

Abstract
T cells are polarized toward regulatory T cells (Tregs) in tumor microenvironment by the shuttling of microRNAs that target T cell–activating signaling pathways. We evaluated the expression of the miR‐182 cluster (miR‐96, 182, and 183) in peripheral blood mononuclear cells (PBMCs) of patients with breast cancer (BC), and T cell polarization by the expression of FOXO1, NFATs, ITK, TCR/CD3 complex, and IL‐2/IL‐2RA. Twenty‐six microRNAs overexpressed in tumor tissues and sera of these patients were extracted by a meta‐analysis. Then, the expression of the miR‐182 cluster was investigated in PBMCs and sera of these patients and correlated with their targets in PBMCs. Finally, miR‐182 was cloned into Jurkat cells to evaluate its effects on T cell polarization. FOXO1, CD3d, ITK, NFATc3, NFATc4, and IL‐2RA were targeted by miR‐182, due to which their expression decreased in PBMCs of patients. Although IL‐6, IL‐17, and TGF‐β increased after miR‐182 transduction, IL‐2 dramatically decreased. We revealed CD4+FOXP3+ T cell differentiation in the miR‐182–transduced group. Although miR‐182 has inhibitory effects on T cells by the inhibition of FOXO1, TCR/CD3 complex, NFATs, and IL‐2/IL‐2RA signaling pathways, it increases FOXP3, TGF‐β, and IL‐17 expression to possibly drive T cell deviation toward the transitional state of IL‐17–producing Tregs and Treg formation in the end.
Keywords: breast cancer, FOXO1, FOXP3, IL‐2/IL‐2RA, Jurkat cells, miR‐182 cluster, NFATs, TCR/CD3
Although miR‐182 has inhibitory effects on T cells by the inhibition of FOXO1, TCR/CD3 complex, NFATs, and IL‐2/IL‐2RA signaling pathways, it increases FOXP3, TGF‐β, and IL‐17 expression to gradually deviate T cells toward a transitional state, namely IL‐17‐producing Tregs and facilitates Treg formation in the end.

Abbreviations
- APC
antigen‐presenting cells
- BC
breast cancer
- CD
cluster of differentiation
- DAG
diacylglycerol
- DMEM
Dulbecco's Modified Eagle Medium
- EYFP
enhanced yellow fluorescent protein
- FBS
fetal bovine serum
- FMO
fluorescence minus one
- FOXO1
forkhead box O1
- FOXP3
forkhead box P3
- IFN‐γ
interferon‐gamma
- IL
interleukin
- IP3
inositol triphosphate
- ITK
insight toolkit
- MAPK
mitogen‐activated protein kinase
- miRNA
microRNA
- NFATs
nuclear factor of activated T cells
- OD
optical density
- PBMCs
peripheral blood mononuclear cells
- PBS
phosphate‐buffered saline
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- RORα
RAR‐related orphan receptor alpha
- RORγt
RAR‐related orphan receptor gamma
- RPMI
Roswell Park Memorial Institute Medium
- STAT
signal transducer and activator of transcription
- TBS
Tris‐buffered saline
- TBST
Tris‐buffered saline with Tween 20
- TCR
T cell receptor
- TGF‐β
transforming growth factor‐beta
- Th
T helper cell
- TNF‐α
tumor necrosis factor‐alpha
- Treg
regulatory T cell
1. INTRODUCTION
Cells interact with other cells to affect their target genes via secretory components. 1 Numerous compartments, such as microRNAs, are found in exosomes and microvesicles as secretory components, which circulate throughout the body. The content of the cargoes secreted from various cells may switch the physiologic state to carcinogenic state. 2 These cargoes are secreted from both cancerous cells or tumor microenvironment, which impair the immune system and accelerate the development of different cancers. 3 , 4 MicroRNAs (miRNAs) are short (18‐23 nucleotide) noncoding RNAs that post‐transcriptionally regulate various complementary mRNAs. Dysregulated microRNA patterns of serum are useful in detecting pathologic conditions, such as cancers. 5
Increased expression of the miR‐182 cluster in breast cancer (BC) has been previously shown. 5 , 6 , 7 , 8 Establishing the miR‐182 cluster (miR‐96, 182, and 183) has effective roles in proliferation, migration, and invasion of BC cells (MCF‐7 and MDA‐MB‐231 cell lines) via targeting the forkhead box O1 (FOXO1). 9 Although the role of FOXO1 has been previously established in the induction of FOXP3 to produce regulatory T cells (Tregs), and thereby to dampen antitumor immune responses, there were some pieces of evidence that increment of some microRNAs, such as miR‐182, which can target FOXO1, facilitates T cell differentiation toward Tregs. 10 , 11 , 12 , 13 , 14 , 15 , 16 However, there was a controversial explanation about targeting FOXO1 by miR‐182, and its effects on differentiation of T cells toward Tregs and Th17 cells, which play prominent roles in BC development.
It should be noted that the main transcription factor which impacts Treg formation is FOXP3, and its binding to RORγt inhibits Th17 polarization, whereas its reduction helps RORγt activation and Th17 polarization. The expression of FOXP3 is regulated by more than 10 transcription factors that bind to the promotor of FOXP3. Some of them are suppressors, such as NFATs (all of NFATs isoforms) and STAT3, and some of them are inducers, eg, FOXO1 and STAT5. Therefore, the presence or absence of these transcription factors determines the fate of FOXP3 transcription and, consequently, Treg or Th17 polarization. 17
Previous studies have considered only the miR‐182‐5p isoform, whereas they did not evaluate miR‐182‐3p and its targets. 15 , 16 Moreover, there is some missing information about the other targets of miR‐183 and miR‐96 associated with T cell differentiation toward Tregs.
Therefore, further investigation is needed to provide insight into possible regulatory roles of the miR‐182‐183‐96 cluster in tumor progression as well as immune cell suppression. Several microRNA‐related databases, such as http://www.targetscan.org and http://www.mirdb.org, can be used to explore the other targets of the miR‐182‐183‐96 cluster associated with signaling pathways in T cell activation, including CD3d, ITK, NFATc3, NFATc4, IL‐2, and IL‐2RA. Hence, their impairment may weaken activated T cell functions. 18 Previous studies revealed that any attenuation, in signal transduction induced by antigen‐presenting cells (APC), may weaken the signaling of the T cell surface receptor/CD3 complex, thereby preventing the activation of MAPK, IP3, and DAG, and deviating T cells’ fates toward Treg cells. 19 , 20 CD3d, as one of the TCR/CD3 complex chain, contributes to triggering T cell signal transduction. 18 The TCR/CD3 complex induces some adaptors, eg, PLCγ1, to recruit some kinases, such as ITK, and, eventually, engage some of the required transcription factors, such as NFATs, AP‐1, and NF‐κB, for T cell activation and proliferation. 18
Nuclear factor of activated T cells plays a key role in lymphocyte proliferation, cytokine and chemokine production, and differentiation of naive T cells into various effector or memory T cells. 21 Also, NFATs reduce the expression of FOXP3 as a required transcription factor for Treg differentiation and function. 22 , 23 The activation and translocation of NFATs are directly controlled by calcineurin‐dependent dephosphorylation through the TCR/CD3 complex. 21 NFAT family comprises four transcription factors, including NFATc1‐c4. 21 , 24 Detailed bioinformatic studies have shown that NFATc2, NFATc3, and NFATc4 are specifically targeted by the miR‐182‐183 cluster (http://www.targetscan.org). NFATc1 is required for Th2 differentiation, while NFATc2 and NFATc3 promote Th1 differentiation and inhibit Th2 differentiation. 21 , 25 , 26 Inhibition of each protein may result in disruption of T cell activation or differentiation pathways. 27 , 28 Moreover, NFATc4 affects thymic development and survival of T cells. 21 NFATc1 and NFATc2 enhance NFATc4 recruitment to bind their target cytokine promoters to induce different cytokines, such as IL‐2, IL‐4, and IFN‐γ. 21
IL‐2 is a cytokine that facilitates the proliferation and promotion of helper CD4+ and cytotoxic CD8+ T cells, 29 and it is also necessary for the stability of Treg cells. Interestingly, the increment of Treg cells has been observed in patients with BC receiving IL‐2 29 , 30 because it can induce the overexpression of the miR‐182‐183 cluster, which impairs FOXO1 expression. 12 , 31 , 32 , 33 Although miR‐182‐183 expression is induced by IL‐2, bioinformatics studies have shown that miR‐182‐183 potentially targets the IL‐2/IL‐2RA pathway and NFATs as IL‐2 inducers (http://www.targetscan.org).
Researchers have investigated a number of onco‐microRNAs using high‐throughput techniques in tumor tissues and sera of patients with BC, which may influence CD3 chains, ITK, NFAT isoforms, and IL‐2/IL‐2RA and are involved in the differentiation of Th1 and Th2 cells. 5 , 7 In this study, we aimed to evaluate the expression levels of miR‐182‐5p, miR‐182‐3p, miR‐183, and miR‐96 in PBMCs of patients with BC and determine the correlation between the miR‐182‐183‐96 cluster and transcripts of CD3d, ITK, NFATc3, NFATc4, and IL‐2/IL‐2RA in PBMCs of patients with BC compared with healthy controls. In addition, we cloned both miR‐182‐5p and ‐3p into Jurkat cells to clarify how T cells are polarized toward Treg or Th17 cells.
2. MATERIALS AND METHODS
This meta‐analysis and experimental study were performed in three phases, including meta‐analysis, clinical studies, and cellular studies from November 2018 to April 2020. First, we selected the microRNAs using the data derived from Gene Expression Omnibus (GEO), which increased in the tumor tissues and serum of patients with BC. Then, the expression of the miR‐182 cluster was investigated in PBMCs and sera of these patients and correlated with their targets in PBMCs. Finally, considering the dominant population of T cells in PBMCs, we aimed to evaluate the expression levels of miR‐182‐5p, miR‐182‐3p, miR‐183, and miR‐96 in PBMCs of patients with BC and determine the correlation between the miR‐182‐183‐96 cluster and transcripts of CD3d, ITK, NFATc3, NFATc4, and IL‐2/IL‐2RA in PBMCs of patients with BC compared with healthy controls. Finally, miR‐182 was cloned into Jurkat cells to evaluate its effects on T cell polarization.
2.1. Bioinformatics analysis
Several microRNAs that had onco‐microRNA properties and immunosuppressive effects on T cells were selected based on microarray results associated with variation of microRNAs. They showed a significant elevation in both BC tissues and serum of these patients in previous studies deposited in https://www.ncbi.nlm.nih.gov/geo by accession numbers of GSE73002, GSE83270, GSE41526, and GSE24509. Then the targets of these microRNAs, which might have a role in inducing the signal transduction in T cells, were predicted using databases such as “http://www.targetscan.org” and “http://www.mirdb.org” and Also, target scan database (http://www.targetscan.org) to predict microRNA‐binding sites on 3′UTR of each gene. Complete predicted position of the target beside complete direct links associated with each microRNA are shown in Table S1. In this regard, we selected miR‐182 cluster, including miR‐182‐5p, miR‐182‐3p, miR‐183, and miR‐96 to evaluate the effect of these microRNAs on the fate of T cells.
2.2. Patients with BC
The first group consisted of 42 patients with BC, aged 22‐75 years who were patients in stages I, II, III without administration of immunosuppressive chemotherapy regimens/radiotherapy. The second group comprised 40 healthy (control) patients aged 27‐70 years who were referred to Shohada Hospital. Written informed consent was obtained from the participants before the study. Pathologic data were gathered using a questionnaire from the pathology department. This study was approved by the Ethics Committee of the Cancer Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran (Ethics code: IR.SBMU.RETECH.REC.1397.562). We confirm that all methods were performed in accordance with the relevant guidelines and regulations. We included patients who were new cases and were in stages I, II, III without administration of immunosuppressive chemotherapy regimens/radiotherapy, glucocorticoid, corticosteroid, or narcotic to avoid any changes in immune cells. Also, we excluded patients who received neoadjuvant chemotherapy before the study. Clinical characteristics and pathologic data associated with the patients are shown in Table S2.
2.3. Cell culture and reagents
The Hek‐293‐T and Jurkat cells were provided by the Pasteur Institute of Iran. They were suspended in a growth medium containing Dulbecco's Modified Eagle Medium (DMEM) and Roswell Park Memorial Institute (RPMI) 1640 medium, supplemented with 10% fetal bovine serum (FBS), 100 mg/mL streptomycin, and 100 U/mL penicillin (all from Gibco). Then the cells were cultured in T12.5 plates and incubated at 37°C in a humidified 5% CO2 incubator for 24 hours. Moreover, a combination of PMA (25 ng/mL) + calcium ionophore ionomycin (1 ug/mL) was used to activate T cells.
2.4. Mir‐182 cloning and transduction
Mir‐182 precursor sequences were derived from human chromosomal DNA by conducting PCR technique. Then, PCR products were cloned in PEZ‐LV105a vector (GeneCopoeia) using XmnI (Sinaclon) and NotI (Sinaclon) restriction enzymes. Lentivirus‐producing vectors, including PEZ‐LV105a, packaging (psPAX2) vectors, and envelope (Pmd2G) vectors were gifted by Dr Bahram Goliaei's lab from the Institute of Biochemistry and Biophysics (IBB). Subsequently, lentivirus was transferred to the plasmids in Hek‐293‐T cells. Finally, the cells were seeded in duplicate in 12‐well plates (1.5 × 105 cells/well) and treated with different volumes of the viruses based on the manufacturer's protocols and incubated at 37°C in a humidified 5% CO2 incubator for 24, 48, and 72 hours. To evaluate mir‐182’s effects on Jurkat cells, we used two control groups, including mock (lentivirus backbone) and nontreated groups.
2.5. Plasmid constructs for luciferase assay
To show the direct repression of the mentioned targets by miR‐182‐3p, we constructed luciferase‐UTR reporter plasmids for 3′UTR of CD3d, ITK, NFATc3, and IL‐2RA by amplification of fragments, and a fragment from 3′UTR of FOXP3 amplified as a negative control for miR‐182. Moreover, we constructed luciferase‐UTR reporter plasmids for mutated 3′UTR of CD3d, ITK, NFATc3, IL‐2RA, and FOXO1 amplified for miR‐182 to show further specificity of miR‐182. Then, the amplified fragments were cloned into psiCHECK‐2 vector using Xho I and Not I restriction enzymes. Finally, miR‐182 and 3′UTR of all CD3d, ITK, NFATc3, IL‐2RA, and FOXO1 were cotransfected using 1 μg DNA containing miR‐182 and 3′UTR of each target gene engulfed in lipofectamine 2000 reagent in 96‐well plates. We used approximately 104 cells per well, and dual luciferase assay was performed after 48 hours of transfection. Also, we used mock and mutated 3′UTR of each target gene, specially FOXP3, as the negative control. A mutation was generated in the binding site of mikR‐182 to provide mutated 3'UTR of each gene using primers mentioned in supplementary file (Tables S3‐S8); subsequently, the amplified fragments were cloned into psiCHECK‐2. It is noteworthy that the accuracy of all of the amplified fragments was confirmed by sequencing, and all the information for plasmid constructs, cloning, sequence alignment, and primers are presented in Figures S1‐S7, Tables S3‐S8.
2.6. Luciferase assay
Direct interaction of miR‐182 with CD3d, ITK, NFATc3, IL‐2RA, FOXO1, and FOXP3 (negative control) was evaluated by transfection of the mentioned plasmids into HEK‐293t cells. Each time PEZ‐LV105a–containing pre‐miR‐182 was cotransfected with one of the psiCHECK‐2–containing Renilla luciferase genes located at the upstream of 3′UTR of CD3d, ITK, NFATc3, IL‐2RA, FOXO1, and FOXP3 genes. Also, an independent firefly luciferase gene was used as an internal control for each experiment in addition to the negative control group. To strengthen the conclusions, we provided 3'UTR‐mutated controls associated with the miR‐182 binding site on the 3'UTR region of CD3d, ITK, NFATc3, and IL‐2RA using specific primers, including one mismatch at the binding site of miR‐182. Also, we cloned them in psiCHECK‐2–containing Renilla luciferase gene, and cotransfected them with PEZ‐LV105a–containing pre‐miR‐182. Luciferase assay was carried out using DualGlo luciferase assay (Promega) 48 hours after cotransfection, according to the manufacturer's instructions. Eventually, luminescence ratios of Renilla/firefly luciferase were assessed for each sample, which was normalized to the control transfection sample that contained each target cassette alone in the condition without transfection of miR‐182.
2.7. Serum isolation and preparation
Five milliliter peripheral blood was collected in tubes, containing EDTA and centrifuged at 1600 g at 4°C for 15 minutes. The sera were stored at −70°C to assess TNF‐α, IL‐1β, IL‐10, and TGF‐β. Moreover, an equal volume of phosphate‐buffered saline (PBS) was added to each blood sample and diluted gently. Ficoll‐Hypaque (Pharmacia) density centrifugation was used to isolate the PBMCs, and the buffy coat containing lymphocyte population was collected.
2.8. RNA extraction and qRT‐PCR
Total RNA was extracted from Jurkat cells and PBMCs using the RiboEx LS reagent (Geneall), and cDNA was synthesized using a first‐strand cDNA synthesis kit (Thermo Fisher Scientific) followed by PCR according to the manufacture's protocol. We used specific hairpin loop primers to synthesize cDNA of microRNAs. The expression of microRNAs (miR‐182‐5p, miR‐182‐3p, miR‐183, and miR‐96) and their targets were evaluated using SYBR Green master mix kit (Genaxxon kit) with MIC qPCR (BioMolecular Systems) instrument. The primer sets are listed in Table 1. Eventually, qRT‐PCR–derived data were analyzed by the and methods.
TABLE 1.
The primer sequences used in the current study
| Stem loops for cDNA synthesis of microRNAs | |
|---|---|
| 182‐5p | 5′‐GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC AGTGTGAG ‐3′ |
| 182‐3p | 5′‐GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC TAGTTGGC ‐3′ |
| miR‐183‐5p | 5′‐GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGTGAATTC‐3′ |
| miR‐96‐5p | 5′‐GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCAAAAATG‐3′ |
| RNU6 | 5′‐GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAAATAT‐3′ |
| Forward | Reverse | |
|---|---|---|
| Primers for qRT‐PCR | ||
| miR ‐182‐5p | 5′‐GTGCAGGGTCCGAGGT‐3′ | 5′‐GATTTGGCAATGGTAGAACTCAC‐3′ |
| miR ‐182‐3p | 5′‐GTGCAGGGTCCGAGGT‐3′ | 5′‐CAATGGTTCTAGACTTGCCAACT‐3′ |
| miR‐183‐5p | 5′‐GTGCAGGGTCCGAGGT‐3′ | 5′‐ATACTATGGCACTGGTAGAATTCACT‐3′ |
| miR‐96‐5p | 5′‐GTGCAGGGTCCGAGGT‐3′ | 5′‐TAATTTGGCACTAGCACATTTTTGC‐3′ |
| RNU6 | 5′‐CGCTTCACGAATTTGCGTGTC‐3′ | 5′‐CGCTTCGGCAGCACATATACT‐3′ |
| Gapdh | 5′‐CCGAGCCACATCGCACAG‐3′ | 5′‐GGCAACAATATCCACTTTACCAG‐3′ |
| Β‐actin | 5′‐AGACGCAGGATGGCATGGG‐3′ | 5′‐GAGACCTTCAACACCCCAGCC‐3′ |
| CD3D | 5′‐AAGTGAGCCCCTTCAAGATACC‐3′ | 5′‐TCTGAGAGCAGTGTTCCCAC‐3′ |
| ITK | F5′‐ACTCCTGAAGACAACAGGCGA‐3′ | 5′ATCCTTCATGCCCATTCCTGTC‐3′ |
| NFATC3 | 5′‐GTGAAGCTCCTGGGCTATAACG‐3′ | 5′‐TATCTCTTGGCTTGCAGTAGCG‐3′ |
| NFATC4 | 5′‐GAAGGGTGAGACGGACATCG‐3′ | 5′‐TTGGAGCCAGTCAGTACCAGT‐3′ |
| IL‐2 | 5′‐AAGGCCACAGAACTGAAACATC‐3′ | 5′‐ATTGCTGATTAAGTCCCTGGGT‐3′ |
| TNF‐α | 5′‐ATGTTGTAGCAAACCCTCAAGC‐3′ | 5′‐AGGACCTGGGAGTAGATGAGG‐3′ |
| TGF‐β | F5′‐ AGTGGACATCAACGGGTTCAC‐3′ | 5′‐TGGAGCTGAAGCAATAGTTGGTG‐3′ |
| IL‐6 | 5′‐ GCACTGGCAGAAAACAACCT‐3′ | 5′‐ CAGGGGTGGTTATTGCATCT‐3′ |
| IL‐2RA | 5′‐ GATGCCAAAAAGAGGCTGACG‐3′ | 5′‐CCACATCAGCAGGTATGAATCCA‐3′ |
| FOXO1 | 5′‐ GAGGGTTAGTGAGCAGGTTACAC‐3′ | 5′‐ TGCTGCCAAGTCTGACGAAAG‐3′ |
| FOXP3 | 5′‐TTCATCTGTGGCATCATCCGAC‐3′ | 5′‐ GTCGCATGTTGTGGAACTTGAAG‐3′ |
| Primers for cloning | ||
| MIR‐182 | 5′‐TTGGAAGGAGTTCGACCCTGCAGGAAGGACCTTG‐3′ | 5′‐GTTGCGGCCGCGGATGCAGGGAAACACAGAGTG‐3′ |
2.9. Cytokine assay
After the transduction of miR‐182, to evaluate the acquisition of regulatory functions by Jurkat cells, the secretion of some cytokines, including TNF‐α, TGF‐β, IL‐2, IL‐1β, IL‐17, IL‐10, and IL‐6 were assessed using ELISA kits (International Brands Limited [IBL]), according to the manufacturer's protocol. To this aim, optical density (OD) was measured at 450 nm by an ELISA reader (ELX800TM).
2.10. Western blotting
Western blotting was conducted to evaluate the expression of FOXO1, NFATc3, CD3d, and GAPDH in transduced Jurkat cells. Proteins were extracted from the mock group, negative control, and miR‐182–transduced groups of Jurkat cells. Therefore, Bradford assay was used to normalize the concentration of the extracted samples. Then 20 μg of each protein sample was subjected to SDS‐PAGE in 14% acrylamide gels and transferred to a PVDF membrane (Millipore). Additionally, 5% of nonfat dry milk and 0.05% Tween‐20 in Tris‐buffered saline (TBS) and Tris‐buffered saline with Tween 20 (TBST) were used for blocking and washing, respectively. The blots were probed with primary antibodies, including rabbit anti‐human CD3d (Abcam), rabbit anti‐human FOXO1 (Abcam), rabbit anti‐human NFATc4 (Merck), and mouse anti‐human GAPDH (Santa Cruz) diluted in TBST (1:1000) and incubated for 2 hours at room temperature (RT). After washing, the blots were incubated with HRP‐conjugated secondary antibodies, including goat anti‐rabbit IgG (Abcam) and anti‐mouse IgG κ light chain binding protein (Santa Cruz). Subsequently, it was developed by using an ECL Western blotting substrate (ECL, Amersham). In the following, the intensity of each band was measured using ImageJ (version 2) software and normalized by the intensity of GAPDH as a housekeeping protein. Effects of miR‐182 overexpression on NFATc3, FOXO1, and CD3d expression and FOXP3+ Tregs are shown in Figure S8.
2.11. Flow cytometry for human FOXP3 and IL17 production by FOXP3+ cells
Two milliliter of freshly prepared human FOXP3 buffer A was added to each sample, pulse vortexed and incubated at RT for 10 minutes. Cells were washed twice with 2 mL of PBS. Cells were permeabilized during 30 minutes of incubation at RT in 500 μL of freshly prepared human FOXP3 buffer C, protected from light. Finally, the cells were washed twice with 2 mL of PBS, and FOXP3 antibody was added. Flow cytometry technique was used for determination of FOXP3+ population. We used three types of control to determine FOXP3+ population (Figure S9). The first one was the untransduced and unstained group for obtaining the population of cells and their size. The second one was the unstained mock group to discriminate transduced cells, and the third one was the mock group with the staining by isotype control antibody coupled with FITC. Each test was performed in duplicate. Moreover, we costained these cells for FOXP3 and IL‐17 (antibody coupled to allophycocyanin [APC]) to confirm IL‐17 secretion within the FOXP3+ Treg population in miR‐182–transduced T cells. Further, we added protein transport inhibitor brefeldin A solution (Invitrogen™) to the harvested cells treated with PMA and ionomycin for 5 hours at 37°C to trap IL17 in the stimulated cells. We used APC‐CY7 live/dead color to discriminate living cells, and fluorescence minus one (FMO) control group was used to determine the cutoff point between background fluorescence and positive populations as well as unstained control. Flowing software 2.5.0 and FlowJo 7.6.1 software were used to analyze flow cytometry data.
2.12. Statistical analysis
R‐Studio 1.0.136 software was used to analyze the high‐throughput data derived from GEO (adjusted P‐value < .05) in both BC tissues and patient's sera. In the current study, P < .05 was considered statistically significant. Finally, multivariate analyses were performed to show the relationship between microRNAs and their targets’ expression levels in the PBMCs of BC patients and demographic data including age, marriage, stage, HER2, estrogen receptor (ER) and progesterone receptor (PR). Relative changes of microRNAs and their targets in PBMCs and the Jurkat cells were assessed using Student’s t‐test. Also, Student’s t‐test was performed to evaluate significant relative changes for luciferase assay in miR‐182–cotransfected groups with other control groups. One‐way ANOVA test was used to assess the rate of changes in each gene expression and cytokine release between the treated groups and followed by a post hoc Bonferroni test. A P‐value < .05 was considered statistically significant.
3. RESULTS
3.1. Alterations in the expression and circulation of microRNAs in tumor tissues and sera of patients with BC
Among 125 studies conducted on microRNA variation in patients with BC, four qualified studies were selected, which were not associated with any interventions and just showed alterations and circulation of microRNAs in tumor tissues and sera. A profile of microRNAs (64 microRNAs) that showed a significant elevation (33 in microRNAs with adjusted P‐value < .05 and 31 microRNAs just with P‐value < .05) for their expression in BC‐associated tumors were determined based on data derived from data sets (GSE41526). Then, their serum level alterations were evaluated by the data derived from the studies with the accession numbers GSE73002, GSE83270, and GSE24509 (Figure 1A).
FIGURE 1.
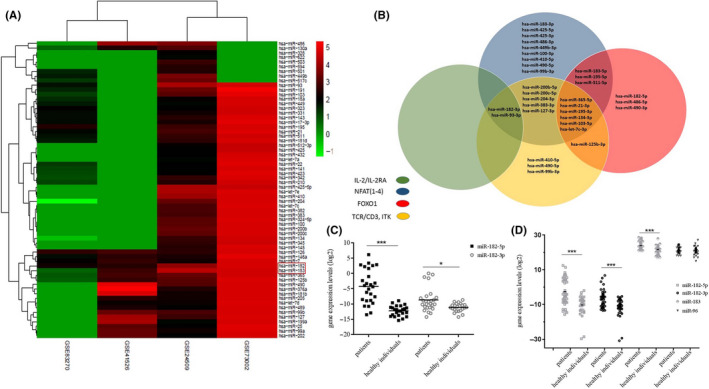
microRNAs’ variation, circulation, and feasible effects in breast cancer (BC) microenvironment as well as variation of the miR‐182‐182‐96 cluster in peripheral blood mononuclear cells (PBMCs) of BC patients. A, A profile of microRNAs, which have a significant alteration in BC patients’ tumor and serum levels (derived from Gene Expression Omnibus [GEO] database). B, microRNAs that predicted targets in the nuclear factor of activated T cells (NFAT) pathway and might be involved in regulatory T cell (Treg) development are shown as Venn diagram (derived from targetscan database). C, Variations of miR‐182‐5p and ‐3p in serum of BC patients (derived from serum data). D, Variations of miR‐182‐5p, ‐3p, miR‐183, and miR‐96 in PBMCs of patients with BC (derived from PBMC data)
The results derived from these meta‐analyses revealed 26 of these microRNAs significantly increased in both BC tissues and serum circulation had a dozen targets in T cells that played crucial roles in T cell suppression. It seems that they may target the transcripts of proteins involved in the NFAT pathway and cause the development of Treg cells by affecting CD3d, CD3g, ITK, NFATc1, NFATc2, NFATc3, NFATc4, IL‐2, IL‐2RA, FOXO1, etc (Figure 1B and Table 2). Also, at least 26 out of these 32 microRNAs highlighted in Figure 1B have the potential to affect both the NFAT pathway and Treg cell development, of which miR‐182, miR‐183, miR‐103, miR‐195, miR‐365, and miR‐511 can be mentioned. It was implied to each accurate target of these microRNAs in each mentioned pathway in Table 2. Of note, the miR‐182 cluster includes miR‐182, miR‐183, and miR‐96. However, despite the elevation in the expression and serum levels of both miR‐182 and miR‐183 in all considered studies, no alteration was observed in miR‐96 rate either in tumor tissues or in the serum of patients with BC.
TABLE 2.
Summary of the microRNAs with at least one predicted target, which play a critical role in T cell activation and differentiation signal IL‐2/IL‐2RA
| Targets | ||
|---|---|---|
| Isoform‐5p | Isoform‐3p | |
| hsa‐miR‐183 | FOXO1 | CD3d, NFATc4, NFAT5 |
| hsa‐miR‐425 | NFATc3 | NFATc2 |
| hsa‐miR‐93 | NFAT5 | NFATc1, NFATc3, NFATc4, NFAT5, CD3d, IL2RA |
| hsa‐miR‐410 | ITK | NFAT5 |
| hsa‐miR‐182 | FOXO1 | NFATc3, NFATc4, CD3d, ITK, IL‐2RA |
| hsa‐miR‐486 | FOXO1, NFAT5 | NFATc2, NFATc4 |
| hsa‐miR‐449b | NFATc4, NFAT5 | |
| hsa‐miR‐200b | NFATc3, NFAT5, ITK | |
| hsa‐miR‐365a/b | FOXO1, CD3d | CD3d, FOXO1, IL‐2RA |
| hsa‐miR‐204 | NFATc3, NFAT5 | CD3d, NFATc3, NFATc4, NFAT5, IL2‐RA |
| hsa‐miR‐200c | CD3d, ITK, NFATc2 | |
| hsa‐miR‐103a | NFATc2, NFATc3, NFATc4, NFAT5, FOXO1, ITK, CD3d, IL‐2RA | NFATc2, NFAT5 |
| hsa‐miR‐125b | FOXO1, NFAT5 | |
| hsa‐miR‐100 | NFATc2, NFAT5 | |
| hsa‐miR‐490 | CD3d, NFAT5, IL‐2RA | FOXO1, NFAT5 |
| hsa‐let‐7c | FOXO1, ITK, NFATc1, NFATc2, NFATc3, NFAT5 | |
| hsa‐miR‐99b | ITK, NFAT5 | |
| hsa‐miR‐383 | NFAT5 | NFATc3, NFAT5, CD3d |
| hsa‐miR‐21 | NFAT5 | CD3d, ITK, NFATc2, NFATc3, NFAT5 |
| hsa‐miR‐127 | NFATc2, ITK | |
| hsa‐miR‐503 | NFATc3, NFAT5 | |
| hsa‐miR‐195 | FOXO1, NFATc2, NFATc3 | FOXO1, CD3d, NFATc2 |
| hsa‐miR‐511 | FOXO1, NFATc3, NFATc4, NFAT5, IL‐2RA | NFATc2, NFAT5 |
| hsa‐miR‐134 | FOXO1, CD3d, NFATc2, NFATc3 | |
| hsa‐miR‐342 | NFATc1, NFATc3 | |
| hsa‐miR‐181d | NFATc2, NFAT5 | |
3.2. Circulating levels of miR‐182‐5p and miR‐182‐3p were increased in sera
Owing to the lack of studies on the expression of miR‐182‐3p in the sera of patients with BC, miR‐182‐5p and miR‐182‐3p were evaluated separately. The expression of miR‐182‐5p and miR‐182‐3p was significantly increased (350.4 folds and 8.66 folds, respectively) (Figure 1C).
3.3. MiR‐182 and 183 were increased in PBMCs
The results revealed that 213.34 folds, 28.4 folds, and 4.7 folds of increase were observed in the expression level of miR‐182‐5p, miR‐182‐3p, and miR‐183 in the PBMCs of the patients, respectively. However, no alteration was seen in the expression of miR‐96 (Figure 1D).
3.4. Target genes for the miR‐182 cluster decreased in PBMCs
The expression levels of FOXO1, CD3d, NFATc3, NFATc4, IL‐2, and IL‐2RA were downregulated by 2.5 folds, 3.9 folds, 13.6 folds, 3.25 folds, 3.38 folds, and 4.35 folds, respectively (Figure 2A). These genes are the targets for miR‐182‐5p/3p and miR‐183, and their expression significantly decreased in the PBMCs of the patients.
FIGURE 2.
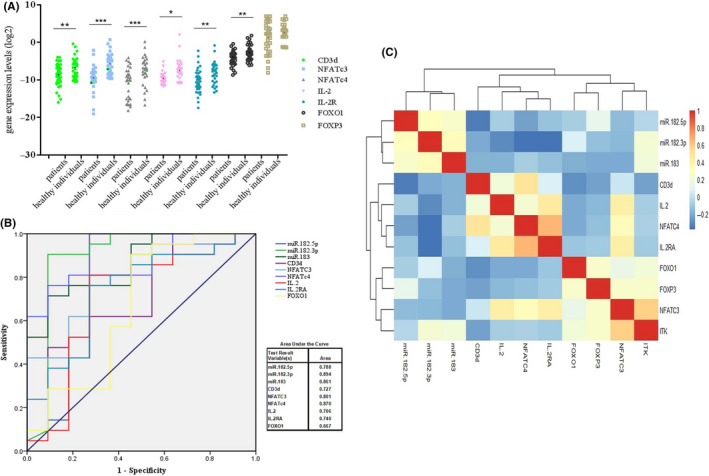
Comparison of the targets of miR‐182 cluster expressions in peripheral blood mononuclear cells (PBMCs) between patients with breast cancer (BC) and healthy individuals. A, Variation of the miR‐182 clusters’ targets in PBMCs of patients with BC. B, Receiver operating characteristic (ROC) curve for diagnosis and prognosis of BC tissues. The areas under the curves are the gene expression level of miR‐182‐5p, miR‐182‐3p, miR‐183, CD3d, NFATC3, NFATc4, IL‐2, IL‐2RA, and FOXO1 in PBMCs of BC patients as compared with PBMCs of healthy individuals. C, Hierarchical clustering analysis showing the relationship between expression variation of miR‐182‐5p/‐3p, miR‐183, CD3D, ITK, NFATc3, NFATc4, IL‐2, IL‐2RA, FOXO1, and FOXP3 in PBMCs of patients with BC
The receiver operating characteristic (ROC) curve showed the behavior of sensitivity and specificity of miR‐182‐5p, miR‐182‐3p, miR‐183, FOXO1, CD3d, NFATc3, NFATc4, IL‐2, and IL‐2RA expression levels by using their expression level in PBMCs of patients with BC. The area under the ROC curve for discrimination and prognosis of BC patients with healthy individuals were 0.788, 0.894, and 0.861 for miR‐182‐5p, miR‐182‐3p, and miR‐183, respectively (Figure 2B). The area under the ROC curve for discrimination and prognosis of BC patients with healthy individuals were 0.727, 0.801, 0.870, and 0.740 for CD3d (P = .037), NFATc3 (P = .06), NFATc4 (P = .01), and IL‐2RA (P = .028), respectively. No significant discrimination was observed for IL‐2 (P = .059) and FOXO1 (P = .127; Figure 2B). Also, statistical analysis showed that there was no significant relationship between miR182‐5p/‐3p, miR‐183, and their targets and any of the pathologic factors in the current study. Clinical data are presented in the supplementary file.
Further statistical analysis in the case of the miR‐182‐183‐96 cluster’s expression changes and their mentioned targets in PBMCs of BC patients showed that there was a negative correlation between miR‐182‐3p and their targets, such as CD3d (r = −.456, P = .029), NFATc4 (r = −.683, P < .001), IL‐2RA (r = −.627, P < .001), and IL‐2 (r = −.444, P ≤ .001). Also, there was a significant association between the reduction of CD3d and NFATC4 (r = .493, P ≤ .0). A significant relationship was observed between NFATc3 reduction and ITK (r = .582, P = .004), IL‐2 (r = .427, P = .042), and IL‐2RA (r = .43, P = .041). NFATc4 decrement correlated with CD3d (r = .493, P ≤ .01) and IL‐2RA (r = .0.641, P ≤ .001). Finally, the change in IL‐2 expression is also relevant to IL‐2RA expression (r = .459, P = .027), beside its relevance to NFATc3 (Figure 2C).
3.5. MiR‐182‐5p and miR‐182‐3p decreased target genes in Jurkat cells
After the transduction of miR‐182 in Jurkat cells (Figure 3A‐D), there were 12.52 folds and 7.41 folds of elevation in the expression of miR‐182‐5p and miR‐182‐3p, respectively (Figure 4A). Subsequently, a significant downregulation was observed in the expression of FOXO1, CD3d, ITK, IL‐2RA, NFATc3, and NFATc4. Most of the reduction associated with FOXO1, CD3d, NFATc3, and IL‐2RA that occurred within 48 hours after the transduction and after 72 hours was related to ITK and NFATc4 (Figure 4B). Furthermore, the expression of some cytokines, such as TNF‐α and IL‐2, along with the targets of miR‐182 showed a significant decrease. The highest reduction of TNF‐α and IL‐2 was observed at 72 and 48 hours, respectively. In contrast, an impressive increase was observed in FOXP3, IL‐6, and TGF‐β expressions in these cells. IL‐6 and TGF‐β showed the highest increase in 48 hours. Moreover, a significant increase was observed in the rate of FOXP3 as a fundamental transcription factor in Jurkat cells upon overexpression of miR‐182. The highest increase of FOXP3 (12.2 folds) was observed at 72 hours (Figure 4C).
FIGURE 3.
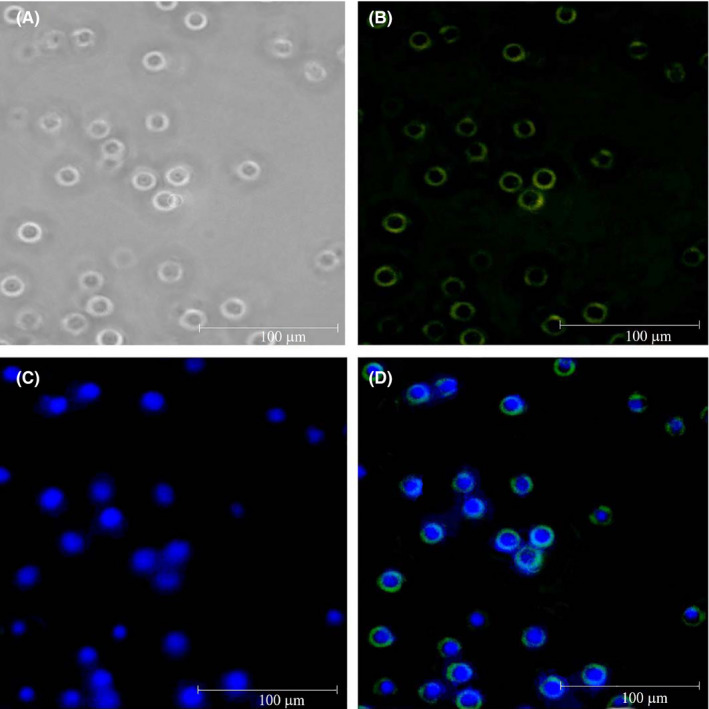
Immune fluorescent microscopy confirmed the successful transduction of the produced lentiviruses into Jurkat cells. A, Jurkat cell population. B, Expression of enhanced yellow fluorescent protein (EYFP) in Jurkat cells. C, Nucleus staining by DAPI. D, Overlaying of the transduced cells with DAPI staining
FIGURE 4.
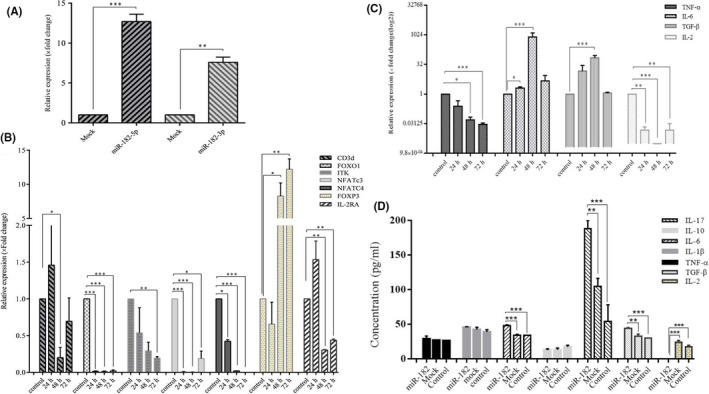
Overexpression of miR‐182 in Jurkat cells and its effects on the expression of different targets and various cytokine secretion. A, Overexpression of miR‐182‐5p and ‐3p in Jurkat cells. B, Effects of miR‐182 on CD3d, FOXO1, ITK, NFATc3, NFATc4, IL‐2RA, and FOXP3 in Jurkat cells. C, Effects of miR‐182 on TNF‐α, IL‐6, TGF‐β, and IL‐2 expression. D, Effects of miR‐182 on TNF‐α, IL‐1β, IL‐6, IL‐10, IL‐17, TGF‐β, and IL‐2. Data represent mean ± SD (error bars) of three independent experiments in triplicate assays (***P < .001, **P < .01 and *P < .05)
3.6. TGF‐β, IL‐17 and IL‐6 increased in Jurkat cells after transduction of miR‐182
Among inflammatory and anti‐inflammatory cytokines, TGF‐β, IL‐17, and IL‐6 showed a significant increase in the supernatant of Jurkat cells in 48 hours after the transduction (Figure 4D). TGF‐β, an anti‐inflammatory cytokine, elevated by 1.46 and 1.34 folds in the miR‐182–transfected group in comparison with mock and nontreated control groups, respectively. IL‐6 increased by 1.42 folds in the miR‐182–transfected group in comparison with both mock and nontreated control groups. Furthermore, IL‐17 increased by about 1.8 folds and 3.48 folds compared with mock and nontreated control groups, respectively. Analysis of cytokine assay showed that IL‐2 release was not detected in the miR‐182–transfected group, while it was observed in both mock and nontreated control groups. However, in spite of a significant decrease in TNF‐α expression, no variation was observed in TNF‐α concentration in the supernatant of Jurkat cells. Also, IL‐1β and IL‐10 showed no changes (Figure 4D).
3.7. CD3d, ITK, NFATc3, IL‐2RA, FOXO1 were directly regulated by miR‐182‐5p and miR‐182‐3p
Cotransfection of PEZ‐LV105a–containing miR‐182 sequence along with psiCHECK‐2 plasmid containing 3′UTR of each CD3d, ITK, NFATc3, and IL‐2RA showed a considerable reduction in Firefly/Renilla luciferase activity in comparison with mock (psiCHECK‐2 vector containing Firefly/Renilla luciferase gene) and control (psiCHECK‐2 plasmid containing 3′UTR of each gene at the end of the luciferase sequence) groups. It was observed that miR‐182 reduced the activity of luciferase by about 2.2, 1.42, 4.9, 1.7, 1.4, and 3.2 folds in the groups which contained 3′UTR sequence of CD3d (P < .01), ITK (P < .01), NFATc3 (P < .001), IL‐2RA (P < .001), and FOXO1 (P < .001) at the end of the luciferase gene. Moreover, it was observed that miR‐182 reduced the activity of luciferase bu about 1.97, 1.3, 4.16, 1.5 and 2.8 folds in the groups which contained 3′UTR‐mutated sequence of CD3d (P < .01), ITK (P < .01), NFATc3 (P < .001), IL‐2RA (P < .001), and FOXO1 (P < .001) at the end of the luciferase gene. So, these results clarified that miR‐182 directly targeted CD3d, ITK, NFATc3, IL‐2RA, and FOXO1 (Figure 5A). Moreover, the binding sites of the seed region of miR‐182‐3p and the way of its interaction on 3′UTR sequences of CD3d, ITK, NFATc3, FOXO1, and IL‐2RA are mentioned in Figure 5B. Also, no changes were observed in luciferase activity associated with miR‐182 function negative control group whenever the experiment was performed on psiCHECK‐2 vector that contained 3′UTR of FOXP3 (Figure 5A).
FIGURE 5.
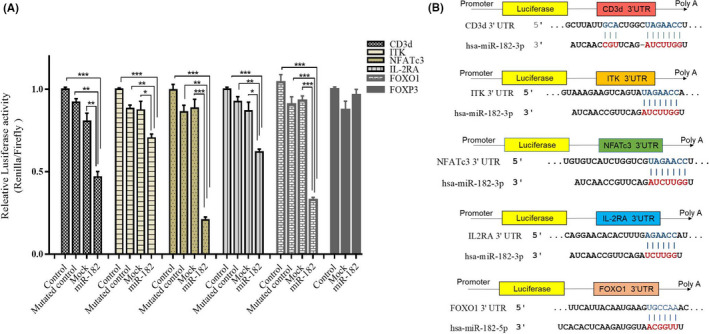
Direct targeting of CD3d, ITK, NFATc3, IL‐2RA, and FOXO1 by overexpression of miR‐182, and polarization of these cells toward FOXP3+ cells (Tregs). A, Comparison between cotransfection of constructs that contain Renilla luciferase reporter gene and 3′UTR of each CD3d, ITK, NFATc3, IL‐2RA, and FOXO1 sequence accompanied by miR‐182–overexpressing vector in HEK‐293t cells. Moreover, miR‐182–overexpressing vector with mutated controls of each gene were used to strengthen the specificity of miR‐182 in targeting of the mentioned targets. The other control group was a mock control which PEZ‐LV105a cotransfected with constructs that contain Renilla luciferase reporter gene and 3′UTR of the mentioned gene. The last control group was psiCHECK‐2 vector that contained Renilla luciferase transfected in HEK293t cells. Luciferase assay represents that miR‐182 is capable of interacting with 3′UTR of CD3d, ITK, NFATc3, IL‐2RA, and FOXO1 sequences cloned downstream of the luciferase ORF and declines luciferase ratios, in contrast to 3′UTR of mutated controls, mock, and FOXP3 (negative control). B, The binding site of CID region of miR‐182‐3p on 3'UTR sequences of CD3d, ITK, NFATc3, and IL‐2RA
3.8. CD3d, NFATc3, and FOXO1 decreased after miR‐182 transduction
Western blot results confirmed that the transduction of miR‐182 can target CD3d, NFATc3, and FOXO1 (Figure 6A). The findings showed that intensities of CD3d in the miR‐182–transduced group decreased by almost 1.8 and 1.5 folds compared with the mock and nontreated control groups, respectively. Likewise, NFATc3 concentration reduced by 4.5 folds and 2.3 folds in the miR‐182–transduced group in comparison with the mock and nontreated control groups, respectively. Similar results were shown regarding FOXO1 inmiR‐182–transduced group versus mock (2.4 folds) and nontreated control (10 folds) groups (Figure 6B).
FIGURE 6.
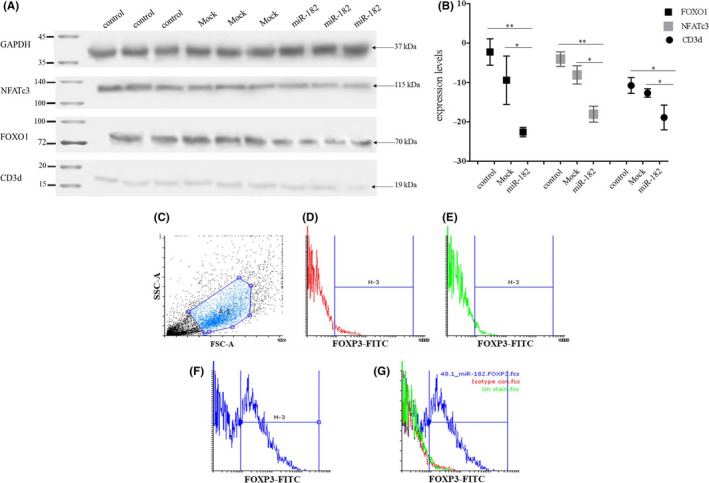
Effects of miR‐182 overexpression on NFATc3, FOXO1, and CD3d expression and FOXP3+ Tregs population differentiation. (A) Western blotting assay shows reduction of NFATc3, FOXO1, and CD3d by miR‐182 in Jurkat cells. B, Quantification: The intensity of each band using ImageJ confirms reduction of NFATc3, FOXO1, and CD3d by miR‐182 in Jurkat cells. In addition, comparison of NFATc3, FOXO1, and CD3d expression levels in the miR‐182–transduced group with mock and nontreated control groups. C, Graph of the cell population by using FSC‐A versus SSC‐A plot on unstained control group (mock). D, Isotype control group. E, Fluorescent intensity in the stained control group (mock). F, Fluorescent intensity in the stained group transduced by miR‐182. G, Comparison of overlaid and comparison of FOXP3 expression between the miR‐182 transduced group and both mock and isotype control groups. Anti‐FOXP3‐FITC; FSC‐A: forward scatter‐A; SSC‐A: side scatter‐A. Data represent mean ± SD (error bars) of three independent experiments in duplicate (***P < .001, **P < .01 and *P < .05)
3.9. FOXP3+ and IL‐17–producing Tregs are highly expressed in the Jurkat cells after miR‐182 transduction
Jurkat cells are CD4+ T cells, and the transduction of miR‐182 in these cells represented a distinctive population of CD4+ T cells that overexpressed FOXP3 in comparison with another control group (mock). The results obtained from flow cytometry showed that about 60% of the gated population expressed FOXP3+ in the cells transduced by miR‐182 (minus isotype control percentage as false positive) and indicated the polarization of T cells toward Treg cells (Figure 6C‐G). However, costaining of these cells for FOXP3 and IL‐17 showed that more than 55% of the population of FOXP3+ cells expressed IL‐17. So, it seems that miR‐182 may induce two different populations composed of FOXP3+ T cells and IL‐17–producing FOXP3+ in CD4+ Jurkat cells (Figure 7A‐H).
FIGURE 7.
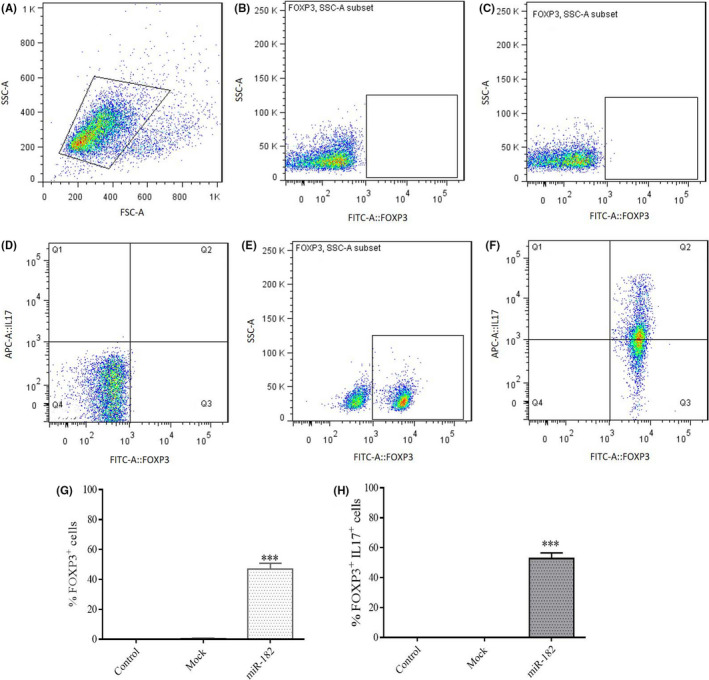
Effects of miR‐182 overexpression on intracellular expression of FoxP3 and IL‐17. A, Graph of the cell population by using FSC‐A versus SSC‐A plot in unstained control group stimulated with PMA/ionomycin and brefeldin A. B, FOXP3+ population in unstained control group stimulated with PMA/ionomycin and brefeldin A that was stained for intracellular FOXP3. C, FOXP3+ population in mock control group stimulated with PMA/ionomycin and brefeldin A that was stained for intracellular FOXP3. D, Quadrant showing the population of FOXP3‐IL17+ T cells (Q1), FOXP3+IL17+ T cells (Q2), FOXP3+IL17‐ T cells (Q3), and FOXP3‐IL17‐ T cells (Q4) in the control group (mock) stimulated with PMA/ionomycin and brefeldin A that was stained for intracellular FOXP3 and IL‐17. E, Population of cells transduced by miR‐182 that expressed intracellular FOXP3. F, Population of cells transduced by miR‐182 and stimulated with PMA/ionomycin and brefeldin A that was stained for intracellular FOXP3 and IL‐17. G, Percentage of FOXP3+ T cells after transduction by miR‐182 and stimulation with PMA/ionomycin and brefeldin A that was stained for intracellular FOXP3 and IL‐17. H, Percentage of FOXP3+ IL17‐producing T cells after transduction by miR‐182 and stimulation with PMA/ionomycin and brefeldin A that were stained for intracellular FOXP3 and IL‐17 (***P < .001). FSC‐A, forward scatter‐A; SSC‐A, side scattering; FITC‐A::FOXP3, Anti‐FOXP3‐FITC‐A; APC‐A::IL‐17, Anti‐IL‐17‐APC‐A
4. DISCUSSION
We tried to show a new landscape of the variation of microRNAs, which have similar targets in the mentioned pathways, by which they affect immune cells, especially T cells to make the tumor suitable for development, while suppressing immune cells or producing suppressor cells. Microarray‐derived results (meta‐analysis) revealed that the levels of some onco‐microRNAs increase in both tumor tissues and sera of patients with BC. Therefore, some of these microRNAs may target various transcripts of different proteins that are implicated in signaling pathways to activate, inhibit, proliferate, or differentiate T lymphocytes. 18 , 21 The targets predicted for these microRNAs are involved in TCR‐associated transduction pathways, including NFATs, IL‐2/IL‐2RA, TCR/CD3 complex, and FOXO1/FOXP3. 10 , 18 , 29
This meta‐analysis showed that most of the mentioned microRNAs have the potential to target at least two isoforms of NFATs, of which 12 simultaneously target one of the CD3 chains and the ITK protein. Accordingly, miR‐93, miR‐410, miR‐200b, miR‐365, miR‐204, miR‐200c, miR‐103, miR‐125b, miR‐490, Let‐7c, miR‐99b, miR‐383, miR‐21, miR‐127, miR‐195, and miR‐134 along with the miR‐182 cluster have the potential to target different chains of CD3 and ITK (http://www.targetscan.org and http://miRwalk.umm.uni-heidelberg.de/search_miRnas). However, the direct effects of a number of microRNAs, eg, miR‐125b, miR‐99b‐3p, miR‐21‐5p, miR‐21‐3p, miR‐181c, and Let‐7c in suppressing and targeting NFATs, which have critical roles in Th1 and Th2 differentiation, have been revealed in previous studies. 34
In addition, given the roles of the TCR/CD3 complex, ITK, and NFATs in T cell signaling, 18 , 21 these microRNAs simultaneously attenuate the signal transduction cascade triggered by the interaction of APCs with the T cell surface receptor/CD3 complex and ITK. Subsequently, this attenuation can prevent the activation of MAPK, IP3, and DAG signal transduction, impair T cell functions, and delineate T cells’ fates whether to be Treg cells. 34 , 35 The overexpression of miR‐182 in the current study showed a dramatic reduction in the gene expression of CD3d, ITK, NFATc3, and NFATc4 in Jurkat cells. Also, a decrease was observed in CD3d and NFATc3 protein levels. As mentioned before, NFATc3 (NFAT4) in addition to NFATc2 promotes the differentiation of Th1 cells, 21 , 26 thereby affecting Th1 differentiation.
Previous studies reported that the miR‐182 cluster and some of the other mentioned microRNAs, ie, miR‐99a, could induce Treg differentiation through targeting the FOXO1 and induce the production of some cytokines, such as IL‐10, IL‐17A, and IL‐6, involved in BC development. 16 , 36 The current study revealed that overexpression of miR‐182 reduces both transcript and protein levels of FOXO1, similar to previous studies, and it supports a population of T cells to secrete IL‐6, IL‐17, and TGF‐β by overexpression of FOXP3. In addition, FOXO1 can be simultaneously targeted by at least 11 microRNAs, such as miR‐183 and miR‐486, confirmed by other studies. 34 , 37 However, there was no report or prediction about targeting FOXP3 by any of the abovementioned microRNAs.
Furthermore, downregulation of FOXO1 and stability of FOXP3 were shown in PBMCs of patients with BC. MiR‐182 downregulates FOXO1, whereas it increases FOXP3 levels required for Treg differentiation in Jurkat cells. 22 Overall, miR‐182 negatively affects Th1 cell differentiation via targeting CD3d, ITK, and NFATc3. MiR‐182 drives Treg differentiation by downregulation of the promoter repressor proteins of FOXP3, such as NFATc3 and NFATc4, as well as targeting the upstream inducer proteins of MAPK, IP3, and DAG signal transduction pathways in the TCR/CD3 complex such as CD3d and ITK. 35 , 36
However, in addition to the overexpression of FOXP3 and production of CD4+ Foxp3+ T cells after overexpression of miR‐182, the current study also showed an elevation of some inflammatory cytokines, such as IL‐6 and IL‐17, along with TGF‐β in these cells. The elevation of IL‐6 and TGF‐β promotes the production of IL‐17A by Th17 cells and a type of Tregs (IL‐17–producing Tregs). 16 , 38 , 39 , 40 IL‐17 has been described as another inflammatory cytokine involved in survival, angiogenesis, invasiveness, and inhibition of immune response in BC development. 39 , 41 Also, proinflammatory cytokines, such as IL‐6 and IL‐1β, are effective in IL‐17A secretion from IL‐17–producing Tregs. 42 The activation of STAT3 by IL‐6 accompanied by RORγt and RORα can stimulate T cells for IL‐17A production. 16 , 43 In addition, there was a relationship between the upregulation of IL‐6 and TGF‐β that their increment can be provoked in Tregs by IL‐1β signaling. 42 IL‐1β is involved in the regulation of the inflammatory tumor microenvironment, proliferation, angiogenesis, and cancer cell invasion in BC. 44 Furthermore, IL‐1β signaling is required for the early phase of Th17 differentiation. In this regard, it can induce differentiation of CD4+ T cells into IL‐17–producing Tregs along with IL‐6 and TGF‐β, which are upregulated in patient's sera with BC. 42 , 45 Like other studies related to Treg function, we did not observe any enhancement in IL‐1β concentration in Jurkat cells transfected by miR‐182, 13 , 21 while there was a significant upregulation in IL‐6 and TGF‐β in both transcription and translation levels. Thus, it seems that increment of IL‐6 and TGF‐β in such Tregs can be associated with other regulatory factors and signal transduction pathways such as miR‐182. 44 , 46
Also, alteration of various cytokines, such as TNF‐α, IL‐10, IL‐1β, IL‐2, IL‐6, IL‐17, and TGF‐β, which plays a critical role in BC development, has been broadly observed. 41 , 44 , 46 , 47 Although a significant reduction was observed in the transcripts of TNF‐α and IL‐2, such a reduction was just established about IL‐2 after overexpression of miR‐182. It has been shown that IL‐2 induced miR‐182 cluster expression and subsequently promoted the clonal expansion of Tregs via activation of STAT5 and FOXO1 targeting, which are in line with the results of the current study. 31 Despite the emphasis on the critical role of the IL‐2/IL‐2RA (CD25) pathway in induction of miR‐182, the results of the current study showed that miR‐182 has the potential to prevent IL‐2/IL‐2RA signaling, which has a pivotal role in controlling differentiation and homeostasis of both Th1 and Th2 cells. Also, considering the role of different NFAT proteins, especially NFATc4 recruited by NFATc1 and NFATc2 to induce IL‐2 production and T cell proliferation and differentiation toward CD4+Th1, 26 CD4+Th2, 25 CD4+Th17, 12 and CD8+cytotoxic T cells, 21 it can be concluded that increasing each microRNA may target NFAT proteins, thereby suppressing different immune responses in effector T cells in BC. 16 , 34
Also, shuttling and overexpression of the onco‐microRNAs such as miR‐182 may be a reason for the fluctuation of IL‐2 rates in BC pathogenesis, especially in patients with advanced BC. 46 , 48 Thus, it seems that the increase of miR‐182‐5p, miR‐182‐3p, and miR‐183 in PBMCs of patients with BC can be associated with both tumor microenvironment secretome and activation of STAT3 and STAT5 induced by other cytokines such as IL‐6, increased in these patients. 16 , 49 However, the current study revealed that IL‐2 /IL‐2RA expression may be affected by the miR‐182‐183 cluster, and overexpression of this cluster of microRNAs dramatically suppresses IL‐2/IL‐2RA signaling through targeting IL‐2RA and different isoforms of NFAT as IL‐2 inducer transcription factor. Therefore, it seems that it is an important controlling way to prevent IL‐2/IL‐2RA signaling and activation of T helper cells. Finally, the results derived from recent studies represented that increase of onco‐microRNAs, especially the miR‐182 cluster through targeting of several proteins involved in the TCR/CD3 complex, IL2/IL‐2RA, ITK, and NFAT isoforms along with FOXO1 may deviate the polarization of T cells toward a transitional T cell bridging Treg and Th17 cells, which simultaneously overexpressed FOXP3 and secreted IL‐6, IL‐17, and TGF‐β.
The current results and meta‐analysis indicate that the microRNAs mentioned in Figure 1 are increased in BC tissues and sera. On the other hand, downregulation of CD3d, ITK, NFATc3, NFATc4, and IL‐2RA targeted by the miR‐182‐183 cluster was accompanied by a significant increase in miR‐182‐5p, miR‐182‐3p, and miR‐183‐5p in the PBMCs of patients with BC, and these data represented a reverse relationship between higher levels of these microRNAs and reduction of their targets. Therefore, considering the ability of tumor microenvironment cells in shuttling some regulatory molecules and the capability of these microRNAs, such as miR‐182, in stimulation and suppression of some cytokines such as IL‐6, IL‐17, and TGF‐β, it seems that increased levels of miR‐182 and other microRNAs with such properties may originate from an exogenous source or may be induced by other signaling pathways found in the PBMCs of these patients. 5 , 6 , 7 , 8 Also, it strengthens the hypothesis that tumor microenvironment secretome disrupts and deviates immune responses by the production of secretory materials and cargoes, including cytokines, microvesicles, and exosomes, which shuttles some immunosuppressive agents, especially the mentioned microRNAs. 3 , 50
In conclusion, it seems that simultaneous increase of onco‐microRNAs (especially miR‐182 and miR‐183) in tumor tissues, sera, and PBMCs of patients with BC have a relationship with each other, and these microRNAs expressed by tumor tissues may exert immunosuppressive functions and deviate effector T cell polarization by induction of FOXP3 expression along with suppression of various targets in several signaling pathways, such as the TCR/CD3 complex chains, IL‐2/IL‐2RA, ITK, FOXO1, and NFAT isoforms. However, simultaneous increased rate of both isoforms of miR‐182 (‐5p and ‐3p) totally has inhibitory effects on T cells by affecting the TCR/CD3 complex and suppressing FOXO1, NFATs, and IL‐2/IL‐2RA signaling pathways. Their overexpression increases FOXP3, TGF‐β, and IL‐17 expression to deviate T cells toward a transitional state, IL‐17–producing Tregs, and eventually may facilitate Treg formation. Finally, further studies associated with the increase of both isoforms of miR‐182 and other microRNAs mentioned in Table 2 along with the targets associated with the TCR/CD3 complex, NFATs, FOXO1, FOXP3, and IL‐2/IL‐2RA signaling pathways in CD4+ T cells are needed to further confirm these findings. Also, conducting further studies on other microRNAs and variation of microRNAs related to the possibility of drug resistance in cancerous patients and evaluation of potential microRNAs, especially miR‐182, miR‐183, and their targets are suggested to determine which patients may be candidates to undergo prophylactic mastectomy, manage appropriate prognosis, and develop personalized medicine.
DISCLOSURE
The authors declare that they have no conflict of interests.
AUTHORS' CONTRIBUTION
MHS, FS, and MP contributed to cloning, Western blot, real time‐PCR, and the ELISA assays. MA was the surgical oncologist and consultant. SG, NH, FPB, and HV collected the samples, isolated PBMCs, extracted RNAs, and performed cDNA synthesis and real time‐PCR. FS critically revised the manuscript for important intellectual content. MP performed flow cytometry, luciferase assay, the statistical analysis, and bioinformatics study. MP and FS conceived the main idea, analyzed the data, depicted the figures, and wrote the first draft. All of the authors read and approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethics Committee of the Cancer Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran with Ethics number IR.SBMU.RETECH.REC.1397.562. Written informed consent was obtained from both groups prior to the initiation of the current study.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Maryam Salari for her useful consultation regarding statistical analysis. This project was supported by grants 3008‐10 and 11628 from the Academic Center for Education, Culture, and Research (ACECR) and the Cancer Research Center (CRC), respectively.
Soheilifar MH, Vaseghi H, Seif F, et al. Concomitant overexpression of mir‐182‐5p and mir‐182‐3p raises the possibility of IL‐17–producing Treg formation in breast cancer by targeting CD3d, ITK, FOXO1, and NFATs: A meta‐analysis and experimental study. Cancer Sci. 2021;112:589–603. 10.1111/cas.14764
Mohammad Hasan Soheilifar and Hajar Vaseghi equally contributed to this work as the first authors.
DATA AVAILABILITY STATEMENT
Please contact the corresponding author for data requests.
REFERENCES
- 1. Mathieu M, Martin‐Jaular L, Lavieu G, Théry C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell‐to‐cell communication. Nat Cell Biol. 2019;21(1):9. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: biogenesis, biologic function and clinical potential. Cell Biosci. 2019;9(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jafarzadeh N, Safari Z, Pornour M, Amirizadeh N, Forouzandeh Moghadam M, Sadeghizadeh M. Alteration of cellular and immune‐related properties of bone marrow mesenchymal stem cells and macrophages by K562 chronic myeloid leukemia cell derived exosomes. J Cell Physiol. 2019;234(4):3697‐3710. [DOI] [PubMed] [Google Scholar]
- 4. Meng Y, Sun J, Wang X, et al. Exosomes: a promising avenue for the diagnosis of breast cancer. Technol Cancer Res Treat. 2019;18:1533033818821421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shimomura A, Shiino S, Kawauchi J, et al. Novel combination of serum microRNA for detecting breast cancer in the early stage. Cancer Sci. 2016;107(3):326‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matamala N, Vargas MT, González‐Cámpora R, et al. Tumor microRNA expression profiling identifies circulating microRNAs for early breast cancer detection. Clin Chem. 2015;61(8):1098‐1106. [DOI] [PubMed] [Google Scholar]
- 7. Zhang K, Wang Y‐W, Wang Y‐Y, et al. Identification of microRNA biomarkers in the blood of breast cancer patients based on microRNA profiling. Gene. 2017;619:10‐20. [DOI] [PubMed] [Google Scholar]
- 8. Heneghan HM, Miller N, Lowery AJ, Sweeney KJ, Newell J, Kerin MJ. Circulating microRNAs as novel minimally invasive biomarkers for breast cancer. Ann Surg. 2010;251(3):499‐505. [DOI] [PubMed] [Google Scholar]
- 9. Yu J, Shen W, Gao B, Zhao H, Xu J, Gong B. MicroRNA‐182 targets FOXF2 to promote the development of triple‐negative breast cancer. Neoplasma. 2017;64(2):209‐215. [DOI] [PubMed] [Google Scholar]
- 10. Hedrick SM, Michelini RH, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. 2012;12(9):649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kelada S, Sethupathy P, Okoye IS, et al. miR‐182 and miR‐10a are key regulators of Treg specialisation and stability during Schistosome and Leishmania‐associated inflammation. PLoS Pathog. 2013;9(6):e1003451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vaeth M, Schliesser U, Müller G, et al. Dependence on nuclear factor of activated T‐cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc Natl Acad Sci USA. 2012;109(40):16258‐16263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Workman CJ, Szymczak‐Workman AL, Collison LW, Pillai MR, Vignali DA. The development and function of regulatory T cells. Cell Mol Life Sci. 2009;66(16):2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR‐27a, miR‐96, and miR‐182 in breast cancer cells. J Biol Chem. 2009;284(35):23204‐23216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wan C, Bi W, Lin P, et al. MicroRNA 182 promotes T helper 1 cell by repressing hypoxia induced factor 1 alpha in experimental autoimmune encephalomyelitis. Eur J Immunol. 2019;49(12):2184‐2194. [DOI] [PubMed] [Google Scholar]
- 16. Ichiyama K, Gonzalez‐Martin A, Kim B‐S, et al. The microRNA‐183‐96‐182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression. Immunity. 2016;44(6):1284‐1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maruyama T, Konkel JE, Zamarron BF, Chen W, editors. The molecular mechanisms of Foxp3 gene regulation. Semin Immunol. 2011;23(6):418‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oh‐hora M. The calcium/NFAT pathway: role in development and function of regulatory T cells. Microbes Infect. 2009;11(5):612‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg‐mediated T cell suppression. Front Immunol. 2012;3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol. 2019;10:3100‐3100. https://www.frontiersin.org/articles/10.3389/fimmu.2019.03100/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee J‐U, Kim L‐K, Choi J‐M. Revisiting the concept of targeting NFAT to control T cell immunity and autoimmune diseases. Front Immunol. 2018;9:2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee W, Lee GR. Transcriptional regulation and development of regulatory T cells. Exp Mol Med. 2018;50(3):e456‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hede K. MicroRNAs as onco‐miRs, drivers of cancer. JNCI: Journal of the National Cancer Institute. 2010;102(17):1306‐1308. 10.1093/jnci/djq349. [DOI] [PubMed] [Google Scholar]
- 24. Daniel C, Gerlach K, Väth M, Neurath MF, Weigmann B. Nuclear factor of activated T cells—A transcription factor family as critical regulator in lung and colon cancer. Int J Cancer. 2014;134(8):1767‐1775. [DOI] [PubMed] [Google Scholar]
- 25. Rengarajan J, Tang B, Glimcher LH. NFATc2 and NFATc3 regulate T H 2 differentiation and modulate TCR‐responsiveness of naive T H cells. Nat Immunol. 2002;3(1):48. [DOI] [PubMed] [Google Scholar]
- 26. Izsepi E, Himer L, Szilagyi O, et al. Membrane microdomain organization, calcium signal, and NFAT activation as an important axis in polarized Th cell function. Cytometry A. 2013;83(2):185‐196. [DOI] [PubMed] [Google Scholar]
- 27. Kaminuma O, Kitamura N, Nishito Y, et al. Downregulation of NFAT3 due to lack of T‐Box transcription factor TBX5 is crucial for cytokine expression in T cells. J Immunol. 2018;200(1):92‐100. [DOI] [PubMed] [Google Scholar]
- 28. Lee M, Park J. Regulation of NFAT activation: a potential therapeutic target for immunosuppression. Mol Cells. 2006;22(1):1‐7. [PubMed] [Google Scholar]
- 29. Ross SH, Cantrell DA. Signaling and function of interleukin‐2 in T lymphocytes. Annu Rev Immunol. 2018;36:411‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y, Zhou L, Sun B, et al. Interleukin‐2 administration after modified radical mastectomy in breast cancer therapy increases peripheral regulatory T cells. Int J Clin Exp Med. 2015;8(5):7816. [PMC free article] [PubMed] [Google Scholar]
- 31. Stittrich A‐B, Haftmann C, Sgouroudis E, et al. The microRNA miR‐182 is induced by IL‐2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol. 2010;11(11):1057. [DOI] [PubMed] [Google Scholar]
- 32. Du R, Zhao H, Yan F, Li H. IL‐17+ Foxp3+ T cells: an intermediate differentiation stage between Th17 cells and regulatory T cells. J Leuko Biol. 2014;96(1):39‐48. [DOI] [PubMed] [Google Scholar]
- 33. Kim KM, Park SJ, Jung SH, et al. miR‐182 is a negative regulator of osteoblast proliferation, differentiation, and skeletogenesis through targeting FoxO1. J Bone Miner Res. 2012;27(8):1669‐1679. [DOI] [PubMed] [Google Scholar]
- 34. Rodríguez‐Galán A, Fernández‐Messina L, Sánchez‐Madrid F. Control of immunoregulatory molecules by miRNAs in T cell activation. Front Immunol. 2018;9:2148 https://www.frontiersin.org/articles/10.3389/fimmu.2018.02148/full. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee GR. The balance of Th17 versus Treg cells in autoimmunity. Int J Mol Sci. 2018;19(3):730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Plitas G, Konopacki C, Wu K, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Puthanveetil P. FoxO1–miRNA interacting networks as potential targets for mitochondrial diseases. Drug Discov Today. 2019;24(1):342‐349. [DOI] [PubMed] [Google Scholar]
- 38. Li MO, Flavell RA. TGF‐β: a master of all T cell trades. Cell. 2008;134(3):392‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jung MK, Kwak J‐E, Shin E‐C. IL‐17A‐producing Foxp3+ regulatory T cells and human diseases. Immune Netw. 2017;17(5):276‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seif F, Ghalehbaghi B, Aazami H, et al. Frequency of CD4+ and CD8+ T cells in Iranian chronic rhinosinusitis patients. Allergy Asthma Clin Immunol. 2018;14(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Welte T, Zhang XH‐F. Interleukin‐17 could promote breast cancer progression at several stages of the disease. Mediators Inflamm. 2015;2015:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li L, Kim J, Boussiotis VA. IL‐1β–mediated signals preferentially drive conversion of regulatory T cells but not conventional T cells into IL‐17–producing cells. J Immunol. 2010;185(7):4148‐4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK‐STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal. 2017;15(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Esquivel‐Velázquez M, Ostoa‐Saloma P, Palacios‐Arreola MI, Nava‐Castro KE, Castro JI, Morales‐Montor J. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res. 2015;35(1):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin‐1 signaling. Immunity. 2009;30(4):576‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jabeen S, Espinoza JA, Torland LA, et al. Noninvasive profiling of serum cytokines in breast cancer patients and clinicopathological characteristics. Oncoimmunology. 2019;8(2):e1537691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kawaguchi K, Sakurai M, Yamamoto Y, et al. Alteration of specific cytokine expression patterns in patients with breast cancer. Sci Rep. 2019;9(1):2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dehqanzada ZA, Storrer CE, Hueman MT, et al. Assessing serum cytokine profiles in breast cancer patients receiving a HER2/neu vaccine using Luminex® technology. Oncol Rep. 2007;17(3):687‐694. [PubMed] [Google Scholar]
- 49. O'Neill LA. Outfoxing Foxo1 with miR‐182. Nat Immunol. 2010;11(11):983. [DOI] [PubMed] [Google Scholar]
- 50. Becker JC, Andersen MH, Schrama D, thor Straten P. Immune‐suppressive properties of the tumor microenvironment. Cancer Immunol Immunother. 2013;62(7):1137‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Please contact the corresponding author for data requests.