing patients with novel effective treatments in a range of different indications. 12
In this study, we investigated the safety, tolerability, PK, and preliminary antitumor activity of i.v. spartalizumab monotherapy in Japanese patients with advanced malignancies.
2. MATERIALS AND METHODS
2.1. Study design
This was a phase I, multicenter, open‐label study (NCT02678260) in Japanese patients with advanced solid tumors, designed and sponsored by Novartis Pharmaceuticals Corporation. The study protocol was approved by an independent ethics committee or institutional review board for each center. The study was carried out according to the principles of the Declaration of Helsinki and was undertaken in compliance with Good Clinical Practice guidelines; written informed consent was obtained from each patient. Patients were treated with increasing doses of spartalizumab. Dose‐escalation decisions were based on data available from all dose levels evaluated including safety, dose‐limiting toxicities (DLTs), and all toxicities of grade 2 or higher during cycle 1 and available PK from evaluable patients.
2.2. Objectives
The primary objective of the study was to estimate the recommended dose and/or maximum tolerated dose (MTD) for spartalizumab in patients with advanced malignancies. Secondary objectives included: characterizing the safety, tolerability, and PK profile of spartalizumab, assessing the emergence of antispartalizumab Abs following i.v. infusions of spartalizumab (data will be reported separately), and evaluating the preliminary antitumor activity of spartalizumab.
2.3. Patient population
This study was open to adult (aged 18 years or older) Japanese patients with advanced/metastatic solid tumors that had progressed despite standard therapy or were intolerant to standard therapy, or for whom no standard therapy existed. Patients were required to have an ECOG performance status of 2 or higher. Patients could not enter the study if they had symptomatic central nervous system (CNS) metastases or CNS metastases requiring local therapy, renal or hepatic dysfunction, impaired cardiac function or clinically significant cardiac disease, a history of hypersensitivity reactions to other mAbs, an active or a history of drug‐induced interstitial lung disease/pneumonitis, or active, known, or suspected autoimmune disease. Other exclusion criteria included patients requiring chronic treatment with systemic steroid therapy, patients currently receiving systemic treatment with immunosuppressive medication or systemic anticancer therapy, radiotherapy, major surgery, or participated in an interventional study within 2 weeks of the study start.
2.4. Treatment plan
Spartalizumab was given as an i.v. infusion over 30 minutes once every 2 weeks (Q2W) until disease progression per immune‐related response criteria (irRC), unacceptable toxicity, or discontinuation at the discretion of the investigator or patient. Each treatment cycle lasted 28 days and patients were followed for safety evaluations 150 days after the last study treatment. The starting dose selected for the study was 1 mg/kg Q2W spartalizumab and was based on the available safety and PK data from the ongoing global dose‐escalation study. Delayed dosing of up to 7 days was allowed for recovery from adverse events (AEs). Dose adjustments and interruptions were allowed for patients who were unable to tolerate the protocol‐specified dose; however, dose reductions below 0.3 mg/kg were not permitted and if more than two consecutive doses of study drug were skipped due to drug‐related AEs, then the patient was discontinued from the study. For the purpose of dose‐escalation decisions, each cohort was comprised of three to six patients who were treated at the specified dose level. Dose‐escalation decisions were based on data available from all dose levels evaluated including safety, DLTs, and all toxicities of grade 2 or higher and available PK from evaluable patients. The recommended dose for the next cohort of patients was guided by the Bayesian logistic regression model (BLRM) with the escalation with overdose control (EWOC) principle. 13 , 14
2.5. Safety assessments
Dose‐limiting toxicities were assessed during the first 28 days of treatment. Regular safety assessments were carried out based on physical examination, laboratory parameters, cardiac assessments, and ECOG performance status. Adverse events were graded according to the NCI’s Common Terminology Criteria for Adverse Events version 4.03 and were assessed continuously throughout the study.
2.6. Response assessments
Tumor response was determined by local investigator’s assessment according to RECIST version 1.1 and irRC. Assessments were undertaken at screening and every two cycles through cycle 11 day 1, then every three cycles until progression of disease per irRC or patient withdrawal and again at the end of treatment. Partial or complete responses per RECIST version 1.1 and irRC were confirmed at least 4 weeks later. Similarly, progressive disease, per irRC, was confirmed after at least 4 weeks.
2.7. Pharmacokinetic assessment
Blood samples for PK analysis were taken predose and at 1, 24, 48, 72, 168, 240, and 336 hours postdose of cycles 1 and 3, predose of cycles 2 and 4, and predose and at 1 hour postdose of cycles 5 and 6, and at the end of treatment. Serum concentrations of spartalizumab were quantified by liquid chromatography‐mass spectrometry with the lower limit of quantification of 0.250 μg/mL. Pharmacokinetic parameters were determined using noncompartmental methods in cycles 1 and 3 using serum concentrations measured during days 1‐15 of each cycle.
2.8. Statistical analysis
Dose escalation was guided by a Bayesian analysis of DLT data during cycle 1. The relationship between the dose level of spartalizumab and the probability of DLT was modelled using the BLRM. 13 Dosing decisions were guided by the EWOC principle. 14 A dose could only be used for newly enrolled patients if the risk of excessive toxicity (DLT rate 33% or higher) at that dose was less than 25%, and the final estimate of the recommended dose and/or MTD also had to satisfy this criterion.
Statistical analysis of study data was descriptive due to the nature of the study. Data analysis was undertaken after the end of the study when all patients had discontinued the study treatment and completed the 150‐day posttreatment safety follow‐up.
3. RESULTS
3.1. Patient population and treatment
A total of 18 patients were enrolled from 18 February 2016; the last patient last visit was 1 September 2017. All 18 patients discontinued the study treatment (two due to AEs [one myositis and one interstitial lung disease], 16 due to progressive disease) and entered the posttreatment safety follow‐up period. Of these, 11 patients (61%) completed the safety follow‐up period and seven (39%) discontinued due to death; all deaths were attributed to disease progression. Patients had a wide range of primary tumor types and were all pretreated; most patients (61%) had received five or more different lines of treatment (Table 1). No patients had received prior treatment with a checkpoint inhibitor. The median duration of exposure to study drug was 10 weeks (range, 4‐48 weeks). Five patients (28%) received treatment for 20 weeks or more with one of those patients (in the 3 mg/kg cohort) having received treatment for 36 weeks or more. One patient (6%) required a single dose interruption due to an AE that was not suspected to be related to the study drug (cholangitis), and no patients required dose reductions.
TABLE 1.
Demographics and baseline characteristics of Japanese patients with advanced malignancies, by spartalizumab treatment group
| 1 mg/kg Q2W n = 6 | 3 mg/kg Q2W n = 6 | 10 mg/kg Q2W n = 6 | All patients N = 18 | |
|---|---|---|---|---|
| Median age, y (range) | 50 (29‐75) | 57 (47‐74) | 54 (32‐70) | 53 (29‐75) |
| Sex, n (%) | ||||
| Female | 5 (83) | 3 (50) | 3 (50) | 11 (61) |
| Male | 1 (17) | 3 (50) | 3 (50) | 7 (39) |
| ECOG performance status, n (%) | ||||
| 0 | 2 (33) | 2 (33) | 1 (17) | 5 (28) |
| 1 | 4 (67) | 4 (67) | 5 (83) | 13 (72) |
| Diagnosis of disease, n (%) | ||||
| Ovarian cancer | 2 (33) | 1 (17) | 0 | 3 (17) |
| Cervical cancer | 0 | 0 | 2 (33) | 2 (11) |
| Head and neck cancer | 1 (17) | 0 | 1 (17) | 2 (11) |
| Triple‐negative breast cancer | 0 | 2 (33) | 0 | 2 (11) |
| Other a | 3 (50) | 3 (50) | 3 (50) | 9 (50) |
| Median time since most recent relapse/progression, mo (range) | 1.3 (0.2‐2.6) | 0.7 (0.2‐2.5) | 0.5 (0.2‐1.3) | 0.9 (0.2‐2.6) |
| Number of prior treatment regimens | ||||
| 2 | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
| 3 | 0 (0) | 0 (0) | 2 (33) | 2 (11) |
| 4 | 0 (0) | 2 (33) | 0 (0) | 2 (11) |
| ≥5 | 5 (83) | 3 (50) | 3 (50) | 11 (61) |
| Therapy type at last treatment, n (%) | ||||
| Chemotherapy | 5 (83) | 5 (83) | 6 (100) | 16 (89) |
| Targeted therapy | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
Q2W, once every 2 wk.
Breast cancer, colorectal cancer, gastric cancer, hepatocellular carcinoma, and transitional cell carcinoma.
3.2. Dose determination and pharmacokinetics
No DLTs were reported in the 18 patients treated in the three dosing cohorts and, hence, dose levels up to 10 mg/kg were considered tolerable. This was supported by assessing other available data of safety, toxicity, and PK, as well as satisfying the EWOC principle: the posterior probability of DLT rate being 33% or higher at 10 mg/kg was 0.4% (less than 25%). The MTD of spartalizumab was not reached at the tested dose levels and it was, therefore, estimated to be 10 mg/kg or more Q2W.
Area under the concentration–time curve calculated to the end of a dosing interval (AUCtau) and maximum serum drug concentration (Cmax) in cycle 1 increased as spartalizumab dose increased from 1 mg/kg to 10 mg/kg (Table 2). A power model on log‐transformed scale was used to assess dose proportionality. The estimated slopes of the power model for AUCtau and Cmax were 1.06 and 1.13, respectively, close to 1, suggesting exposure increased in a dose‐proportional manner. Pharmacokinetic variability in Cmax and AUCtau between patients was low to moderate in cycle 1, with geometric coefficient of variation (%) ranging from 8.9% to 40.4%. The half‐life estimated in cycle 1 ranged from 12.1 to 22.0 days (Table 2). Following repeated doses of 1 mg/kg and 3 mg/kg, in which AUCtau was estimable in both cycle 1 and cycle 3, AUC accumulation (cycle 3 / cycle 1) was approximately twofold.
TABLE 2.
Primary pharmacokinetic (PK) parameters of spartalizumab in Japanese patients with advanced malignancies
| Primary PK parameter | 1 mg/kg Q2W n = 6 | 3 mg/kg Q2W n = 6 | 10 mg/kg Q2W n = 6 |
|---|---|---|---|
| Cycle 1 | |||
| Patients, n a | 6 | 6 | 6 |
| Geo‐mean AUCtau, day*μg/mL (Geo‐CV%) | 123 (40.4) | 392 (11.9) | 1380 (19.7) |
| Geo‐mean Cmax, μg/mL (Geo‐CV%) | 17.1 (37.3) | 59.1 (8.9) | 187 (18.1) |
| Median Tmax, h (range) | 1.66 (1.50‐1.92) | 1.68 (1.50‐2.13) | 1.58 (1.52‐1.83) |
| Geo‐mean T1/2, day (Geo‐CV%) | 22.0 (104.9) | 12.1 (21.8) | 14.0 (29.4) |
| Cycle 3 | |||
| Patients, n a | 2 | 4 | 3 |
| Geo‐mean AUCtau, day*μg/mL (Geo‐CV%) | 273 (36.0) | 744 (36.2) | b |
| Geo‐mean Cmax, μg/mL (Geo‐CV%) | 27.7 (19.8) | 90.3 (20.2) | 238 (26.0) |
| Median Tmax, h (range) | 1.57 (1.53‐1.60) | 1.70 (1.53‐1.97) | 71.2 (1.57‐333) |
| Geo‐mean T1/2, day (Geo‐CV%) | 27.5 c | 15.7 (20.4) | b |
Cmax, maximum serum drug concentration; Geo‐CV%, geometric coefficient of variation (%); Geo‐mean, geometric mean; Q2W, once every 2 wk; Tmax, time of maximum serum drug concentration.
Number of patients with evaluable PK parameters.
No patients were evaluable for area under the concentration–time curve calculated to the end of a dosing interval (AUCtau) or half‐life (T1/2).
Only one patient was evaluable for T1/2.
3.3. Safety
Adverse events suspected to be drug‐related were reported in 11 patients (61%), including grade 3/4 AEs in two patients (11%; Table 3). The most common drug‐related AEs were maculopapular rash (four patients [22%]), followed by malaise and increased serum alkaline phosphatase (two patients [11%] each). The two grade 3/4 AEs were increased serum alkaline phosphatase and increased serum creatine phosphokinase (one patient [6%] each). The incidence of AEs suspected to be related to the study drug was generally comparable across all doses. Serious adverse events (SAEs) suspected to be drug‐related were reported in two patients (11%) and both led to discontinuation of study drug according to the protocol; these were grade 2 myositis, in a patient with thymoma presenting with dysphagia, and grade 1 interstitial lung disease. One SAE (grade 3 enterocolitis) developed during the extended safety follow‐up period (154 days after the last dose of treatment) and was reported as suspected to be drug related; it should be noted, however, that this SAE resolved without treatment after 18 days.
TABLE 3.
All grade and grade 3/4 adverse events (AEs) suspected to be related to study drug treatment in the phase I trial of spartalizumab in Japanese patients with advanced malignancies
| AE, n (%) | 1 mg/kg Q2W n = 6 | 3 mg/kg Q2W n = 6 | 10 mg/kg Q2W n = 6 | All patients N = 18 | ||||
|---|---|---|---|---|---|---|---|---|
| All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | All grades | Grade 3/4 | |
| Total | 5 | 2 | 3 | 0 | 3 | 0 | 11 (61) | 2 (11) |
| Maculopapular rash | 0 | 0 | 2 | 0 | 2 | 0 | 4 (22) | 0 (0) |
| Malaise | 1 | 0 | 0 | 0 | 1 | 0 | 2 (11) | 0 (0) |
| Increased blood alkaline phosphatase | 2 | 1 | 0 | 0 | 0 | 0 | 2 (11) | 1 (6) |
| Hyperthyroidism | 0 | 0 | 0 | 0 | 1 | 0 | 1 (6) | 0 (0) |
| Hypothyroidism | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Dysphagia | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Increased ALT | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Increased AST | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Increased blood creatine phosphokinase | 1 | 1 | 0 | 0 | 0 | 0 | 1 (6) | 1 (6) |
| Decreased blood thyroid‐stimulating hormone | 0 | 0 | 1 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Increased free thyroxine | 0 | 0 | 1 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Decreased appetite | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Hyperglycemia | 0 | 0 | 1 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Hypoalbuminemia | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Hypokalemia | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Hypophosphatemia | 0 | 0 | 1 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Myalgia | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Myositis | 1 | 0 | 0 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Interstitial lung disease | 0 | 0 | 0 | 0 | 1 | 0 | 1 (6) | 0 (0) |
| Onychomadesis | 0 | 0 | 1 | 0 | 0 | 0 | 1 (6) | 0 (0) |
| Pruritus | 0 | 0 | 0 | 0 | 1 | 0 | 1 (6) | 0 (0) |
| Rash | 0 | 0 | 1 | 0 | 0 | 0 | 1 (6) | 0 (0) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; Q2W, once every 2 wk.
Adverse events regardless of cause are reported in Table S1. Serious AEs, regardless of cause, were reported in seven patients (39%); three patients (17%) experienced grade 3/4 SAEs including ileus, esophageal varices hemorrhage, and cholangitis. The SAE of cholangitis required a temporary dose interruption of spartalizumab.
Immune‐related AEs of special interest were reported in 11 patients (61%; Table 4). The most common were maculopapular rash (four patients [22%]), pruritus, increased serum alkaline phosphatase, increased aspartate aminotransferase, and diarrhea (three patients [17%] each). Three patients experienced grade 3/4 AEs of special interest; these were increased serum alkaline phosphokinase (two patients [11%]) and hyperglycemia (one patient [6%]), the other AEs of special interest were grade 1/2 in severity. One patient with thymoma (29‐year‐old woman) experienced drug‐related grade 2 myositis. Myositis was diagnosed on day 43 and the study drug was temporarily interrupted. The patient was treated with prednisolone and pyridostigmine bromide. The patient was positive for acetylcholine Abs; however, she did not have any symptoms of myasthenia gravis. The decision was ultimately taken for the patient to permanently discontinue from the study. The myositis improved to grade 1 and finally resolved. The myositis was considered to be an immune‐related toxicity of spartalizumab.
TABLE 4.
Adverse events (AEs) of special interest in the phase I trial of spartalizumab in Japanese patients with advanced malignancies
| AE, n (%) | 1 mg/kg Q2W n = 6 | 3 mg/kg Q2W n = 6 | 10 mg/kg Q2W n = 6 | All patients n = 18 |
|---|---|---|---|---|
| Any AE of special interest | 5 (83) | 3 (50) | 3 (50) | 11 (61) |
| Colitis | ||||
| Diarrhea | 1 (17) | 1 (17) | 1 (17) | 3 (17) |
| Endocrinopathies | ||||
| Decreased blood thyroid‐stimulating hormone | 0 (0) | 1 (17) | 0 (0) | 1 (6) |
| Hyperglycemia | 0 (0) | 1 (17) a | 0 (0) | 1 (6) |
| Hyperthyroidism | 0 (0) | 0 (0) | 1 (17) | 1 (6) |
| Hypothyroidism | 1 (17) | 0 (0) | 0 (0) | 1 (6) |
| Increased free thyroxine | 0 (0) | 1 (17) | 0 (0) | 1 (6) |
| Liver enzyme increases | ||||
| Increased AST | 2 (33) | 0 (0) | 1 (17) | 3 (17) |
| Increased blood alkaline phosphatase | 2 (33) a | 0 (0) | 1 (17) | 3 (17) |
| Increased ALT | 1 (17) | 0 (0) | 1 (17) | 2 (11) |
| Increased blood bilirubin | 1 (17) | 0 (0) | 0 (0) | 1 (6) |
| Nephritis | ||||
| Increased blood creatine | 0 (0) | 0 (0) | 1 (17) | 1 (6) |
| Other immune‐related events: muscular disorder | ||||
| Myositis | 1 (17) | 0 (0) | 0 (0) | 1 (6) |
| Pneumonitis | ||||
| Interstitial lung disease | 0 (0) | 0 (0) | 1 (17) | 1 (6) |
| Rash | ||||
| Maculopapular rash | 0 (0) | 2 (33) | 2 (33) | 4 (22) |
| Pruritus | 2 (33) | 0 (0) | 1 (17) | 3 (17) |
| Rash | 0 (0) | 1 (17) | 0 (0) | 1 (6) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; Q2W, once every 2 wk.
Grade 3/4 AEs; all other AEs are grade 1/2.
3.4. Preliminary antitumor activity
No patients achieved a complete response per investigator assessment according to RECIST version 1.1 criteria (Table 5, Figure 1; post‐hoc analysis, Figure S1). Two patients (11%), one with transitional cell carcinoma and the other with hepatocellular carcinoma, achieved a partial response resulting in an overall response rate of 11% (95% confidence interval [CI], 1‐35). The duration of response for the patient with transitional cell carcinoma (3 mg/kg cohort) was 107 days and for the patient with hepatocellular carcinoma (10 mg/kg cohort) was 40 days (based on a censored observation as the patient discontinued due to an AE [grade 1 interstitial lung disease]). A further four patients (22%) had stable disease as their best overall response, leading to a disease control rate of 33% (95% CI, 13‐59). The duration of stable disease in these four patients was 52 (censored observation as the patient discontinued due to an AE [grade 2 myositis]), 112 and 191 days for the patients in the 1 mg/kg cohort and 110 days for the patient in the 3 mg/kg cohort. No patient experienced an additional response by irRC criteria. Four of the 16 patients evaluable for measurement of percentage change as per RECIST 1.1 had some reduction in tumor size from baseline; tumor shrinkage was seen in all dose groups (Figures 1 and S1).
TABLE 5.
Best overall response per investigator assessment based on RECIST version 1.1 by spartalizumab treatment group in Japanese patients with advanced malignancies
| 1 mg/kg Q2W n = 6 | 3 mg/kg Q2W n = 6 | 10 mg/kg Q2W n = 6 | All patients N = 18 | |
|---|---|---|---|---|
| Best overall response, n (%) | ||||
| CR | 0 | 0 | 0 | 0 |
| PR | 0 | 1 (17) | 1 (17) | 2 (11) |
| SD | 3 (50) | 1 (17) | 0 | 4 (22) |
| Progressive disease | 3 (50) | 4 (67) | 5 (83) | 12 (67) |
| Overall response rate (CR + PR), % (95% CI) | 0 (0‐46) | 17 (0‐64) | 17 (0‐64) | 11 (1‐35) |
| Disease control rate (CR + PR + SD), % (95% CI) | 50 (12‐88) | 33 (4‐78) | 17 (0‐64) | 33 (13‐59) |
CI, confidence interval; CR, complete response; PR, partial response; SD, stable disease; Q2W, once every 2 wk.
FIGURE 1.
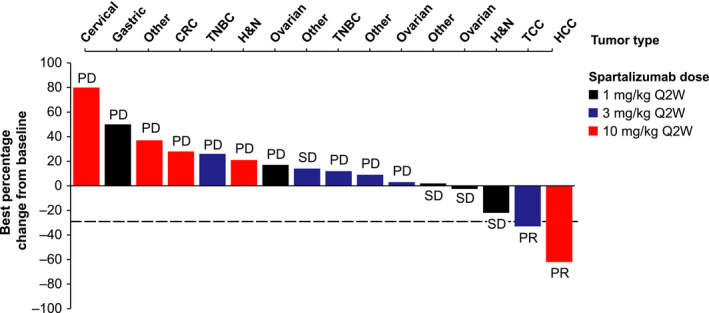
Waterfall plot of best percentage change from baseline in sum of measured diameters per investigator assessment based on RECIST version 1.1 in phase I trial of spartalizumab in Japanese patients with advanced malignancies. Only patients with measurable disease at baseline and at least one valid postbaseline assessment were included in this plot (n = 16). CRC, colorectal cancer; H&N, head and neck cancer; HCC, hepatocellular carcinoma; PD, progressive disease; PR, partial response; Q2W, once every 2 wk; SD, stable disease; TCC, transitional cell cancer; TNBC, triple‐negative breast cancer
4. DISCUSSION
Spartalizumab treatment was generally well‐tolerated in this Japanese population of heavily pretreated patients with advanced malignancies. Incidences of drug‐related AEs (61%) were consistent with those reported for spartalizumab in the parallel global study of patients with advanced tumors (59%). 9 The most common treatment‐related AEs in the current study included maculopapular rash (22%), malaise (11%), and increased serum alkaline phosphatase (11%) compared with fatigue (22%), diarrhea (17%), pruritus (14%), hypothyroidism (10%), and nausea (10%) in the dose‐escalation phase of the phase I/II global study. 9 The similarity of safety profiles between these two studies is important for the global development of spartalizumab. No new safety concerns were noted for Japanese patients. It should be noted that direct comparisons between this study and the global study should be treated with caution as the patient populations are not matched in terms of tumor types and prior treatment lines. The safety profile for spartalizumab treatment was also consistent with other approved anti‐PD‐1 mAbs. 7 , 8 Immune‐related AEs were observed in 11 patients (61%); the majority of these were mild in severity with only three patients (17%) reporting grade 3/4 AEs of special interest. Grade 2 drug‐related myositis was reported in one patient with thymoma. Myositis and myasthenia gravis are rare autoimmune disorders that cause muscular weakness. 15 Anti‐PD‐1 mAbs used to treat patients with various solid tumors, including thymoma, can trigger autoimmune complications such as myositis and myasthenia gravis. 16 , 17 , 18 The observation of myositis in the patient in this study is suggestive of the immunotherapeutic mechanism of action of spartalizumab.
No DLTs were reported in this study; the MTD was not reached and was estimated to be 10 mg/kg or more Q2W. Similarly, no DLTs were documented in the global phase I study of spartalizumab. 9 Based on similar binding affinities to the PD‐1 receptor and pharmacodynamic activity of spartalizumab and marketed inhibitors of PD‐1, 9 , 19 , 20 the criteria for dose determination used a PK end‐point to define a dose of spartalizumab that achieved an AUC0–336h of approximately 1000 μg*day/mL after cycle 3. 9 This exposure was achieved with 3 mg/kg Q2W or 5 mg/kg Q4W, hence 300 mg Q3W or 400 mg Q4W were recommended. 9 Accordingly, the same doses would also be appropriate for Japanese patients based on similarities between the safety and PK profiles of this study and the global study, and the decision was taken to terminate dose escalation in this study.
An overall response rate of 11% was reported based on investigator assessment using RECIST version 1.1. It should be noted that this is a heavily pretreated population with the majority of patients having received at least five lines of prior therapy. Furthermore, patients in this study had a range of primary tumor types; it is known that response rates vary across tumor types, and not all patients benefit from anti‐PD‐1 monotherapy. 21 Moreover, PD‐L1 expression was not a requirement for study entry; however, previous data have suggested that PD‐L1 expression is associated with response to anti‐PD‐1 therapies. 22 Consequently, this response rate was in line with expectations for this population based on data from the global spartalizumab study and early‐phase studies of other anti‐PD‐1 Abs. 23 , 24
In conclusion, spartalizumab was well‐tolerated in heavily pretreated Japanese patients with advanced malignancies. The MTD was not reached and, as no DLTs were observed across all dose levels tested, it was estimated to be 10 mg/kg or more Q2W. The study confirmed that the recommended dose determined in the global study of 400 mg Q4W or 300 mg Q3W would also be appropriate for patients from Japan. These regimens are currently being further investigated as a single agent in phase II of the global study in patients with anaplastic thyroid cancer, triple‐negative breast cancer, non‐small‐cell lung cancer, and unresectable or metastatic melanoma. 10 , 11 Spartalizumab is also being studied as a combination partner for various targeted therapies and other immunotherapies.
DISCLOSURE
H. Minami has received lecture fees, honoraria, or other fees from Ono Pharmaceutical and Daiichi‐Sankyo, research funds from Chugai Pharmaceutical, Ono Pharmaceutical, Pfizer, Novartis, and Bristol‐Myers Squibb, and scholarship endowments from Taiho Pharmaceutical, Chugai Pharmaceutical, Kyowa‐Kirin, Astellas, Daiichi‐Sankyo, Eli Lilly, Ono Pharmaceutical, Takeda Pharmaceutical, and Bayer Yakuhin. T. Doi has received fees as an advisor from Rakuten Medical, lecture fees, honoraria, or other fees from AbbVie, Bristol‐Myers Squibb, and Taiho Pharmaceutical, and research funds from Bristol‐Myers Squibb, Taiho Pharmaceutical, MSD, Novartis, Merck Serono, Eli Lilly, AbbVie, Boehringer Ingelheim, Eisai, Kyowa Hakko Kirin, IQVIA, Pfizer, Dainippon Sumitomo, and Daiichi‐Sankyo. M. Toyoda has received lecture fees, honoraria, or other fees from Taiho Pharmaceutical. Y. Naito has received lecture fees, honoraria, or other fees from Pfizer, Chugai Pharmaceutical, and Novartis, and research funds from Roche. N. Matsubara has received lecture fees, honoraria, or other fees from Janssen, and research funds from Janssen, AstraZeneca, MSD, and Roche. Y. Ando has received lecture fees, honoraria, or other fees from Chugai Pharmaceutical, and research funds from Chugai Pharmaceutical and Yakult Honsha. T. Tajima, K. Tokushige, K. Ishihara, and S. Cameron are employees of Novartis. The study was designed and sponsored by Novartis Pharmaceuticals Corporation. Novartis Pharmaceuticals Corporation collected and analyzed the data and contributed to the interpretation of the study. All authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication. Y. Imamura, N. Kiyota, and T. Shimokata have no conflicts of interest to disclose.
Supporting information
Figure S1
Table S1
ACKNOWLEDGMENTS
The authors would like to thank the patients who participated in the trial and their families, and the staff at each site who assisted with the study. Medical writing assistance was provided by Zoe Crossman, PhD, of Articulate Science Ltd and was funded by Novartis Pharmaceuticals Corporation (the study sponsor).
Minami H, Doi T, Toyoda M, et al. Phase I study of the antiprogrammed cell death‐1 Ab spartalizumab (PDR001) in Japanese patients with advanced malignancies. Cancer Sci. 2021;112:725–733. 10.1111/cas.14678
ClinicalTrials.gov registration number: NCT02678260
Funding information
Novartis Pharmaceuticals Corporation.
DATA AVAILABILITY STATEMENT
Novartis is committed to sharing with qualified external researchers, access to patient‐level data, and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided is anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described on www.clinicalstudydatarequest.com
REFERENCES
- 1. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riley JL. PD‐1 signaling in primary T cells. Immunol Rev. 2009;229(1):114‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. KEYTRUDA (pembrolizumab) . Highlights of Prescribing Information, MSD International GmbH, (revised 06/2018); 2014. https://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Accessed June 2020.
- 6. Opdivo (nivolumab) . Highlights of Prescribing Information, Bristol‐Myers Squibb Company, 2014. (revised 07/2018). https://packageinserts.bms.com/pi/pi_opdivo.pdfhttps://packageinserts.bms.com/pi/pi_opdivo.pdf. Accessed June 2020.
- 7. Brahmer JR, Hammers H, Lipson EJ. Nivolumab: targeting PD‐1 to bolster antitumor immunity. Future Oncol. 2015;11(9):1307‐1326. [DOI] [PubMed] [Google Scholar]
- 8. Khoja L, Butler MO, Kang SP, Ebbinghaus S, Joshua AM. Pembrolizumab. J Immunother Cancer. 2015;3(1):78‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Naing A, Gainor JF, Gelderblom H, et al. A first‐in‐human phase 1 dose escalation study of spartalizumab (PDR001), an anti‐PD‐1 antibody, in patients with advanced solid tumors. J Immunother Cancer. 2020;8(1):e000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin CC, Taylor M, Boni V, et al. Phase I/II Study of Spartalizumab (PDR001), an Anti‐PD‐1 mAb, in patients with advanced melanoma or non‐small cell lung cancer. Ann Oncol. 2018;29:1159P. [Google Scholar]
- 11. Wirth LJ, Eigendorff E, Capdevila J, et al. Phase I/II study of spartalizumab (PDR001), an anti‐PD1 mAb, in patients with anaplastic thyroid cancer. J Clin Oncol. 2018;36:6024. [Google Scholar]
- 12. ClinicalTrials.gov.www.clinicaltrials.gov. Accessed June 2020.
- 13. Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27(13):2420‐2439. [DOI] [PubMed] [Google Scholar]
- 14. Rogatko A, Schoeneck D, Jonas W, Tighiouart M, Khuri FR, Porter A. Translation of innovative designs into phase I trials. J Clin Oncol. 2007;25(31):4982‐4986. [DOI] [PubMed] [Google Scholar]
- 15. Paik JJ, Corse AM, Mammen AL. The co‐existence of myasthenia gravis in patients with myositis: a case series. Semin Arthritis Rheum. 2014;43(6):792‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fox E, Dabrow M, Ochsner G. A Case of Nivolumab‐Induced Myositis. Oncologist. 2016;21(12):e3‐0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez NL, Puwanant A, Lu A, Marks SM, Zivkovic SA. Myasthenia triggered by immune checkpoint inhibitors: new case and literature review. Neuromuscul Disord. 2017;27(3):266‐268. [DOI] [PubMed] [Google Scholar]
- 18. Yoshioka M, Kambe N, Yamamoto Y, Suehiro K, Matsue H. Case of respiratory discomfort due to myositis after administration of nivolumab. J Dermatol. 2015;42(10):1008‐1009. [DOI] [PubMed] [Google Scholar]
- 19. European Medicines A . CHMP Assessment Report ‐ Keytruda. https://www.ema.europa.eu/en/documents/assessment‐report/keytruda‐epar‐public‐assessment‐report_en.pdf. Published 2015. Accessed June 2020.
- 20. European Medicines A . CHMP Assessment Report ‐ Opdivo. https://www.ema.europa.eu/en/documents/assessment‐report/opdivo‐epar‐public‐assessment‐report_en.pdf. Published 2015. Accessed June 2020.
- 21. Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin Cancer Res. 2016;22(8):1865‐1874. [DOI] [PubMed] [Google Scholar]
- 22. Khunger M, Hernandez AV, Pasupuleti V, et al. Programmed cell death 1 (PD‐1) ligand (PD‐L1) expression in solid tumors as a predictive biomarker of benefit from PD‐1/PD‐L1 axis inhibitors: a systematic review and meta‐analysis. JCO Precis Oncol. 2017;1:1‐15. [DOI] [PubMed] [Google Scholar]
- 23. Patnaik A, Kang SP, Rasco D, et al. Phase I study of pembrolizumab (MK‐3475; Anti‐PD‐1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015;21(19):4286‐4293. [DOI] [PubMed] [Google Scholar]
- 24. Brahmer JR, Horn L, Antonia S, Spigel DR, Gandhi L, Sequist LV. Clinical activity and safety of anti‐PD1 (BMS‐936558, MDX‐1106) in patients with advanced non‐small‐cell lung cancer (NSCLC). J Clin Oncol. 2012;30(suppl):Abstract 7509. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Data Availability Statement
Novartis is committed to sharing with qualified external researchers, access to patient‐level data, and supporting clinical documents from eligible studies. These requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided is anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described on www.clinicalstudydatarequest.com