

Abstract
The aim of this study was to develop a noninvasive serological diagnostic approach in identifying and evaluating a panel of candidate autoantibodies to tumor‐associated antigens (TAAs) based on protein microarray technology for early detection of ovarian cancer (OC). Protein microarray based on 154 proteins encoded by 138 cancer driver genes was used to screen candidate anti‐TAA autoantibodies in a discovery cohort containing 17 OC and 27 normal controls (NC). Indirect enzyme‐linked immunosorbent assay (ELISA) was used to detect the content of candidate anti‐TAA autoantibodies in sera from 140 subjects in the training cohort. Differential anti‐TAA autoantibodies were further validated in the validation cohort with 328 subjects. Subsequently, 112 sera from the patients with ovarian benign diseases with 104 OC sera and 104 NC sera together were recruited to identify the specificity of representative autoantibodies to OC among ovarian diseases. Five TAAs (GNAS, NPM1, FUBP1, p53, and KRAS) were screened out in the discovery phase, in which four of them presented higher levels in OC than controls (P < .05) in the training cohort, which was consistent with the result in the subsequent validation cohort. An optimized panel of three anti‐TAA (GNAS, p53, and NPM1) autoantibodies was identified to have relatively high sensitivity (51.2%), specificity (86.0%), and accuracy (68.6%), respectively. This panel can identify 51% of OC patients with CA125 negative. This study supports our assumption that anti‐TAA autoantibodies can be considered as potential diagnostic biomarkers for detection of OC; especially a panel of three anti‐TAA autoantibodies could be a good tool in immunodiagnosis of OC.
Keywords: autoantibody, early detection, ovarian cancer, protein microarray, tumor‐associated antigen
We developed a noninvasive serological diagnostic approach of ovarian cancer (OC). Autoantibodies can be considered as potential biomarkers for the detection of OC. A panel of three anti‐TAA (GNAS, p53, and NPM1) autoantibodies was identified.

Abbreviations
- AUC
area under the ROC
- BSA
bovine sera albumin
- CI
confidence interval
- ELISA
enzyme‐linked immunosorbent assay
- FIGO
International Federation of Gynecology and Obstetrics
- IgG
immunoglobulin G
- +LR
positive likelihood ratio
- −LR
negative likelihood ratio
- NHS
normal human sera
- NPV
negative predictive value
- OC
ovarian cancer
- OD
optical density
- PBST
phosphate‐buffered saline Tween 20
- PPV
positive predictive value
- ROC
receiver operating characteristic curve
- RT
room temperature
- SNR
signal to noise ratio
- TAA(s)
tumor‐associated antigen(s)
- YI
Youden's index
1. INTRODUCTION
Ovarian cancer (OC) is one of the most common malignancies in women's reproductive organs. There were 295 414 new cases and 184 799 deaths in 2018 worldwide. 1 As the ovaries are deep in the pelvic cavity, and there is no early effective screening diagnostic method, 70%‐80% of OC patients are already in advanced stage with poor prognosis and 5‐year survival rate lower than 20% when they are diagnosed. Only 20% of OC patients are diagnosed in stage I or II. 2 Histopathological biopsy is thought to be the gold standard in diagnosing OC, 3 not applicable for screening. Currently, transvaginal ultrasound and detection of CA125 and HE4 in sera are mainly used to identify benign and malignant pelvic mass. However, the problem is that CA125 and HE4 are not sensitive enough to detect early‐stage OC. Moreover, CA125 and HE4 tests have a very low predictive value and a high false positive rate. 4 , 5 Therefore, more novel biomarkers are urgently needed for early diagnosis of OC.
Genetic information changes or abnormal expression of gene products caused by a series of gene mutations can drive normal cells to tumor cells, these mutated genes are called cancer driver genes. They can threaten life and health, leading to occurrence and development of tumors. 6 , 7 , 8 Expression of abnormal proteins is often accompanied by the development of tumor tissues, some of which can enter the blood circulation system and be recognized by the host immune system. These proteins are named tumor‐associated antigens (TAAs). 9 , 10 , 11 The immune system of tumor patients can recognize these abnormally expressed proteins and produce corresponding anti‐TAA autoantibodies, which can exist in the blood for a long time, even months or years prior to clinical diagnosis. Meanwhile, they are at low levels in normal human sera (NHS); therefore, the anti‐TAA autoantibodies may have a great potential as tumor biomarkers for cancer detection. 9 , 12 , 13
Protein microarray is a commonly used high‐throughput technology for identification of novel TAAs. This approach only needs a very small amount of serum samples and reagents to detect multiple TAAs or anti‐TAA autoantibodies simultaneously on a chip, with the advantages of automation, speediness, and high sensitivity. 14 , 15 Therefore, this approach has gradually become the mainstream technology for screening and identifying new tumor biomarkers. Enzyme‐linked immunosorbent assay (ELISA) is one of the most extensively used methods for evaluating TAAs or anti‐TAA autoantibodies as tumor biomarkers. The present study was the first to use customized protein microarray based on proteins encoded by 138 tumor driver genes 16 in order to screen potential TAAs in OC. The selected anti‐TAA autoantibodies were further confirmed by indirect ELISA with two testing cohorts. In the training cohort, differential expressions of anti‐TAA autoantibodies were verified. Following the evaluation in the training cohort, a validation cohort was used to confirm the results from the training cohort and to construct a new panel of anti‐TAA autoantibodies for early diagnosis of OC with high sensitivity and specificity. The diagnostic specificity of the panel in OC diagnosis among ovary diseases was also evaluated by using the sera from three groups. Figure 1 is the flow‐chart of the study design. Our ultimate goal was to establish a new noninvasive method to diagnose OC.
Figure 1.

Study design
2. MATERIALS AND METHODS
2.1. Study subjects
Three independent cohorts consisting of 251 OC patients and 261 normal controls (NCs) were included in this study. All NCs were confirmed without malignant diseases. The discovery cohort consisted of 17 OC patients and 27 NCs. The training cohort contained 70 OC patients and 70 NCs. The validation cohort included 164 OC patients and 164 NCs. In addition, 112 benign control sera (including 95 ovarian cysts and 17 adenomas) were used to evaluate the specificity of the panel for the diagnosis of OC in ovarian diseases. All the subjects in the OC and NC groups were matched by age. In the present study, 17 OC sera and 27 NHS in the discovery phase were derived from the serum bank of the Tumor Epidemiology Laboratory of Zhengzhou University . The OC sera in the training and validation cohorts and benign control sera were collected from three affiliated hospitals of Zhengzhou University in Henan, China from July 2017 to May 2018, while NHS were collected from the physical examination department in the same hospitals in the same time period. All blood samples were centrifuged at 1500 g for 10 minutes after collection, and then the sera were stored in a freezer at − 80°C for further use.
All subjects participating in the study have signed the informed consent form. The study has been approved by the Medical and Health Research Ethics Committee of Zhengzhou University.
2.2. Protein microarray
The customized protein microarray included 154 proteins: 143 of them were encoded by 138 cancer driver genes and bought from CDI Labs ; the other 11 proteins were from the Tumor Epidemiology Laboratory of Zhengzhou University. The names and arrangement of the 138 cancer driver genes are listed in Table S1. This customized protein microarray was made by BC Biotechnology Co., LTD. Autoantibodies in the sera bond to proteins coated on the microarray and then reacted with fluorescence‐labeled anti‐human IgG secondary antibodies, forming fluorescent complexes whose signals could be captured and read on a fluorescent scanner. In brief, the chip was removed from the − 80°C freezer and placed in 4°C for 30 minutes, and then it was reheated at room temperature (RT) for 15 minutes. After the chip was fixed in the side‐swing shaker, 10% bovine sera albumin (BSA) was added to block the chip for 3 hours at RT. Following the blocking step, a serum dilution of 1:50 was quickly added to the chip for overnight incubation at 4°C. After being washed three times with phosphate‐buffered saline (PBS) containing 0.05% Tween (PBST), the chip was incubated with 3 ml of Cy5‐labeled goat anti‐human IgG antibody in a dark room at RT for 1 hour. Subsequently, the chip was washed three times with PBST and twice with double‐distilled H2O. Finally, the chip was scanned with the LuxScanTM 10K Microarray Scanner (BioCapital), and the probe signal was acquired using the GenePix Pro 6.0 software (Molecular Devices). In order to minimize the deviation caused by inconsistent backgrounds, the ratio of the foreground value to the background value (SNR, signal to noise ratio) of each protein was used in the following analysis. In detail, the median value of the signal foreground value under the 532 nm channel was defined as F532 median, and the median of the background value under the 532 nm channel was defined as B532. The SNR, F median/B median, was defined to eliminate the deviation caused by the inconsistency of background values between different samples. In order to evaluate the stability of different chips operating at different times, the samples were repeatedly tested according to different times, different chips, and different positions in the experiment up to 30 times. Based on SNR, the results were preliminarily screened and analyzed for 154 tumor‐related antigens.
2.3. Enzyme‐linked immunosorbent assay
Five recombinant proteins including GNAS, NPM1, FUBP1, p53, and KRAS were used in ELISA. GNAS, FUBP1, and KRAS were purchased from Cloud‐Clone Corporation. p53 and NPM1 were provided by the Tumor Epidemiology Laboratory of Zhengzhou University . Each recombinant protein was diluted in PBS to a final concentration of 0.5 μg/ml for coating on the 96‐well plates overnight at 4°C. Subsequently all plate wells were blocked with 2% BSA dissolved in PBST for 16 hours at 4°C. After washing three times with PBST, a serum dilution of 1:100 was added to each well at 37°C for 1 hour. The washed plates were then incubated with horseradish peroxidase (HRP)‐conjugated goat anti‐human IgG diluted at 1:10 000 at 37°C for another 1 hour, followed by washing with PBST. The substrate mixed by 50% of 3,3’,5,5’‐tetramethylbenzidine and 50% hydrogen peroxide (100 μL) was added to each well at 37°C for 5 minutes. In the last step, 50 μL termination solution (2M H2SO4) was used to stop the reaction. All plates were put on the microplate reader to read the optical density (OD) value at double wavelength of 450 and 620 nm. Each run of ELISA included eight NHS for normalization of the OD value among the plates as well as two negative and two positive sera for quality control.
2.4. Statistical analysis
All statistical analyses were performed using SPSS 21.0 and GraphPad 6.0. Chi‐square test or Fisher's exact test was used for statistical analysis of classified data. The Mann‐Whitney U test was used to compare the continuous variables between two groups. The Kruskal‐Wallis H test was used to compare continuous variables among the three groups. Receiver operating characteristic (ROC) curves were generated. Area under the ROC curve (AUC), sensitivity, specificity, Youden’s index (YI), accuracy rate with negative likelihood ratio (−LR), positive likelihood ratio (+LR), positive predictive value (PPV), and negative predictive value (NPV) together were used to evaluate the diagnostic value of all anti‐TAA antibodies. P values <.05 were considered significant.
3. RESULTS
The 512 participants were classified into three phases. The basic information and clinical characteristics of all OC patients in the three phases are shown in Table 1.
Table 1.
Characteristics of ovarian cancer (OC) patients in the discovery, training, and validation cohort
| Variables | Discovery cohort (%) (n = 17) | Training cohort (%) (n = 70) | Validation cohort (%) (n = 164) |
|---|---|---|---|
| Age (year) | |||
| Mean ± SD | 48.00 ± 17.90a | 51.79 ± 13.09a | 53.01 ± 10.84a |
| Family history | |||
| No | 17 (100.00) | 57 (75.71) | 128 (78.05) |
| Yes | 0 (0.00) | 13 (18.57) | 36 (21.95) |
| FIGO | |||
| I | 3 (17.65) | 13 (18.57) | 27 (16.46) |
| II | 3 (17.65) | 12 (17.14) | 20 (12.20) |
| III | 7 (41.18) | 28 (40.00) | 73 (44.51) |
| IV | 2 (11.76) | 14 (20.00) | 40 (24.39) |
| Unknown | 2 (11.76) | 3 (4.29) | 4 (2.44) |
| Histologic type, n (%) | |||
| Epithelial tumor | 14 (82.35) | 61 (87.12) | 133 (81.10) |
| Sexual cord interstitial tumor | 1 (5.88) | 2 (2.86) | 11 (6.71) |
| Germ cell tumor | 0 (0.00) | 3 (4.29) | 6 (3.66) |
| Unknown | 2 (11.76) | 4 (5.71) | 14 (8.54) |
| Lymph node metastasis | |||
| No | 9 (52.94) | 33 (47.14) | 71 (43.29) |
| Yes | 8 (47.06) | 37 (52.86) | 93 (56.71) |
| Distant metastasis | |||
| No | 15 (88.24) | 53 (75.71) | 124 (75.61) |
| Yes | 2 (11.76) | 17 (24.29) | 40 (24.39) |
Abbreviation: FIGO, International Federation of Gynecology and Obstetrics.
aCompared with the control group.
P value on age was greater than 0.05 in three phases.
3.1. Protein microarray technology in the discovery phase and ELISA verification in the training phase
In this phase, five anti‐TAA (GNAS, NPM1, FUBP1, p53, and KRAS) autoantibodies were screened out based on AUCs (>0.5) and P values (<.05). The SNR values of five anti‐TAA autoantibodies in OC sera were significantly higher than those in NHS. The ROC curves and scatter plots are shown in Figure 2. The range of AUC of a single anti‐TAA autoantibody was 0.686‐0.712.
Figure 2.

Receiver operating characteristic curve (ROC) and scatter plots for five anti–tumor‐associated antigen (TAA) autoantibodies in the discovery cohort. A, C, E, G, I, Analysis of ROC with area under curve (AUC) and 95% confidence interval (CI). B, D, F, H, J, Serological levels in signal to noise ratio (SNR) of five anti‐TAA autoantibodies in ovarian cancer (OC) and normal human sera (NHS) in scatter plots. The longest line means median, and the 25th and 75th percentiles are represented by the shorter lines
Five anti‐TAA autoantibodies identified by protein microarray were further verified by ELISA in the training cohort. Their OD values and ROC curves are presented in Table 2 and Figure 3. The OD values of four anti‐TAA (NPM1, GNAS, p53, and KRAS) autoantibodies in the OC sera were significantly higher than those in the NHS (P < .05). The range of AUC of four anti‐TAA autoantibodies was 0.611‐0.708. These four anti‐TAA autoantibodies were subsequently confirmed in the following validation phase.
Table 2.
Five anti–tumor‐associated antigens’ (TAAs) autoantibody levels between ovarian cancer (OC) and normal human sera (NHS) in training and validation
| Anti‐TAA autoantibody | Training cohort | Validation cohort | ||||||
|---|---|---|---|---|---|---|---|---|
| OC (OD) | NHS (OD) | Z | P | OC (OD) | NHS (OD) | Z | P | |
| NPM1 | 0.391(0.334‐0.456)* | 0.358(0.310‐0.411) | ‐2.095 | .021 | 0.391(0.334‐0.437) | 0.342 (0.293‐0.397) | ‐4.384 | <.001 |
| GNAS | 0.186 (0.139‐0.246) | 0.153 (0.125‐0.182) | ‐4.115 | <.001 | 0.190 (0.148‐0.237) | 0.154 (0.121‐0.185) | ‐6.222 | <.001 |
| FUBP1 | 0.332 (0.203‐0.461) | 0.286 (0.234‐0.368) | ‐0.751 | .610 | ‐ | ‐ | ‐ | ‐ |
| p53 | 0.325 (0.237‐0.445) | 0.232 (0.170‐0.319) | ‐5.570 | <.001 | 0.352 (0.240‐0.423) | 0.240 (0.177‐0.316) | ‐6.794 | <.001 |
| KRAS | 0.319 (0.264‐0.385) | 0.247 (0.208‐0.299) | ‐3.886 | <.001 | 0.321 (0.269‐0.397) | 0.250 (0.185‐0.339) | ‐5.586 | <.001 |
Abbreviation: OD, optical density.
Median (25 percentile to 75 percentile).
Figure 3.
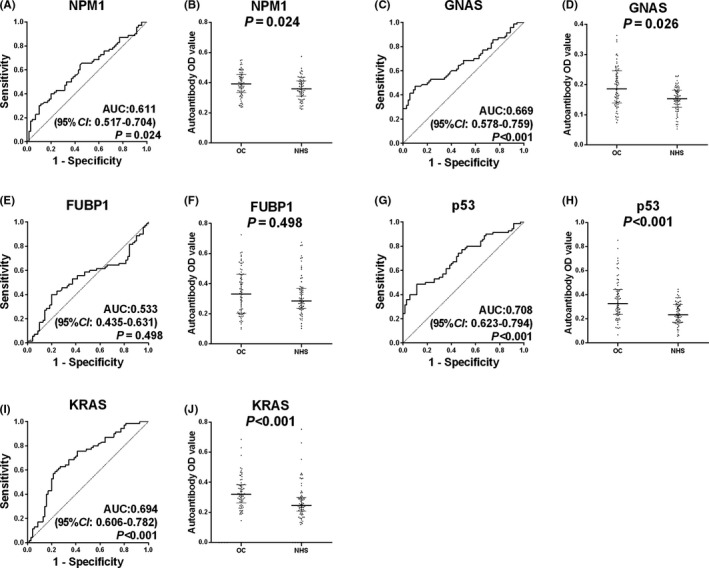
Receiver operating characteristic curve (ROC) and scatter plots for five anti–tumor‐associated antigen (TAA) autoantibodies in the training cohort. A, C, E, G, I, ROC analysis with area under curve (AUC) and 95% confidence interval (CI). B, D, F, H, J, Scatter plots; the longest line means median, and the 25th and 75th percentiles are represented by the shorter lines
3.2. Further confirmation in the validation phase
Four anti‐TAA (NPM1, GNAS, p53, and KRAS) autoantibodies with differences between groups in the training cohort were further confirmed in the validation cohort. The ROC curves and scatter plots for four individual anti‐TAA autoantibodies are shown in Figure 4. The results also confirmed that the OD values of four anti‐TAA autoantibodies in the OC sera were significantly higher than those in the NHS (P < .05). The range of AUC of four anti‐TAA autoantibodies was 0.640‐0.717.
Figure 4.

Performance of four anti–tumor‐associated antigen (TAA) autoantibodies in the validation cohort. A, C, E, G, Receiver operating characteristic curve (ROC) analysis with area under curve (AUC) and 95% confidence interval (CI). B, D, F, H, Scatter plots; the longest line means median, and the 25th and 75th percentiles are represented by the shorter lines
As the OD values of anti‐TAA autoantibodies did not meet the normal distribution, the 95th percentile of NHS was selected as the cutoff value. The sera with anti‐TAA autoantibodies higher than or equal to the cutoff value were judged as positive sera, otherwise were defined as normal. The positive rate and diagnostic value of each anti‐TAA autoantibody are shown in Table 3. The sensitivities ranging from 4.88% to 34.76% with specificity at 95.12% were significantly higher in the OC group than those in the NC group except the anti‐KRAS autoantibody. Among them, the diagnostic value of the anti‐p53 autoantibody was the highest with a diagnostic sensitivity of 34.76%, specificity of 95.12%, and YI of 0.2988.
Table 3.
Diagnostic value of four anti–tumor‐associated antigen (TAA) autoantibodies in the validation cohort
| Anti‐TAA autoantibody | Positive (%) |
χ2 |
P | Sensitivity (%) | Specificity (%) | YI | Cutoff value | +LR | −LR | PPV (%) | NPV (%) | Accuracy (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OC (n = 164) | NHS (n = 164) | ||||||||||||
| NPM1 | 27 (16.46) | 8 (4.88) | 11.55 | .001 | 16.46 | 95.12 | 0.1159 | 0.4685 | 3.38 | 0.88 | 77.14 | 53.24 | 55.79 |
| GNAS | 47 (28.66) | 8 (4.88) | 33.23 | <.001 | 28.66 | 95.12 | 0.2378 | 0.2248 | 5.88 | 0.75 | 85.45 | 57.14 | 61.89 |
| p53 | 57 (34.76) | 8 (4.88) | 46.07 | <.001 | 34.76 | 95.12 | 0.2988 | 0.3927 | 7.13 | 0.69 | 87.69 | 59.32 | 64.94 |
| KRAS | 8 (4.88) | 8 (4.88) | <0.01 | 1.000 | 4.88 | 95.12 | 0.0000 | 0.5234 | 1.00 | 1.00 | 50.00 | 50.00 | 50.00 |
Cutoff value: the 95th percentile of NHS was set up as the cutoff value.
Abbreviations: +LR, positive likelihood ratio; −LR, negative likelihood ratio; NHS, normal health sera; NPV, negative predictive value; OC, ovarian cancer; PPV, positive predictive value; YI, Youden’s index.
3.3. Stepwise increase in the sensitivity of anti‐TAA autoantibodies with successive addition of TAAs
As described above in the validation stage, the sensitivity of an individual anti‐TAA autoantibody as a marker in OC is always very low. When we added anti‐TAA autoantibodies in turn to the panel starting with p53, which had the highest frequency of autoantibody in OC, there was a stepwise increase in sensitivity up to 51.83%, which was significantly higher than that in NHS (P < .05) (Table 4). Obviously, with the increasing number of anti‐TAA autoantibodies, the sensitivity increased gradually, and the specificity was gradually reduced. When the number of combined anti‐TAA autoantibodies reached three, the YI at 0.372 and the accuracy at 68.60% were already able to achieve the maximum. Therefore, a panel of three anti‐TAA (p53, GNAS, and NPM1) autoantibodies is most likely an optimal combination in the detection of OC (Table 4 and Figure 5).
Table 4.
Evaluation of diagnostic value of autoantibodies to different combinations of tumor‐associated antigens (TAAs)
| TAA panel | Positive (%) | Sensitivity (%) | Specificity (%) | YI | ++LR | −LR | PPV (%) | NPV (%) | Accuracy (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| OC (n = 164) | NHS (n = 164) | |||||||||
| p53 | 57 (34.76) | 8 (4.88) | 34.76 | 95.12 | 0.2988 | 7.13 | 0.69 | 87.69 | 59.32 | 64.94 |
| p53 or GNAS | 74 (45.12) | 15 (9.15) | 45.12 | 90.85 | 0.3598 | 4.93 | 0.60 | 83.15 | 62.34 | 67.99 |
| p53 or GNAS or NPM1 | 84 (51.22) | 23 (14.02) | 51.22 | 85.98 | 0.3720 | 3.65 | 0.57 | 78.50 | 63.80 | 68.60 |
| p53 or GNAS or NPM1 or KRAS | 85 (51.83) | 27 (16.46) | 51.83 | 83.54 | 0.3537 | 3.15 | 0.58 | 75.89 | 63.43 | 67.68 |
Abbreviations: +LR, positive likelihood ratio; −LR, negative likelihood ratio; NHS, normal health sera; NPV, negative predictive value; OC, ovarian cancer; PPV, positive predictive value; YI, Youden’s index.
Figure 5.
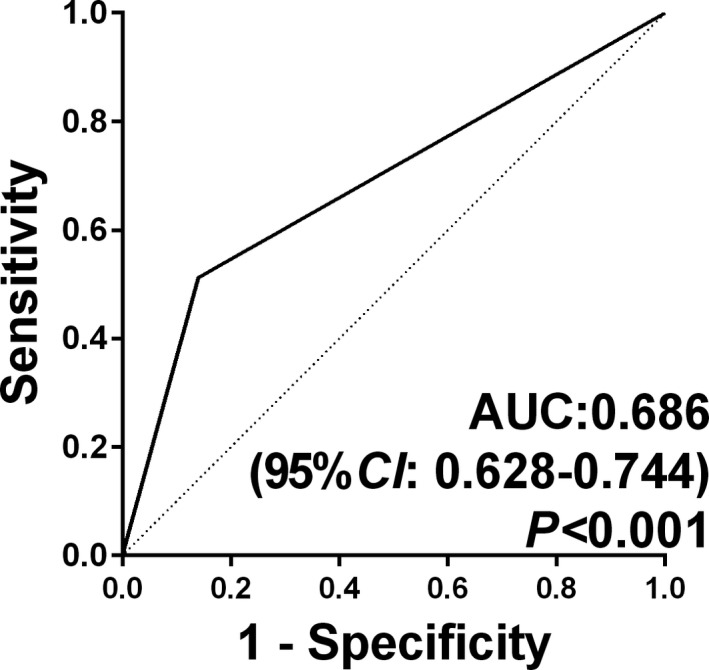
Receiver operating characteristic curve (ROC) analysis of ovarian cancer OC versus normal human sera (NHS) for the panel of three anti–tumor‐associated antigen (TAA) autoantibodies using prediction probability value. area under curve (AUC) and 95% confidence interval (CI) were based on the sensitivity and specificity of the panel
3.4. The diagnostic value of the optimal combination of anti‐TAA autoantibodies
In the validation group, OC subjects were divided into two subgroups (early‐stage and late‐stage). The patients at stage I and II were defined as early‐stage, the patients at stage III and IV were defined as late‐stage. As shown in Table 5, the positive rates of this optimal panel in both early and late stages in the OC group were significantly higher than those in the NC group. The sensitivity, specificity, YI, and accuracy of the panel were 57.45%, 85.98%, 0.4342, and 79.62%, respectively, in early‐stage OC, and 48.67%, 85.98%, 0.3465, and 70.76%, respectively, in late‐stage OC. The ability of the optimal panel to distinguish OC patients from NCs reached 57.45% in early stage while 48.67% in late stage. No significant differences were seen in the subgroups with other clinical features.
Table 5.
Comparison of diagnostic value of the panel for different FIGO stages of OC
| Group | n | Positive (%) | P * | P # | Sensitivity (%) | Specificity (%) | YI | +LR | −LR | PPV (%) | NPV (%) | Accuracy (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Early‐stage (I‐II) | 47 | 27 (57.45) | .312 | <.001 | 57.45 | 85.98 | 0.4342 | 1.17 | 0.14 | 54.00 | 87.58 | 79.62 |
| Late‐stage (III‐IV) | 113 | 55 (48.67) | <.001 | 48.67 | 85.98 | 0.3465 | 2.39 | 0.41 | 70.51 | 70.85 | 70.76 | |
| NHS | 164 | 23 (14.02) |
Abbreviations: +LR, positive likelihood ratio; −LR, negative likelihood ratio; NHS, normal health sera; NPV, negative predictive value; OC, ovarian cancer; PPV, positive predictive value; YI, Youden’s index.
P refers to the comparison between early‐stage and late‐stage.
P refers to the comparison between OC and NHS.
3.5. The specificity of the optimal combination of anti‐TAA autoantibodies in the diagnosis of OC among ovarian diseases
In order to explore whether these autoantibodies can differentiate OC from ovarian benign diseases, we tested the autoantibodies against p53, GNAS, and NPM1 in the best combination panel in three groups’ sera, in which 112 sera were from patients with ovarian benign diseases, 104 were OC sera, and 104 NHS were randomly drawn from the datasets used for ELISA training and validation. The average ages of subjects for benign diseases, NHS, and OC groups were 35.83 ± 11.05, 48.72 ± 12.10, and 51.17 ± 12.31, respectively. As shown in Figure 6, all three individual autoantibody levels were significantly higher in OC sera than in benign tumor and NC sera, without difference between benign tumor sera and NC sera. When the NHS were used as controls, the AUC of p53, GNAS, and NPM1 autoantibodies reached 0.661, 0.644, and 0.715, respectively, while when benign disease sera were used as controls, the AUC reached 0.622, 0.680, and 0.618 respectively, showing that these autoantibodies were capable of distinguishing not only OC from normal but also OC from benign ovarian diseases.
Figure 6.

Performance of three anti–tumor‐associated antigen (TAA) autoantibodies in three groups. B‐D, F‐H, J‐L, Receiver operating characteristic curve (ROC) analysis with area under curve (AUC) and 95% confidence interval (CI) between three groups. A, E, I, Scatter plots; the longest line means median, and the 25th and 75th percentiles are represented by the shorter lines
3.6. three anti‐TAA autoantibodies in combination with CA125
In the present study, CA125 for 108 of 164 OC patients was available for us to determine the relationship of CA125 to anti‐TAA autoantibodies. As shown in Table 6, 34 of 67 OC patients (50.75%) with normal level of CA125 (<35 U/ml) can be detected and diagnosed as positive using the panel of three anti‐TAA autoantibodies.
Table 6.
Sensitivity of combined use of both CA125 and a panel of three anti–tumor‐associated antigen (TAAs).
| Panel | CA125 | Total | |
|---|---|---|---|
| ≥35 U/mL | <35 U/mL | ||
| + | 18 (A) | 34 (B) | 52 |
| − | 23 (C) | 33 (D) | 56 |
| Total | 41 | 67 | 108 |
Sensitivity (%) of CA125 = (A + C) / (A + B + C + D) = 41/108 = 37.96%.
Sensitivity (%) of a panel of three anti‐TAAs = (A + B)/ (A + B + C + D) = 52/108 = 48.15%.
Sensitivity (%) of combined both CA125 and a panel of three anti‐TAAs = (A + B + C)/ (A + B + C + D) = 75/108 = 69.44%.
As shown in Table 7, the differences in the positive rates between a single parameter (anti‐TAA autoantibody panel or CA125) and the combination of the anti‐TAA autoantibody panel and CA125 were statistically significant, respectively (P < .05). If both the panel of three anti‐TAA autoantibodies and CA125 were simultaneously used as diagnostic markers, 75 of 108 (69.44%) OC patients could be correctly detected. CA125 and anti‐TAA autoantibody panel appear to be independent but supplementary serological biomarkers for the detection of OC.
Table 7.
Positive rates of the panel, CA125 and the combination of CA125 and panel
The comparison of positive rates between panel and the combination of CA125 and panel.
The comparison of positive rates between CA125 and the combination of CA125 and panel.
4. DISCUSSION
Early diagnosis of OC is of great significance for improving clinical treatment effect and prognosis. 4 CA125, HE4, OVA1 tests, and ROMA algorithm have been used clinically to assess the risk of malignancy of female pelvic tumors, but these indicators or tests did not meet the requirements for early screening. 17 , 18 Noninvasive serological screening was a better choice, and many studies have confirmed that anti‐TAA autoantibodies can be used as effective biomarkers to diagnose tumors early. 2 , 19 , 20 , 21 , 22 Anti‐TAA autoantibody is a noninvasive biomarker with the advantage that it can stay stable and persists longer in human sera than TAA itself. 23 On the other hand, anti‐TAA autoantibody can be detected months or even years before the onset of major clinical symptoms. 9 , 13 , 24 Protein microarray is a new technology developed at the beginning of this century to study protein interaction, especially for proteomic research. 25 Currently, it is extremely efficient and reliable in searching for potential tumor biomarkers. Multiple tumor‐related antigens or anti‐TAA autoantibodies can be detected quickly and accurately on a fixed chip, 26 which plays a major role in the diagnosis of tumors. 27 , 28 , 29
The protein microarray used in this study was constructed based on the proteins encoded by 138 cancer driver genes reported by Vogelstein et al, 16 which can make our screening very efficient and targeted. Using the protein microarray approach, five anti‐TAAs (NPM1, GNAS, FUBP1, KRAS, and p53) were screened out as candidate biomarkers to evaluate their immunodiagnostic value in the detection of OC. These five TAAs were found to be expressed in different tumors and to play important roles in tumorigenesis. GNAS is an important protein participating in several cancer signaling pathways. 30 Recent studies have shown that abnormal proliferation of many tumor cells was associated with abnormal functions of GNAS, such as pancreatic cancer, 31 lung cancer, 32 breast cancer, 33 etc. Tominaga et al 34 showed that the amplification of GNAS may be a biomarker for predicting the prognosis of OC. NPM1 is involved in ribosome biosynthesis, 35 mRNA processing, 36 and chromatin remodeling, 37 and it also plays an important role in DNA repair and regulation of apoptosis. 38 Studies have shown that NPM1 can promote tumor growth by inactivating the acting pathway of tumor suppressor p53 and ARF. 39 FUBP1 is an RNA‐binding protein which can regulate mRNA translation or gene stability, 40 promote cell proliferation during the cell cycle, 41 and it is overexpressed in various tumors, such as liver cancer, 42 , 43 non–small cell lung cancer, 44 gastric cancer, 45 etc. As a tumor suppressor protein, p53 exerts its biological activity by blocking cell cycle and inducing apoptosis. 46 Distortion in p53 is the most common genetic mutation in OC, which can be seen in almost 96% of high‐grade serous OCs. 47 Anti‐p53 autoantibody could be detected in 21%‐30% of serum samples from patients with OC. Moreover, the elevated titer of anti‐p53 autoantibody was detected at 22.9 months before cancer diagnosis. 22 KRAS plays a role in promoting cell proliferation and carcinogenesis, and it is also involved in signal transduction. 48 As described above, many studies have indicated that these five TAAs may have a close correlation with cancers. Three (KRAS, GNAS, and p53) of them were identified as the most frequently mutated genes in mucinous OC. 49 , 50 , 51 The mutations in KRAS and GNAS were identified in a series of mucinous ovarian tumors, and the presence of concurrent KRAS and GNAS mutations was also observed. Both of these mutations were associated with oncogenesis. 50 Ryland et al 49 also demonstrated that there was a significant difference in p53 mutation frequency among the three tumor subtypes (P = .003), suggesting that aberrant p53 contributes to the invasive phenotype in a proportion of these OCs. By querying the International Cancer Genome Consortium (ICGC) Data Portal, the mutation rates of p53, GNAS, KRAS, NPM1, and FUBP1 were detected in 93 OCs to be 98.92%, 27.96%, 12.9%, 6.45%, 6.45%, respectively. Studies demonstrated that mutations in cancer driver genes can cause corresponding protein function changes, and these abnormal proteins may be recognized as foreign antigens by the immune system, and thus corresponding autoantibodies will be produced. 16 , 52 According to the suggestion from these studies, it is inferred that high mutation frequencies in p53, KRAS, and GNAS in OC may be related to the occurrence of OC and may be one of the reasons why the corresponding autoantibodies to these target proteins were elevated in OC patients’ sera observed in our study.
The expressions of the identified five TAAs (GNAs, p53, NPM1, KRAS, and FUBP1) were investigated using the Human Protein Atlas (HPA) dataset. It was found that the protein expressions of GNAs, KRAS, and p53 could not be detected in normal ovary tissues, while they were detected at different expression levels in half or more than half of the tested OC tissues. NPM1 and FUBP1 proteins were highly expressed in both the tested normal ovary tissues and OC tissues, suggesting that the autoantibodies against GNAs, KRAS, and p53 that were elevated in OC sera in our study may be correlated with the differential expression of these proteins in normal ovary and OC tissues. High levels of anti‐NPM1 and FUBP1 autoantibodies in OC patients’ sera may be related to other features of immunogens. However, there were other studies showing that high expression of NPM1 protein (73.4%) in OC tissues was a marker of poor prognosis. 53 , 54 High p53 expression in OC tissues was significantly correlated with poor prognosis. 55 , 56 , 57 There are few reports on the expression of GNAS protein in OC.
As the most commonly studied target, the results of p53 at gene, protein, and autoantibody levels all gave important information and suggestions. In a study comprising 82 mucinous ovarian tumors, 49 the frequency of p53 mutations were detected at 9.1% (2/22), 13.8% (4/29), and 51.6% (16/31) in benign tumors, borderline tumors, and carcinomas, respectively, with a significant difference between carcinomas and benign and borderline tumors. The positive rates of p53 protein expression were 0%, 5.7%, and 54.2% in normal ovarian tissues, epithelial ovarian benign tumor, and epithelial OC, respectively. 55 Karen et al 58 reported that p53 autoantibody was detected in 41.7% (25/60) of serous OCs, 13.3% (4/30) of nonserous OCs, 10% (3/30) of benign disease, and 8.3% (10/120) of NCs (combined P = .0002). Our current study indicated that the positive rates of p53, GNAs, and NPM1 autoantibodies were significantly elevated in sera from OC patients compared with sera from ovarian benign disease patients and NCs, which is in line with Karen et al's report as mentioned above. Both Karen et al's and our present results on autoantibody levels are also consistent with the results of other reports. 58 It is inferred from this that the high frequencies of mutations in p53, GNAs, and NPM1 and the high expression of these proteins in OC might be the reason why these corresponding autoantibodies were elevated in sera of patients with OC.
In our study, while evaluating the diagnostic value of anti‐TAA autoantibodies in OC in the validation cohort, the sensitivity range of a single anti‐TAA autoantibody was 4.88%‐34.76%, with specificity at 95.12%, indicating that the diagnostic value of a single anti‐TAA autoantibody is limited. Many previous studies have demonstrated that tumorigenesis is caused by multiple gene mutations, and one type of tumor may produce multiple anti‐TAA autoantibodies. By the detection of a single anti‐TAA autoantibody it is difficult to meet the requirements for early diagnosis in clinical practice; therefore, an optimized panel consisting of several anti‐TAA autoantibodies can significantly increase the sensitivity in the detection of tumor at early stage. 56 , 57 , 59
According to our testing results from the validation dataset, when we added three anti‐TAA autoantibodies one by one to a panel, there was a stepwise increase of sensitivity. Among multiple combinations, a panel of three anti‐TAA (p53, GNAS, and NPM1) autoantibodies presented up to 51.22% of sensitivity, 85.98% specificity, and maximum of YI, which is most likely an optimal combination in the detection of OC. Moreover, the panel could distinguish 57.45% of early‐stage OC patients and 48.67% of late‐stage OC patients from NCs.
A previous study has used the protein microarray approach and identified a panel of eleven anti‐TAA autoantibodies (ICAM3, CTAG2, p53, STYXL1, PVR, POMC, NUDT11, TRIM39, UHMK1, KSR1, and NXF3) as potential biomarkers for diagnosis of OC with sensitivity and specificity at 45% and 98%, respectively. 60 The concern was that in this study the sample size was small, and there was no further verification. Wang et al 19 identified an optimal panel of nine anti‐TAA autoantibodies (RalA, p62, p53, koc, p90, p16, c‐myc, AHSG, and 14‐3‐3zeta) for OC, in which the sensitivity reached 61.4% with 85.0% of specificity, which was much higher than any individual anti‐TAA autoantibody (<20%). If it is further combined with CA125, the sensitivity, specificity, and AUC could reach 94.7%, 78.2%, and 0.914, respectively. Sun et al 61 detected 15 anti‐TAA autoantibodies in OC and NHS and found that the combined detection of MDM2, PLAT, NPM1, 14‐3‐3zeta, p53, and RalA had the highest diagnostic value, with a sensitivity and specificity reaching 72.7% and 96%, respectively. Qin's study 62 also demonstrated that the diagnostic value of the combination of a single anti‐TAA autoantibody with clinical markers was better than any individual marker. The drawback of the above studies was that the TAAs used for autoantibody screening were either selected from scattered literature reports or without a large sample size, especially lacking a large‐scale coverage of cancer driver genes.
In the current study, we constructed a protein microarray based on proteins encoded by cancer driver genes and further used this high‐throughput technology to screen out potential anti‐TAA autoantibodies in OC sera from patients. In the subsequent study, we used two cohorts of subjects to verify and validate the findings from the initial microarray screenings. Compared with other similar studies in OC, the customized protein microarray used in our study includes more potential TAAs, which enabled the screening with high efficacy. We have also developed an optimized panel of three anti‐TAA autoantibodies for the detection of OC. If both the panel of three anti‐TAA autoantibodies and CA125 were simultaneously used as diagnostic markers, almost 70% of OC patients could be correctly detected. CA125 and anti‐TAA autoantibodies appear to be independent but supplementary serological biomarkers for the detection of OC. In brief, the advantages of our study were that the protein chip based on 138 cancer driver genes could efficiently screen out target proteins closely related to OC, and that the expression levels of autoantibodies against p53, GNAS, and NPM1 in sera from the patients with OC, benign ovarian diseases, and normal persons more clearly showed the changes of these biomarkers in the process of ovarian disease development. Moreover, by comparing their levels among the three groups, these three autoantibodies were proved to be specific to OC patients among the patients with different ovarian diseases. In a future study, serial bleeding serum samples at the different time‐points will be collected from OC patients for clinical follow‐up evaluation.
In conclusion, this study supports our assumption that anti‐TAA autoantibodies can be identified as potential diagnostic biomarkers in the diagnosis of OC. An optimal combination of three anti‐TAA autoantibodies can be applied as a good tool for early immunodiagnosis of OC especially in patients with CA125 negative.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
Table S1
ACKNOWLEDGMENTS
This work was supported by the Major Project of Science and Technology in Henan Province [grant number 161100311400] and was also supported in part by the National Institutes of Health (grant numbers R01CA193511 and R01CA232587).
Ma Y, Wang X, Qiu C, et al. Using protein microarray to identify and evaluate autoantibodies to tumor‐associated antigens in ovarian cancer. Cancer Sci. 2021;112:537–549. 10.1111/cas.14732
Yan Ma and Xiao Wang contributed equally to the work.
Contributor Information
Bing‐Hua Jiang, Email: bing-hua-jiang@uiowa.edu.
Jianying Zhang, Email: jianyingzhang@hotmail.com.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Elias KM, Guo J, Bast RC Jr. Early detection of ovarian cancer. Hematol Oncol Clin North Am. 2018;32:903‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perez‐Lopez FR, Chedraui P, Troyano‐Luque JM. Peri‐ and post‐menopausal incidental adnexal masses and the risk of sporadic ovarian malignancy: new insights and clinical management. Gynecol. Endocrinol. 2010;26:631‐643. [DOI] [PubMed] [Google Scholar]
- 4. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376‐1388. [DOI] [PubMed] [Google Scholar]
- 5. Partridge E, Kreimer AR, Greenlee RT, et al. Results from four rounds of ovarian cancer screening in a randomized trial. Obstet. Gynecol. 2009;113:775‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895‐904. [DOI] [PubMed] [Google Scholar]
- 7. Sparano JA, Ostrer H, Kenny PA. Translating genomic research into clinical practice: promise and pitfalls. Am Soc Clin Oncol Educ Book. 2013;15‐23. [DOI] [PubMed] [Google Scholar]
- 8. Zhu Q, Liu M, Dai L, et al. Using immunoproteomics to identify tumor‐associated antigens (TAAs) as biomarkers in cancer immunodiagnosis. Autoimmun Rev. 2013;12:1123‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan EM, Zhang J. Autoantibodies to tumor‐associated antigens: reporters from the immune system. Immunol Rev. 2008;222:328‐340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu W, Peng B, Lu Y, et al. Autoantibodies to tumor‐associated antigens as biomarkers in cancer immunodiagnosis. Autoimmun Rev. 2011;10:331‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lachmann PJ. Tumour immunology: a review. J R Soc Med. 1984;77:1023‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bracci PM, Zhou M, Young S, Wiemels J. Serum autoantibodies to pancreatic cancer antigens as biomarkers of pancreatic cancer in a San Francisco Bay Area case‐control study. Cancer. 2012;118:5384‐5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitchell P. A perspective on protein microarrays. Nat Biotechnol. 2002;20:225‐229. [DOI] [PubMed] [Google Scholar]
- 15. Sreekumar A, Chinnaiyan AM. Using protein microarrays to study cancer. Biotechniques. 2002;46‐53. [PubMed] [Google Scholar]
- 16. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi JX, Qin JJ, Ye H, et al. Tumor associated antigens or anti‐TAA autoantibodies as biomarkers in the diagnosis of ovarian cancer: a systematic review with meta‐analysis. Expert Rev Mol Diagn. 2015;15:829‐852. [DOI] [PubMed] [Google Scholar]
- 18. Daly MB, Ozols RF. The search for predictive patterns in ovarian cancer: proteomics meets bioinformatics. Cancer Cell. 2002;1:111‐112. [DOI] [PubMed] [Google Scholar]
- 19. Wang P, Qin J, Ye H, et al. Using a panel of multiple tumor‐associated antigens to enhance the autoantibody detection in the immunodiagnosis of ovarian cancer. J Cell Biochem. 2019;120:3091‐3100. [DOI] [PubMed] [Google Scholar]
- 20. Mustafa MZ, Nguyen VH, Le Naour F, et al. Autoantibody signatures defined by serological proteome analysis in sera from patients with cholangiocarcinoma. J Transl Med. 2016;14:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin LH, Xu YW, Huang LS, et al. Serum proteomic‐based analysis identifying autoantibodies against PRDX2 and PRDX3 as potential diagnostic biomarkers in nasopharyngeal carcinoma. Clin Proteomics. 2017;14:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang WL, Gentry‐Maharaj A, Simmons A, et al. Elevation of TP53 autoantibody before CA125 in preclinical invasive epithelial ovarian cancer. Clin Cancer Res. 2017;23:5912‐5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anderson KS, LaBaer J. The sentinel within: exploiting the immune system for cancer biomarkers. J. Proteome Res. 2005;4:1123‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang JY, Tan EM. Autoantibodies to tumor‐associated antigens as diagnostic biomarkers in hepatocellular carcinoma and other solid tumors. Expert review of molecular diagnostics. 2010;10:321‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Braun P, Hu Y, Shen B, et al. Proteome‐scale purification of human proteins from bacteria. Proc Natl Acad Sci USA. 2002;99:2654‐2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall DA, Ptacek J, Snyder M. Protein microarray technology. Mech Ageing Dev. 2007;128:161‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wingren C, Sandstrom A, Segersvard R, et al. Identification of serum biomarker signatures associated with pancreatic cancer. Cancer Res. 2012;72:2481‐2490. [DOI] [PubMed] [Google Scholar]
- 28. Huang CS, George S, Lu M, et al. Application of photonic crystal enhanced fluorescence to cancer biomarker microarrays. Anal Chem. 2011;83:1425‐1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rho JH, Mead JR, Wright WS, et al. Discovery of sialyl Lewis A and Lewis X modified protein cancer biomarkers using high density antibody arrays. J Proteomics. 2014;96:291‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bastepe M. GNAS mutations and heterotopic ossification. Bone. 2018;109:80‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ideno N, Ohtsuka T, Matsunaga T, et al. Clinical significance of GNAS mutation in intraductal papillary mucinous neoplasm of the pancreas with concomitant pancreatic ductal adenocarcinoma. Pancreas. 2015;44:311‐320. [DOI] [PubMed] [Google Scholar]
- 32. Ritterhouse LL, Vivero M, Mino‐Kenudson M, et al. GNAS mutations in primary mucinous and non‐mucinous lung adenocarcinomas. Mod. Pathol. 2017;30:1720‐1727. [DOI] [PubMed] [Google Scholar]
- 33. Jin X, Zhu L, Cui Z, et al. Elevated expression of GNAS promotes breast cancer cell proliferation and migration via the PI3K/AKT/Snail1/E‐cadherin axis. Clin Transl Oncol. 2019;21(9):1207‐1219. [DOI] [PubMed] [Google Scholar]
- 34. Tominaga E, Tsuda H, Arao T, et al. Amplification of GNAS may be an independent, qualitative, and reproducible biomarker to predict progression‐free survival in epithelial ovarian cancer. Gynecol Oncol. 2010;118:160‐166. [DOI] [PubMed] [Google Scholar]
- 35. Yu Y, Maggi LB, Brady SN Jr, et al. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol. 2006;26:3798‐3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murano K, Okuwaki M, Hisaoka M, Nagata K. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol Cell Biol. 2008;28:3114‐3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okuwaki M, Matsumoto K, Tsujimoto M, Nagata K. Function of nucleophosmin/B23, a nucleolar acidic protein, as a histone chaperone. FEBS Lett. 2001;506:272‐276. [DOI] [PubMed] [Google Scholar]
- 38. Box JK, Paquet N, Adams MN, et al. Nucleophosmin: from structure and function to disease development. BMC Mol Biol. 2016;17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493‐505. [DOI] [PubMed] [Google Scholar]
- 40. Zheng Y, Miskimins WK. Far upstream element binding protein 1 activates translation of p27Kip1 mRNA through its internal ribosomal entry site. Int J Biochem Cell Biol. 2011;43:1641‐1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding Z, Liu X, Liu Y, et al. Expression of far upstream element (FUSE) binding protein 1 in human glioma is correlated with c‐Myc and cell proliferation. Mol Carcinog. 2015;54:405‐415. [DOI] [PubMed] [Google Scholar]
- 42. Rabenhorst U, Beinoraviciute‐Kellner R, Brezniceanu ML, et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology. 2009;50:1121‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Malz M, Weber A, Singer S, et al. Overexpression of far upstream element binding proteins: a mechanism regulating proliferation and migration in liver cancer cells. Hepatology. 2009;50:1130‐1139. [DOI] [PubMed] [Google Scholar]
- 44. Singer S, Malz M, Herpel E, et al. Coordinated expression of stathmin family members by far upstream sequence element‐binding protein‐1 increases motility in non‐small cell lung cancer. Cancer Res. 2009;69:2234‐2243. [DOI] [PubMed] [Google Scholar]
- 45. Zhang F, Tian Q, Wang Y. Far upstream element‐binding protein 1 (FUBP1) is overexpressed in human gastric cancer tissue compared to non‐cancerous tissue. Onkologie. 2013;36:650‐655. [DOI] [PubMed] [Google Scholar]
- 46. Ebata T, Hirata H, Kawauchi K. Functions of the Tumor Suppressors p53 and Rb in Actin Cytoskeleton Remodeling. Biomed Res Int. 2016;2016:9231057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cancer Genome Atlas Research N . Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sideris M, Emin EI, Abdullah Z, et al. The role of KRAS in endometrial cancer: a mini‐review. Anticancer Res. 2019;39:533‐539. [DOI] [PubMed] [Google Scholar]
- 49. Ryland GL, Hunter SM, Doyle MA, et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015;7:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Matson DR, Xu J, Huffman L, et al. KRAS and GNAS Co‐mutation in metastatic low‐grade appendiceal mucinous neoplasm (LAMN) to the ovaries: a practical role for next‐generation sequencing. Am J Case Rep. 2017;18:558‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bulun SE, Wan Y, Matei D. Epithelial mutations in endometriosis: link to ovarian cancer. Endocrinology. 2019;160:626‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan EM. Autoantibodies as reporters identifying aberrant cellular mechanisms in tumorigenesis. J Clin Invest. 2001;108:1411‐1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Londero AP, Orsaria M, Tell G, et al. Expression and prognostic significance of APE1/Ref‐1 and NPM1 proteins in high‐grade ovarian serous cancer. Am J Clin Pathol. 2014;141:404‐414. [DOI] [PubMed] [Google Scholar]
- 54. Fan X, Wen L, Li Y, et al. The expression profile and prognostic value of APE/Ref‐1 and NPM1 in high‐grade serous ovarian adenocarcinoma. APMIS. 2017;125:857‐862. [DOI] [PubMed] [Google Scholar]
- 55. Liang M, Zhao J. Protein expressions of AIB1, p53 and Bcl‐2 in epithelial ovarian cancer and their correlations with the clinical pathological features and prognosis. Eur Rev Med Pharmacol Sci. 2018;22:5134‐5139. [DOI] [PubMed] [Google Scholar]
- 56. Zhang JY, Casiano CA, Peng XX, et al. Enhancement of antibody detection in cancer using panel of recombinant tumor‐associated antigens. Cancer Epidemiol Biomarkers Prev. 2003;12:136‐143. [PubMed] [Google Scholar]
- 57. Qin J, Wang S, Shi J, et al. Using recursive partitioning approach to select tumor‐associated antigens in immunodiagnosis of gastric adenocarcinoma. Cancer Sci. 2019;110:1829‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Anderson KS, Wong J, Vitonis A, et al. p53 autoantibodies as potential detection and prognostic biomarkers in serous ovarian cancer. Cancer Epidemiol. Biomarkers Prev. 2010;19:859‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang S, Qin J, Ye H, et al. Using a panel of multiple tumor‐associated antigens to enhance autoantibody detection for immunodiagnosis of gastric cancer. Oncoimmunology. 2018;7:e1452582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Katchman BA, Chowell D, Wallstrom G, et al. Autoantibody biomarkers for the detection of serous ovarian cancer. Gynecol. Oncol. 2017;146:129‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sun H, Shi JX, Zhang HF, et al. Serum autoantibodies against a panel of 15 tumor‐associated antigens in the detection of ovarian cancer. Tumour Biol. 2017;39:1010428317699132. [DOI] [PubMed] [Google Scholar]
- 62. Qin J, Wang S, Wang P, et al. Autoantibody against 14‐3‐3 zeta: a serological marker in detection of gastric cancer. J Cancer Res Clin Oncol. 2019;145:1253‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1