

Abstract
High‐mobility group protein A2 (HMGA2) is highly expressed in hepatocellular carcinoma (HCC) cells and contributes to tumor metastasis and poor patient survival. However, the molecular mechanism through which HMGA2 is transcriptionally regulated in HCC cells remains largely unclear. Here, we showed that the expression HMGA2 was upregulated in HCC, and that elevated HMGA2 could promote tumor metastasis. Incubation of HCC cells with epidermal growth factor (EGF) could promote the expression of HMGA2 mRNA and protein. Mechanistic studies suggested that EGF can phosphorylate p300 at Ser1834 residue through the PI3K/Akt signaling pathway in HCC cells. Knockdown of p300 can reverse EGF‐induced HMGA2 expression and histone H3‐K9 acetylation, whereas a phosphorylation‐mimic p300 S1834D mutant can stimulate HMGA2 expression as well as H3‐K9 acetylation in HCC cells. Furthermore, we identified that p300‐mediated H3‐K9 acetylation participates in EGF‐induced HMGA2 expression in HCC. In addition, the levels of H3‐K9 acetylation positively correlated with the expression levels of HMGA2 in a chemically induced HCC model in rats and human HCC specimens.
Keywords: EGFR, hepatocellular carcinoma, histone H3, HMGA2, p300
We first showed that EGF can induce p300 phosphorylation at the Ser1834 site through activation of the PI3K/AKT signaling pathway in hepatocellular carcinoma; p300 subsequently catalyzes histone H3 acetylation at K9 residues, leading to HMGA2 transcription in HCC
Abbreviations
- Ac‐H3
acetylation of histone H3
- DEN
diethylnitrosamine
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐mesenchymal transition
- HAT
histone acetyltransferase
- HCC
hepatocellular carcinoma
- HDAC
histone deacetylase
- HMGA2
high‐mobility group protein A2
- HMGA2‐KD
HMGA2‐knockdown
- HMGA2‐OE
HMGA2 overexpression
- IHC
immunohistochemistry
- lncRNAs
long non‐coding RNAs
- NSCLC
non‐small cell lung cancer
- PKA
protein kinase A
1. INTRODUCTION
Hepatocellular carcinoma is one of the malignant tumors with poor prognosis, and ranked as the second leading cause of cancer‐related deaths worldwide. 1 The most important cause of death in HCC patients is the high metastatic capacity of HCC cell. 2 , 3 Therefore, exploring the mechanisms of HCC metastasis will be helpful for the prevention and treatment of HCC.
HMGA2 is a kind of non‐histone chromatin protein that is widely found in eukaryotes. 4 It contains three special at‐hook structures, which can bind to at‐sequence‐enriched regions on DNA, promote or inhibit the expression of related target genes, and thus affect the development of embryos. 5 Generally, HMGA2 is only highly expressed at the early stage of fetal development, and the expression of HMGA2 can hardly be detected in normal adult tissues. 6 , 7 A large number of studies have shown that HMGA2 is highly expressed in most malignant tumors, and its expression level is closely related to the degree of malignancy and metastasis, including breast, melanoma, colorectal and bladder cancer. 8 , 9 More importantly, accumulating evidence has shown that HMGA2 is highly expressed in HCC, and that elevated HMGA2 predicts a high incidence of metastasis and poor patient survival in HCC. 10 , 11 , 12 For example, overexpression of HMGA2 is an important contributor to the invasion of HCC cells, while HMGA2 knockdown impaired the migratory capacity of tumor cells in HCC. 12 , 13 However, the specific molecular mechanism of HMGA2 transcriptional regulation in HCC cells is still unclear.
Acetylation of histone H3 is one of the most frequent epigenetic modifications that affects chromatin structure and gene expression. 14 , 15 It has been shown that histone deacetylase inhibition results in decreased HMGA2 transcription level and reduced HMGA2 promoter binding to acetylated histone H3 in HeLa cells. 16 This raised the possibility that histone H3 acetylation may be involved in regulating HMGA2 transcription in HCC.
Epidermal growth factor receptor is a transmembrane tyrosine kinase and frequently overexpressed in HCC. 17 , 18 Persistent activation of the EGFR signaling system has been implicated in liver responses to injury and carcinogenesis. 19 , 20 Several studies have shown that histone H3 acetylation in response to the activation of EGFR was associated with gene transcription and tumorigenesis. 21 , 22 However, it remains unclear whether EGFR‐mediated H3 acetylation is involved in the transcription of HGMA2 in HCC. Here, our study demonstrated that EGFR activation induces p300 phosphorylation at Ser1834 via PI3K/Akt signaling pathway, then p300 can acetylate histone H3 at residue K9, leading to high expression of HMGA2 in HCC.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
The inhibitors used in this study were purchased from Cell Signaling Technology. Rabbit polyclonal anti‐HMGA2 antibody (ab97276), rabbit polyclonal anti‐Histone H3 antibody (ab1791), rabbit polyclonal anti‐Histone H3 (acetyl K9) antibody (ab10812), rabbit polyclonal anti‐Lamin B1 antibody (ab65986), HRP‐conjugated goat anti‐rabbit IgG (ab6721) and goat anti‐mouse IgG (ab205719) were all purchased from Abcam. Rabbit monoclonal anti‐Akt antibody (#4691) and rabbit monoclonal anti‐Akt (phosphor Ser473) antibody were purchased from Cell Signaling Technology. Rabbit polyclonal anti‐p300 (phosphor Ser1834) antibody (PA5‐64531) was purchased from Thermo Fisher Scientific, and all antibodies were diluted in accordance with 1:1000. Besides, Recombinant Human EGF Protein (236‐EG) was purchased from R&D systems.
2.2. Cell lines and cell culture
SNU‐368, SNU‐739, Huh‐7 and HepG2 cells were obtained from the Cell Bank of the Chinese Academy of Science. Cells were cultured in DMEM supplemented with 10% FBS (HyClone) at 37°C in a humidified incubator containing 5% CO2.
2.3. Plasmid construction, mutagenesis and transfection
Histone H3‐depleted cells were generated as previously described. 23 Briefly, pGIPZ Histone H3 shRNA was generated with CCT ATG AAA GGA TGC AATA. The pGIPZ control was generated with control oligonucleotide GCT TCT AAC ACC GGA GGT CTT. Then, SNU‐368 cells were plated at a density of 4 × 105/60‐mm dish 18 hours prior to transfection. Transfection was carried out using HyFect reagents according to the vendor’s instructions. Two days after infection, Histone H3‐depleted cell populations were selected with 2 µg/mL puromycin (Sigma‐Aldrich) for 14 days.
pcDNA 3.1/hygro (+)‐Histone H3 K9R was made by using the QuikChange site‐directed mutagenesis kit (Stratagene). Cells were then transfected with WT Histone H3 or Histone H3 K9R mutant. Twenty‐four hours after transfection, these cells were incubated in the presence or absence of EGF (100 ng/mL) for 12 hours.
2.4. Western blot analysis
Cells or liver tissue were extracted using ice‐chilled RIPA lysis buffer containing 50 mmol/L Tris‐HCl (pH 8.0), 150 mmol/L NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP‐40, 5 mmol/L EDTA, 0.25 mmol/L PMSF and protease inhibitor cocktail. The concentration of protein was determined using the BCA kit (Pierce). Protein samples were denatured and separated by SDS‐PAGE. When the protein is completely separated, it is transferred to a methanol‐activated PVDF membrane. The membrane was then blocked in 5% skim milk powder or 3% BSA for 1 hour at room temperature. After blocking, the PVDF membrane was then incubated with the corresponding primary and HRP‐conjugated secondary antibodies. Finally, ECL plus reagent was used in the FluorChem E (Protein Simple) imager to detect blotting.
2.5. Real‐time PCR
RNA from cancer cells and rat liver tissues was extracted using TRIzol reagent (Invitrogen). Complementary DNA was synthesized by using random hexamers and MMLV reverse transcriptase according to the manufacturer’s instructions (Takara). Real‐time PCR was carried out using 2× SYBR Green PCR Master Mix (Promega) on an ABI7500 sequence detection system (Applied Biosystems). The setting procedures were as follows: 95°C, predenaturation for 5 minutes, 95°C for 15 seconds, 60°C for 1 minute, a total of 40 cycles. GAPDH was used as an internal control. The relative expression level of HMGA2 was calculated by the 2−ΔΔCt method. The primer sequences used were as follows: human HMGA2, 5′‐TCC CTC TAA AGC AGC TCA AAA‐3′ (sense) and 5′‐ACT TGT TGT GGC CAT TTC CT‐3′ (antisense); human GAPDH, 5′‐GAC CCC TTC ATT GAC CTC AAC‐3′ (sense) and 5′‐CTT CTC CAT GGT GGT GAA GA‐3′ (antisense); rat HMGA2, 5′‐ATC CCG TCT CCC GAA AGGT‐3′ (sense) and 5′‐CTC GGT TGG ACA CTC GGGA‐3′ (antisense); rat GAPDH, 5′‐GAC AAC TTT GGC ATC GTG GA‐3′ (sense) and 5′‐ATG CAG GGA TGA TGT TCT GG‐3′ (antisense).
2.6. Chromatin immunoprecipitation
A ChIP assay was carried out using the SimpleChIP Plus Enzymatic Chromatin IP Kit (Agarose Beads) (#9004S; Cell Signaling Technology) according to the manufacturer’s protocols. DNA was eluted and purified from the complexes and then amplified by PCR using primers specific for the HMGA2 promoters. The following primers were used for real‐time quantitative PCR after ChIP assay: region 1 (−288 to −164), 5′‐GCA CGA TTG GCA GTT AGG AA‐3′ (sense) and 5′‐GAG GGA GGG GTC TAA CAG GA‐3′ (antisense); region 2 (−936 to −838), 5′‐AGG CAG AAG GTT AAA TCG‐3′ (sense) and 5′‐ATG TTC CCA GGG TAA AGG‐3′ (antisense); region 3 (−1380 to −1252), 5′‐GAG GCG GAG TAC GGC TGT CA‐3′ (sense) and 5′‐TCT ACC GTG CCG AGC CAA CA‐3′ (antisense). Three replicates for each experimental point were carried out. Results were normalized using the internal control IgG.
2.7. Animals and experimental design
Male Sprague‐Dawley rats (6‐week‐old) and male athymic BALB/c nu/nu mice (5‐week‐old) were purchased from Beijing Weitong Lihua Animal Co. All animal procedures were carried out in accordance with the guidelines of the Institutional Animal Care and Use Committee of Henan University. During the experiment, all animals were housed at 20‐24°C with a relative humidity of 50 ± 10% and a light/dark cycle of 12 hours. The animals were fed with sufficient water and food every day. Before experimental procedures, all animals were allowed to adapt to the environment for a week.
Rat model of HCC was induced by DEN. Briefly, the rats were intraperitoneally injected with 50 mg/kg DEN once a week for 16 weeks. Rats in the control group were intraperitoneally injected with normal saline. At the end of the experiment, all animals were killed and their livers were removed. Some livers were fixed with 4% paraformaldehyde and used for immunohistochemistry. The rest was stored at −80°C for western blot and real‐time PCR.
In the lung metastasis experiment, we randomly divided BALB/c mice into five groups with 10 mice in each group: H22/sh‐vector, H22/HMGA2‐KD1 (KD, knockdown), H22/HMGA2‐KD2, H22/vector, H22/HMGA2‐OE (HMGA2 overexpression). Then 1 × 106 cells transfected with the plasmids of vector, HMGA2 shRNA or HMGA2 overexpression were injected into the lateral caudal vein of each mouse. To ensure that the mice had prominent nodules in their lungs, pulmonary metastasis was allowed to develop for 6 weeks. Then the mice were killed and their lungs were removed. After 1 day of fixation with 4% paraformaldehyde, the number of nodules on the lung surface of the mice in each group was counted.
2.8. Human tissue specimens
Between 2014 and 2018, we obtained matched pairs of HCC and adjacent non‐tumor tissues from 39 patients from the First Affiliated Hospital of Henan University. Use of patient tissues was approved by the hospital research ethics committee, and informed consent was obtained from all enrolled patients.
2.9. Immunohistochemical staining
Tissues were dehydrated and embedded in paraffin after fixation with paraformaldehyde. The paraffin‐embedded tissues were then cut into pieces (4 μm) and placed on polylysine‐coated slides. IHC was incubated overnight with HGMA2 antibody (1:100) or acetyl‐histone H3 antibody (1:100) at 4°C overnight. Then the slides were washed 3 times with PBS and streptavidin‐bound HRP antibody was incubated for 1 hour at room temperature. Subsequently, DAB was used as chromogen and DAB was developed. Immunoreactivity was examined under a light microscope. Two independent pathologists analyzed the expression of the target protein by visualizing the brown‐stained section. Staining intensity was scored on a scale of 0‐3:0 (negative), 1 (weak), 2 (moderate), or 3 (strong). Staining extent was scored as follows: 0, if no tumor sections were stained; 1, if <1% of sections were stained; 2, if 2%‐10% of sections were stained; 3, if 11%‐30% of sections were stained; 4, if 31%‐70% of sections were stained; 5, if 71%‐100% of sections were stained. The staining intensity and staining extent scores were then added to calculate the final stain score (0‐8) for each tissue.
2.10. Statistical analysis
Statistical analyses were carried out using GraphPad Prism 5.0 (GraphPad Software, Inc.). All data are presented as means ± SEM. Statistical significance of the difference between the two groups was tested by an unpaired, two‐tailed Student’s t test. One‐way analysis of variance (ANOVA) followed by Tukey or Dunnett’s post‐tests was used to compare means of multiple experimental groups. P < .05 was considered to be significant.
3. RESULTS
3.1. HMGA2 is overexpressed and promotes tumor metastasis in HCC
Before exploring the molecular mechanism of HMGA2 transcriptional regulation in HCC cells, we first collected 39 matched HCC tissues and adjacent non‐tumor tissues and examined the expression of HMGA2. As the results of IHC shown in Figure 1A, the expression of HMGA2 in human HCC tumor tissues was higher than that in adjacent tissues. Moreover, we then established a HCC model by injecting DEN into rats and detected the expression of HMGA2. Through real‐time PCR and western blotting, we found that mRNA and protein expression of HMGA2 in the livers of DEN‐treated rats were significantly upregulated (Figure 1B, C). The results of IHC further showed overexpression of HMGA2 protein in the livers of DEN‐treated rats (Figure 1D). In order to explore the role of HMGA2 in the metastasis of HCC, we established HMGA2‐overexpressing and HMGA2‐knockdown H22 cells (Figure 1E). These cells were then inoculated into the lateral tail vein of BALB/c mice. After 6 weeks, we counted the lung metastases nodules. As the results showed, overexpression of HMGA2 increased the number of tumor metastatic nodules, while knockdown of HMGA2 significantly reduced the number of lung metastasis (Figure 1F, G). Obviously, these results suggested that overexpression of HMGA2 in HCC can promote the metastasis of HCC.
Figure 1.
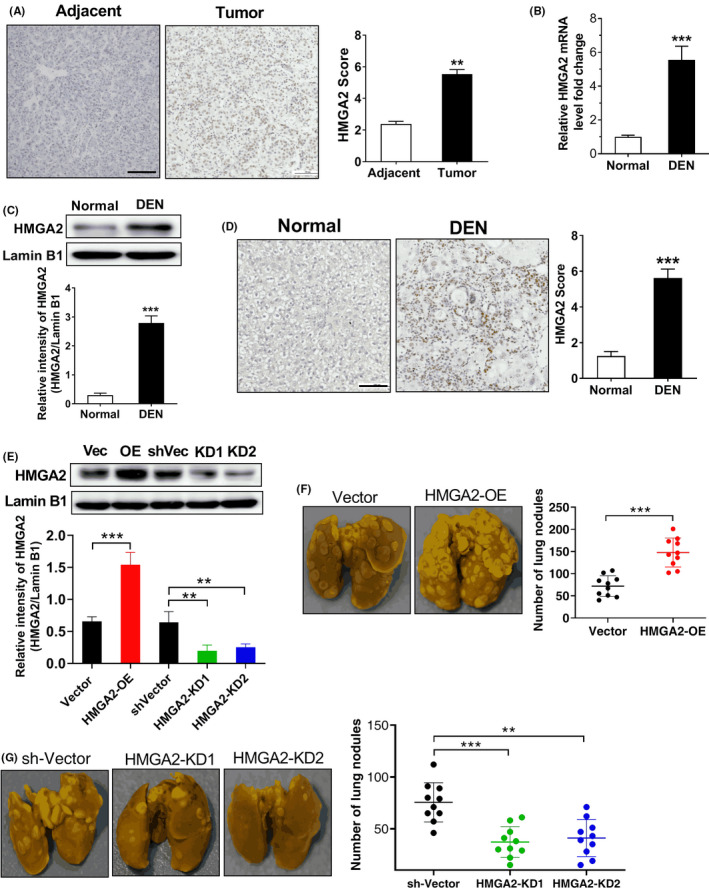
High‐mobility group protein A2 (HMGA2) is upregulated and promotes tumor metastasis in hepatocellular carcinoma (HCC). A, IHC suggests that HMGA2 expression was increased in tumor tissues compared to adjacent non‐tumor tissues, **P < .01, two‐tailed unpaired t test, n = 39 per group. Scale bar, 100 µm. B, Real‐time PCR assay shows that mRNA expression of HMGA2 was upregulated in the livers of diethylnitrosamine (DEN)‐treated rats. ***P < .001 compared to normal rats, two‐tailed unpaired t test, n = 8 per group. C, Western blot analysis shows that the protein expression of HMGA2 was upregulated in the livers of DEN‐treated rats. ***P < .001 compared to normal rats, two‐tailed unpaired t test, n = 6 per group. Lamin B1 was used as an internal control. D, IHC suggests that HMGA2 expression in the livers of DEN‐rats was enhanced compared to normal rats, ***P < .001, two‐tailed unpaired t test, n = 8 per group. Scale bar, 100 µm. E, Western blot analysis of HMGA2 protein level in HMGA2‐overexpressing and HMGA2‐knockdown H22 cells. Vec, vector; OE, overexpression; KD, knockdown; **P < .01, ***P < .001, one‐way ANOVA, n = 5 independent experiments per group. Lamin B1 was used as an internal control. F, Overexpression of HMGA2 increased the number of tumor metastatic nodules in tumor‐bearing mice. Left, representative photos of metastatic lung nodules; right, quantification of the lung nodules. ***P < .001, two‐tailed unpaired t test, n = 10 per group. G, Knockdown of HMGA2 reduced the number of lung metastases in tumor‐bearing mice. Left, representative photos of metastatic lung nodules; right, quantification of the lung nodules. **P < .01, ***P < .001, one‐way ANOVA, n = 10 per group
3.2. EGF promotes HMGA2 transcription in HCC through the PI3K/AKT signaling pathways
Next, we investigated whether EGF promoted the expression of HMGA2. We stimulated SNU‐368, SNU‐739, Huh‐7 and HepG2 cells with EGF, and detected the protein level of HMGA2. We found that the mRNA and protein expression levels of HMGA2 in HCC cells were significantly upregulated after treatment with 100 ng/mL EGF (Figure 2A,B). To determine whether EGF‐induced HMGA2 overexpression is dependent on EGFR activation, we pretreated SNU‐368 cells with 20 µmol/L AG1478 (an inhibitor of EGFR tyrosine kinase) 30 minutes prior to coincubation with 100 ng/mL EGF for 12 hours. Through real‐time PCR and western blot, we found AG1478 could reverse the upregulation of HMGA2 mRNA and protein levels induced by EGF (Figure 2C, D).
Figure 2.
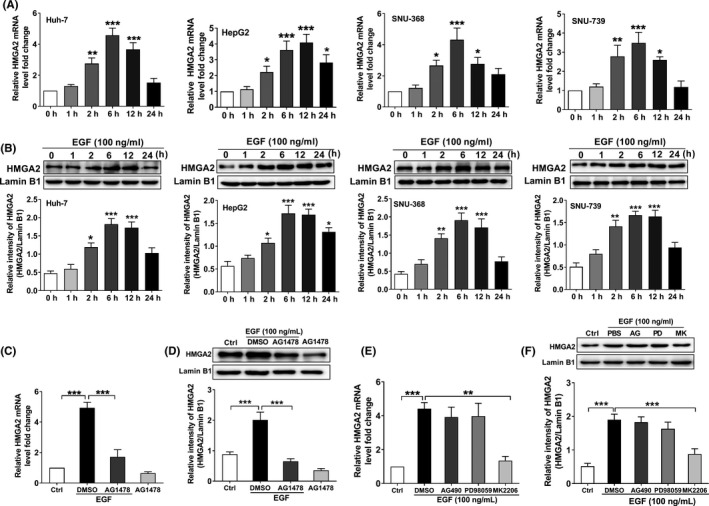
Epidermal growth factor (EGF) promotes high‐mobility group protein A2 (HMGA2) expression through the PI3K/AKT signaling pathway in hepatocellular carcinoma (HCC). EGF induced a significant increase in HMGA2 mRNA (A) and protein expression (B) in SNU‐368, SNU‐739, Huh‐7 and HepG 2 cells. *P < .05, **P < .01, ***P < .001 compared to control group, one‐way ANOVA, n = 6 per group. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 (20 µmol/L) could reverse EGF‐mediated upregulation of HMGA2 mRNA (C) and protein (D) expression in SNU‐368 cells. ***P < .001 compared to control group, one‐way ANOVA, n = 6 per group. EGF‐induced upregulation of HMGA2 mRNA (E) and protein (F) can be blocked by Akt inhibitor MK2206 (10 µmol/L) in SNU‐368 cells, whereas JAK inhibitor AG490 (20 µmol/L) and MEK inhibitor PD98059 (20 µmol/L) have no effects on EGF‐induced HMGA2 mRNA and protein expression. **P < .01, ***P < .001, one‐way ANOVA, n = 6 per group
EGFR carries out its biological functions through activation of the Src‐JAK‐STAT3, Ras‐Raf‐MEK‐ERK, and PI3K‐Akt‐mTOR pathways. 17 , 24 To explore the pathways by which EGF induces HMGA2 expression in HCC cells, we examined the effects of AG490 (20 µmol/L, a JAK inhibitor), PD98059 (20 µmol/L, a MEK inhibitor), and MK2206 (10 µmol/L, an Akt inhibitor) on EGF‐induced HMGA2 mRNA and protein expression in HCC cells. As the results showed, pretreatment with MK2206 can reverse EGF‐induced HMGA2 mRNA and protein expression in SNU‐368 cells (Figure 2C, D) after coincubation with 100 ng/mL EGF for 12 hours, while AG490 and PD98059 had no significant effect on EGF‐induced HMGA2 mRNA and protein expression (Figure 2E, F). These results clearly demonstrate that EGF promoted HMGA2 expression through PI3K/Akt pathways.
3.3. Akt‐dependent p300 phosphorylation mediates the EGF‐induced increase in HMGA2 transcription in HCC
Previous literature has shown that EGFR phosphorylates the p300 Ser1834 residue through the PI3K/AKT pathway, thereby promoting the expression of the relevant gene. 25 Therefore, we then investigated whether Akt‐dependent phosphorylation of p300 is involved in EGF‐induced HMGA2 transcriptional expression. First, we incubated SNU‐368 cells using EGF and detected the phosphorylation level on Akt Ser473 and p300 Ser1834 residues. As shown in Figure 3A,B, after exposure of SNU‐368 cells to 100 ng/mL EGF, the phosphorylation levels of Akt Ser473 and p300 Ser1834 residues in SNU‐368 cells were significantly increased. Then, the effects of AG1478 (an inhibitor of EGFR tyrosine kinase) and MK2206 (10 µmol/L, an Akt inhibitor) on EGF‐induced phosphorylation of p300 Ser1834 residues were evaluated in SNU‐368 cells, respectively. The results showed that when SNU‐368 cells were pretreated with AG1478 or MK2206 for 30 minutes followed by coincubation with 100 ng/mL EGF for 2 hours, EGF‐induced increase in the phosphorylation of p300 Ser1834 could be reversed by both of these two inhibitors (Figure 3C). These data indicate that EGF phosphorylates p300 at S1834 site by the PI3K/Akt signaling pathway in HCC.
Figure 3.
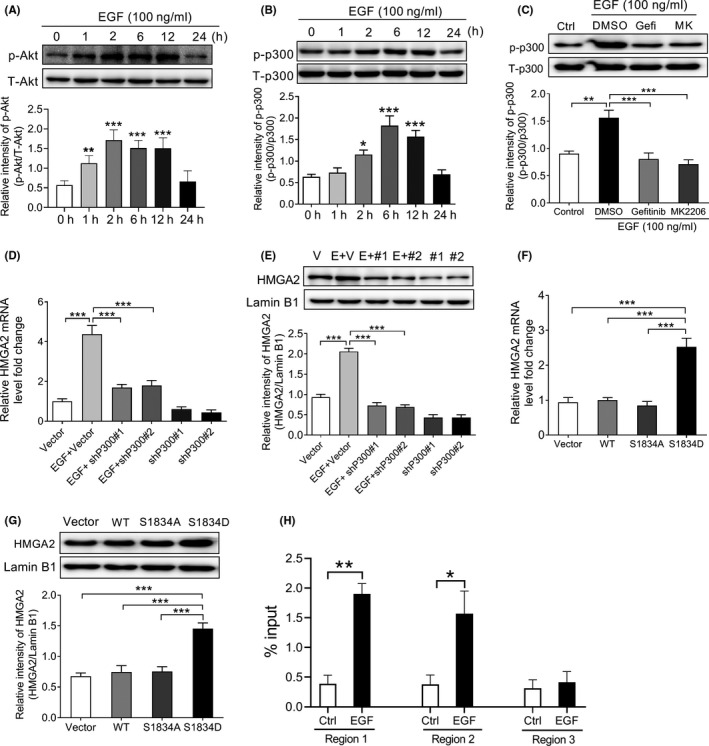
Akt‐dependent p300 phosphorylation mediates epidermal growth factor (EGF)‐induced increase in high‐mobility group protein A2 (HMGA2) transcription in SNU‐368 cells. Western blot analysis suggests that EGF induced an increase in the level of Akt Ser473 (A) and p300 Ser1834 (B) phosphorylation in SNU‐368 cells. *P < .05, **P < .01, ***P < .001, one‐way ANOVA, n = 6 per group. C, EGF‐induced increased phosphorylation levels of p300 at Ser1834 was attenuated by 20 µmol/L AG1478 (an EGFR tyrosine kinase inhibitor) and MK2206 (an Akt inhibitor) after 2 h coincubation in SNU‐368 cells. Gefi, Gefitinib; MK, MK2206. **P < .01, ***P < .001, one‐way ANOVA, n = 5 per group. Knockdown of p300 with two specific shRNAs could reverse EGF‐induced HMGA2 mRNA (D) and protein (E) expression in SNU‐368 cells, ***P < .001, one‐way ANOVA, n = 6 per group. Expression of a phosphorylation‐mimic p300 S1834D mutant could induce HMGA2 mRNA (F) and protein (G) in SNU‐368 cells. ***P < .001, one‐way ANOVA, n = 6 per group. H, ChIP analysis shows the amounts of p300 recruited to the HMGA2 promoter regions in SNU‐368 cells treated with or without 100 ng/mL EGF for 12 h. *P < .05, **P < .01, two‐tailed unpaired t test, n = 5 per group
Subsequently, we examined whether p300 participates in EGF‐induced upregulation of HMGA2 expression in HCC. As the results showed, knockdown of P300 with shRNA could reverse EGF‐enhanced HMGA2 mRNA and protein in SNU‐368 cells (Figure 3D, E). To verify that p300 phosphorylation at the Ser1834 site is involved in EGF‐induced expression of HMGA2 protein and mRNA levels, we constructed a Flag‐tagged WT p300, a phosphorylation‐defective p300 S1834A mutant and a phosphorylation‐mimic p300 S1834D mutant was then transfected into SNU‐368 cells. After transfection for 24 hours, we detected the expression of HMGA2 mRNA and protein. Results showed that only a phosphorylation‐mimic p300 S1834D mutant can stimulate HMGA2 mRNA and protein expression in SNU‐368 cells (Figure 3F, G). To gain insight into the mechanism underlying HGMA2 transcription, we then designed 3 paired primers covering the 1.5 kb promoter region and carried out qPCR to examine whether p300 binds to HGMA2 promoter. Through chromatin immunoprecipitation assay, we found that EGF stimulation resulted in p300 enrichment at the HMGA2 promoter region 1 (−288 to −164) and region 2 (−936 to −838) in SNU‐368 cells (Figure 3H).
3.4. P300‐dependent acetylation of histone H3 K9 is required for EGFR‐mediated HMGA2 transcription in HCC
Acetylation of histone H3 K9 in response to EGFR activation was associated with gene expression and tumorigenesis. 22 As an acetyltransferase, p300 can enhance the acetylation of histones H3 K9, leading to activation of target gene expression. 26 , 27 Therefore, we supposed that p300‐dependent acetylation of histone H3 K9 is required for EGFR‐mediated HMGA2 transcription in HCC. To test this hypothesis, we first stimulated SNU‐368 cells with EGF to see if it could induce the acetylation of histone H3 K9. As shown in Figure 4A, the acetylation level at the K9 site was significantly upregulated when EGF was applied to SNU‐368 cells. Then, we knocked down p300 using shRNA and examined the level of H3 K9 acetylation induced by EGF treatment. As expected, knockdown of p300 can attenuate the EGF‐induced increased level of H3 K9 acetylation in SNU‐368 cells (Figure 4B). Moreover, we also transfected a Flag‐tagged WT p300, a phosphorylation‐defective p300 S1834A mutant and a phosphorylation‐mimic p300 S1834D mutant into SNU‐368 cells and examined the level of histones H3 K9 acetylation. As the results showed in Figure 4C, only a phosphorylation‐mimic p300 S1834D mutant can stimulate histones H3 K9 acetylation in SNU‐368 cells. Together, these results suggested that histones H3 K9 acetylation depends on EGF‐mediated p300 phosphorylation at the S1834 site.
Figure 4.
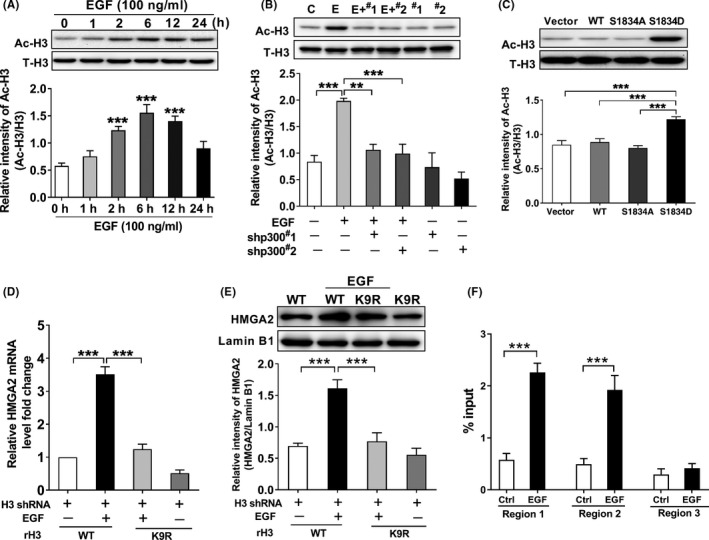
p300‐Dependent acetylation of histone H3 is required for epidermal growth factor receptor (EGFR)‐mediated high‐mobility group protein A2 (HMGA2) transcription in SUN‐368 cells. A, Epidermal growth factor (EGF) treatment induced a significant increase in H3‐K9 acetylation in SNU‐368 cells, ***P < .001, one‐way ANOVA, n = 6 per group. B, Knockdown of p300 reversed EGF‐induced H3‐K9 acetylation in SNU‐368 cells. C, control; E, EGF; #1, shp300#1; #2, shp300#2. **P < .01, ***P < .001, one‐way ANOVA, n = 5 per group. C, Expression of the phosphorylation‐mimic p300 S1834D mutant induced a higher expression of H3‐K9 acetylation in SNU‐368 cells. ***P < .001, one‐way ANOVA, n = 5 per group. Reconstituted expression of RNAi‐resistant histone H3‐K9R abrogated EGF‐induced mRNA (D) and protein (E) expression of HMGA2 in endogenous H3‐depleted SNU‐368 cells. ***P < .001, one‐way ANOVA, n = 5 per group. F, ChIP analysis shows that EGF treatment resulted in enhanced H3‐K9 acetylation at the HMGA2 promoter region 1 and region 2 in SNU‐368 cells. ***P < .01, two‐tailed unpaired t test, n = 5 per group
To explore the significance of histone H3‐K9 acetylation in EGF‐induced HMGA2 expression, we replaced histone K9 site with arginine to construct the H3 K9R mutant and expressed it into endogenous H3‐depleted SNU‐368 cells to detect the role of H3‐K9 acetylation in EGF‐induced HMGA2 expression. As the results showed, mutation of K9 of histone H3 into arginine abrogated EGF‐induced mRNA and protein expression of HMGA2 (Figure 4D, E). Meanwhile, chromatin immunoprecipitation results also showed that EGF stimulation can lead to increased acetylation of H3 K9 at HMGA2 promoter in SNU‐368 cells (Figure 4F). In in vivo experiments, we detected the acetylation level of histone H3 K9 in DEN‐induced rat liver tissues using western blot and IHC. Through these two methods, we observed an enhanced acetylation level of histone H3 K9 in the livers of DEN‐treated rats (Figure 5A, B). Subsequently, to explore whether HMGA2 expression correlates with H3K9Ac level in the rat model of HCC, we carried out immunohistochemistry analyses of the livers of DEN‐treated rats with antibodies against H3‐K9 acetylation and HMGA2. As expected, the expression of HMGA2 was significantly positively correlated with H3‐K9 acetylation (Figure 5C, D). To define the clinical relevance of our finding that H3‐K9 acetylation regulates HMGA2 transcription, we also carried out immunohistochemistry analyses of 39 human HCC tumor specimens. As the results showed, the levels of H3‐K9 acetylation were also positively correlated with the expression levels of HMGA2, quantification of the staining showed that these correlations were significant (Figure 5E, F). Taken together, these results suggest that p300‐dependent acetylation of histone H3 K9 is necessary for EGF‐induced HMGA2 expression in HCC.
Figure 5.
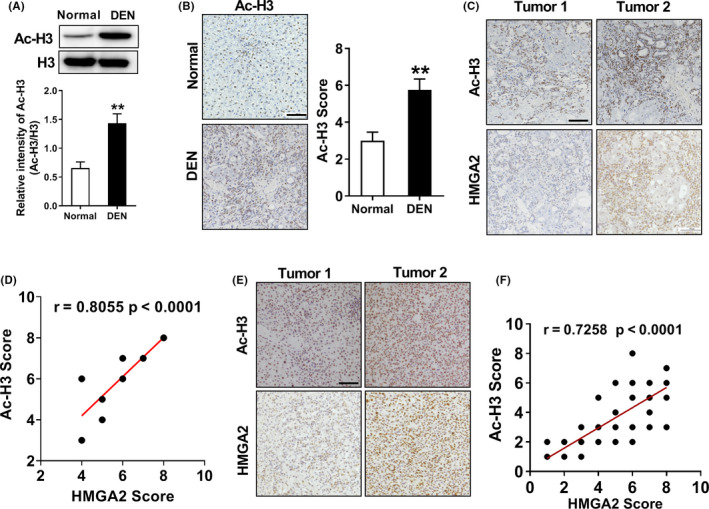
Histone H3 K9 acetylation is positively correlated with high‐mobility group protein A2 (HMGA2) expression in hepatocellular carcinoma (HCC). A, Western blot analysis shows that acetyl‐H3 K9 expression was upregulated in the livers of diethylnitrosamine (DEN)‐treated rats. **P < .01 compared to normal rats, two‐tailed unpaired t test, n = 8 per group. B, Immunohistochemistry (IHC) suggested that acetyl‐H3 expression in the livers of DEN‐rats was enhanced compared to normal rats, **P < .01, two‐tailed unpaired t test, n = 8 per group. Scale bar, 100 µm. C, Representative photos of IHC of DEN‐treated rat livers with anti‐acetyl‐H3‐K9 and HMGA2 antibodies. Scale bar, 100 µm. D, Level of H3‐K9 acetylation correlated with the expression levels of HMGA2 in DEN‐treated rats; Pearson product moment correlation test. E, Representative photos of IHC of 39 HCC specimens with anti‐acetyl‐H3‐K9 and HMGA2 antibodies. Scale bar, 100 µm. F, Level of H3‐K9 acetylation correlated with the expression levels of HMGA2. Pearson product moment correlation test; note that some of the dots on the graphs represent more than one specimen (ie, some scores overlapped)
4. DISCUSSION
Overexpression of HMGA2 is present in many epithelial malignancies, such as pancreatic cancer, breast cancer, ovarian cancer, oral squamous cell carcinoma and lung cancer. 8 Accumulating evidence has suggested HMGA2 functions as a tumor promoter, which can promote tumor metastasis and enhance the formation of EMT. 12 , 28 For example, HMGA2 can induce enhanced transcription of Twist1 and Snail by directly binding to the critical element of endogenous target promoters, resulting in EMT; 29 , 30 the silencing of HMGA2 expression inhibited glioblastoma invasion by repression of the transcription of the MMP2 gene. 31 Similarly, highly expressed HMGA2 was also observed in HCC cells, and the elevated expression of HMGA2 could promote HCC cell migration and invasion. 10 , 12 In support of these studies, we also observed an abnormal expression of HMGA2 in the livers of DEN‐treated rats; overexpression of HMGA2 increased the numbers of tumor metastatic nodules in a mouse model of lung metastasis.
Recently, several lines of reports focused on the regulation of HMGA expression have emerged. For example, long non‐coding RNA VPS9D1 antisense RNA 1 (VPS9D1‐AS1) promotes HMGA2 expression by sponging miR‐532‐3p in NSCLC cells. 32 Oxidized low‐density lipoprotein receptor 1 (OLR1) increased HMGA2 transcription by upregulating c‐Myc expression in pancreatic cancer cells. 33 However, the mechanism underlying which regulates the transcriptional expression of HMGA2 in HCC cells remains largely unclear. Here, our findings suggested EGF stimulation can promote HMGA2 expression at the transcriptional level via activation of PI3K/Akt signaling pathways in HCC cells. In support of our results, Voon et al found that exposure of murine gastric epithelial (GIF‐14) cells to EGF led to increased HMGA2 transcription. 34 Certainly, we cannot rule out other mechanisms involved in the regulation of HMGA2 expression in HCC. In fact, several lncRNAs, such as HULC, LSINCT5, LINC00473, SNHG16 and EGOT, have been found to promote HMGA2 expression in HCC. 11 , 35 , 36 , 37 , 38 In addition, a series of microRNAs (miRs or miRNAs) including miR‐663a, miR‐107, microRNA‐337 and microRNA‐9 could inhibit HCC progression through suppression of HMGA2 expression. 39 , 40 , 41
Abnormal expression of p300 has been identified in esophageal squamous carcinoma, NSCLC cells and HCC. 42 , 43 , 44 Numerous studies have demonstrated p300 was associated with enhanced EMT and correlates with poor prognosis in these cancers. 42 , 44 Overexpression of p300 promotes cell proliferation, migration, and invasion in NSCLC cells; 43 knockdown of p300 inhibited EMT, invasion and other malignant events of HCC cells. 44 However, we must note that p300 may act as a suppressor of tumor metastasis in some cancers. For example, Wang et al 45 have reported that p300 can acetylate JHDM1A to inhibit the growth and metastasis of osteosarcoma. Acting as a transcriptional coactivator, p300 performs these functions through altering chromatin structure to stimulate tumor‐related gene transcription. 46 , 47 In detail, p300 controls this gene expression by transferring an acetyl group to lysine residues on histones and non‐histone proteins. 46 Consistent with these reports, we demonstrated that p300 could acetylate histone H3 at K9 and promote HMGA2 transcription in HCC. To the best of our knowledge, we disclosed for the first time that p300 can stimulate HMGA2 expression at the transcriptional level. As an acetyltransferase, the catalytic activity of p300 can be regulated by post‐translational modification including methylation, acetylation as well as phosphorylation at some critical sites. 48 For instance, a wide array of protein kinases, such as PKA, PKB, PKC, and mitogen‐activated protein kinases can catalyze p300 phosphorylation at specific sites, resulting in activation or repression of target gene expression. 48 , 49 Here, we also demonstrated that Akt could phosphorylate p300 at the S1834 site to enhance the activity of p300, which promotes HMGA2 transcription in HCC cells. In support of our findings, Srivastava et al also demonstrated that Akt could directly phosphorylate p300 at S1834 in response to EGF stimulation in lung cancer cells. 25
Histone H3 acetylation has been shown to participate in the regulation of transcriptional activity by altering the structure of chromatin. 50 In generally, hyper‐acetylation is largely associated with the activation of gene transcription, while hypo‐acetylation means repression of gene expression. For example, altered levels of acetylation of H3 at lysine (K) 4, 9, 14, 27 have been correlated with tumor progression and metastasis in a variety of cancers. 51 Histone H3 acetylation can be achieved by two groups of enzymes with opposing functions: HAT and HDAC. 50 Here, we identified that p300, one member of the HAT family, can add acetyl moieties to K9 residues upon EGFR activation in HCC, resulting in the activation of HMGA2 transcription. Certainly, we cannot exclude that other histone H3 lysine residues, including H3K18, H3K27, and H3K56, participate in p300‐induced HMGA2 transcription, because H3K18, K27, and K56 are also reported as the targets of p300. In addition, there is another limitation that other HAT or HDAC are involved in catalyzing H3 K9 acetylation that existed in the present study, because Yang et al 23 suggested that EGF stimulation promotes HDAC3 removal from histone H3 and facilitates subsequent H3‐Lys9 acetylation. Indeed, these limitations need to be further explored.
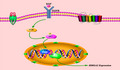
In conclusion, we first showed that EGF can induce p300 phosphorylation at the Ser1834 site through activation of the PI3K/AKT signaling pathway in HCC; p300 subsequently catalyzes histone H3 acetylation at K9 residues, leading to HMGA2 transcription in HCC (Figure 6).
Figure 6.
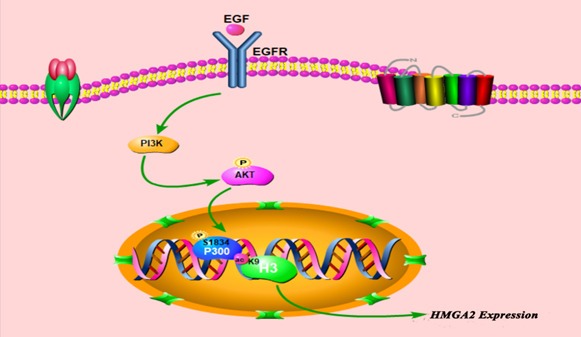
Schematic representation of epidermal growth factor (EGF)‐mediated high‐mobility group protein A2 (HMGA2) transcription in hepatocellular carcinoma (HCC) cells. EGF can induce p300 phosphorylation at the Ser1834 site through activation of the PI3K/AKT signaling pathway in HCC; p300 subsequently catalyzes histone H3 acetylation at K9 residues, leading to HMGA2 transcription in HCC. EGFR, epidermal growth factor receptor
DISCLOSURE
The authors declare that they have no competing interest.
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (Grant Nos. 81701110, 81772832) and the Innovative Research Team (in Science and Technology) in University of Henan Province (Grant No. 19IRTSTHN004), Scientific Research Cultivating Program for Young Talents in Henan Medical School (Grant No. 2019016), Program for Science and Technology of the Department of Education of Henan Province (21A310004), Guangdong Provincial Key Laboratory of Drug Non‐clinical Evaluation and Research (Grant No. 2018B030323024).
Liang C, Niu J, Wang X, et al. P300‐dependent acetylation of histone H3 is required for epidermal growth factor receptor‐mediated high‐mobility group protein A2 transcription in hepatocellular carcinoma. Cancer Sci. 2021;112:679–690. 10.1111/cas.14729
Chao Liang and Jie Niu have contributed equally to this work.
Contributor Information
Dong Fang, emailfangdong@163.com.
Song‐Qiang Xie, Email: xiesq@henu.edu.cn.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017;173:106‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65(4):798‐808. [DOI] [PubMed] [Google Scholar]
- 4. Pfannkuche K, Summer H, Li O, Hescheler J, Droge P. The high mobility group protein HMGA2: a co‐regulator of chromatin structure and pluripotency in stem cells? Stem Cell Rev Rep. 2009;5(3):224‐230. [DOI] [PubMed] [Google Scholar]
- 5. Hammond SM, Sharpless NE. HMGA2, microRNAs, and stem cell aging. Cell. 2008;135(6):1013‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vignali R, Marracci S. HMGA genes and proteins in development and evolution. Int J Mol Sci. 2020;21(2):654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parisi S, Piscitelli S, Passaro F, Russo T. HMGA proteins in stemness and differentiation of embryonic and adult stem cells. Int J Mol Sci. 2020;21(1):362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang S, Mo Q, Wang X. Oncological role of HMGA2 (Review). Int J Oncol. 2019;55(4):775‐788. [DOI] [PubMed] [Google Scholar]
- 9. Sgarra R, Pegoraro S, Ros G, et al. High Mobility Group A (HMGA) proteins: molecular instigators of breast cancer onset and progression. Biochim Biophys Acta Rev Cancer. 2018;1869(2):216‐229. [DOI] [PubMed] [Google Scholar]
- 10. Zhao YC, Jiao Y, Li YQ, Fu Z, Yang ZY, He M. Elevated high mobility group A2 expression in liver cancer predicts poor patient survival. Rev Esp Enferm Dig. 2020;112(1):27‐33. [DOI] [PubMed] [Google Scholar]
- 11. Li S, Peng F, Ning Y, et al. SNHG16 as the miRNA let‐7b‐5p sponge facilitates the G2/M and epithelial‐mesenchymal transition by regulating CDC25B and HMGA2 expression in hepatocellular carcinoma. J Cell Biochem. 2020;121(3):2543‐2558. [DOI] [PubMed] [Google Scholar]
- 12. Luo YZ, Li WF, Liao H. HMGA2 induces epithelial‐to‐mesenchymal transition in human hepatocellular carcinoma cells. Oncol Lett. 2013;5(4):1353‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hengjuan LV, Liu G, Kun LI, Mingqiu LI, Zhang D. Angiogenin regulates epithelial‐mesenchymal transition of hepatocellular carcinoma through upregulation of HMGA2. Pharmazie. 2019;74(5):301‐304. [DOI] [PubMed] [Google Scholar]
- 14. Pradeepa MM, Grimes GR, Kumar Y, et al. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat Genet. 2016;48(6):681‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo P, Chen W, Li H, Li M, Li L. The histone acetylation modifications of breast cancer and their therapeutic implications. Pathol Oncol Res. 2018;24(4):807‐813. [DOI] [PubMed] [Google Scholar]
- 16. Ferguson M, Henry PA, Currie RA. Histone deacetylase inhibition is associated with transcriptional repression of the Hmga2 gene. Nucleic Acids Res. 2003;31(12):3123‐3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang P, Xu X, Wang L, Zhu B, Wang X, Xia J. The role of EGF‐EGFR signalling pathway in hepatocellular carcinoma inflammatory microenvironment. J Cell Mol Med. 2014;18(2):218‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye QH, Zhu WW, Zhang JB, et al. GOLM1 modulates EGFR/RTK cell‐surface recycling to drive hepatocellular carcinoma metastasis. Cancer Cell. 2016;30(3):444‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. 2014;49(1):9‐23. [DOI] [PubMed] [Google Scholar]
- 20. Komposch K, Sibilia M. EGFR signaling in liver diseases. Int J Mol Sci. 2015;17(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheung P, Tanner KG, Cheung WL, Sassone‐Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell. 2000;5(6):905‐915. [DOI] [PubMed] [Google Scholar]
- 22. Yang W, Xia Y, Ji H, et al. Nuclear PKM2 regulates beta‐catenin transactivation upon EGFR activation. Nature. 2011;480(7375):118‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang W, Xia Y, Hawke D, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150(4):685‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li W, Hou JZ, Niu J, et al. Akt1 inhibition promotes breast cancer metastasis through EGFR‐mediated beta‐catenin nuclear accumulation. Cell Commun Signal. 2018;16(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Srivastava S, Mohibi S, Mirza S, Band H, Band V. Epidermal growth factor receptor activation promotes ADA3 acetylation through the AKT‐p300 pathway. Cell Cycle. 2017;16(16):1515‐1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou W, Jiang D, Tian J, et al. Acetylation of H3K4, H3K9, and H3K27 mediated by p300 regulates the expression of GATA4 in cardiocytes. Genes Dis. 2019;6(3):318‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qiao Y, Wang R, Yang X, Tang K, Jing N. Dual roles of histone H3 lysine 9 acetylation in human embryonic stem cell pluripotency and neural differentiation. J Biol Chem. 2015;290(16):9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hawsawi O, Henderson V, Burton LJ, Dougan J, Nagappan P, Odero‐Marah V. High mobility group A2 (HMGA2) promotes EMT via MAPK pathway in prostate cancer. Biochem Biophys Res Commun. 2018;504(1):196‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sun J, Sun B, Sun R, et al. HMGA2 promotes vasculogenic mimicry and tumor aggressiveness by upregulating Twist1 in gastric carcinoma. Sci Rep. 2017;7(1):2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan EJ, Thuault S, Caja L, Carletti T, Heldin CH, Moustakas A. Regulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial‐to‐mesenchymal transition. J Biol Chem. 2012;287(10):7134‐7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang S, Zhang H, Yu L. HMGA2 promotes glioma invasion and poor prognosis via a long‐range chromatin interaction. Cancer Med. 2018;7(7):3226–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han X, Huang T, Han J. Long noncoding RNA VPS9D1‐AS1 augments the malignant phenotype of non‐small cell lung cancer by sponging microRNA‐532‐3p and thereby enhancing HMGA2 expression. Aging. 2020;12(1):370‐386. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33. Yang G, Xiong G, Feng M, et al. OLR1 promotes pancreatic cancer metastasis via increased c‐Myc expression and transcription of HMGA2. Mol Cancer Res. 2020;18(5):685–697. [DOI] [PubMed] [Google Scholar]
- 34. Voon DC, Wang H, Koo JK, et al. EMT‐induced stemness and tumorigenicity are fueled by the EGFR/Ras pathway. PLoS One. 2013;8(8):e70427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y, Chen F, Zhao M, et al. The long noncoding RNA HULC promotes liver cancer by increasing the expression of the HMGA2 oncogene via sequestration of the microRNA‐186. J Biol Chem. 2017;292(37):15395‐15407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang H, Li Y, Li J, et al. Long noncoding RNA LSINCT5 promotes endometrial carcinoma cell proliferation, cycle, and invasion by promoting the Wnt/beta‐catenin signaling pathway via HMGA2. Ther Adv Med Oncol. 2019;11:1758835919874649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mo J, Li B, Zhou Y, et al. LINC00473 promotes hepatocellular carcinoma progression via acting as a ceRNA for microRNA‐195 and increasing HMGA2 expression. Biomed Pharmacother. 2019;120:109403. [DOI] [PubMed] [Google Scholar]
- 38. Wu S, Ai H, Zhang K, Yun H, Xie F. Long non‐coding RNA EGOT promotes the malignant phenotypes of hepatocellular carcinoma cells and increases the expression of HMGA2 via down‐regulating miR‐33a‐5p. OncoTargets Ther. 2019;12:11623‐11635. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39. Huang W, Li J, Guo X, Zhao Y, Yuan X. miR‐663a inhibits hepatocellular carcinoma cell proliferation and invasion by targeting HMGA2. Biomed Pharmacother. 2016;81:431‐438. [DOI] [PubMed] [Google Scholar]
- 40. Wang Y, Chen F, Zhao M, et al. MiR‐107 suppresses proliferation of hepatoma cells through targeting HMGA2 mRNA 3'UTR. Biochem Biophys Res Commun. 2016;480(3):455‐460. [DOI] [PubMed] [Google Scholar]
- 41. Xu X, Zou H, Luo L, Wang X, Wang G. MicroRNA‐9 exerts antitumor effects on hepatocellular carcinoma progression by targeting HMGA2. FEBS Open Bio. 2019;9(10):1784‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bi Y, Kong P, Zhang L, et al. EP300 as an oncogene correlates with poor prognosis in esophageal squamous carcinoma. J Cancer. 2019;10(22):5413‐5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hou X, Gong R, Zhan J, et al. p300 promotes proliferation, migration, and invasion via inducing epithelial‐mesenchymal transition in non‐small cell lung cancer cells. BMC Cancer. 2018;18(1):641. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44. Ma C, Huang S, Xu L, Tian L, Yang Y, Wang J. Transcription co‐activator P300 activates Elk1‐aPKC‐iota signaling mediated epithelial‐to‐mesenchymal transition and malignancy in hepatocellular carcinoma. Oncogenesis. 2020;9(3):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Y, Sun B, Zhang Q, Dong H, Zhang J. p300 Acetylates JHDM1A to inhibit osteosarcoma carcinogenesis. Artificial Cells Nanomed Biotechnol. 2019;47(1):2891‐2899. [DOI] [PubMed] [Google Scholar]
- 46. Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci. 2013;70(21):3989‐4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage‐specific tumours. Nature. 2017;550(7674):128‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol. 2007;213(3):663‐671. [DOI] [PubMed] [Google Scholar]
- 49. Vo N, Goodman RH. CREB‐binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276(17):13505‐13508. [DOI] [PubMed] [Google Scholar]
- 50. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harbor Perspect Biol. 2016;8(4):a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kurdistani SK. Histone modifications as markers of cancer prognosis: a cellular view. Br J Cancer. 2007;97(1):1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]