

Abstract
Lung adenocarcinoma (LAC) is the most prevalent form of lung cancer. Epithelial cell transforming sequence 2 (ECT2) is a guanine nucleotide exchange factor that has been implicated in oncogenic and malignant phenotypes of LAC. Here, we identified an oncogenic role of ECT2 in the extracellular matrix (ECM) dynamics of LAC cells. We showed that suppression of ECT2 decreased adhesion and spreading of LAC cells on ECM components. Morphologically, ECT2‐depleted cells exhibited a rounded shape and cytoskeletal changes. Examination of transcriptional changes by RNA sequencing revealed a total of 1569 and 828 genes whose expressions were altered (absolute fold change and a difference of >2 fold) in response to suppression of ECT2 in two LAC cells (Calu‐3 and NCI‐H2342), respectively, along with 298 genes that were common to both cell lines. Functional enrichment analysis of common genes demonstrated a significant enrichment of focal adhesions. In accord with this observation, we found that ECT2 suppression decreased the expression level of proteins involved in focal adhesion signaling including focal adhesion kinase (FAK), Crk, integrin β1, paxillin, and p130Cas. FAK knockdown leads to impaired cell proliferation, adhesion, and spreading of LAC cells. Moreover, in LAC cells, ECT2 binds to and stabilizes FAK and is associated with the formation of the focal adhesions. Our findings provide new insights into the underlying role of ECT2 in cell‐ECM dynamics during LAC progression and suggest that ECT2 could be a promising therapeutic avenue for lung cancer.
Keywords: ECT2, extracellular matrix, focal adhesion, lung adenocarcinoma, RNA sequencing
In this study, we demonstrated that epithelial cell transforming sequence 2 (ECT2) is an essential mediator of cell‐extracellular matrix (ECM) dynamics in the malignant progression of lung adenocarcinoma. We found that suppression of ECT2 decreased adhesion and spreading of lung adenocarcinoma cells on the ECM and demonstrated that ECT2 has a regulatory role in focal adhesion signaling, being associated with the focal adhesion formation that contains both focal adhesion kinase (FAK) and ECT2.

1. INTRODUCTION
Lung cancer is the main cause of cancer‐related death worldwide, and adenocarcinoma is the most common histological subtype of lung cancer. 1 , 2 Patients with lung adenocarcinoma (LAC) harbor genetic alterations including mutations and translocations in EGFR, EML4‐ALK, and other oncogenes that have been identified in advanced cases. 3 , 4 , 5 Currently, however, no effective targeted therapies are available, as intrinsic and acquired resistance to targeting drugs frequently arise at the advanced stage. Therefore, for improved diagnosis and treatment, additional molecular signatures involved in the progression of adenocarcinoma need to be identified. Previously, we have examined the genetic alterations occurring in early‐stage adenocarcinoma and shown that epithelial cell transforming sequence 2 (ECT2) is amplified and its protein overexpressed in early invasive adenocarcinoma. 6 Subsequently, we clarified that aberrant cytoplasmic expression of ECT2 is a specific characteristic of LAC cells associated with poor patient outcomes. 7 ECT2 is a guanine nucleotide exchange factor for Rho family small GTPases proteins, especially Rac1, RhoA, and Cdc42. 8 ECT2 consists of the DH/PH/C domain, which catalyzes its guanine exchange factor (GEF) activity, three breast cancer gene 1 carboxyl‐terminal domain (BRCT) domains that regulate its GEF activity and localization, and a central S domain that has two nuclear localization signals (NLSs) required for ECT2 nuclear localization. 9 , 10 , 11 ECT2 was originally identified as a proto‐oncogene capable of transforming NIH 3T3 mouse fibroblasts. This original form, not found in human cancers, was N‐terminally truncated and lacked the BRCT domains and first NLS sequence. 12 On the other hand, overexpression of full‐length ECT2 has been reported in various types of cancer including those of the lung, esophagus, ovary, breast, and brain, as well as in osteosarcoma cells. Overexpression of ECT2 is correlated with poor patient outcomes. 13 , 14 , 15 , 16 , 17 However, the best‐characterized function of ECT2 is regulation of cytokinesis. In normal cells, ECT2 is localized in the nucleus during interphase and becomes distributed to the cytoplasm upon breakdown of the nuclear membrane. Subsequently, ECT2 becomes condensed at the central spindle and then in the cleavage furrow, where it activates RhoA and stimulates cytokinesis. 8 , 18 , 19 However, ECT2 is localized in both the nucleus and the cytoplasm of cancer cells, and its oncogenic activity has been correlated with Rac1 activation. 7 , 14 , 20 , 21 , 22 In non‐small cell lung carcinoma (NSCLC) cells, the PKCι‐Par6 oncogenic complex binds to ECT2 and regulates both the cytoplasmic localization of ECT2 and Rac1 activity. 20 Moreover, ECT2 suppression has been shown to significantly reduce the growth of glioblastoma and also to cause growth arrest of oral squamous cell carcinoma in G1 phase. 23 , 24 Our previous functional analysis has also shown that ECT2 depletion led to a significant reduction in the growth, migration, and invasion of LAC cells. 7 Nevertheless, ECT2 can support cell‐cell interaction. The localization and expression of ECT2 at cell‐cell contacts of Madin‐Darby canine kidney (MDCK) cells is regulated by calcium, an essential regulator of cell‐cell adhesion. 25 In MCF‐7 human breast cancer cells, ECT2 supports cell‐cell interactions, especially at adherens junctions, and during interphase ECT2 stabilizes E‐cadherin through RhoA and myoIIA to preserve the integrity of the junctions. 26 More recent studies have suggested that ECT2 can induce epithelial‐mesenchymal transition of osteosarcoma and LAC cells. 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 Consistent with its putative role in regulation of cell‐cell adhesion, siRNA screening targeting genes most closely related to cell adhesion and cytoskeletal function has recently revealed that ECT2 suppression significantly decreases focal adhesion size and impairs the migration of MCF‐7 cells. 28 Despite these previous advances, the biological function of ECT2 and its precise mechanisms in cell‐extracellular matrix (ECM) dynamics in relation to tumor progression remain unclear. Here, using LAC cells, we investigated the role of ECT2 in cell‐ECM interaction in the context of cancer cell adhesion, spreading, and morphology, and demonstrated that ECT2 has a regulatory role in focal adhesion signaling.
2. MATERIALS AND METHODS
2.1. ECT2 knockdown with siRNA
Calu‐3, PC‐9, and NCI‐H2342 cells were transfected with two siRNAs targeting the ECT2 gene (siECT2#1, siECT2#2) for 48, 24, and 48 hours, respectively. As a negative control, siCON (Stealth RNAi Negative Control Medium GC; Thermo Fisher Scientific) was employed. The transfection was performed with Lipofectamine RNAiMAX (Thermo Fisher Scientific) in OPTI‐MEM reduced serum medium (Thermo Fisher Scientific) for 20 minutes at room temperature. The Lipofectamine complex was transferred to 2.5 mL of medium containing 2 × 105 cells in each well of a six‐well plate. Calu‐3 and NCI‐H2342 were transfected with siRNAs targeting the focal adhesion kinase (FAK) gene (siFAK#1, siFAK#2) for 48 hours. Thereafter, the procedures employed were identical to those for the knockdown experiment described above. The specific siRNA targeting ECT2 or FAK were purchased from Thermo Fisher Scientific (Materials and Methods in Appendix S1).
2.2. Cell proliferation assay
Calu‐3 and NCI‐H2342 cells were transfected with siECT2#1 or siECT2#2, and seeded on 96‐well plates coated with collagen type l (COL1) (Iwaki Biosciences) at a density of 8 × 104 cells/ml. After 48 hours, cell proliferation was assessed using a cell counting kit‐8 (Dojindo). Absorbance was measured at 450 nm using a Microplate Reader (Bio‐Rad Laboratories). The procedures employed were identical to the cells transfected with siFAK#1 and siFAK#2.
2.3. Assay of adherent cell viability
Calu‐3, PC‐9, and NCI‐H2342 were transfected with siECT2#1, and 8 × 104 cells/mL were seeded on 24‐well plates coated with fibronectin (FN) (Corning Incorporated) or COL l (Iwaki Biosciences). The cells were then washed once with phosphate‐buffered saline (PBS) (Cell Signaling) and stained with 0.2% crystal violet containing 20% methanol for 30 minutes. The crystal violet was released using 1% SDS (Sigma Aldrich) in PBS, and the absorbance was read on a Microplate Reader (Bio‐Rad Laboratories) at 595 nm.
2.4. Cell adhesion assay
Calu‐3, PC‐9, and NCI‐H2342 were transfected with siECT2#1 or siCON in medium containing 1% FBS. The cells were then treated with 0.05% trypsin and washed twice with serum‐free medium. For 1 hour before culture, 24‐well plates that had been untreated or coated with COL l and FN were blocked with 1% bovine serum albumin (BSA) (Sigma‐Aldrich). The plates were then washed once with PBS and allowed to air dry before being seeded with the above cells at a density of 5‐7 × 105/mL in the serum‐free medium. After 90 minutes, nonadherent cells were removed and the adherent cells were washed twice with PBS, then stained for 1 hour with 2% crystal violet containing 20% methanol. The cells were then washed five times with deionized water, and the stain was released using 1% SDS before the absorbance was read on a Microplate Reader (Bio‐Rad Laboratories) at 560 nm. The procedures employed were identical to those employed for the cells transfected with siFAK#1.
2.5. Cell spreading assay
Calu‐3, PC‐9, and NCI‐H2342 cells were transfected with siECT2#1 or siCON. The cells were then trypsinized, washed, and resuspended in medium, before being seeded at 1‐2 × 105 cells/ml on 24‐well plates coated with FN and COL l and incubated for 12 and 24 hours. Images of the cells were taken by microscopy and the areas of cell spreading were evaluated from images of four separate wells using ImageJ. Calu‐3 and NCI‐H2342 cells were transfected with siFAK#1 or siCON for 48 hours. The procedures employed were identical to the cells transfected with siFAK#1.
2.6. RNA isolation and quantitative real‐time PCR
Total RNA was extracted from the Calu‐3, PC‐9, and NCI‐H2342 cells using a RNeasy Mini Plus Kit (Qiagen) in accordance with the manufacturer's instructions. Complementary DNA (cDNA) was synthesized from total RNA (1 μg) using a high‐capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Quantitative real‐time (RT‐PCR) was carried out with SYBR Premix EX Taq (Perfect Real Time; Takara Bio) on a GeneAmp 7300 sequence detection system (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. The 18S ribosomal RNA gene was used for normalization. The specific primer pairs used for RT‐PCR were purchased from Takara (Materials and Methods in Appendix S1).
2.7. Western blotting
Calu‐3, PC‐9, and NCI‐H2342 cells were dissolved in cell lysis buffer using M‐PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific) containing protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The protein levels were measured using bicinchoninic acid assay (BCA) protein assay kits (Thermo Fisher Scientific). Polypeptides were electrophoresed on Mini‐PROTEAN TGX Precast Gels (Bio‐Rad Laboratories) and transferred to polyvinylidene difluoride membranes using the iBlot gel transfer system (Thermo Fisher Scientific). The membranes were incubated with each primary antibody overnight at 4°C, washed with PBS containing 0.1% Tween‐20 (Thermo Fisher Scientific), and then incubated with an appropriate secondary antibody. The proteins were visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific), and images were captured using a ChemiDoc Touch Imaging system (Bio‐Rad Laboratories).
2.8. DNA constructs
The full‐length human Flag‐ECT2 vector was donated by Professor Toru Miki (Nagoya University). The full‐length human Myc‐Flag‐FAK vector was purchased from OriGene Technologies.
2.9. Transfection with plasmid DNA
Plasmid DNA was purified using a Plasmid Maxi kit (QIAGEN). OPTIMEM (Thermo Fisher Scientific) and Fugene HD (Promega) were used for plasmid transfection in accordance with the manufacturer's instructions.
2.10. Co‐immunoprecipitation (Co‐IP)
Calu‐3, NCI‐H2342, PC‐9, and A549 were lysed in Pierce IP Lysis Buffer (Thermo Fisher Scientific). The lysate from each cell line was incubated with appropriate antibodies and protein A or G magnetic beads (Bio‐Rad Laboratories) overnight at 4°C on a rotator. The samples were further analyzed by Western blotting using the appropriate primary and secondary antibodies.
2.11. Cycloheximide chase assay
Calu‐3 cells were transfected with siECT2#1 or siCON for 48 hours and then incubated in eagle’s minimum essential medium (EMEM) with 50 μg/mL cycloheximide (CHX; Sigma‐Aldrich). After treatment with CHX, the total cell lysates were collected at different times and analyzed by Western blotting. For statistical analysis in this study, two‐tailed Student's t test was used to determine the significance of differences between groups. Additional information about materials and methods is available in the Materials and Methods section of Appendix S1.
3. RESULTS
3.1. Loss of ECT2 expression impairs cellular proliferation and the viability of adherent cells
We transfected Calu‐3 and NCI‐H2342 with siECT2#1 or siECT2#2 for 48 hours. It was found that the growth of LAC cells transfected with siECT2#1 and siECT2#2 was significantly lower than that of control cells (Figure 1A). As siECT2#1 showed the strongest suppression of cellular proliferation, we selected it and used it for further analysis. We next evaluated whether ECT2 regulates the viability of adherent cells. We found that ECT2 suppression significantly impaired the viability of adherent cells on the ECM components COL l and FN compared with control cells in Calu‐3, NCI‐H2342, and PC9 (Figure 1B and Figure S1A‐D).
FIGURE 1.
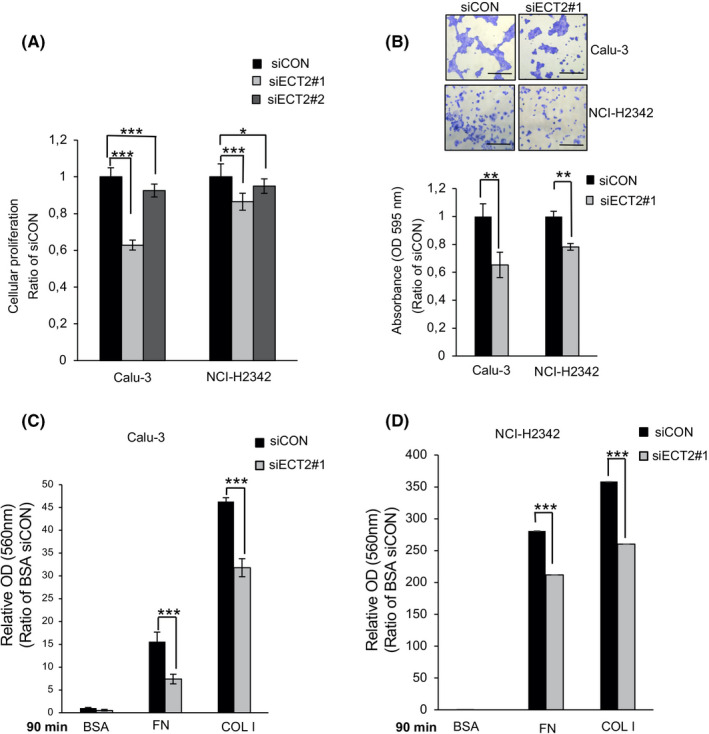
Epithelial cell transforming sequence 2 (ECT2) suppression reduces cellular proliferation, the viability of adherent cells, and cell adhesion. A, Cellular proliferation was examined after transfection of Calu‐3 and NCI‐H2342 cells using siECT2#1 or siECT2#2 for 48 h. Error bars represent mean ± SD, n = 11. B, Calu‐3 and NCI‐H2342 cells were transfected with siECT2#1 or siCON and seeded on COLl‐coated plates. The viability of adherent cells was measured and quantified. Error bars represent mean ± SD from more than four independent experiments. Scale bar 100 μm. Calu‐3 (C) and NCI‐H2342 (D) cells were transfected with siECT2#1 or siCON. After siRNA treatment, the cells were plated on fibronectin (FN)‐, COLl‐, or 1% BSA‐coated plates for 90 min. Error bars represent mean ± SD, n = 4, *P < .05, **P < .01, ***P < .001
3.2. ECT2 suppression inhibits cell adhesiveness on FN and collagen type I
As cell‐ECM interaction is a critical process involved in cancer development and progression, 29 here we investigated the effect of ECT2 silencing on the adhesiveness of LAC cells. As shown in Figure 1C,D and Figure S1E, ECT2 suppression markedly decreased the adhesive ability of Calu‐3, NCI‐H2342, and PC‐9 cells 90 minutes after seeding them on FN and COL l. BSA was used as a negative control.
3.3. ECT2 silencing decreases cell spreading, changes cell morphology, and alters actin cytoskeleton
Spreading is an essential step of cell motility that is tightly controlled by interaction with the ECM. 30 We investigated the influence of ECT2 silencing on the spreading of LAC cells. Twelve hours after plating, ECT2‐depleted cells exhibited a significant reduction of the cell spreading area on COL l and FN relative to the control cells. The same effect was seen at 24 hours (Figure 2A,B and Figure S2). We further examined whether ECT2 silencing would affect cell shape and actin structure. Although Calu‐3 and NCI‐H2342 showed a substantial reduction in the area of cell spreading at 12 hours on both COL1 and noncoated coverslips, ECT2‐depleted cells showed a significant reduction in the area of spread on COL1, with more than a 2‐fold difference between siECT2#1 and siCON (Figure 3A,B). In addition, the actin cytoskeleton was more noticeably intense in control cells than in ECT2‐depleted cells on COL1‐coated coverslips (Figure 3C). These results suggested that ECT2 in LAC cells altered cell spreading and morphology more robustly through a change in ECM interaction.
FIGURE 2.
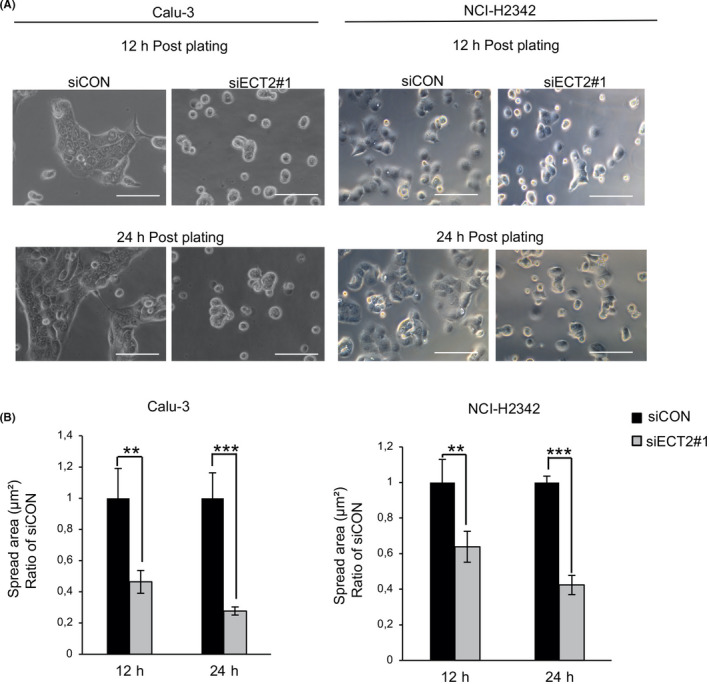
Suppression of epithelial cell transforming sequence 2 (ECT2) inhibits the spreading of lung adenocarcinoma (LAC) cells. Calu‐3 and NCI‐H2342 cells transfected with siECT2#1 or siCON were spread on COLl‐coated plates for 12 and 24 h. A, Phase‐contrast micrographs show that the cells treated with siECT2#1 exhibit delayed or absent spreading relative to control cells. B, The area of spread of transfected cells was analyzed and quantified using ImageJ software. Error bars represent mean ± SD, n = 4, scale bar 100 μm, **P < .01, ***P < .001
FIGURE 3.
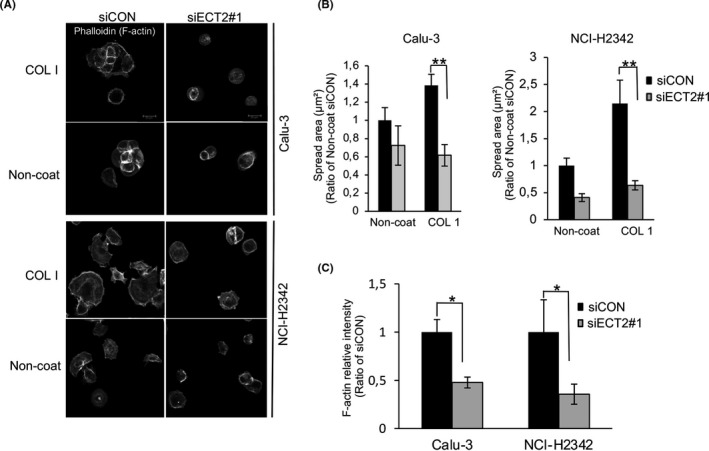
Changes of morphology and actin organization in epithelial cell transforming sequence 2 (ECT2)‐depleted cells. A, Confocal images of Calu‐3 and NCI‐H2342 cells transfected with siECT2#1 or siCON and then left to spread on COLl or noncoated coverslips for 12 h, Scale bar 20 μm. B, The area of spread of transfected cells was analyzed and quantified from three images using ImageJ software. Error bars represent mean ± SD, n = 3. C, Histogram shows fluorescence intensity measurement of F‐actin after transfection with siECT2#1 compared with siCON at 12 h post plating on COL1. Error bars represent mean ± SD, n = 3, *P < .05, **P < .01
3.4. ECT2 targeting of focal adhesion signaling in LAC cells
To gain insight into the mechanisms underlying the role of ECT2 in altering LAC cell‐ECM interaction, we assessed transcriptional changes in two LAC cells (Calu‐3 and NCI‐H2342) in response to siECT2#1 and siCON after 48 hours using RNA‐seq. The Calu‐3 and NCI‐H2342 showed overexpression of ECT2 protein and gene amplification. 7 We found that ECT2 altered 1569 and 828 differentially expressed genes (absolute fold change and a difference of >2 fold) in Calu‐3 and NCI‐H2342, respectively (Figure 4A and Table S1). We further confirmed the transcriptional changes in Calu‐3 by RNA‐seq after 72 hours of siECT2#1. Comparison of changes in gene expression at two time points of siRNA transfection (48 and 72 hours) revealed a total of 2913 differentially expressed genes (absolute fold change and a difference of >2 fold). A similar expression pattern was observed in siECT2#1‐ and siCON‐transfected cells at 48 and 72 hours (Figure S3 and Table S2). Venn diagram analysis showed that 298 genes (considered to be common genes) were expressed in both Calu‐3 and NCI‐H2342 (Figure 4B). The Gene Ontology (GO) biological process for common genes showed strong enrichment for terms related to tumor progression, including the ephrin receptor signaling pathway, negative regulation of apoptosis, cell‐cell adhesion, and cell migration (Figure S4). Moreover, GO cellular component terms of common genes were significantly enriched in the cytosol, extracellular exosome, focal adhesion, cell‐cell adherens junction, cytoplasm, nucleoplasm, mitochondria, membrane, endoplasmic reticulum, and endoplasmic reticulum lumen (Figure 4C). Additionally, kyoto encyclopedia of genes and genomes (KEGG) pathway analysis of common genes demonstrated significant enrichment of multiple pathways including microRNAs in cancer, bladder cancer, p53 signaling, proteoglycans in cancer, focal adhesion, hippo signaling, viral carcinogenesis, T cell receptor signaling, and regulation of the actin cytoskeleton (Figure 4D). As focal adhesions are known to have a potentially important influence on cell‐ECM dynamics, we focused on this form of signaling and further analyzed its impact on LAC progression. We found several transcripts involved in focal adhesions in Calu‐3 or/and NCI‐H2342 were downregulated, including MET, HRAS, ACTB, PAK4, EPHA2, CRK, ITGB1, ITGB6, AKT3, LAMC2, with normalized fold expression values of >2, and an upregulated gene, Vav3, with a normalized fold expression value of <0.5 (Table S1). Moreover, KEGG analysis demonstrated that the cell cycle pathway was the most significantly rated network, consistent with the broad physiological roles of ECT2 in the regulation of cytokinesis (Figure 4D and Figure S4).
FIGURE 4.
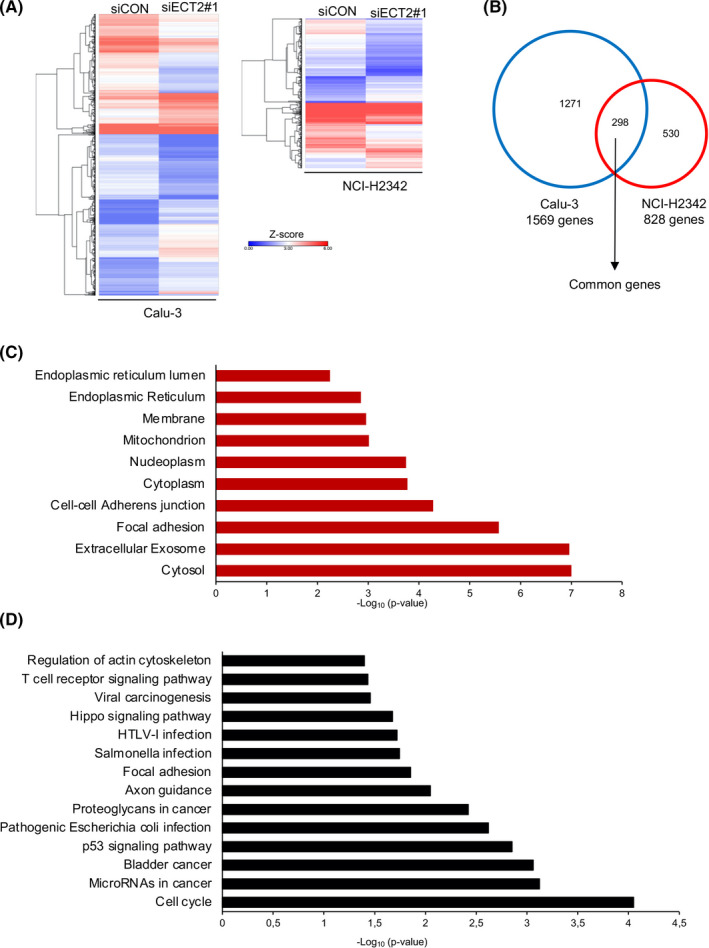
RNA‐seq profiling of epithelial cell transforming sequence 2 (ECT2) silencing in lung adenocarcinoma (LAC) cells. A, Clustered heatmap shows a total of 1569 and 828 differentially expressed genes for Calu‐3 and NCI‐H2342 cells, respectively. Data represent n = 1 sample per condition. B, Venn diagram showing the overlap between genes differentially expressed in Calu‐3 and NCI‐H2342 cells; 298 genes were defined as common to both cell lines. C, Gene Ontology (GO) terms enrichment analysis of common genes showing the top 10 significantly enriched cellular component terms. D, KEGG pathway enrichment analysis of common genes. Significant enrichment of KEGG pathways was defined as P < .05 and more than five genes
3.5. ECT2 silencing affects the expression of proteins involved in focal adhesion signaling
First, we confirmed the RNA‐seq data for two genes, CRK and ITGB1, by RT‐PCR. This revealed that cells transfected with siECT2#1 had significantly reduced levels of mRNA expression for both genes (Figure S5). We further confirmed that the protein expression levels of Crk and integrin β1 were markedly decreased after siECT2#1 in Calu‐3 and NCI‐H2342 (Figure 5A,B). We next examined whether ECT2 suppression was able to change the expression of other focal adhesion proteins. We found that the expression of FAK, paxillin, and p130Cas was reduced after siECT2#1 (Figure 5A,C). Moreover, ECT2 suppression substantially reduced the degree of phosphorylation at Tyr221 of Crk, Tyr397, Tyr925, Tyr576, and Tyr577 of FAK, Tyr416 of Src, Tyr118 of paxillin, and Tyr410 of p130Cas (Figure S6A). The same effect was seen with siECT2#2 (Figure S6B,C). ECT2 suppression was confirmed by Western blotting, immunofluorescence, and quantitative RT‐PCR (Figure 5A, Figures S6B,C and S7).
FIGURE 5.
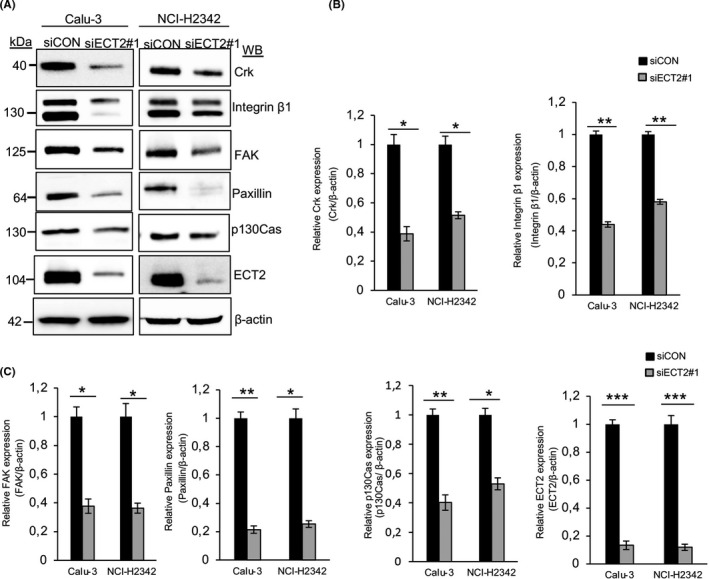
Knockdown of epithelial cell transforming sequence 2 (ECT2) attenuates the proteins involved in focal adhesion signaling. Equal amounts of whole‐cell lysates were collected from Calu‐3 and NCI‐H2342 cells and probed for various proteins involved in focal adhesion signaling. A, Immunoblots of Crk, integrin β1, focal adhesion kinase (FAK) , paxillin, and p130Cas showed a reduction in their expression after transfection with siECT2#1 relative to siCON. B, C, For quantitative Western blot analysis, the density of bands was measured using ImageJ and normalized to β‐actin. Error bars represent mean ± SD from more than three independent experiments, *P < .05, **P < .01, ***P < .001
3.6. Knockdown of FAK decreases the proliferation, adhesion, and spreading of LAC cells
In order to investigate the influence of focal adhesion molecules on ECM dynamics, we focused on FAK, which acts as a major mediator of adhesion and motility in normal cells and tumor cells. 30 We transfected Calu‐3 and NCI‐H2342 with specific siFAK#1 or siFAK#2. As shown in Figure 6A,B, both siRNAs successfully suppressed the expression of FAK mRNA and protein and significantly reduced their proliferation (Figure 6C). This knockdown of FAK also significantly impaired cell adhesion and spreading of both LAC cells (Figure 6D,E).
FIGURE 6.
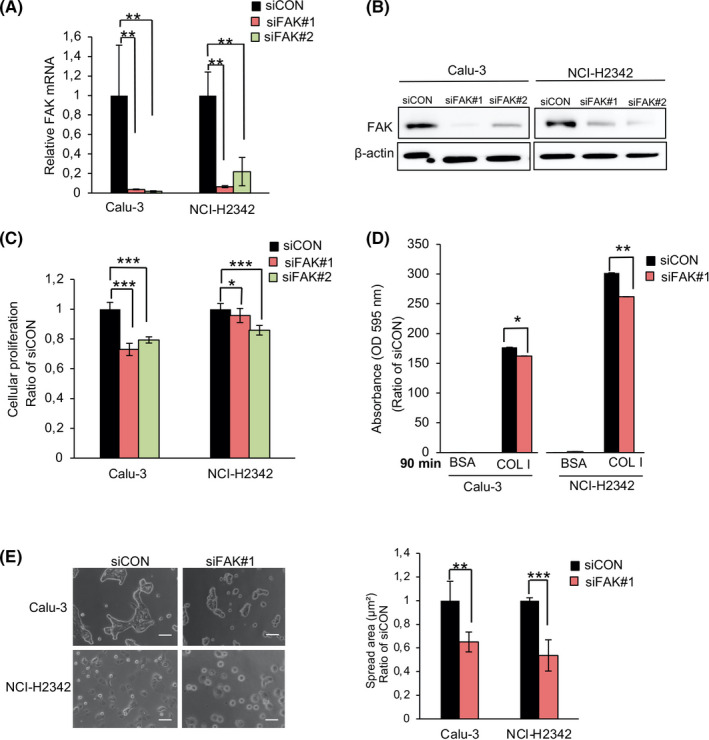
Knockdown of focal adhesion kinase (FAK) regulates the proliferation, adhesion, and spreading of lung adenocarcinoma (LAC) cells. Calu‐3 and NCI‐H2342 were transfected with siFAK#1 and siFAK#2. Knockdown efficiency was confirmed at both the mRNA and protein levels A, B. Error bars represent mean ± SD, n = 3. C, Cell proliferation of the Calu‐3 and NCI‐H2342 cells. Error bars represent mean ± SD from more than nine independent experiments. D, Calu‐3 and NCI‐H2342 cells were transfected with siFAK#1 or siCON and then plated on COLl‐ or 1% BSA‐coated plates for 90 min. Error bars represent mean ± SD, n = 3. E, The area of spread of transfected Calu‐3 and NCI‐H2342 cells was analyzed and quantified using ImageJ software. Error bars represent mean ± SD, n = 4, *P < .05, **P < .01, ***P < .001
3.7. Identification of FAK, a novel ECT2‐binding protein in LAC cells
A previous study had identified ECT2 as a FAK binding partner in mouse squamous cell carcinoma cells. 31 Here, we examined the potential interaction between ECT2 and FAK in LAC cells. The results showed that Co‐IP of endogenous ECT2 or endogenous FAK coprecipitated endogenous FAK and ECT2, respectively, in Calu‐3 (Figure 7A) and NCI‐H2342 (Figure 7B). Moreover, we transiently expressed Flag‐tagged full‐length ECT2 or Myc‐tagged full‐length FAK in A549 (Figure 7C) and PC‐9 (Figure 7D) and used Co‐IP with anti‐Flag or anti‐Myc. It was found that ECT2 became bound to FAK in LAC cells.
FIGURE 7.
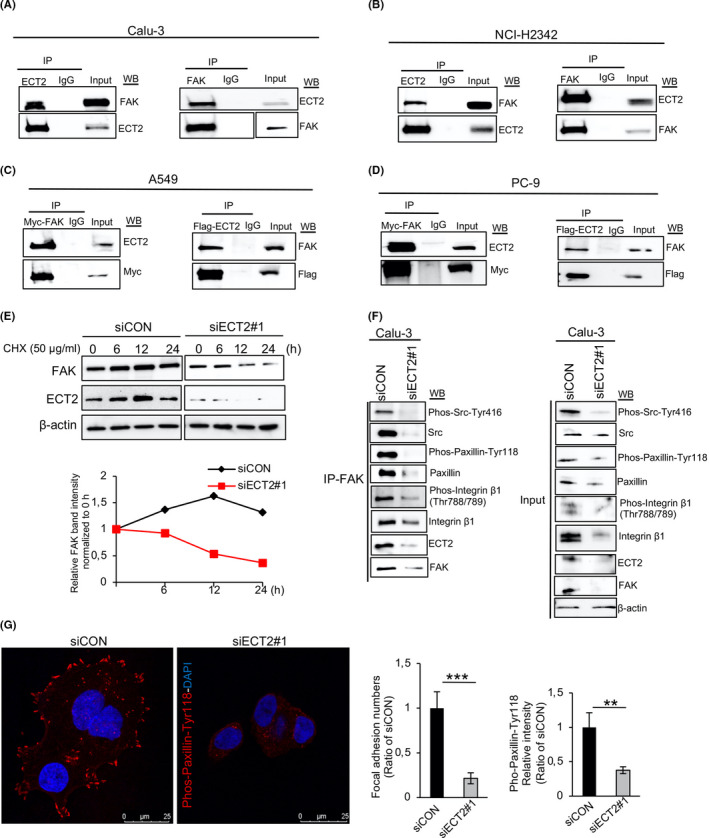
Epithelial cell transforming sequence 2 (ECT2) binds to and stabilizes focal adhesion kinase (FAK) and is associated with focal adhesion assembly. A, B, Interaction of endogenously expressed ECT2 with FAK examined using total cell lysates from Calu‐3 (A) and NCI‐H2342 (B) cells. A549 (C) and PC‐9 (D) cells were transfected with full‐length Flag‐ECT2 or full‐length Myc‐FAK, then immunoprecipitated with anti‐Flag or anti‐Myc antibodies, and analyzed by Western blotting for Myc, Flag, ECT2, and FAK. IgG was used as a negative control. E, The half‐life of FAK in Calu‐3 cells was assessed by Western blotting after transfection with siECT2#1. The cells were treated with cycloheximide (CHX; a protein synthesis inhibitor) for the indicated times. F, Equal amounts of whole‐cell lysate from Calu‐3 cells transfected with siECT2#1 or siCON for 48 h were immunoprecipitated with anti‐FAK antibody and analyzed by Western blotting for Pho‐Src‐Tyr416, Src, Phos‐Paxillin‐Tyr118, paxillin, Phos‐integrin β1 (Thr788/789), integrin β1, ECT2, FAK, and β‐actin. G, NCI‐H2342 cells were transfected with siECT2#1 or siCON for 48 h. Fixed cells were stained with Phos‐paxillin‐Tyr118 antibody and DAPI. Phos‐paxillin‐Tyr118‐positive focal adhesions were quantified from 20 cells in four pictures using ImageJ software. Error bars represent mean ± SD, scale bar 25 μm, **P < .01, ***P < .001
3.8. ECT2 expression is important for the formation of the focal adhesion complex
Next, we investigated whether ECT2 affected the protein stability of FAK. The CHX chase assay indicated that the half‐life of FAK after knockdown of ECT2 was shorter at 12 hours of CHX treatment than in the control cells, suggesting that ECT2 might regulate the half‐life or stability of FAK (Figure 7E). As it is known, the assembly of focal adhesion molecules leads to cell‐ECM remodeling and alteration. 32 Therefore, we next investigated whether ECT2 expression is necessary for the formation of the focal adhesion complex by performing a Co‐IP assay with FAK antibody in Calu‐3 cells after treatment with siECT2#1. We found that ECT2 suppression impaired FAK interaction with Src, paxillin, and integrin β1. Similar effects were observed with Pho‐Src‐Tyr416, Phos‐paxillin‐Tyr118, and Phos‐integrin β1 (Thr788/789). Consistent with this, we found that silencing of ECT2 in NCI‐H2342 cells led to a decrease of focal adhesion assembly; ECT2‐depleted cells showed a significant decrease in the number of focal adhesions and a reduction of fluorescence intensity quantified as Phos‐paxillin‐Tyr118 (Figure 7G). The Rho family GTPases is known to play a critical role in the control of ECM remodeling and alteration. 32 , 33 , 34 Here, we further confirmed that the activation of Rac1 (Rac1‐GTP) was reduced in ECT2‐depleted cells, whereas ECT2 suppression had almost no effect on the activation of Cdc42 (Cdc42‐GTP) (Figure S8).
4. DISCUSSION
Cell‐ECM interaction involves both transmembrane adhesion receptors and intracellular signaling molecules, which promote a wide range of cellular processes including adhesion, spreading, migration, invasion, and metastasis. 35 , 36
In this study, we identified ECT2 as an essential mediator of cell‐ECM interaction in the malignant progression of LAC. Loss of ECT2 expression inhibits LAC cell adhesion and spreading on the ECM. We suspected that the mechanistic basis of these effects lies in focal adhesion signaling, being associated with the focal adhesion complex that contains both FAK and ECT2.
It is known that cell spreading requires cytoskeletal reorganization and is mediated by several cytoplasmic proteins. Therefore, changes in the actin cytoskeleton are a prerequisite for remodeling and alteration of cell‐ECM interaction. 37 , 38 Previously, Solski et al showed that expression of mutated ECT2 comprising the Db1‐homology domain/pleckstrin homology domain/C‐terminus sequence (DH/PH/C) domain led to the development of lamellipodia and altered actin organization through regulation of Rho family GTPases. 9 The present study confirmed that actin formation and lamellipodia, as well as filopodia extensions, were decreased in ECT2‐depleted cells (Figure 3). Our previous study showed that ECT2 is mislocalized to the cytoplasm of LAC cells. Aberrant cytoplasmic mislocalization of ECT2 becomes evident in early invasive adenocarcinoma and increases during cancer progression. 7 Here, we found that suppression of ECT2 effected cell adhesion, spreading, and morphology, and that the impact was more robust in the presence of ECM components than in their absence (Figures 1, 2, 3 and Figures S1 and S2). Although ECT2 may mediate cell adhesion, spreading, and morphology independently, our findings suggest that ECT2 is likely localized to the cytoplasm and exerts a strong influence on the behavior of cancer cells in relation to the ECM.
Focal adhesions are macromolecular complexes that mediate cell‐ECM interaction and consist of integrin receptors linked to the actin cytoskeleton through focal adhesion–associated proteins, including paxillin, FAK, Src, p130Cas, and Crk, which act in a coordinated manner to regulate cell‐ECM responses. 39 , 40 These molecules are localized predominantly in the cytoplasm and cytoplasmic membrane, often exhibiting altered expression in cancer cells and facilitating cell adhesion and spreading on the ECM. 41 Interestingly, phosphorylated ECT2 at Thr790 is cancer‐specifically localized in the cytoplasm, and in the membrane of LAC cells, suggesting that cytoplasmic and membrane ECT2 may acquire some oncogenic function that facilitates tumor progression. 7 In the present study, the molecular functions of ECT2 were investigated by RNA‐seq analysis of Calu‐3 and NCI‐H2342. Interestingly, our functional enrichment data revealed that ECT2 was localized to focal adhesion complex and affected the focal adhesion signaling pathway. We think that focal adhesion signaling plays an important role in cell‐ECM dynamics. Although other effectors might also contribute to alterations of cell‐ECM interaction, our results strongly suggest that ECT2 regulates cell‐ECM dynamics and alters focal adhesion signaling. Western blot analysis showed that the expression levels of focal adhesion proteins were decreased in ECT2‐depleted cells (Figure 5 and Figure S6), suggesting that ECT2 regulates the expression and/or stability of certain focal adhesion molecules. However, the effects of ECT2 overexpression and whether ECT2 directly regulates focal adhesion molecules in LAC cells remain to be determined.
It has been reported that ECT2 is capable of modulating cell‐cell interaction in both normal cells and cancer cells, 25 , 26 and our present functional enrichment analysis demonstrated a clear association of ECT2 with cell‐cell adhesion (Figure 4C and Figure S4), suggesting that ECT2 is an important mediator of both cell‐ECM and cell‐cell interactions in LAC.
FAK, a nonreceptor tyrosine kinase, functions as a key regulator of cell‐ECM dynamics. Integrin‐ligand interactions stimulate FAK tyrosine autophosphorylation, and in turn FAK binds to Src to facilitate recruitment of the p130Cas‐Crk complex to FAK. However, the assembly of focal adhesion molecules ultimately leads to activation of various biological cellular responses involved in ECM dynamics. 41 Here, we found that FAK can regulate cell proliferation, adhesion, and spreading of LAC cells (Figure 6). Moreover, we found that the level of FAK protein and its phosphorylation sites, and the number of focal adhesion molecules, were reduced in ECT2‐depleted cells (Figure 5 and Figure S6), suggesting that the reduced expression of focal adhesion‐related proteins by knockdown of ECT2 is likely to cause a decrease in LAC cell adhesion and spreading and, consequently, affects ECM dynamics. However, our data showed that ECT2 suppression has a strong influence on cellular responses compared with cells that have been transfected with siFAK. We believe this likely reflects an essential requirement of ECT2 for completion of cytokinesis. It is known that FAK‐mediated regulation of cell adhesion and spreading involves activation of small GTPases including Rac1. 42 We found that ECT2 interacts with and stabilizes FAK in LAC cells (Figure 7A‐E) and that ECT2 serves as a GEF for Rac1 in LAC cells (Figure S8). On the basis of these facts, we speculated that ECT2 might influence the formation of the focal adhesion. Therefore, we examined physical interactions of FAK with molecules involved in the focal adhesions in ECT2‐depleted cells. After knockdown of ECT2, immunoprecipitated FAK impaired the binding of Src, paxillin, and integrin β1, as well as their phosphorylation sites, suggesting that ECT2 acts as an effector of focal adhesion formation by modulating the total amounts of specific proteins and their phosphorylation during ECM dynamics. Phosphorylation of paxillin is known to be necessary for focal adhesion assembly. Our findings indicate that ECT2 suppression results in a reduction in the number of focal adhesions and the intensity of Phos‐Pax‐Ty118 (Figure 7G), suggesting that ECT2 may mediate focal adhesion assembly and direct the cell‐ECM interaction.
In conclusion, we propose that ECT2 is a functional component of cell‐ECM interaction, exerting regulatory effects on cell‐ECM dynamics and possibly regulating the focal adhesion cascade during LAC progression. Our present findings suggest that ECT2 plays an essential role in the pathological steps of LAC progression and could be a potential molecular target for cancer therapy.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
Supporting information
Appendix S1
Fig S1‐S8
Table S1
Table S2
ACKNOWLEDGEMENTS
We thank Professor Masafumi Muratani for his support with RNA‐seq analysis using NextSeq 500 (Illumina) at Tsukuba i‐Laboratory LLP (Tsukuba, Ibaraki, Japan).
Kosibaty Z, Murata Y, Minami Y, Noguchi M, Sakamoto N. ECT2 promotes lung adenocarcinoma progression through extracellular matrix dynamics and focal adhesion signaling. Cancer Sci. 2021;112:703–714. 10.1111/cas.14743
REFERENCES
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. Ca‐Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 2. Nakamura H, Saji H. A worldwide trend of increasing primary adenocarcinoma of the lung. Surg Today. 2014;44:1004‐1012. [DOI] [PubMed] [Google Scholar]
- 3. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497‐1500. [DOI] [PubMed] [Google Scholar]
- 4. Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4‐ALK non‐small cell lung cancer. Eur J Cancer. 2010;46:1773‐1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Noguchi M. Stepwise progression of pulmonary adenocarcinoma‐clinical and molecular implications. Cancer Metast Rev. 2010;29:15‐21. [DOI] [PubMed] [Google Scholar]
- 6. Murata Y, Minami Y, Iwakawa R, et al. ECT2 amplification and overexpression as a new prognostic biomarker for early‐stage lung adenocarcinoma. Cancer Sci. 2014;105:490‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kosibaty Z, Murata Y, Minami Y, et al. Cytoplasmic expression of epithelial cell transforming sequence 2 in lung adenocarcinoma and its implications for malignant progression. Lab Invest. 2019;99:551‐567. [DOI] [PubMed] [Google Scholar]
- 8. Tatsumoto T, Xie XZ, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147:921‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Solski PA, Wilder RS, Rossman KL, et al. Requirement for C‐terminal sequences in regulation of Ect2 guanine nucleotide exchange specificity and transformation. J Biol Chem. 2004;279:25226‐25233. [DOI] [PubMed] [Google Scholar]
- 10. Saito S, Liu XF, Kamijo K, et al. Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation. J Biol Chem. 2004;279:7169‐7179. [DOI] [PubMed] [Google Scholar]
- 11. Kim JE, Billadeau DD, Chen JJ. The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J Biol Chem. 2005;280:5733‐5739. [DOI] [PubMed] [Google Scholar]
- 12. Miki T, Smith CL, Long JE, Eva A, Fleming TP. Oncogene Ect2 is related to regulators of small Gtp‐binding proteins. Nature. 1993;364:737. [DOI] [PubMed] [Google Scholar]
- 13. Hirata D, Yamabuki T, Miki D, et al. Involvement of epithelial cell transforming sequence‐2 oncoantigen in lung and esophageal cancer progression. Clin Cancer Res. 2009;15:256‐266. [DOI] [PubMed] [Google Scholar]
- 14. Huff LP, Decristo MJ, Trembath D, et al. The role of Ect2 nuclear RhoGEF activity in ovarian cancer cell transformation. Genes Cancer. 2013;4:460‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang HK, Liang JF, Zheng HX, Xiao H. Expression and prognostic significance of ECT2 in invasive breast cancer. J Clin Pathol. 2018;71:442‐445. [DOI] [PubMed] [Google Scholar]
- 16. Sano M, Genkai N, Yajima N, et al. Expression level of ECT2 proto‐oncogene correlates with prognosis in glioma patients. Oncol Rep. 2006;16:1093‐1098. [PubMed] [Google Scholar]
- 17. Chen ZG, Liu JT, Zhang YH. Role of epithelial cell transforming sequence 2 (ECT2) in predicting prognosis of osteosarcoma. Med Sci Monitor. 2017;23:3861‐3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hara T, Abe M, Inoue H, et al. Cytokinesis regulator ECT2 changes its conformation through phosphorylation at Thr‐341 in G2/M phase. Oncogene. 2006;25:566‐578. [DOI] [PubMed] [Google Scholar]
- 19. Yuce O, Piekny A, Glotzer M. An ECT2‐centralspindlin complex regulates the localization and function of RhoA. J Cell Biol. 2005;170:571‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Justilien V, Fields AP. Ect2 links the PKCiota‐Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009;28:3597‐3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weeks A, Okolowsky N, Golbourn B, Ivanchuk S, Smith C, Rutka JT. ECT2 and RASAL2 mediate mesenchymal‐amoeboid transition in human astrocytoma cells. Am J Pathol. 2012;181:662‐674. [DOI] [PubMed] [Google Scholar]
- 22. Justilien V, Ali SA, Jamieson L, et al. Ect2‐dependent rRNA synthesis is required for KRAS‐TRP53‐driven lung adenocarcinoma. Cancer Cell. 2017;31:256‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salhia B, Tran NL, Chan A, et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol. 2008;173:1828‐1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iyoda M, Kasamatsu A, Ishigami T, et al. Epithelial cell transforming sequence 2 in human oral cancer. PLoS One. 2010;5(11):e14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu XF, Ishida H, Raziuddin R, Miki T. Nucleotide exchange factor ECT2 interacts with the polarity protein complex Par6/Par3/protein kinase C zeta (PKC zeta) and regulates PKC xi activity. Mol Cell Biol. 2004;24:6665‐6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ratheesh A, Gomez GA, Priya R, et al. Centralspindlin and alpha‐catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012;14:818‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tan H, Wang X, Yang X, Li H, Liu B, Pan P. Oncogenic role of epithelial cell transforming sequence 2 in lung adenocarcinoma cells. Exp Ther Med. 2016;12:2088‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Fokkelman M, Balcioglu HE, Klip JE, et al. Cellular adhesome screen identifies critical modulators of focal adhesion dynamics, cellular traction forces and cell migration behaviour. Sci Rep. 2016;6:31707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berrier AL, Yamada KM. Cell‐matrix adhesion. J Cell Physiol. 2007;213:565‐573. [DOI] [PubMed] [Google Scholar]
- 30. Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56‐68. [DOI] [PubMed] [Google Scholar]
- 31. Schoenherr C, Byron A, Sandilands E, et al. Ambra1 spatially regulates Src activity and Src/FAK‐mediated cancer cell invasion via trafficking networks. Elife. 2017;6:e23172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metast Rev. 2009;28:35‐49. [DOI] [PubMed] [Google Scholar]
- 33. Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509‐514. [DOI] [PubMed] [Google Scholar]
- 34. Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23‐32. [DOI] [PubMed] [Google Scholar]
- 35. Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209:139‐151. [DOI] [PubMed] [Google Scholar]
- 36. Jinka R, Kapoor R, Sistla PG, Raj TA, Pande G. Alterations in cell‐extracellular matrix interactions during progression of cancers. Int J Cell Biol. 2012;2012:219196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Broussard JA, Webb DJ, Kaverina I. Asymmetric focal adhesion disassembly in motile cells. Curr Opin Cell Biol. 2008;20:85‐90. [DOI] [PubMed] [Google Scholar]
- 39. Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol. 2011;3:a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagano M, Hoshino D, Koshikawa N, Akizawa T, Seiki M. Turnover of focal adhesions and cancer cell migration. Int J Cell Biol. 2012;2012:310616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee BY, Timpson P, Horvath LG, Daly RJ. FAK signaling in human cancer as a target for therapeutics. Pharmacol Therapeut. 2015;146:132‐149. [DOI] [PubMed] [Google Scholar]
- 42. Tomar A, Schlaepfer DD. Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr Opin Cell Biol. 2009;21:676‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Fig S1‐S8
Table S1
Table S2