

Abstract
In patients with impaired renal function, S‐1–related toxicities increase due to higher exposure of 5‐fluorouracil (5‐FU). Our previous pharmacokinetic study in 16 cancer patients with various renal functions developed an S‐1 dosage formula based on individual creatinine clearance (CLcr) and body surface area (BSA). To evaluate and refine the formula, this prospective study was conducted. Thirty‐three patients with various renal functions received S‐1 for 4 weeks at doses determined by the nomogram derived from the previously developed formula. A series of blood samples were collected after the first dose to calculate the area under the concentration‐time curve (AUC) of 5‐FU. Thirty patients with BSA of 1.14‐1.84 m2 and CLcr of 23.8‐96.4 mL/min were assessable for pharmacokinetics. The observed daily AUC ranged from 712.6 to 2868.7 ng·h/mL, and 18 patients achieved the target AUC (1447.8 ± 545.4 ng·h/mL). Three patients experienced S‐1–related grade 3 adverse events during the first course. In the population pharmacokinetic analysis from the combined data of 46 patients in this study and the previous study, sex was identified as a statistically significant covariate for 5‐FU clearance. Hence, the refined formula includes sex as an additional factor: Recommended daily dose = target AUC × (14.5 + 8.23 × SEX [0 for female and 1 for male] + 0.301 × CLcr) × BSA. Revised nomograms for recommended daily doses derived from the refined formula can be used in clinical practice to achieve the target AUC ensuring efficacy and safety of S‐1.
Keywords: dosage formula, nomogram, renal function, S‐1, target AUC of 5‐FU
Our previously developed S‐1 dosage formula based on renal function was prospectively evaluated. Revised nomograms for recommended daily doses derived from the refined formula can be used in clinical practice to achieve the target AUC ensuring efficacy and safety of S‐1.

1. INTRODUCTION
An oral anticancer drug S‐1 was first approved in Japan in 1999 and has been widely used in Asia and Europe. S‐1 contains tegafur, a 5‐fluorouracil (5‐FU) prodrug; 5‐chloro‐2,4‐dihydroxypyridine (CDHP); and potassium oxonate. 1 , 2 CDHP contributes to maintaining high concentration of 5‐FU by inhibiting dihydropyrimidine dehydrogenase (DPD), which is a rate‐limiting enzyme catabolizing 5‐FU. Potassium oxonate is distributed in the epithelium of the intestine that inhibits 5‐FU phosphorylation to reduce gastrointestinal toxicity. The approved dose of S‐1 in its monotherapy is 40 mg/m2 as tegafur, twice daily, for 4 weeks, followed by 2 weeks rest in Asia; 3 , 4 while in Europe the dose is 25 mg/m2 as tegafur, twice daily, for 3 weeks, followed by 1 week rest in combination with cisplatin at 75 mg/m2 every 4 weeks, but its monotherapy is not approved. 5 S‐1–containing regimens have been reported to improve clinical outcomes of various cancers including gastric cancer, 6 , 7 , 8 , 9 , 10 non–small cell lung cancer, 11 , 12 , 13 colorectal cancer, 14 , 15 breast cancer, 16 pancreatic cancer, 17 and biliary tract cancer. 18
As >50% of CDHP is excreted in the urine, 19 renal dysfunction increases exposure of CDHP leading to excessive inhibition of DPD, and results in a sustained high concentration of 5‐FU. 20 , 21 A postmarketing survey on S‐1 demonstrated that the incidences of adverse reactions were high in patients with impaired renal function. 22 Therefore, S‐1 is contraindicated to patients with severe renal impairment (creatinine clearance [CLcr] <30 mL/min). The European summary of product characteristics (SmPC) of S‐1 indicates a reduced dose for patients with moderate renal impairment (CLcr = 30‐50 mL/min) from a Monte Carlo simulation of virtual patients with renal dysfunction. 5 In contrast, the Asian package insert of S‐1 does not refer to any dose modification for patients with renal impairment. Hence, no reliable recommendation on S‐1 dose reduction for patients with renal dysfunction has been provided.
Our previous pharmacokinetic study in 16 cancer patients with various degrees of renal function developed a population pharmacokinetic (PPK) model and proposed an S‐1 dosage formula based on renal function and body surface area (BSA): Recommended daily dose = target AUC × (21.9 + 0.375 × CLcr) × BSA, where AUC is the area under the concentration‐time curve. 21 We also proposed nomograms for the recommended daily dose of S‐1 to achieve the target AUC (1447.8 in Asia 19 and 1177.2 ng·h/mL in Europe 23 ) according to individual BSA and CLcr by taking into account the approved dosage strengths (20 and 25 mg as tegafur in Asia, 15 and 20 mg as tegafur in Europe). As the next step, this prospective pharmacokinetic study in cancer patients with various renal functions was conducted to evaluate and refine the formula.
2. PATIENTS AND METHODS
This study consists of two stages. In part I, a prospective pharmacokinetic study in cancer patients with various degrees of renal function was conducted at seven institutions in Japan. In part II, the formula was refined by PPK analysis using the combined data of the current and previous studies. Study procedures were in accordance with the ethical standards of the Declaration of Helsinki. Written informed consent was obtained from each patient before enrollment. This study does not fall under the specified clinical trials of the Clinical Trials Act. Therefore, the study was approved by the institutional review board at each institution and registered at the University Hospital Medical Information Network Clinical Trials Registry (UMIN000023880).
2.1. Part I
2.1.1. Study design and patients
The primary endpoint in part I was to evaluate the observed daily AUC of 5‐FU in patients receiving S‐1 at doses determined by our previously developed formula. The secondary endpoint was the toxicity during the first course (6 weeks).
The eligibility criteria for part I were as follows: histologically confirmed solid tumor in patients planned to receive S‐1 monotherapy, CLcr estimated by the Cockcroft‐Gault equation 24 ≥15 mL/min, age ≥20 years, Eastern Cooperative Oncology Group performance status 0‐1, adequate organ function except for renal function (white blood cell count 3500‐12 000/mm3, neutrophil count ≥2000/mm3, platelet count ≥100 000/mm3, total bilirubin ≤1.5 mg/dL, aspartate transaminase [AST] and alanine transaminase [ALT] concentrations ≤100 U/L), and ability to take oral medications.
2.1.2. Treatment
Patients received S‐1 for 4 weeks at doses determined by the nomogram derived from our previously developed formula for the Asian‐approved daily dose of 80 mg/m2. 21 Accordingly, S‐1 was given at 80, 70, 60, 50, and 40 mg twice daily (morning and evening) when the determined daily dose was ≥80 mg, or at 60 and 50 mg once daily (morning) when the daily dose was ≤60 mg. On the first day, S‐1 was administered only in the morning (within 30 minutes after breakfast) to assess pharmacokinetics over 24 hours following a single dose. Adverse events were graded by the US National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. 25
2.1.3. Pharmacokinetic sampling and assay
Blood samples were collected before and 2, 4, 7, 12, and 24 hours after the first administration of S‐1. Peripheral blood (3 mL) was drawn into heparinized tubes at each sampling time and was centrifuged at 3000 rpm for 10 minutes at room temperature. Plasma was frozen and stored at −20°C until analysis. 5‐FU in collected samples was protected from ex vivo catabolism by DPD 26 because of existing CDHP.
Plasma concentrations of 5‐FU were determined using an ultraperformance liquid chromatography (UPLC)–tandem mass spectrometry method developed specifically for this study as previously reported, 21 according to the US Food and Drug Administration Guidance for Industry Bioanalytical Method Validation. 27
2.1.4. Evaluation of the previously developed formula
The AUC of 5‐FU following a single dose of S‐1 was calculated using the linear trapezoidal rule. The observed daily AUC of 5‐FU was obtained by doubling the actually calculated AUC of 5‐FU following a single dose in patients whose determined daily dose was ≥80 mg because of receiving S‐1 at half of daily dose in the morning on the first day.
2.2. Part II
2.2.1. Population pharmacokinetic analysis
To refine the formula, PPK analysis was performed using the combined data of 30 patients in this study and 16 patients in the previous study with Phoenix NLME (version 7.0; Certara LP) on Microsoft Windows 7. As a pharmacokinetic structural model for 5‐FU, one‐compartment linear model with first‐order absorption (subroutines ADVAN2 and TRANS2) with the absorption lag time was used. Moreover, the first‐order estimation method (FO) was used for the analysis. The basic pharmacokinetic parameters of 5‐FU following administration of S‐1 included clearance (CL), distribution volume (V), first‐order absorption rate constant (Ka), and absorption lag time (Tlag). The CL and V were interpreted as the ratio of clearance to bioavailability (CL/F) and the ratio of the distribution volume to bioavailability (V/F), respectively.
A log‐normal distribution was assumed for an interindividual variability of pharmacokinetic parameters as follows:
where ηj is the random effect for individual j, θ is the population mean parameter, and η is a random variable with mean zero and variance ω2.
Residual variability was described by a proportional error model as follows:
where denotes the concentration predicted by ith model for patient j, Ci,j denotes the measured concentration, and εi,j denotes the residual intraindividual random error. Demographic variables of age, sex, serum albumin level, AST, ALT, total bilirubin, chemotherapy setting (advanced or adjuvant), and history of gastrectomy were examined to identify whether these variables could explain the observed substantial interindividual variability. They were included one at a time through as stepwise selection based on the likelihood ratio test. The minimum value of the NLME objective function was used as a statistic to choose suitable models during the model‐building process. The potentially significant covariates affecting the CL were identified as factors that, when added to the basic model individually, resulted in decreased objective function of ≥3.84 (P < .05).
To evaluate the validity and robustness of the current developed PPK model, a nonparametric bootstrap resampling method was used. A total of 1000 bootstrap resampled datasets were generated, each containing the same number of patients as the original dataset, and each of them was individually fitted to the final PPK model. The consistency between the median values and 95% confidence intervals for parameter estimates calculated by bootstrap and parameter estimates obtained from the original dataset was assessed.
2.2.2. Estimation of the AUCs of 5‐FU in patients receiving S‐1 at determined doses
Because the pharmacokinetics of 5‐FU following S‐1 administration was reported to be linear, 23 the daily AUC in each 30 patients receiving a certain dose can be estimated using their actually observed daily AUC in part I. Accordingly, the AUCs at doses determined by the refined formula and by BSA (approved doses) in these patients were estimated.
2.3. Statistical analyses
Statistical analyses were performed using Stata/SE 12.1 for Mac (StataCorp). Mann‐Whitney U test was used to assess the correlation between observed daily AUC and occurrence of neutropenia. Wilcoxon’s signed‐rank test was used to compare absolute deviations of observed or estimated AUCs from the target AUC in 30 patients receiving S‐1 at determined doses. A P‐value of <.05 was considered indicative of statistical significance.
3. RESULTS
3.1. Part I: Prospective evaluation of the previously developed dosage formula
3.1.1. Patient characteristics
Between March 2017 and February 2018, 33 patients were enrolled. Three patients were excluded: two patients because of incorrect dosing and one patient because of an outlier (observed daily AUC of 5‐FU, 108.4 ng·h/mL). Characteristics of the remaining 30 patients in this prospective evaluation study are shown in Table 1. Two, nine, sixteen, and three patients had normal renal function (CLcr ≥ 80 mL/min), mild dysfunction (CLcr = 60‐79 mL/min), moderate dysfunction (CLcr = 30‐59 mL/min), and severe dysfunction (CLcr < 30 mL/min), respectively. Median BSA was 1.52 m2 (range, 1.14‐1.84 m2). All patients including off‐label use were treated with S‐1 under health insurance, based on evidence demonstrating its effectiveness to each specified cancer type.
TABLE 1.
Characteristics of patients
| Characteristics | Formula establishment a (Previous study) | Prospective evaluation (This study) |
|---|---|---|
| Number | 16 | 30 |
| CLcr, mL/min | ||
| Median (range) | 63.0 (15.9‐108.8) | 51.4 (23.8‐96.4) |
| BSA, m2 | ||
| Median (range) | 1.66 (1.18‐2.02) | 1.52 (1.14‐1.84) |
| Sex | ||
| Male | 11 | 23 |
| Female | 5 | 7 |
| Age, y | ||
| Median (range) | 65.5 (45‐78) | 74.5 (52‐92) |
| Serum creatinine, mg/dL | ||
| Median (range) | 1.01 (0.48‐1.66) | 0.89 (0.52‐1.39) |
| Height, cm | ||
| Median (range) | 167.5 (148.0‐177.4) | 160.8 (139.9‐181.4) |
| Weight, kg | ||
| Median (range) | 56.3 (30.2‐87.0) | 51.3 (32.7‐70.9) |
| ECOG performance status | ||
| 0 | 12 | 18 |
| 1 | 2 | 12 |
| 2 | 2 | 0 |
| Cancer type | ||
| Gastric | 11 | 11 b |
| Pancreatic | 0 | 9 b |
| Esophagus | 0 | 6 |
| Esophagogastric | 0 | 3 |
| Lung | 2 | 1 |
| Biliary tract | 1 | 1 |
| Colorectal | 1 | 0 |
| Thymic | 1 | 0 |
| Gastrectomy | ||
| Yes | 9 | 12 |
| No | 7 | 18 |
| Chemotherapy setting | ||
| Adjuvant | 7 | 16 |
| Advanced | 9 | 14 |
Abbreviations: BSA, body surface area; CLcr, creatinine clearance estimated by the Cockcroft‐Gault equation.
Pharmacokinetic sampling times were before and 1, 2, 4, 7, 12, and 24 h after administration of S‐1.
One patient had double cancer of stomach and pancreas.
3.1.2. Evaluation of the observed daily AUC of 5‐FU
Daily S‐1 doses were 50 mg in one, 60 mg in two, 80 mg in 12, 100 mg in eight, 120 mg in five, and 140 mg in two patients. Figure 1 shows the plasma 5‐FU concentration‐time profiles following a single dose of S‐1 on the first day in 30 patients. The observed daily AUC ranged from 712.6 to 2868.7 ng·h/mL, and 18 patients (60%) achieved the target range (1447.8 ± 545.4 ng·h/mL). 19 Eleven patients had higher AUC, and one patient had lower AUC than the target range (Figures 2 and 4B).
FIGURE 1.
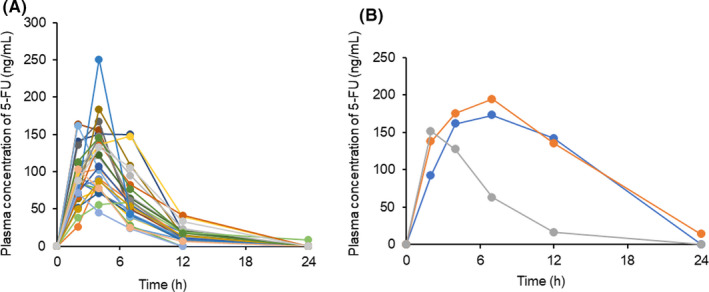
Plasma concentration‐time profiles of 5‐FU after a single dose of S‐1 on the first day (A) in 27 patients receiving S‐1 at half of daily dose (daily dose ≥80 mg) and (B) in three patients receiving S‐1 at full daily dose (daily dose ≤60 mg)
FIGURE 2.
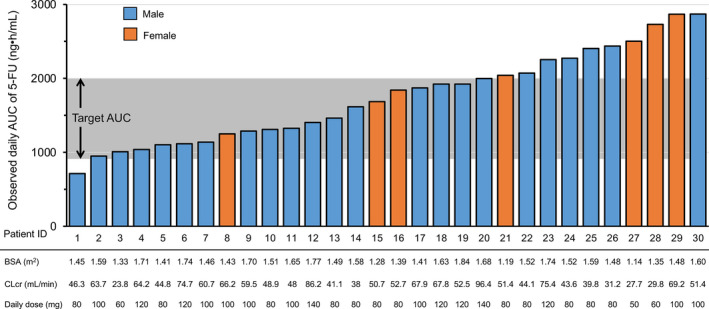
Observed daily area under the concentration‐time curve (AUC) of 5‐FU. Body surface area (BSA), creatinine clearance estimated by the Cockcroft‐Gault equation (CLcr), and daily dose in each of the 30 patients
FIGURE 4.
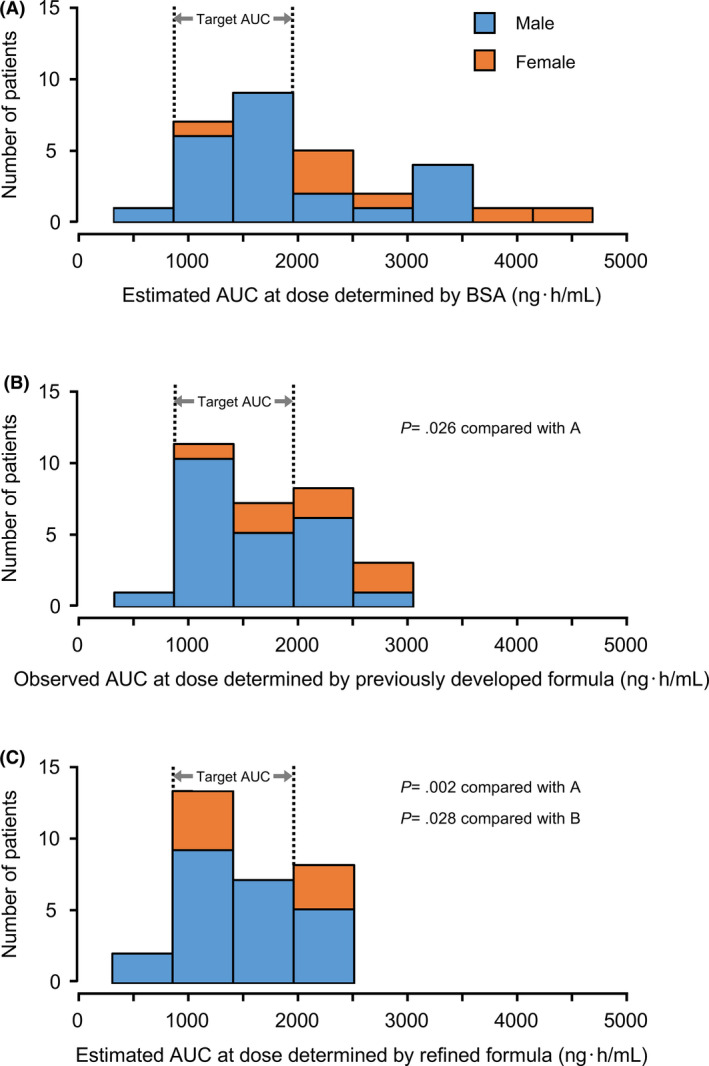
Distributions of areas under the concentration‐time curve (AUCs) of 5‐FU. (A) Estimated AUCs of 5‐FU at doses determined by body surface area (BSA) (approved doses), (B) Observed AUCs of 5‐FU at doses determined by the previously developed formula in part I, (C) Estimated AUCs of 5‐FU at doses determined by the refined formula
3.1.3. Toxicity
No patients experienced S‐1–related grade ≥4 adverse events during the first course. The most frequent S‐1–related adverse events were white blood cell decreased, neutrophil count decreased, and anorexia (13 patients, 43.3%), and anemia (11 patients, 36.7%) as shown in Table 2. Four patients discontinued S‐1 because of grade 3 neutropenia (Patient 30), grade 3 erythema multiform (Patient 6), grade 3 anorexia (Patient 18), and grade 2 anorexia (Patient 8), respectively. Exposure‐toxicity analysis demonstrated that higher AUC of 5‐FU was significantly associated with the occurrence of neutropenia (grade 0 in 17 patients vs grade 1/2/3 in 13 patients, P = .025) (Figure S1).
TABLE 2.
Adverse events related to S‐1
| Any grade | Grade 3 | |
|---|---|---|
| N (%) | N (%) | |
| White blood cell decreased | 13 (43.3) | 0 |
| Neutrophil count decreased | 13 (43.3) | 1 (3.3) |
| Anemia | 11 (36.7) | 0 |
| Platelet count decreased | 7 (23.3) | 0 |
| Anorexia | 13 (43.3) | 1 (3.3) |
| Nausea | 5 (16.7) | 0 |
| Diarrhea | 7 (23.3) | 0 |
| Fatigue | 5 (16.7) | 0 |
| Mucositis oral | 1 (3.3) | 0 |
| Pigmentation | 3 (10) | 0 |
| Watering eyes | 2 (6.7) | 0 |
| Erythema multiforme | 1 (3.3) | 1 (3.3) |
| AST increased | 3 (10) | 0 |
| ALT increased | 2 (6.7) | 0 |
| Creatinine increased | 7 (23.3) | 0 |
| Total bilirubin increased | 4 (13.3) | 0 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
3.2. Part II: Refinement of the dosage formula
3.2.1. Population pharmacokinetic analysis
Sex was identified as a statistically significant covariate (P < .05) for CL with female patients having lower CL in the PPK analysis from the combined data of 46 patients, whereas age, serum albumin level, AST, ALT, total bilirubin, chemotherapy setting, and history of gastrectomy did not affect the CL (Table S1). Therefore, the estimated population mean and variance of the pharmacokinetic parameters including Ka, V, and CL were described as shown in Table 3.
TABLE 3.
Estimated population mean and variance of the pharmacokinetic parameters of 5‐FU
| Parameter | Population mean | Interindividual variability (as variance ω2) |
|---|---|---|
| Ka (h−1) | 0.548 | 1.42 |
| V/F (L/m2) | 290 × BSA | 0.638 |
| CL/F (L/h/m2) | (14.5 + 0.301 × CLcr + 8.23 × SEX [0 for female and 1 for male]) × BSA | 0.0714 |
| Tlag (h) | 0.489 | 2.94 |
| Residual variability (%) | 37.3 |
Abbreviations: BSA, body surface area; CL, clearance; CLcr, creatinine clearance; F, bioavailability; Ka, first‐order absorption rate constant; Tlag, absorption lag time; V, distribution volume.
In the bootstrap resampling for the PPK model validation, >99% of the generated datasets were successfully converged. The median values for bootstrap simulation were very consistent with the final parameter estimates obtained from the dataset of 46 patients. It demonstrated that the final model is robust and reliable for describing the pharmacokinetics of 5‐FU following S‐1 administration. A good liner correlation was shown between observed concentrations and predicted concentrations by the final model (Figure S2).
3.2.2. Refinement of the S‐1 dosage formula
The 5‐FU clearance was described as related to CLcr, BSA, and sex (Table 3);
| (1) |
where SEX is 0 for female and 1 for male.
As AUC = F × Dose/CL, Equation (1) was rearranged to
| (2) |
Equation (2) was further rearranged to
| (3) |
Hence, the recommended daily dose as tegafur to achieve the target AUC was provided as follows:
| (4) |
Nomograms for the recommended daily dose to achieve the target AUC of 1447.8 ng·h/mL in Asia 19 and 1177.2 ng·h/mL in Europe 23 were derived from Equation (4) by taking into account the approved dosage strengths (Figure 3).
FIGURE 3.
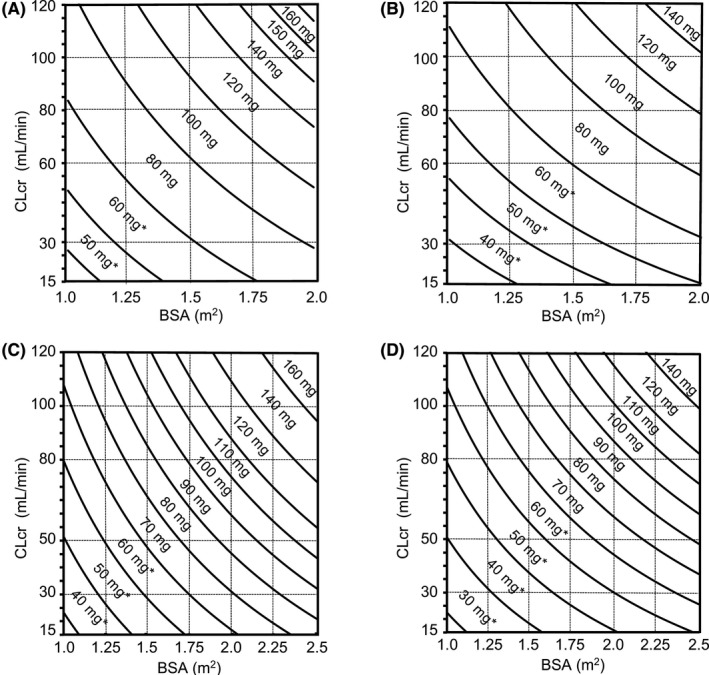
Nomograms derived from refined S‐1 dosage formula to determine the recommended daily doses of S‐1 as tegafur (A) for males in Asia, (B) for females in Asia, (C) for males in Europe, and (D) for females in Europe. *Once‐daily administration regarding approved dosage strengths. BSA, body surface area; CLcr, creatinine clearance estimated by the Cockcroft‐Gault equation
3.2.3. Prediction of the AUCs of 5‐FU in patients receiving S‐1 at doses determined by the refined formula
The estimated AUCs at doses determined by the revised nomograms derived from the refined formula (Figure 3A,B) in these patients ranged from 712.6 to 2403.2 ng·h/mL, which were expected to achieve the target range in 20 patients (Figure 4C). Furthermore, the estimated AUCs at doses determined by BSA (approved doses) in these patients ranged from 890.8 to 4550.8 ng·h/mL, which were expected to achieve the target range in 16 patients (Figure 4A). The absolute deviations of the observed or estimated AUCs from the target AUC (1477.8 ng·h/mL) in 30 patients were calculated. The absolute deviations of the estimated AUCs at doses determined by the refined formula were found to be significantly lower than those of the observed AUCs at doses determined by the previous formula in part I (mean, 424.6 ng·h/mL vs 548.0 ng·h/mL; P = .028) and those of the estimated AUC determined by BSA (859.2 ng·h/mL; P = .002). Similar tendency was observed in female patients (at doses determined by the previous formula, P = .043; at doses determined by BSA, P = .028).
4. DISCUSSION
In a previous pharmacokinetic study in 16 cancer patients with CLcr in the range of 15.9‐108.8 mL/mL, an S‐1 dosage formula based on individual CLcr and BSA values was developed. 21 For practical application, this study was conducted to evaluate and refine the formula. The numbers of patients in our studies were defined by referring to the development of a carboplatin dosage formula by Calvert et al. 28 They derived the initial formula from a retrospective analysis of carboplatin pharmacokinetics in 18 patients with various renal functions and evaluated it prospectively in 31 patients. The Calvert formula refined by the combined data of 49 patients has been used worldwide in the clinical practice.
Of 31 patients receiving S‐1 at doses determined by the nomogram derived from the previously developed formula, one patient with the observed daily AUC of 108.4 ng·h/mL was regarded as an outlier. Although the reason for this extraordinary value, such as wrong dose or inappropriate procedure, was not identified, it was considered appropriate to exclude this outlier to evaluate and refine the formula. In the remaining 30 patients, the received dose of S‐1 was higher, equal, and lower compared with the doses determined by BSA (approved dose) in five, 10, and 15 patients, respectively. Although S‐1 at a lower dose than the approved dose was given to 10 patients, only one of them had lower observed daily AUC than the target range. This demonstrates that our previous formula did not propose excessive dose reduction.
Our concept of developing an S‐1 dosage formula was to achieve the target AUCs of 5‐FU, which had been observed at the approved doses in Asia and Europe, ensuring efficacy and safety confirmed in the pivotal registration studies of S‐1, 19 , 23 as well as the concept of the Calvert formula for carboplatin. Goto et al 29 reported the pharmacokinetics following administration of S‐1 in patients with impaired renal function at modified doses based on BSA and the renal function of glomerular filtration rate (GFR) estimated by the Japanese GFR equation. They compared the pharmacokinetic parameters including AUC of 5‐FU between four groups classified by renal function (normal, mild, moderate, severe), but did not apply the concept of the target AUC.
The 5‐FU clearance has a large interindividual variability, which cannot be reduced by drug dosing only based on BSA. 30 The clearance of injected 5‐FU has been reported to be lower in females than that in males, 31 , 32 , 33 although the mechanism of this sex‐specific elimination of 5‐FU is unclear. Our PPK analysis from the combined data of 46 patients comprising 34 males and 12 females also identified that sex was a significant covariate for 5‐FU clearance with female patients having lower clearance. It suggests that higher observed daily AUC in female patients was attributed to not including sex as a factor in the previous formula. The refined formula significantly reduced the deviations of the estimated AUC from the target AUC; accordingly, the number of patients achieving the target range increased (Figure 4B,C). The evaluation and refinement of the previously developed formula were performed for the Asian‐approved daily dose of 80 mg/m2 only; however, the recommended daily doses for male patients proposed by the nomogram in Europe (Figure 3C) are consistent with the reduced doses for the approved daily dose of 50 mg/m2 stated in the European SmPC. 5
The package insert of S‐1 in Asia states that the approved daily dose of 80 mg/m2 is 80 mg for patients with BSA of <1.25 m2, 100 mg for those with BSA of ≤1.25 to <1.50 m2, and 120 mg for those with BSA of ≥1.50 m2, according to BSA with only three classifications. In contrast, the European SmPC describes that the approved daily dose of 50 mg/m2 is classified into seven doses according to BSA (≤1.29 m2, 60 mg; 1.3‐1.49 m2, 70 mg; 1.5‐1.69 m2, 80 mg; 1.7‐1.89 m2, 90 mg; 1.9‐2.09 m2, 100 mg; 2.1‐2.29 m2, 110 mg; ≥2.30 m2, 120 mg). Asian oncologists are concerned about undertreatment due to insufficient dose in patients with larger BSA (≥1.75 m2) and normal renal function, as previously reported. 34 The revised nomograms in Asia (Figure 3A,B) propose maximum daily doses of 160 mg for male and 140 mg for female patients, whereas the approved maximum daily starting dose is 120 mg in Asia. Hence, it is expected that the revised nomograms in Asia can also improve the efficacy of S‐1 for these patients. Furthermore, the revised nomograms recommend doses even for patients with severe renal impairment (CLcr < 30 mL/min) for whom S‐1 is contraindicated. It is also expected that the revised nomograms can provide a new treatment option for patients with severe renal impairment. However, close monitoring for toxicity due to change in pharmacodynamics of S‐1 affected by renal impairment is required.
In conclusion, of 30 patients receiving S‐1 at doses determined by the previously developed formula on the basis of individual BSA and renal function, 18 patients achieved the target range. The refined formula includes sex as an additional factor based on PPK analysis using the combined data of this study and the previous study. The revised nomograms for recommended daily doses derived from the refined formula can be used in clinical practice to achieve the target AUC ensuring efficacy and safety of S‐1.
DISCLOSURE
Chiyo K. Imamura has received research fund from Otsuka Pharmaceutical Co., Ltd. Hiroya Takeuchi has received scholarship endowments from Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Factory Inc, Ono Pharmaceutical Co., Ltd., and Eli Lilly Japan KK. Takuro Mizukami has received research funds from Taiho Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., and Eli Lilly Japan KK. Taro Funakoshi belongs to an endowed chair funded by Chugai Pharmaceutical Co., Ltd. and Yakult Honsha Co., Ltd. Narikazu Boku has received honoraria from Taiho Pharmaceutical Co., Ltd. and Bristol‐Myers Squibb KK, and research funds from Ono Pharmaceutical Co., Ltd. and Takeda Pharmaceutical Co., Ltd. Yusuke Tanigawara has received honoraria from Chugai Pharmaceutical Co., Ltd.; research funds from Taiho Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., and Asahi Kasei Pharma Corp.; and consulting fee from Fujimoto Pharmaceutical Co. Yuko Kitagawa has received honoraria from Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Asahi Kasei Pharma Corp., and Otsuka Pharmaceutical Factory Inc, and scholarship endowments from Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Co., Ltd., Otsuka Pharmaceutical Factory Inc, Ono Pharmaceutical Co., Ltd., Asahi Kasei Pharma Corp., Yakult Honsha Co., Ltd., Takeda Pharmaceutical Co., Ltd., Eisai Co., Ltd., Tsumura Co., and Medicon Inc. The other authors declare that they have no conflict of interest.
Supporting information
Fig S1
Fig S2
Table S1
ACKNOWLEDGMENTS
We thank the patients and their families, as well as the investigators and study teams, for their participation in this trial. This study was supported by the JSPS KAKENHI Grant Number JP17K08961.
Takeuchi M, Imamura CK, Booka E, et al. Prospective evaluation and refinement of an S‐1 dosage formula based on renal function for clinical application. Cancer Sci. 2021;112:751–759. 10.1111/cas.14758
REFERENCES
- 1. Shirasaka T, Nakano K, Takechi T, et al. Antitumor activity of 1 M tegafur‐0.4 M 5‐chloro‐2,4‐ dihydroxypyridine‐1 M potassium oxonate (S‐1) against human colon carcinoma orthotopically implanted into nude rats. Cancer Res. 1996;56:2602‐2606. [PubMed] [Google Scholar]
- 2. Shirasaka T, Shimamato Y, Ohshimo H, et al. Development of a novel form of an oral 5‐fluorouracil derivative (S‐1) directed to the potentiation of the tumor selective cytotoxicity of 5‐fluorouracil by two biochemical modulators. Anticancer Drugs. 1996;7:548‐557. [DOI] [PubMed] [Google Scholar]
- 3. Taiho Pharmaceutical . TS‐1 prescribing information in Japan; TS‐1 combination OD tablet. Ver. 4; July 2014.
- 4. Taiho Pharmaceutical . TS‐1 prescribing information in Singapore. Revised: TS‐ONE capsule; 2013.
- 5. European summary of product characteristics, Teysuno. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/001242/WC500104415.pdf. Accessed April 20, 2020
- 6. Sakuramoto S, Sasako M, Yamaguchi T, et al. Adjuvant chemotherapy for gastric cancer with S‐1, an oral fluoropyrimidine. N Engl J Med. 2007;357(18):1810‐1820. [DOI] [PubMed] [Google Scholar]
- 7. Sasako M, Sakuramoto S, Katai H, et al. Five‐year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S‐1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol. 2011;29:4387‐4393. [DOI] [PubMed] [Google Scholar]
- 8. Koizumi W, Narahara H, Hara T, et al. S‐1 plus cisplatin versus S‐1 alone for first‐line treatment of advanced gastric cancer (SPIRITS trial): a phase III trial. Lancet Oncol. 2008;9:215‐221. [DOI] [PubMed] [Google Scholar]
- 9. Yamada Y, Higuchi K, Nishikawa K, et al. Phase III study comparing oxaliplatin plus S‐1 with cisplatin plus S‐1 in chemotherapy‐naive patients with advanced gastric cancer. Ann Oncol. 2015;26:141‐148. [DOI] [PubMed] [Google Scholar]
- 10. Ajani JA, Rodriguez W, Bodoky G, et al. Multicenter phase III comparison of cisplatin/S‐1 with cisplatin/infusional fluorouracil in advanced gastric or gastroesophageal adenocarcinoma study: the FLAGS trial. J Clin Oncol. 2010;28:1547‐1553. [DOI] [PubMed] [Google Scholar]
- 11. Nokihara H, Lu S, Mok TSK, et al. Randomized controlled trial of S‐1 versus docetaxel in patients with non‐small‐cell lung cancer previously treated with platinum‐based chemotherapy (East Asia S‐1 Trial in Lung Cancer). Ann Oncol. 2017;28:2698‐2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshioka H, Okamoto I, Morita S, et al. Efficacy and safety analysis according to histology for S‐1 in combination with carboplatin as first‐line chemotherapy in patients with advanced non‐small‐cell lung cancer: updated results of the West Japan Oncology Group LETS study. Ann Oncol. 2013;24:1326‐1331. [DOI] [PubMed] [Google Scholar]
- 13. Kubota K, Sakai H, Katakami N, et al. A randomized phase III trial of oral S‐1 plus cisplatin versus docetaxel plus cisplatin in Japanese patients with advanced non‐small‐cell lung cancer: TCOG0701 CATS trial. Ann Oncol. 2015;26:1401‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamada Y, Takahari D, Matsumoto H, et al. Leucovorin, fluorouracil, and oxaliplatin plus bevacizumab versus S‐1 and oxaliplatin plus bevacizumab in patients with metastatic colorectal cancer (SOFT): an open‐label, non‐inferiority, randomised phase 3 trial. Lancet Oncol. 2013;14:1278‐1286. [DOI] [PubMed] [Google Scholar]
- 15. Yamada Y, Denda T, Gamoh M, et al. S‐1 and irinotecan plus bevacizumab versus mFOLFOX6 or CapeOX plus bevacizumab as first‐line treatment in patients with metastatic colorectal cancer (TRICOLORE): a randomized, open‐label, phase III, noninferiority trial. Ann Oncol. 2018;29:624‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takashima T, Mukai H, Hara F, et al. Taxanes versus S‐1 as the first‐line chemotherapy for metastatic breast cancer (SELECT BC): an open‐label, non‐inferiority, randomised phase 3 trial. Lancet Oncol. 2016;17:90‐98. [DOI] [PubMed] [Google Scholar]
- 17. Uesaka K, Boku N, Fukutomi A, et al. Adjuvant chemotherapy of S‐1 versus gemcitabine for resected pancreatic cancer: a phase 3, open‐label, randomised, non‐inferiority trial (JASPAC 01). Lancet. 2016;388:248‐257. [DOI] [PubMed] [Google Scholar]
- 18. Morizane C, Okusaka T, Mizusawa J, et al. Combination gemcitabine plus S‐1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA‐BT (JCOG1113) randomized phase III clinical trial. Ann Oncol. 2019;30:1950‐1958. [DOI] [PubMed] [Google Scholar]
- 19. Hirata K, Horikoshi N, Aiba K, et al. Pharmacokinetic study of S‐1, a novel oral fluorouracil antitumor drug. Clin Cancer Res. 1999;5:2000‐2005. [PubMed] [Google Scholar]
- 20. Ikeda M, Furukawa H, Imamura H, et al. Pharmacokinetic study of S‐1, a novel oral fluorouracil antitumor agent in animal model and in patients with impaired renal function. Cancer Chemother Pharmacol. 2002;50(1):25‐32. [DOI] [PubMed] [Google Scholar]
- 21. Booka E, Imamura CK, Takeuchi H, et al. Development of an S‐1 dosage formula based on renal function by a prospective pharmacokinetic study. Gastric Cancer. 2016;19:876‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagashima F, Ohtsu A, Yoshida S, et al. Japanese nationwide post‐marketing survey of S‐1 in patients with advanced gastric cancer. Gastric Cancer. 2005;8:6‐11. [DOI] [PubMed] [Google Scholar]
- 23. van Groeningen CJ, Peters GJ, Schornagel JH, et al. Phase I clinical and pharmacokinetic study of oral S‐1 in patients with advanced solid tumors. J Clin Oncol. 2000;18:2772‐2779. [DOI] [PubMed] [Google Scholar]
- 24. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31‐41. [DOI] [PubMed] [Google Scholar]
- 25. US Department of Health and Human Services . Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. 2010. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010‐06‐14_QuickReference_8.5x11.pdf. Accessed April 20, 2020
- 26. Murphy RF, Balis FM, Poplack DG. Stability of 5‐fluorouracil in whole blood and plasma. Clin Chem. 1987;33:2299‐2300. [PubMed] [Google Scholar]
- 27. U.S. Department of Health and Human Services Food and Drug Administration . Bioanalytical method. Validation guidance for industry. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical‐Method‐Validation‐Guidance‐for‐Industry.pdf. Accessed April 20, 2020
- 28. Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol. 1989;7:1748‐1756. [DOI] [PubMed] [Google Scholar]
- 29. Goto K, Fujiwara Y, Isobe T, et al. Pharmacokinetic study of the fluorouracil antitumor agent S‐1 in patients with impaired renal function. Cancer Sci. 2019;110:1987‐1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gamelin E, Boisdron‐Celle M, Guérin‐Meyer V, et al. Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5‐FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: a potential interest for predicting 5‐FU toxicity and determining optimal 5‐FU dosage. J Clin Oncol. 1999;17:1105‐1110. [DOI] [PubMed] [Google Scholar]
- 31. Milano G, Etienne MC, Cassuto‐Viguier E, et al. Influence of sex and age on fluorouracil clearance. J Clin Oncol. 1992;10:1171‐1175. [DOI] [PubMed] [Google Scholar]
- 32. Gusella M, Crepaldi G, Barile C, et al. Pharmacokinetic and demographic markers of 5‐fluorouracil toxicity in 181 patients on adjuvant therapy for colorectal cancer. Ann Oncol. 2006;17:1656‐1660. [DOI] [PubMed] [Google Scholar]
- 33. Mueller F, Buchel B, Koberle D, et al. Sex‐specific elimination of continuous‐infusional 5‐fluorouracil in patients with gastrointestinal malignancies: results from a prospective population pharmacokinetic study. Cancer Chemother Pharmacol. 2013;71:361‐370. [DOI] [PubMed] [Google Scholar]
- 34. Fujita K, Ichikawa W, Yamamoto W, et al. Fixed dosing and pharmacokinetics of S‐1 in Japanese cancer patients with large body surface areas. Ann Oncol. 2009;20:946‐949. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1