

Abstract
Recent studies of the cancer genome have identified numerous patients harboring multiple mutations (MM) within individual oncogenes. These MM (de novo MM) in cis synergistically activate the mutated oncogene and promote tumorigenesis, indicating a positive epistatic interaction between mutations. The relatively frequent de novo MM suggest that intramolecular positive epistasis is widespread in oncogenes. Studies also suggest that negative and higher‐order epistasis affects de novo MM. Comparison of de novo MM and MM associated with drug‐resistant secondary mutations (secondary MM) revealed several similarities with respect to allelic configuration, mutational selection and functionality of individual mutations. Conversely, they have several differences, most notably the difference in drug sensitivities. Secondary MM usually confer resistance to molecularly targeted therapies, whereas several de novo MM are associated with increased sensitivity, implying that both can be useful as therapeutic biomarkers. Unlike secondary MM in which specific secondary resistant mutations are selected, minor (infrequent) functionally weak mutations are convergently selected in de novo MM, which may provide an explanation as to why such mutations accumulate in cancer. The third type of MM is MM from different subclones. This type of MM is associated with parallel evolution, which may contribute to relapse and treatment failure. Collectively, MM within individual oncogenes are diverse, but all types of MM are associated with cancer evolution and therapeutic response. Further evaluation of oncogenic MM is warranted to gain a deeper understanding of cancer genetics and evolution.
Keywords: drug sensitivity, epistasis, multiple mutations, precision medicine, synergy
Recent studies have identified that multiple mutations (MM) within individual oncogenes in cis synergistically activate the mutated oncogene and promote tumorigenesis. In this review, we discuss the function, frequency, and classification of MM, as well as their association with epistasis, drug sensitivity, and cancer evolution.
1. INTRODUCTION
Significant advances in cancer genomics in recent years have enabled the identification and characterization of many cancer genes and the associated mutations driving each patient’s tumor. 1 , 2 In contrast to tumor suppressor genes (TSG), which are generally disrupted by copy number deletions and loss‐of‐function (nonsense and frameshift) mutations, oncogenes are mainly activated by focal amplifications and missense mutations. TSG are often biallelically inactivated, as described in Knudson’s two‐hit theory. 2 In contrast, oncogenes are usually activated by dominant‐acting heterozygous single events. Mutational distribution is different for oncogenes and TSG. In TSG, loss‐of‐function mutations are distributed throughout their length, whereas in oncogenes, missense mutations preferentially affect specific hotspots. 3 These hotspots include few highly recurrent (major) positions (such as KRAS G12, G13, and Q61) and a much larger number of rare (minor) positions. Minor hotspot mutations are functionally weak and rare as individuals but account for a substantial fraction of the accumulated mutations. It has been reported that 12%‐87% of hotspot mutations are in minor positions in oncogenes. 4 However, the reasons why such minor mutations are frequently observed in cancer despite their weak function have not been well investigated.
Several recent large‐scale genetic studies have reported that numerous patients are affected by “multiple mutations (MM) within individual oncogenes,” demonstrating that MM synergistically activate the mutated oncogenes (Figure 1). 4 , 5 , 6 , 7 , 8 These MM are different from single hotspot mutations with respect to mutational pattern, function, and sensitivity to molecularly targeted therapy. Herein, we discuss what has been learned about oncogenic MM and, more importantly, what this novel mechanism has taught us about cancer genomics and its future application in precision medicine.
Figure 1.
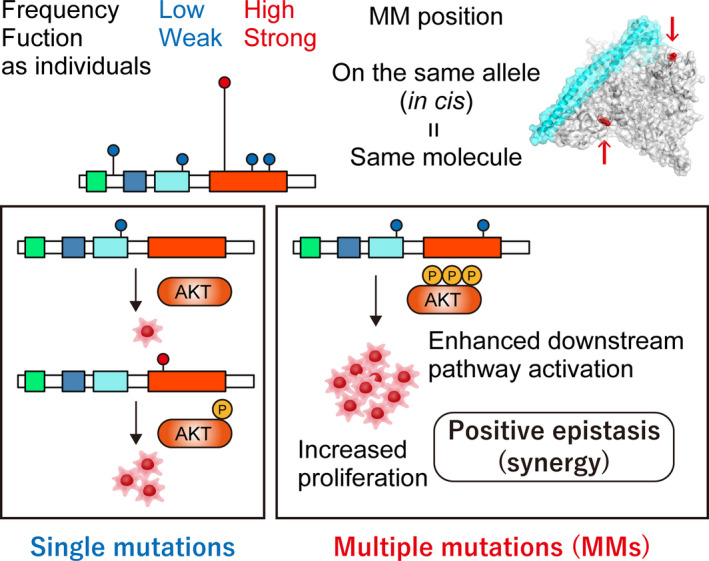
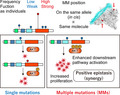
Characteristics of de novo multiple mutations (MM) within individual oncogenes. Functionally weak, minor (infrequent) mutations are selected in de novo MM. MM in cis synergistically enhance downstream pathway activation and tumor growth, suggesting positive epistasis
2. FREQUENT DE NOVO MULTIPLE MUTATIONS IN UNTREATED TUMORS
A recent remarkable advance in this field is the comprehensive characterization of oncogenic MM from a pan‐cancer genetic study of more than 60 000 tumors. 4 This study showed that approximately one‐quarter of oncogenes examined (14 oncogenes out of 60) were significantly affected by MM. Surprisingly, 9% of samples with at least one mutation in these genes harbored MM. In particular, PIK3CA and EGFR had the highest frequency of MM (10%) (Figure 2). This study also identified several oncogenes showing significant enrichment of MM in a specific cancer type, such as NOTCH1 in T‐cell acute lymphoblastic leukemia (ALL) and CARD11 in non‐Hodgkin’s lymphoma. These MM are frequently observed in primary untreated tumors, suggesting that they are “de novo” MM unrelated to cancer therapy. These de novo MM have several genetic characteristics (Figure 1 and Table 1). First, the MM are mostly present in cis (two mutations on the same allele) with concordant variant allele frequencies (Figure 3A), 4 , 5 , 8 , 9 suggesting that MM arise from the same alleles of the same clones. This is in contrast to TSG; although MM in cis are occasionally observed in TSG, 8 the majority of them are in trans (two mutations on different alleles), suggesting biallelic inactivation. Second, both of the mutations in each MM are under positive selection equal to the selection on single hotspot mutations, implying that MM are a combination of driver‐driver mutations. Third, the mutational patterns of MM are skewed from single hotspot mutations with respect to type (missense and in‐frame), position, and amino acid change. MM generally arise from a pair of mutations that are uncommon as single mutations. In other words, mutations from “minor” hotspots are frequently observed in MM. 4 , 8 , 9 This result suggests the presence of differences in the mutational selection between single mutations and MM.
Figure 2.
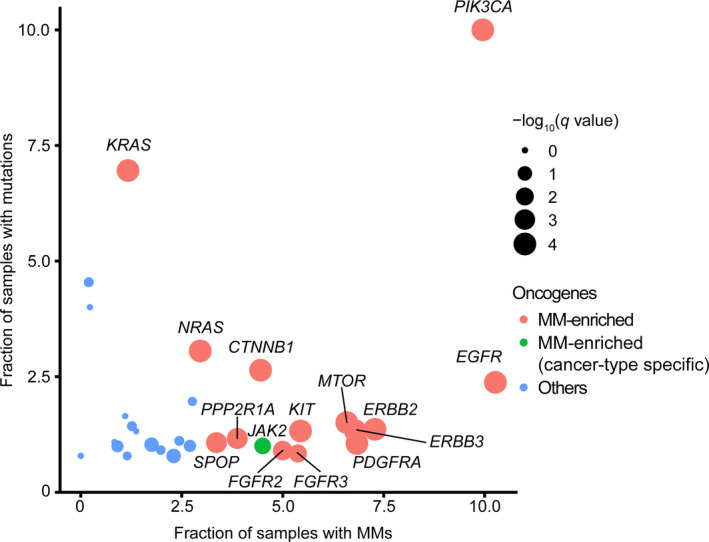
The prevalence of multiple mutations (MM) across oncogenes. Fraction of samples with MM (out of the total number of mutated samples; x‐axis) and fraction of samples with mutations (y‐axis) across 30 oncogenes. Reanalysis of data from Saito et al (n = 11 043). 4 Circle sizes indicate q‐values of the permutation test. Oncogenes with significant enrichment of MM in pan‐cancer and cancer‐type‐specific manners are indicated in red and green, respectively
Table 1.
Differences and similarities between three types of oncogenic multiple mutations (MM)
| De novo MM | Secondary MM | MM from different subclones | |
|---|---|---|---|
| Allelic configuration | Mostly cis |
Cis/trans (mainly cis) |
Trans |
| Mutational positions |
Minor (infrequent) mutations MM from different domains |
Combinations of hotspot and specific resistant mutations | Combination of hotspot mutations |
| Sensitivities to molecularly targeted therapy |
Highly sensitive (at least in PIK3CA) |
Resistant | N/A. Maybe associated with relapse and treatment failure. |
N/A, not available.
Figure 3.
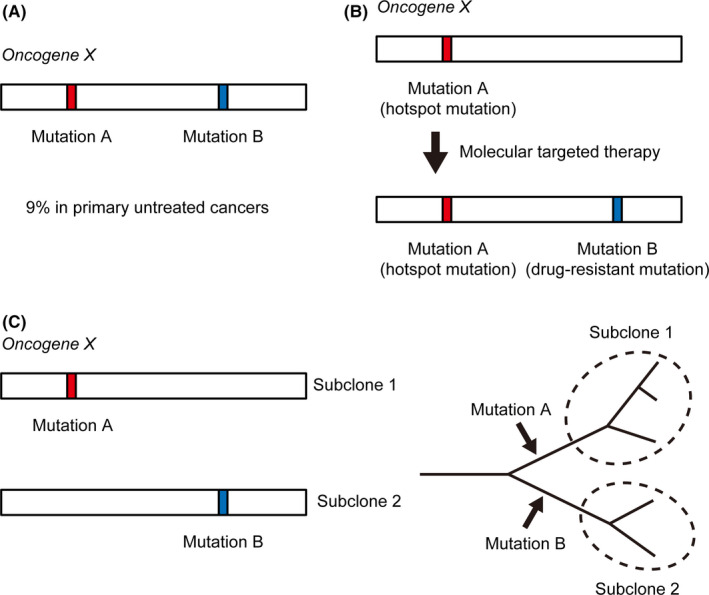
Types of multiple mutations (MM) within individual oncogenes. A, De novo MM. Two mutations (mutations A and B) within individual oncogenes are frequent (9% in primary untreated cancers) and are mostly located in cis. B, Secondary MM associated with drug‐resistant mutations. Tumors with hotspot mutations (mutation A) treated with molecularly targeted therapy often acquire secondary mutations (mutation B), which confer resistance to the therapy. C, MM from different subclones (left). Evolutionary trees illustrating MM causing parallel evolution (right). Two subclonal populations of tumor cells (subclones 1 and 2) independently acquire mutations (mutations A and B) in oncogene X, resulting in independent evolution
3. EPISTATIC EFFECT OF DE NOVO MULTIPLE MUTATIONS
The functional consequences of two driver mutations can be synergistic, additive, or suppressed. 10 , 11 If two mutations are independently driving tumors, they are expected to be additive. 12 However, the functionality of two mutations can be higher (synergistic) or lower (suppressed) than the simple addition, suggesting a genetic interaction between mutations. These genetic interactions are best described using the term “epistasis.” 13 Epistasis was first used in the early twentieth century to describe the effect of a variant masking another variant from manifesting its effect 14 ; however, currently, it is commonly defined as the deviation from the expected outcome when combining mutations. 15 Epistasis has been experimentally shown in several organisms, such as phages, 16 yeast, 17 and, more recently, human cell lines. 18 , 19 Epistasis can occur in two different ways; namely, between variants in the same gene (“intramolecular epistasis”) and between variants in different genes (“intermolecular epistasis”). 20 Compensatory mutational screening in the phage identified that approximately half are intramolecular and others are intermolecular. 16 However, in cancer genomics, most studies have focused on intermolecular epistasis. For example, several genomic studies looked for mutual exclusivity among mutations from different genes, such as BRAF and NRAS mutations in cutaneous melanoma. 21 , 22 , 23 Mutually exclusive driver mutations are not advantageous if present in the same tumor and are, thus, thought to be negatively selected during cancer evolution.
In contrast to mutations from different genes, oncogenic MM in cis are representative of intramolecular epistasis. In fact, various recurrent MM have synergistic functions, indicative of positive epistasis (Figure 1). For example, in PIK3CA, several recurrent combinations of MM, especially those involving minor mutations, are more frequently observed than expected. The functional synergy of such MM, including E545K‐E726K and R88Q‐H1047R, has been validated using several experimental approaches. Synergistic upregulation of AKT phosphorylation and enhanced cell proliferation in vitro have been validated in mutant transduced cell lines by western blotting and cell growth assays, respectively. Subcutaneous xenograft models have shown that these double mutant transduced cell lines proliferate more than single mutant transduced cell lines in vivo. 4 , 5 Importantly, the effect of MM on proliferation (eg, R88Q‐H1047R) is higher than the summed effect of individual mutations (eg, R88Q plus H1047R) in these assays, confirming the positive epistasis between mutations. For NOTCH1 MM in T‐cell ALL, combinations of mutations from different specific functional domains (ie, HD domain and PEST domain) are significantly enriched. Functional synergy has also been experimentally shown in such combinations, including the augmented transcriptional activity of NOTCH1 MM in T‐cell ALL, as revealed by the luciferase assay. 4 , 24 Although individual functional assays may capture only limited aspects of cancer phenotypes, these findings suggest the positive epistasis between mutations (at least in some quantified traits).
As mentioned above, one‐quarter of the oncogenes showed significant enrichment of MM 4 ; most MM in these genes show positive epistasis, resulting in a strong fitness increase that exceeds the additive effects of individual mutations. However, it is noteworthy that not all combinations of MM result in positive epistasis. For example, in PIK3CA, the combinations of major hotspots (major‐major) are significantly less common than expected, and combinations of mutations from the same functional domain are particularly depleted. E542K and E545K are the major hotspots in the helical domain, but these combinations were rarely observed, and if any were present, all of them were in trans. 4 In fact, the functional evaluation revealed that the E542K‐E545K combination does not show a synergistic effect; the effect on proliferation is equal to or even less than those of single mutations. Similarly, major‐major combinations are significantly less common than expected in other oncogenes (such as EGFR and KRAS), and those in close proximity are almost always located in trans. 4 Although the statistical power is limited, it is estimated that major‐major combinations in cis, particularly those in close proximity (approximately equal to the combinations of mutations from the same functional domain), are under negative epistasis (suppression).
What are the molecular mechanisms of intramolecular epistasis? Several excellent reviews have discussed the possible mechanism of epistasis. 10 , 15 , 20 For example, synergistic epistasis can occur when a conformation change caused by one mutation is required for a second mutation to realize its effect on protein function (conformational epistasis). In such cases, the secondary mutated residue may contact a novel substrate only when the first mutation causes a conformational change. Another explanation includes the stability thresholds in which the mutation has a detrimental effect only when it reduces the protein stability below a critical threshold. In this scenario, single mutations have little effect, but two mutations reaching the critical threshold have a very deleterious effect. The structural mechanism of oncogenic MM‐mediated activation has been experimentally investigated in PIK3CA. Biochemical analysis revealed that several double mutant PIK3CA proteins easily detach from the regulatory subunit and show increased binding to anionic and PIP2 liposomes. 5 In addition, molecular‐dynamic simulation demonstrated that PIK3CA R88Q‐H1047R coordinately promotes the conformational change to the active state by rendering the salt bridge between the ABD and kinase domains unstable. 4 These investigations suggest conformational epistasis as a mechanism of oncogenic MM‐mediated positive epistasis, although the molecular mechanisms causing epistasis may be diverse among combinations.
An interesting observation of oncogenic MM is the relationship with other mutations. For example, PIK3CA mutations tend to be mutually exclusive with PIK3R1 mutations but co–occur with PTEN mutations in uterine corpus endometrial carcinoma. 22 These associations are augmented by PIK3CA MM, 4 suggesting that intermolecular epistasis with PIK3R1 and PTEN is affected by intramolecular epistasis by MM. These findings suggest that epistasis involves not only two mutations (pairwise epistasis) but also more mutations (higher‐order epistasis). 15 , 17 Additional evidence of higher‐order epistasis in cancer is the lineage specificity of oncogenic MM. For instance, MM in PDGFRA are recurrently observed in glioblastoma but not in gastrointestinal stromal tumor (GIST). MM in NOTCH1 are highly prevalent in T‐cell ALL but not in chronic lymphocytic leukemia. 4 These observations suggest that the epistatic interactions of MM are themselves context‐dependent; the interactions may depend on other factors, such as additional genetic variants of the tumor.
The positive epistasis of oncogenic MM may provide an explanation as to why functionally weak mutations or variants of unknown significance (VUS) accumulate in cancer. Rare mutations are functionally weak as individual mutations, 25 but they may show strong oncogenic potential in combination. Previous systematic approaches of VUS annotation were focused on the evaluation of mutations individually, 26 , 27 which does not address the functional role of VUS in combination.
4. SIMILARITIES AND DIFFERENCES BETWEEN DE NOVO MULTIPLE MUTATIONS AND DRUG‐RESISTANT SECONDARY MULTIPLE MUTATIONS
This review focuses on “de novo” MM, but another well‐known type of MM is associated with drug‐resistant secondary mutations (hereafter referred to as secondary MM) (Figure 3B). Tyrosine kinase inhibitors (TKI) are widely used for lung cancer patients harboring EGFR mutations; however, despite their initial response, most patients eventually acquire drug resistance. In 2005, T790M was identified as a drug‐resistant secondary mutation in patients treated with first‐generation TKI. 28 This means that these patients acquire MM (primary mutation plus T790M) in EGFR. Patients harboring T790M mutations are sensitive to third‐generation EGFR‐TKI, but C797S is reported to cause resistance to even third‐generation EGFR‐TKI. 29 Drug‐resistant secondary mutations are not specific to EGFR‐TKI, but they are also common in patients receiving various other TKI. Several KIT mutations are reported as imatinib‐resistant in GIST patients. 30 , 31 FLT3 mutations in the tyrosine kinase domains (TKD) cause resistance to FLT3 inhibitors in acute myeloid leukemia (AML) patients harboring FLT3 internal tandem duplication (FLT3‐ITD). 32 , 33 , 34 Intriguingly, FLT3‐ITD cells acquiring both TKD1 and TKD2 mutations show greater resistance than those acquiring individual mutations. 34 These resistance mechanisms are also observed in fusion genes, including BCR‐ABL for imatinib resistance, 35 , 36 CCDC6‐RET for vendatanib, 37 and EML4‐ALK for crizotinib. 38 , 39 These resistant mutations are not specific to TKI. A striking example is the IDH2 inhibitor enasidenib for AML. The Q316E or I319M mutation together with the R140Q hotspot mutation causes resistance. 40 Importantly, resistance is caused only when MM exist; the Q316E or I319M mutation alone does not cause resistance to the therapy.
De novo MM and secondary MM have several shared characteristics (Table 1). First, both types of MM occur in cis. 31 , 41 This suggests that both mutations need to be in the same molecule to exert functional effects. One exception is the secondary MM in IDH2. They are located in trans 40 but confer resistance by blocking the drug interaction with the IDH2 dimer. In addition, EGFR C797S can occur in the trans position of the T790M allele, although the majority of them are in cis. 42 Second, mutations that are rarely observed as single mutations are selected in both de novo and secondary MM. Drug‐resistant acquired mutations in secondary MM, such as EGFR T790M and KIT V654A, do not usually occur as single mutations, similarly to minor mutations in de novo MM. In addition, the drug‐resistant mutations are weakly oncogenic (as in EGFR T790M) 41 or are of unknown oncogenic potential as single mutations, again similar to minor hotspot mutations in de novo MM. These common characteristics between de novo and secondary MM may suggest a shared molecular mechanism between them. Consistent with these findings, it has been reported that MM associated with drug‐resistant mutations can occur as de novo MM in untreated cancers. Several cases have been described where patients carry both the T790M and L858M mutations in EGFR prior to EGFR‐TKI therapy. 4 , 43 While FLT3 TKD‐resistant mutations can be acquired during FLT3 inhibitor therapy in AML patients with FLT3‐ITD, approximately 1%‐2% of AML patients carry both ITD and TKD mutations at the time of diagnosis. 44 , 45 These observations reinforce the idea that de novo MM and secondary MM have common mechanisms.
However, there are differences between de novo MM and secondary MM. The prominent difference is the distribution of mutations. Drug‐resistant mutations generally occur at specific amino acids; most of these mutations are present at residues within or around drug‐binding pockets or gatekeeper residues, resulting in a weakened affinity with specific inhibitors. 30 , 33 , 46 Conversely, there are numerous recurrent combinations in de novo MM, although mutations from different domains or those involving minor hotspot mutations are overrepresented. This may be due to the difference in selective pressure between untreated and treated tumors.
5. DRUG SENSITIVITIES OF DE NOVO MULTIPLE MUTATIONS
As secondary MM are associated with drug resistance, it is reasonable to speculate that de novo MM are also associated with the response to specific inhibitors (Table 1). In fact, several groups have reported that PIK3CA MM are associated with increased sensitivities to PI3K inhibitors. Cell lines harboring PIK3CA MM were reported to be more sensitive to PI3K inhibitors than those harboring single mutations. 4 Consistently, these PIK3CA MM‐harboring cell lines were more dependent on PIK3CA itself. In addition, patients with PIK3CA MM detected by circulating tumor DNA showed increased sensitivity to the PI3K inhibitor taselisib compared with those harboring single mutations. 5 These data suggest that de novo MM (at least in PIK3CA) are the opposite of secondary MM with respect to sensitivities to molecularly targeted therapy. The drug sensitivities of MM were also evaluated in EGFR and ERBB2 using high‐throughput screening assays. 47 , 48 Intriguingly, the drug sensitivities of MM were different between mutational combinations and types of drugs. For instance, Ba/F3 cells expressing EGFR L858R and E709A/G in cis were resistant to gefitinib and erlotinib but responded to afatinib. 47 The drug sensitivities of MM may differ between genes, and individual evaluations are needed.
6. MULTIPLE MUTATIONS FROM DIFFERENT SUBCLONES
Next, we discuss the MM from different subclones (Figure 3C). Although most de novo MM and secondary MM occur in cis within the same clone, several MM are thought to be present in trans. In particular, almost all major‐major combinations in proximity (such as combinations of KRAS/NRAS G12 and G13 missense mutations) are located in trans. 4 These MM show discordant and relatively low allele frequencies, suggesting that they arise as different subclones. Single‐cell sequencing technologies confirmed the presence of MM arising in different subclones, including NRAS G12‐G13 MM in AML. 49 Recently, due to the advances of several technologies, intratumor heterogeneity has been well studied in the context of cancer evolution. Several studies suggest the model of “parallel evolution,” which refers to the independent evolution of similar traits from a single ancestral clone. 50 If two subclones independently acquire mutations to the same oncogene, parallel evolution results. Importantly, the parallel evolution may contribute to relapse and treatment failure (Table 1), as represented by a case of B‐cell ALL harboring different JAK2 R683 missense mutations in founder and relapsed clones. 51 The MM from different subclones may provide insights into the genetic basis of clonal heterogeneity, and careful attention should be paid to this type of MM.
7. CLINICAL APPLICATIONS OF MULTIPLE MUTATIONS
These findings have several clinical implications. First, de novo MM, as well as secondary MM, can be used as biomarkers for predicting the sensitivities to molecularly targeted therapy. Several MM show higher sensitivities to specific inhibitors (as in the case of PIK3CA), but others may confer resistance to them. Comprehensive evaluation of drug sensitivities for individual MM using systematic screening methods 26 , 27 , 47 , 48 may be important for precision medicine. Second, the findings suggest the importance of evaluating minor hotspot mutations. Although a substantial proportion of hotspot mutations are in minor positions, 4 most of them are not evaluated by PCR or hotspot‐region‐only sequencing. In fact, the frequency of PIK3CA MM is 10% or more of the mutated samples, 4 , 5 but commercially available diagnostic tests underestimate the relative frequency (0.7%). 52 This leads to the overlooking of patients who may benefit from PI3K inhibitors such as alpelisib. 53 The observations suggest the importance of comprehensive mutational evaluation, at least in MM‐enriched oncogenes.
8. CONCLUSION
Although it has long been believed that oncogenes gain tumor‐promoting functions by acquiring single hotspot mutations, several recent studies suggest that MM within individual oncogenes are alternative mechanisms of oncogene activation. MM are observed in several oncogenes across different cancer types, suggesting that MM are universal genetic mechanisms of cancer pathogenesis. Oncogenic MM are individually rare, but collectively common, affecting as many as 9% of patients harboring mutations in oncogenes (Figure 1). These MM are observed in primary untreated cancers, indicating that they are “de novo” MM. De novo MM and secondary MM associated with drug‐resistant acquired mutations have several common characteristics, which may imply a shared molecular mechanism between them. However, they have several differences, most prominently the difference in drug sensitivities to the specific inhibitors. De novo MM and secondary MM are usually in cis within the same clone, which should be distinguished from MM from parallelly evolved independent subclones.
De novo MM are different from single hotspot mutations with respect to mutation distribution. Interestingly, minor (infrequent) mutations are preferentially selected in de novo MM, which are functionally weak individually but exhibit stronger oncogenic potential in combination. This provides an explanation of why functionally weak minor mutations and VUS accumulate in cancer. In addition, this suggests the importance of epistasis in cancer. Recurrent MM are under positive epistasis, but the study of de novo MM also implies the existence of negative epistasis and higher‐order epistasis. Further evaluation of MM using novel technologies, including single‐cell sequencing or high‐throughput functional screening, is warranted to gain a deeper understanding of cancer genetics and evolution.
DISCLOSURE
KK holds stock in Asahi Genomics and has received research funding from Otsuka Pharmaceutical, Takeda Pharmaceutical, Chugai Pharmaceutical, Chordia Therapeutics, and Bristol‐Myers Squibb. The other authors declare no competing financial interests.
ACKNOWLEDGMENTS
This work was supported by a Grant‐in‐Aid for Challenging Research (Exploratory) (17K19592) and the National Cancer Center Research and Development Funds (30‐A‐1). We thank all laboratory members for the discussions regarding the manuscript.
Saito Y, Koya J, Kataoka K. Multiple mutations within individual oncogenes. Cancer Sci. 2021;112:483–489. 10.1111/cas.14699
REFERENCES
- 1. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371‐385.e318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campbell PJ, Getz G, Korbel JO, et al. Pan‐cancer analysis of whole genomes. Nature. 2020;578:82‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saito Y, Koya J, Araki M, et al. Landscape and function of multiple mutations within individual oncogenes. Nature. 2020;582:95‐99. [DOI] [PubMed] [Google Scholar]
- 5. Vasan N, Razavi P, Johnson JL, et al. Double <em>PIK3CA</em> mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science. 2019;366:714‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Madsen RR, Knox RG, Pearce W, et al. Oncogenic PIK3CA promotes cellular stemness in an allele dose‐dependent manner. Proc Natl Acad Sci USA. 2019;116:8380‐8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3‐kinase induce gain of function by different mechanisms. Proc Natl Acad Sci. 2008;105:2652‐2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gorelick AN, Sánchez‐Rivera FJ, Cai Y, et al. Phase and context shape the function of composite oncogenic mutations. Nature. 2020;582:100‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Z, Feng J, Saldivar JS, Gu D, Bockholt A, Sommer SS. EGFR somatic doublets in lung cancer are frequent and generally arise from a pair of driver mutations uncommonly seen as singlet mutations: one‐third of doublets occur at five pairs of amino acids. Oncogene. 2008;27:4336‐4343. [DOI] [PubMed] [Google Scholar]
- 10. Pérez‐Pérez JM, Candela H, Micol JL. Understanding synergy in genetic interactions. Trends Genet. 2009;25:368‐376. [DOI] [PubMed] [Google Scholar]
- 11. Mackay TF. Epistasis and quantitative traits: using model organisms to study gene‐gene interactions. Nat Rev Genet. 2014;15:22‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martienssen R, Irish V. Copying out our ABCs: the role of gene redundancy in interpreting genetic hierarchies. Trends Genet. 1999;15:435‐437. [DOI] [PubMed] [Google Scholar]
- 13. van de Haar J, Canisius S, Yu MK, Voest EE, Wessels LFA, Ideker T. Identifying epistasis in cancer genomes: a delicate affair. Cell. 2019;177:1375‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bateson D. Discussion on the influence of heredity on disease, with special reference to tuberculosis, cancer, and diseases of the nervous system: introductory address. Proc R Soc Med. 1909;2:22‐30. [PMC free article] [PubMed] [Google Scholar]
- 15. Domingo J, Baeza‐Centurion P, Lehner B. The causes and consequences of genetic interactions (Epistasis). Annu Rev Genomics Hum Genet. 2019;20:433‐460. [DOI] [PubMed] [Google Scholar]
- 16. Poon A, Chao L. The rate of compensatory mutation in the DNA bacteriophage φX174. Genetics. 2005;170:989‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Domingo J, Diss G, Lehner B. Pairwise and higher‐order genetic interactions during the evolution of a tRNA. Nature. 2018;558:117‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horlbeck MA, Xu A, Wang M, et al. Mapping the genetic landscape of human cells. Cell. 2018;174:953‐967.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rauscher B, Heigwer F, Henkel L, Hielscher T, Voloshanenko O, Boutros M. Toward an integrated map of genetic interactions in cancer cells. Mol Syst Biol. 2018;14:e7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lehner B. Molecular mechanisms of epistasis within and between genes. Trends Genet. 2011;27:323‐331. [DOI] [PubMed] [Google Scholar]
- 21. Sanchez‐Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome Atlas. Cell. 2018;173:321‐337.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levine DA, Getz G, Gabriel SB, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269‐271. [DOI] [PubMed] [Google Scholar]
- 25. Dogruluk T, Tsang YH, Espitia M, et al. Identification of variant‐specific functions of PIK3CA by rapid phenotyping of rare mutations. Cancer Res. 2015;75:5341‐5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berger AH, Brooks AN, Wu X, et al. High‐throughput phenotyping of lung cancer somatic mutations. Cancer Cell. 2017;32:884. [DOI] [PubMed] [Google Scholar]
- 27. Kim E, Ilic N, Shrestha Y, et al. Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 2016;6:714‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352:786‐792. [DOI] [PubMed] [Google Scholar]
- 29. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182‐4190. [DOI] [PubMed] [Google Scholar]
- 31. Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764‐4774. [DOI] [PubMed] [Google Scholar]
- 32. Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn‐676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293‐300. [DOI] [PubMed] [Google Scholar]
- 33. Smith CC, Wang Q, Chin C‐S, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang W, Gao C, Konopleva M, et al. Reversal of acquired drug resistance in FLT3‐mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin Cancer Res. 2014;20:2363‐2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI‐571 cancer therapy caused by BCR‐ABL gene mutation or amplification. Science. 2001;293:876‐880. [DOI] [PubMed] [Google Scholar]
- 36. Cortes J, Jabbour E, Kantarjian H, et al. Dynamics of BCR‐ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood. 2007;110:4005‐4011. [DOI] [PubMed] [Google Scholar]
- 37. Nakaoku T, Kohno T, Araki M, et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat Commun. 2018;9:625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Choi YL, Soda M, Yamashita Y, et al. EML4‐ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734‐1739. [DOI] [PubMed] [Google Scholar]
- 39. Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK‐rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer‐interface mutations. Nature. 2018;559:125‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suda K, Onozato R, Yatabe Y, Mitsudomi T. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol. 2009;4:1‐4. [DOI] [PubMed] [Google Scholar]
- 42. Ricordel C, Friboulet L, Facchinetti F, Soria JC. Molecular mechanisms of acquired resistance to third‐generation EGFR‐TKIs in EGFR T790M‐mutant lung cancer. Ann Oncol. 2019;30:858. [DOI] [PubMed] [Google Scholar]
- 43. Toyooka S, Kiura K, Mitsudomi T. EGFR mutation and response of lung cancer to gefitinib. N Engl J Med. 2005;352:2136. Author reply 2136. [DOI] [PubMed] [Google Scholar]
- 44. Chen W, Jones D, Medeiros LJ, Luthra R, Lin P. Acute myeloid leukaemia with FLT3 gene mutations of both internal tandem duplication and point mutation type. Br J Haematol. 2005;130:726‐728. [DOI] [PubMed] [Google Scholar]
- 45. Moreno I, Martín G, Bolufer P, et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica. 2003;88:19‐24. [PubMed] [Google Scholar]
- 46. Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070‐2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kohsaka S, Nagano M, Ueno T, et al. A method of high‐throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci Transl Med. 2017;9:eaan6566. [DOI] [PubMed] [Google Scholar]
- 48. Nagano M, Kohsaka S, Ueno T, et al. High‐throughput functional evaluation of variants of unknown significance in ERBB2. Clin Cancer Res. 2018;24:5112‐5122. [DOI] [PubMed] [Google Scholar]
- 49. Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high‐throughput single‐cell genomics. Nat Commun. 2020;11:5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613‐628. [DOI] [PubMed] [Google Scholar]
- 51. Ma X, Edmonson M, Yergeau D, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B‐acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497‐5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andre F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA‐mutated, hormone receptor‐positive advanced breast cancer. N Engl J Med. 2019;380:1929‐1940. [DOI] [PubMed] [Google Scholar]