

Abstract
Ion channels are polymorphic membrane proteins whose high-resolution structures offer images of individual conformations, giving us starting points for identifying the complex and transient allosteric changes that give rise to channel physiology. Here, we report live-cell imaging of voltage-dependent structural changes of voltage-gated Kv2.1 channels using peptidyl tarantula toxins labeled with an environment-sensitive fluorophore, whose spectral shifts enable identification of voltage-dependent conformation changes in the resting voltage sensing domain (VSD) of the channel. We synthesize a new environment-sensitive, far-red fluorophore, julolidine phenoxazone (JP) azide, and conjugate it to tarantula toxin GxTX to characterize Kv2.1 VSD allostery during membrane depolarization. JP has an inherent response to polarity of its immediate surroundings, offering site-specific structural insight into each channel conformation. Using voltage clamp spectroscopy to collect emission spectra as a function of membrane potential, we find that they vary with toxin labeling site, the presence of Kv2 channels, and changes in membrane potential. With a high-affinity conjugate in which the fluorophore itself interacts closely with the channel, the emission shift midpoint is 50 mV more negative than the Kv2.1 gating current midpoint. This suggests that substantial conformational changes at the toxin-channel interface are associated with early gating charge transitions, and these are not concerted with VSD motions at more depolarized potentials. These fluorescent probes enable study of conformational changes that can be correlated with electrophysiology, putting channel structures and models into a context of live-cell membranes and physiological states.
Keywords: potassium channel, fluorophore, toxin, allostery, structure, spectroscopy
INTRODUCTION
Voltage-gated potassium (Kv) channels regulate electrical activity in neurons and other electrically active cells, making them attractive targets for both venomous predators and human therapeutics.1 Like most other membrane proteins, individual Kv channels are highly polymorphic (i.e., able to adopt many functional conformations), and electrophysiological studies have long shown that changes in channel structure underlie voltage-gated K+ flux. Kv channels are tetramers of subunits each containing 6 transmembrane helices (termed S1-S6) in which the voltage-sensing domain (S1-S4, VSD) S4 helix contains multiple cationic sidechains (typically Arg) that move in response to changes in the membrane electrical field and give rise to a gating current.2–4 The VSD is coupled to the channel pore (S5-S6), and voltage-dependent movement of the S4 helix leads to its opening to allow K+ flux. Attempts to understand dynamic changes in Kv channel structures have spanned decades, and have included electrophysiology,5–7 site-directed mutagenesis,8, 9 sidechain reactivity studies,10–12 voltage-clamp fluorometry,10, 13 computational modeling,10, 14 and most recently, determination of full channel structures.4, 15–17
Major advances in membrane protein X-ray crystallography and direct detector cryo-EM have offered a wealth of structural detail about Kv channels. These structures are determined in the absence of cell membranes or intact lipid bilayers that can hold a membrane potential, but studies of K+ channels gated by chemical modulators have shown multiple conformational states of those channels, and reveal a complex array of related structures even within the closed state.17 Because these structures are determined outside of live-cell membranes under specific membrane potentials, the relationships between these static structures and channel allostery remains poorly understood. Live-cell structural techniques that can be correlated with voltage-dependent activity are needed for a full understanding of Kv channel allostery under physiological conditions.
Venomous species commonly target ion channels with peptidyl gating-modifying toxins (GMTs) that preferentially bind to specific channel conformations, trapping them in particular physiological states.18 This conformational specificity has made GMTs critical tools for understanding channel allostery and physiology. GxTX-1E, a lipophilic cystine-knot peptide from the Chinese hissing bronze wolf tarantula C. guangxiensis,19–21 targets the Kv2.1 closed state VSD with 5400-fold greater affinity than its open state,22 shifting channel opening to more depolarized potentials and effectively closing the channel without directly occluding its pore (Figure 1). Synthetic analogs of GxTX have been functionalized with optical probes and preferentially bind to closed, wild-type Kv2.1 channels in cells and tissue.23, 24 With standard fluorescent reporters, labeled GxTX can be used to image endogenous Kv2.1 gating, but offers no molecular insight into the toxin-VSD structure or channel allostery.
Figure 1.
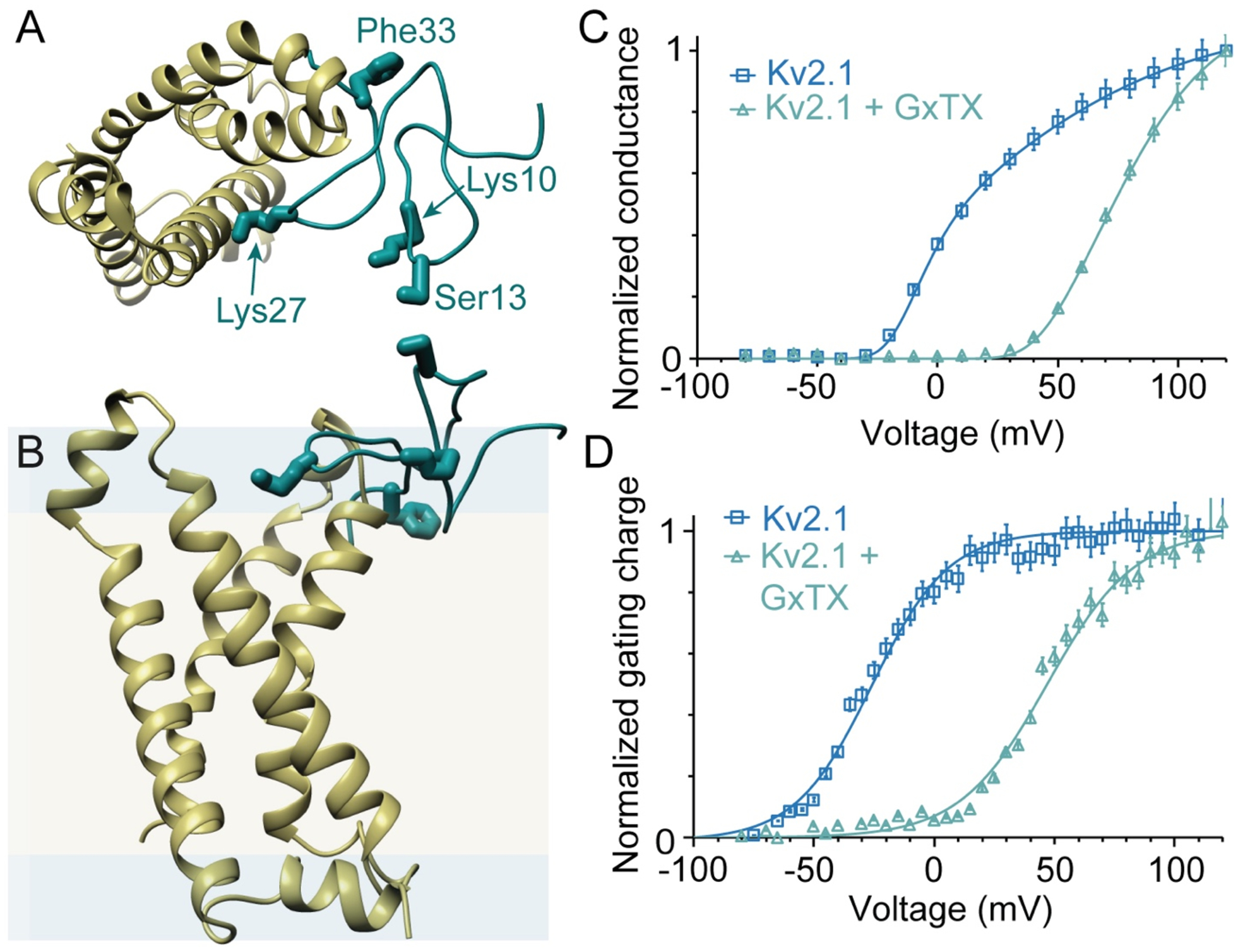
Structural models and activity of GxTX with Kv channels. (A, B) Top and side views of a model of GxTX (green) bound to a Kv2.1 voltage sensor (gold) in its active state. The GxTx-Kv2.1 VSD model was generated from a Rosetta-based model of the Kv2.1 VSD20 and X-ray structure of the ProTx-II-hNav1.7-NavAb complex.32 The 4 positions of propargylglycine (Pra) substitution and fluorophore conjugation are highlighted. Membrane is shown as hydrophobic interior (yellow) and polar interface (blue), with dimensions calculated using the RosettaMembrane energy function.33 (C) Normalized conductance, G/Gmax, of Kv2.1 with and without 100 nM GxTX, in Kv2.1-transfected CHO cells. (D) Normalized gating charge, Qoff, with and without 1 μM GxTX in Kv2.1-transfected CHO cells. Gating currents measured from Qoff at −140 mV and reported as mean ± standard error of the mean (SEM). Solid lines in (C) and (D) are two-state Boltzmann functions. Data replotted from ref22.
Fluorescence has long been used to characterize protein dynamics, including voltage-clamp fluorometry (VCF) studies of Kv channels using cysteine-tethered probes that are (de)quenched with changing protein conformation.10, 13, 25, 26 Environment-sensitive fluorophores are distinct from those typically used in VCF in that they have asymmetric structures designed to respond to the polarity of their immediate surroundings with large shifts in excitation and emission peaks,27–29 and are particularly well-suited for reporting protein allostery.30, 31 Here, we introduce a new environment sensitive far-red fluorophore, julolidine phenoxazone (JP) and conjugate it to GxTX to characterize Kv2.1 voltage sensor allostery during membrane depolarization. JP has an inherent response to thepolarity of its immediate surroundings, offering site-specific structural insight into each channel conformation. We use voltage clamp spectroscopy (VCS) to collect full emission spectra of JP-GxTX conjugates as a function of membrane potential and find that they vary with labeling site on the toxin, the presence of Kv2 channels, and in response to Kv2.1 voltage activation. With Lys27Pra(JP) GxTX, a high-affinity conjugate in which the JP interacts directly with the VSD, emission shifts occur at voltages almost 50 mV more negative than measured for Kv2.1 channel opening or gating currents. This suggests that conformational changes around Lys27Pra(JP) GxTX are associated with early gating charge transitions, and these are not concerted with later VSD motions.
RESULTS
To measure voltage-dependent Kv structural changes in live cell membranes, we synthesized conjugates of the cystine knot tarantula toxin GxTX-1E (Figure 1; called “GxTX” throughout) with a novel far red fluorophore whose emission is responsive to changes in the polarity of its environment. GxTX was synthesized by solid-phase peptide synthesis and oxidatively refolded (see Methods).23 Single substitutions with the alkynyl amino acid propargylglycine (Pra) were introduced at Lys10, Ser13, Lys27, or Phe33 (Figure 1A) for fluorophore conjugation by Cu-catalyzed click chemistry.34
To conjugate to these alkynyl toxins, azide-functionalized fluorophores were synthesized (Figure 2) based on an aminophenoxazone (AP) ring system, which has shown large polarity-based spectral shifts both in vitro and when conjugated to membrane proteins.31 To reduce rotation of the exocyclic nitrogen, which reduces fluorophore brightness and stability, the dimethylamine of AP was replaced with the julolidine ring system found in Texas Red.35 Azide-functionalized julolidine phenoxazone (JP-N3, Figures 2A and S1) was synthesized by Lewis Acid-catalyzed condensation of nitroso-julolidine and resorcinol precursors and azidification of its hydroxmethyl substituent. Characterization of JP-N3 in simple solvents show a strong solvatochromic emission shift based on solvent polarity, from 609 nm in hydrocarbon to 640 nm in MeOH (Figure 2B), similar to the behavior of AP.31 Comparisons of AP to JP conjugates to GxTX by epifluoroescence microscopy of Kv2.1-expressing CHO cells show the JP ring system increases brightness by 1.6-fold and photostability by 2-fold (Figure 2C) under standard microscopy conditions. The large Stokes shifts of JP (Figure 2B) enable simultaneous excitation (typically, λexc = 543 nm) in different environments and acquisition of full emission spectra, both of which facilitate the real-time characterization of protein conformational changes described below.
Figure 2.
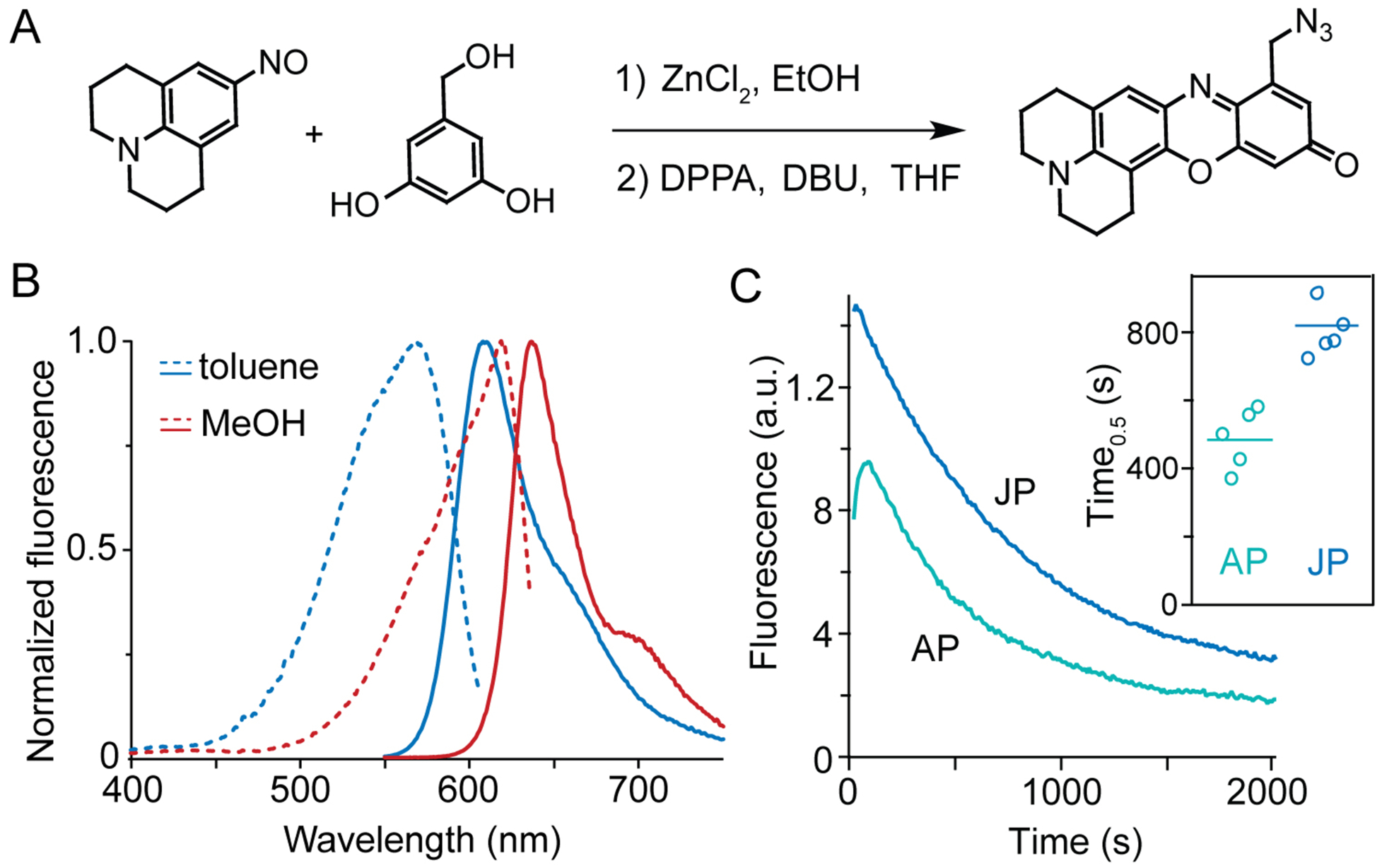
Synthesis and fluorescence of a far-red environment sensor. (A) Synthesis of JP-N3 by condensation of nitroso julolidine and hydroxymethyl resorcinol. DPPA: Diphenyl phosphorazidate; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. (B) JP environment sensitivity. Excitation (dashed) and emission (solid lines) spectra of JP-N3 in nonpolar and polar solvents. (C) Enhanced brightness and stability (inset) conferred by julolidine rings. Emission measured by epifluorescence microscopy with 550-nm excitation of AP (ref31) or JP adducts to Lys27Pra on Kv2.1-expressing CHO cells.
To assess whether JP-GxTX conjugates are able to selectively label endogenous Kv2 channels, we stained dissociated rat hippocampal neurons with Lys27Pra(JP) GxTX (Figure 3A). JP fluorescence is observed in clusters in the plasma membrane surrounding neuronal cell bodies and proximal dendrites, consistent with known patterns of Kv2.1 localization.36 In Kv2.1-expressing Chinese hamster ovary (CHO) cells co-plated with Kv2-free CHO cells, Kv2.1-specific JP fluorescence is observed with JP conjugated at Lys10, Ser13, and Lys27, with little apparent staining of Kv2-free cells (Figures 3B and S2). JP emission appears predominantly as clusters at the glass-adhered basal surface, which is a hallmark of Kv2.1 channel localization in neurons and other mammalian cells grown on glass.37, 38 JP conjugation at Phe33 leads to weak JP emission from both Kv2+ and Kv2- cell surfaces, suggesting a loss of specific Kv2 binding but not plasma membrane localization. Spectra of resting cells stained with JP-GxTX conjugates were fit using split pseudo-Voigt functions, which are commonly used to fit asymmetric peak shapes.39, 40 Full JP emission spectra from resting CHO cells (Figure 3C) show that JP conjugates at Lys10 and Ser13 have emission peaks of ~650 nm, consistent with JP-GxTX conjugates in buffer (Figure 3D) and aqueous localization of AP fluorophores on membrane proteins.31
Figure 3.
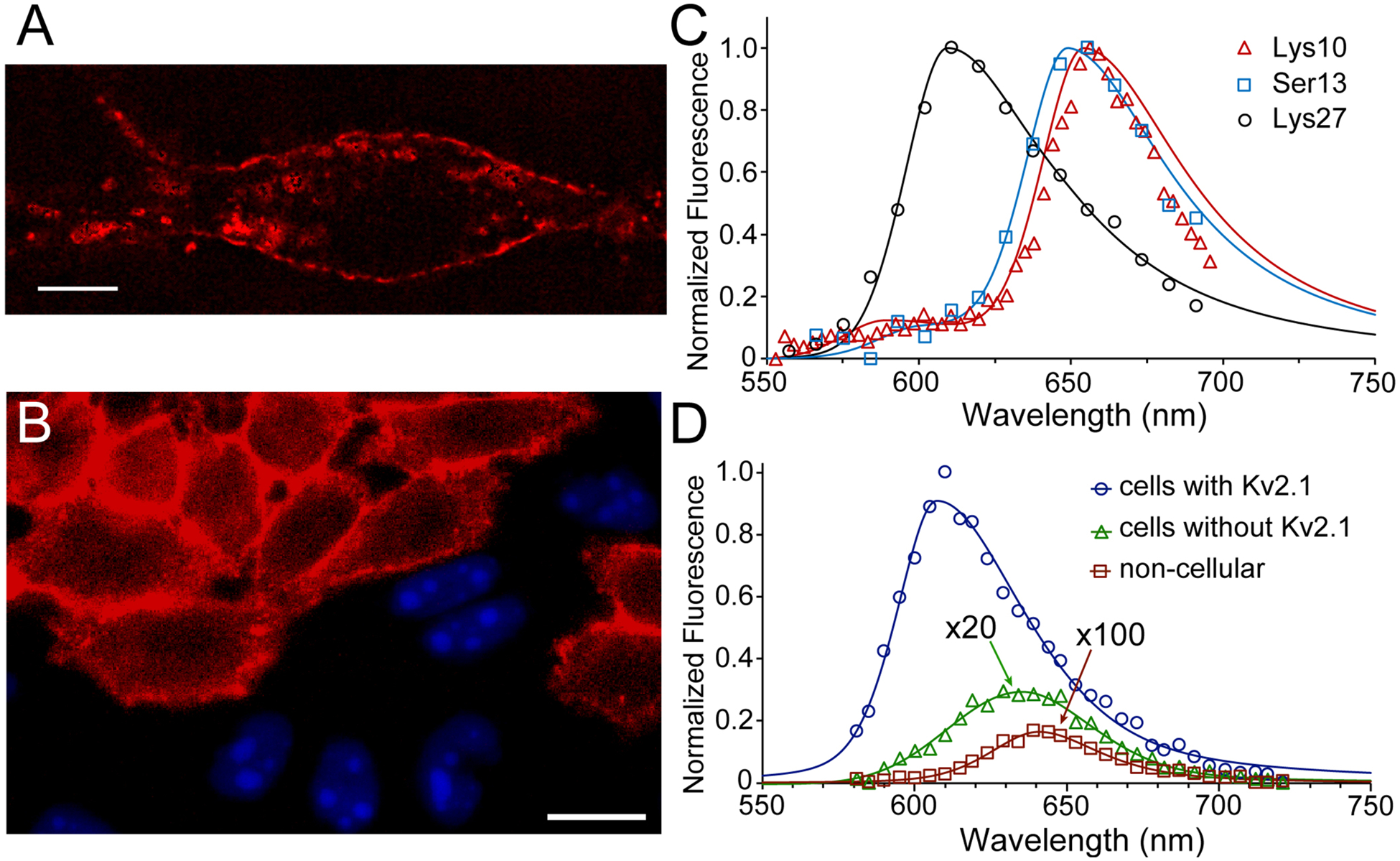
Fluorescence of JP-GxTX conjugates on live cells. (A) Compressed z-stack confocal image of a live rat hippocampal neuron stained with 100 nM Lys27Pra(JP) GxTX. The cell is excited at 561 nm and its emission isolated around 625 nm. Scalebar is 10 μm. (B) Specificity of JP-GxTX conjugates for Kv2-expressing cells. Confocal image of co-plated CHO cells with or without Kv2.1, stained with 100 nM Lys27Pra(JP) GxTX (red membranes). Only cells without Kv2 channels express nuclear BFP. (C) Spectral imaging of JP-GxTX conjugates on Kv2.1 CHO cells. (D) Emission spectra of Lys27Pra(JP) GxTX imaged from CHO cells with (blue line) or without Kv2.1 (green line, magnified 20x), or from extracellular regions (red line, magnified 100x). Spectra are fit with 2-component pseudo-split Voigt functions, with shape values and root-mean-squared deviations (R2) listed in Table S1.
Relative to these aqueous JP conjugates, the emission peak from Lys27Pra(JP) GxTX is blueshifted by 40 nm, similar to non-polar environments like hydrocarbon solvents (Figure 2B). In cell membranes without Kv2 channels, Lys27Pra(JP) GxTX emission is ~50-fold weaker and significantly red-shifted compared to membranes with Kv2.1 (Figure 3D). We also observed that Lys27Pra(JP) GxTX washes out far more slowly than unlabeled GxTX from resting Kv2.1-expressing cells. To quantify interactions of Lys27Pra(JP) GxTX with Kv2.1, we calculated microscopic binding and dissociation rates (Table 1). At a holding potential of −100 mV, the channels are in resting states, and the affinity of this JP conjugate is 3-fold higher than that of unlabeled GxTX. A Rosetta-generated homology model of the Kv2.1 VSD in its activated state in complex with GxTX20 (Figure 1) shows the Lys27 sidechain residing in the polar region of the membrane adjacent to S4, sitting within roughly 8 Å of Arg296 and Arg299 (termed R2 and R3, based on homology to Shaker Kv channels).
Table 1.
Association/dissociation rates and affinities of GxTX and Lys27Pra(JP) GxTX for Kv2.1 channels.
| GxTX | Lys27Pra(JP) GxTX | |||
|---|---|---|---|---|
| −100 mV | 0 mV | −100 mV | 0 mV | |
| kon (s−1 nM−1) | 3.30 ± 1.10 × 10−4 | 2.5 ± 1.2 × 10−5 | 2.16 ± 0.14 × 10−4 | 1.64 ± 0.90 × 10−5 |
| koff (s−1) | 0.84 ± 0.18 × 10−3 | 38 ± 9 × 10−3 | 1.80 ± 0.65 × 10−4 | 8.13 ± 1.39 × 10−3 |
| Kd (nM) | 2.55 ± 1.01 | 1520 ± 810 | 0.83 ± 0.30 | 496 ± 285 |
To determine the voltage-dependent changes in the GxTX-VSD structure, we measured full emission spectra from Lys27Pra(JP) on Kv2.1-expressing cells at membrane potentials from −100 to +100 mV (Figures 4 and S3). Unlike GxTX conjugates with lipophobic fluorophores,23 we observed no substantial dissociation of Lys27Pra(JP) from cell membranes in response to the 1–5 second depolarization steps for spectral measurement, consistent with its slow koff rate (Table 1). Raw emission data were fitted with split pseudo-Voigt functions, with 2-component fits more accurate (R2 ≥ 0.98; see Table S1 and Methods) than single component fits or other lineshape functions. At all voltages, fit functions consistently show non-polar (~610 nm) and polar (~640 nm) peaks, with gradual increases in the polar component as the VSD moves from its resting to activated state (Table S1). This suggests multiple JP-GxTX-VSD species are present, and that depolarization changes the dominant JP species from a lipid/protein environment to a more polar one (although less polar than the fully aqueous positions in Figure 3). In contrast to Lys27Pra(JP), emission spectra of JP-GxTX conjugates substituted at Lys10 or Ser13 show no significant voltage dependence (Table S1 and Figure S4). Control cells not expressing Kv2 channels showed weaker Lys27Pra(JP) emissions and that are also independent of voltage. These emissions are not satisfactorily be fit by 2-component split pseudo-Voigt functions, suggesting nonspecific interactions with cell membranes.20 We emphasize that the 2-component fits are optimal lineshapes for the data but do not necessarily mean there are exactly 2 species, or that the weightings of the fit components reflect percentages of particular conformations.
Figure 4.
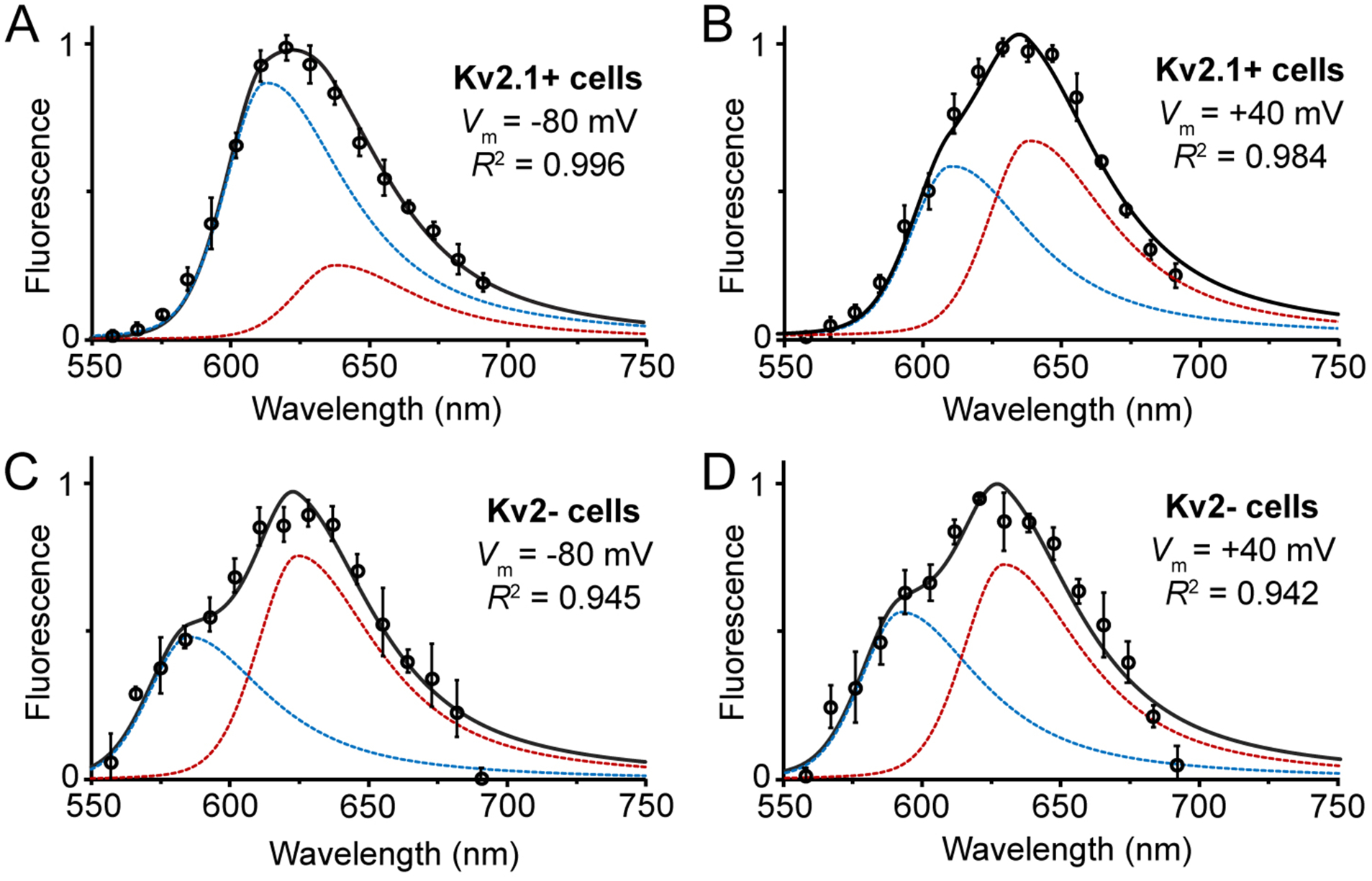
GxTX-JP conjugate fluorescence from cell membranes depends on Kv2.1 channels and membrane potential. Normalized fluorescence of Lys27Pra(JP) GxTX on CHO cells (A) expressing Kv2.1 channels at −80 mV, (B) expressing Kv2.1 channels at +40 mV, (c) without Kv2 channels at −80 mV, and (b) without Kv2 channels at +40 mV. All cells were excited at 543 nm, and component peaks shorter than 600 nm in (C) and (D) likely represent cellular autofluorescence rather than JP emission. Data points are mean ± SEM of normalized emission from 4 cells. All spectra are shown with 2-component split pseudo-Voigt fits (black) with individual components (blue, red). Fit parameters and R2 values are listed Table S1.
To better understand the conformational changes of the GxTX-VSD complex, we compared voltage-dependent changes in Lys27Pra(JP) GxTX fluorescence with Kv2.1 gating currents (Figure 5). Mean emission spectra calculated from integrations of 2-component fittings show a redshift fit well by a two-state Boltzmann function with a midpoint (F1/2) of −15 mV, and that plateaus above +40 mV. This 2-state Boltzmann function may hide more subtle transitions within the data but is better constrained than more complex functions. By comparison, gating currents (Qoff) of the Kv2.1-Lys27Pra(JP) GxTX complex, which reflect the activation of S4 and movement of its 3 Arg through the membrane electric field, have a midpoint of +36 mV, similar to those of Kv2.1 with unlabeled GxTX (Figure 1D). At the F1/2 of −15 mV, Qoff is at ~5% of its maximal value. This large discrepancy between voltage-dependent fluorescence shifts and gating currents suggests a complex VSD allostery.
Figure 5.
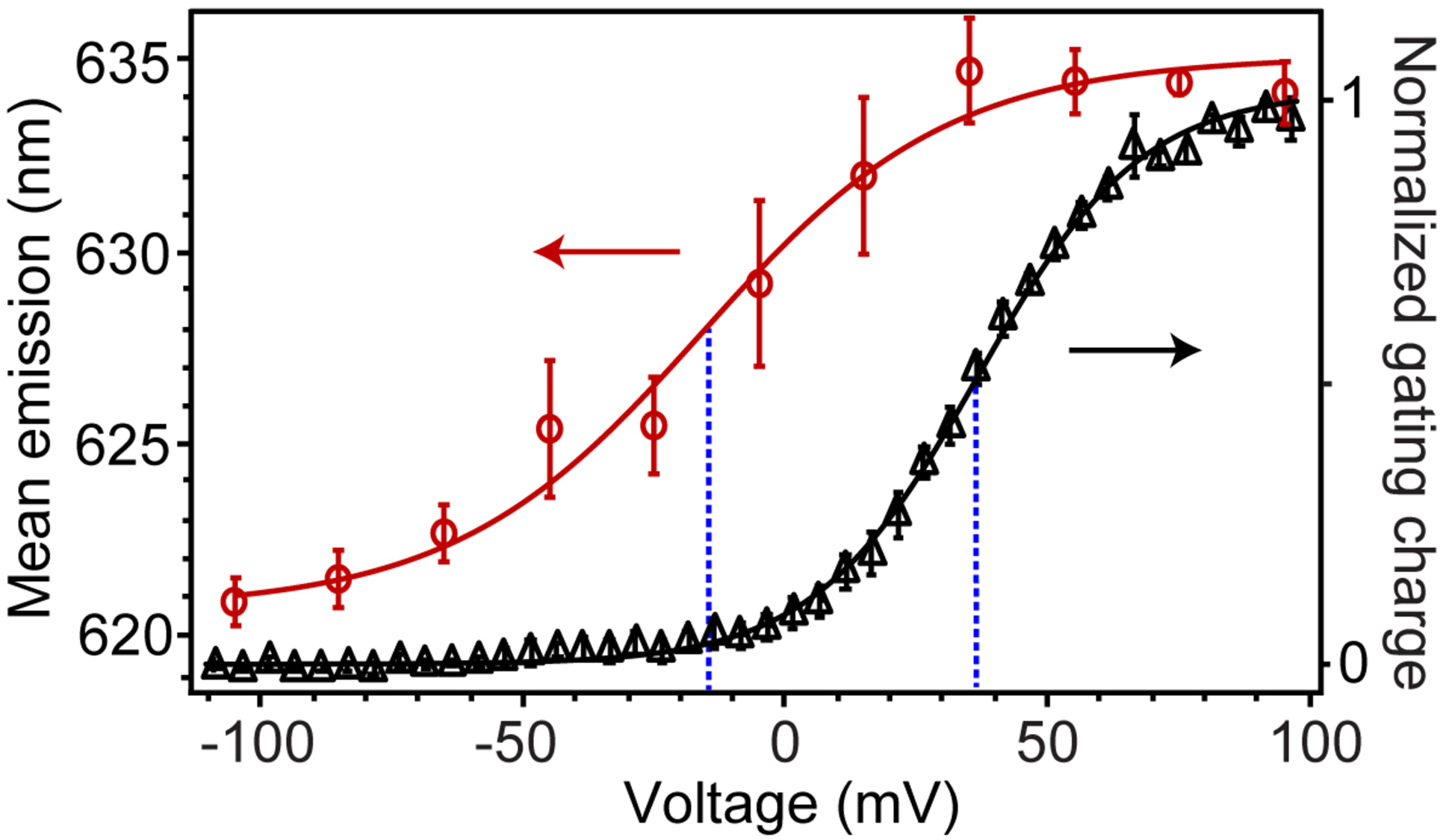
Fluorescence shifts and gating currents of Kv2.1 with Lys27Pra(JP) GxTX, recorded from Kv2.1-expressing CHO cells. Mean fluorescence emission of Lys27Pra(JP) as a function of membrane potential (red circles), calculated from integrations of 2-component split pseudo-Voigt fittings of spectral data. Normalized Kv2.1 gating charge (black triangles), with 100 nM Lys27Pra(JP) GxTX, shown as Q/Qmax and measured as Qoff from −140 mV. For both plots, data are mean ± SEM and solid lines are two-state Boltzmann functions. Blue dashed lines are mid-point voltages of fluorescence shift (−15 mV) and Qoff (+37 mV).
DISCUSSION
Since the original Hodgkin and Huxley description of voltage-gated K+ channel gating in squid giant axons, channels have been proposed to adopt multiple conformations before opening in response to depolarizing voltage stimuli.41 Over half a century later, advances in membrane protein X-ray crystallography and single particle cryo-EM have enabled determination of full structures of voltage-gated K+ channels15, 16, 42, 43 and analogous voltage-gated channels,32, 44, 45 which have offered insight into the allostery of voltage gating. While structures of channels that are gated or modulated by ligands can be determined in apo and ligand-bound states, channels that are gated primarily by voltage present an added challenge of determining the corresponding physiological state of a particular structure in the absence of functional cell membranes with clearly defined potentials.46, 47 For example, in cryo-EM analysis of Nav1.7 bound to the GMT ProTX2 (Figure S5), the “deactivated” structure may show a partially or fully down S4,32 but these cannot be distinguished on the basis of structure alone. For these and Kv structures, making sense of conformational heterogeneity and dynamics requires novel techniques able to characterize channel allostery with correlated electrophysiological recording in live cell membranes.
The observed voltage-dependent solvatochromic shifts (Figures 4 and S3) demonstrate that JP-conjugated GxTX detects conformational changes in the Kv2.1-GxTX complex. When complexed with the VSD, the JP of GxTX conjugate Lys27Pra(JP) moves from hydrocarbon-type surroundings in the resting state to an aqueous environment in the activated state. This is specific to JP substituted at Lys27 and is the only of the 4 conjugates whose fluorescence changes with depolarization (Figures S3 and S4). This voltage-dependent spectral shift also requires the presence of Kv2 channels in the membrane (Figure 4: compare panels A-B with C-D). These experiments show that JP-GxTX fluorescence is specific, depending on the position of JP conjugation, the presence of Kv2 channels, and the membrane potential.
The lack of high-resolution resting state Kv2.1 structures makes pinpointing specific interactions between the JP at Lys27 and the VSD more speculative. Homologous Nav1.7-ProTx2 cryoEM structures32 provide some guidance, showing the toxin-VSD interface undergoes only subtle structural changes moving between deactivated and activated states (Figure S5). A homology model of active Kv2.1-GxTX (Figure 1) points Lys27 into the center of 4-helix bundle between S2 and S3, with the ammonium group residing in the polar part of the membrane. The added size of the Pra(JP) (Figure S1) makes placement of the JP at this position uncertain, although emission peaks place the JP in an aqueous environment when Lys27Pra(JP) is bound to the active VSD, and in hydrocarbon when bound to the resting VSD. This suggests that VSD activation moves the JP at Lys 27 deep into the membrane to become surrounded by hydrophobic VSD sidechains, lipid tails, or both. More specific interactions between Kv2.1 and JP-GxTX conjugates will need to be examined with precise modeling or structural studies.
Gating currents are a direct measure of the S4 Arg moving through the membrane electric field, so the 50 mV gap between gating current and Lys27Pra(JP) GxTX fluorescence midpoints (Figure 5) is at first puzzling. Careful studies of Kv gating currents have shown distinct early and late VSD conformational changes as voltages are stepped to more positive voltages.5, 7, 48 Specific VSD conformations may be accentuated with point mutations that segregate individual gating charge movements,2, 7, 11, 13 although these mutations do not establish the structural changes that give rise to altered physiology. While high-resolution resting state Kv channel structures have not been reported, one structural hypothesis of VSD allostery is that negatively charged Kv residues residing in the center of the membrane electric field stabilize distinct S4 conformations in the transition from resting to active states.4 Models of the Kv2 VSD resting (i.e., closed) state suggest the S4 Arg are shifted toward the cell interior by 2–4 helical turns compared to the active state.4, 10, 14, 32, 49
We find that the solvatochromic shift of Lys27Pra(JP) GxTX emission occurs at voltages far more negative than the majority of Kv2.1 gating currents (Figure 5) or subsequent channel opening, suggesting that the conformational changes that alter the chemical environment of the JP are early VSD motions in the closed channel. The mean emission wavelength plateaus by +40 mV, giving no indication of a component corresponding to the +37 mV midpoint of Qoff and suggesting the conformational changes at the GxTX-VSD interface are distinct from later S4 motions. Studies of gating charge movement in GxTX-Kv2.1 complexes have shown that GxTX acts by inhibiting early, fast gating charge transitions in a voltage range similar to the fluorescence shifts measured here.22 It is also possible that some allosteric changes are too subtle to be seen in Qoff measurements, or that the emission shifts occur more slowly than the fast charge movements that give rise to gating currents, which may allow the VSD to relax into alternate conformations that occur after extended activation.13 Each of these possibilities suggest that the earliest VSD-GxTX motions are measurable and are not concerted with the S4 motions at more depolarized potentials.
Attachment of an environment-sensitive probe to a GMT rather than to a cysteine substitution in the channel itself offers a method to place the probe in closer contact with the channel, as all common conjugation chemistries for modifying cysteine sidechains invariably introduce distance and flexibility between channel and probe.31 For other channels, an impressive natural library of channel- and state-specific peptidyl neurotoxins18 may similarly be conjugated to environment-sensitive probes for live-cell analysis of protein allostery correlated with physiology. While the addition of julolidine rings to the red fluorescent aminophenoxazone ring system increases its brightness and photostability, (Figure 2), newer ring systems50, 51 may be useful for further optical improvements or near infrared emission.
In addition to studying Kv allostery, voltage clamp spectroscopy offers insight into the mechanism of action of GxTX and other GMTs. GMTs have been shown to require intact lipid bilayers to bind with high affinity to VSDs,47 but their precise mechanism of action has remained the subject of some debate.18, 20, 22, 32, 52 For JP-GxTX conjugates, we observe that cell membranes with Kv2.1 channels have ~50-fold higher JP emission than those without Kv2 channels in co-cultures of both cell types, and the surrounding buffer has much lower (but still measurable) concentrations of JP-GxTX (Figure 3D). Each of these shows a distinct JP emission spectrum, a result of the 3 different environments surrounding the fluorophore, and the emission spectra of cells with Kv2.1 show no appreciable contribution from unbound toxin. For JP-GxTX mutants in which Kv2 affinity has been substantially weakened, we observe similar levels of labeling in Kv2.1-expressing and Kv2-free cells (Figure S1, Phe33 mutant).
This shows that the toxins partition non-specifically to cell membranes but disproportionate to cells that contain high-affinity binding sites, suggesting a rapid equilibrium in and out of membranes that is biased by channel binding.53 This leads to a model in which rapid entry and exit from the around channel binding sites while allowing the toxin to escape from cells or subcellular regions without Kv2 channels.
CONCLUSION
We have synthesized a novel far-red environment-sensitive fluorophore, conjugated it to the gating-modifying toxin GxTX, and used the conjugate to study voltage-dependent conformational changes of Kv2.1 channels in the cell membrane. Fluorescence is both position and state-specific for GxTX conjugates bound to Kv2.1 channels, enabling us to use fluorescence shifts to characterize channel allostery. For a JP-GxTX conjugate in which the JP interacts directly with the channel, emission shifts occur at voltages almost 50 mV more negative than measured for Kv2.1 channel opening or gating currents, suggesting substantial conformational changes at the toxin-channel interface associated with early gating charge transitions distinct from VSD motions at more depolarized potentials. These probes and voltage clamp spectroscopy place channel structures and models into the context of cell membranes and physiological states for detailed study of channel allostery.
METHODS
Safety Concerns.
Tarantula toxin GxTX-1E is a component of a mixture of neurotoxins in P. guangxiensis venom, but on its own shows no toxicity up to 10 mg/kg injected into mice. Some reagents for solid-phase peptide synthesis (e.g., DIC, piperidines, TFA) are hazardous and should be treated with caution using proper personal protective equipment.
Fluorophore synthesis.
JP-N3, 10-(azidomethyl)-julolidino-phenoxazinone. p-Nitroso-julolidine (250 mg, 1.2 mmol, prepared by nitrosylation of julolidine-HCl with NaNO2 and HCl) was dissolved in 10 mL of dry iPrOH with of 5-hydroxylmethylresorcinol (Aldrich, 191 mg, 1.4 mmol). ZnCl2 (84 mg, 0.62 mmol) was added and the reaction heated to 85° C for 3h. It was cooled and concentrated to a solid, which was rinsed with 2 × 20 mL of ether, and then 2 × 20 mL of H2O. It was purified on a Biotage silica column around 10:1 CH2Cl2/MeOH to 255 mg (64% yield). Of this, 60 mg (0.19 mmol) was dissolved in 5 mL of dry THF under N2. Diphenyl phosphorazidate (DPPA, 60 μL, 0.28 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 31 μL, 0.21 mmol) were added via syringe and the reaction stirred overnight. Purification by C18 HPLC with a linear 30–50% CH3CN gradient over 40 min gave major blue peak at 32 min (42% CH3CN), which was lyophilized to 32 mg (50%). For 1H NMR (acetone-d6, 500 MHz), shifts are: δ 7.43 (s, 1 H), 7.07 (s, 1 H), 6.94 (s, 1 H), 4.81 (s, 2 H), and 3.84 (m, 4 H), 3.03 (m, 2H), 2.98 (m, 2 H), 2.16 (m, 4H). MS for C19H18N5O2 (MH)+ calculated: 348.4; found: 348.1.
Fluorophore characterization.
Emission and excitation spectra were recorded from 200 nM solutions of HPLC-purified JP-N3 with a Fluoromax-4 fluorometer (Horiba Jobin Yvon). Samples were prepared in anhydrous solvents (or water) from a 200x acetone stock solution Spectra were corrected for variations in detector efficiency with files provided by the manufacturer. Excitation spectra were collected as S/R, where S is the emission signal and R the lamp intensity reference.
Toxin synthesis and refolding.
All guangxitoxin-1E mutants were synthesized with Met35 replaced by its isostere norleucine (Nle) to avoid problems associated with Met oxidation; Met35Nle guangxitoxin-1E is used in all experiments in this study and is referred to as “GxTX” throughout. Linear GxTX peptides were synthesized on an AAPTEC Apex 396 peptide synthesizer using standard Fmoc/DIC/HOBt methodology, with single propargylglycine (Pra, Sigma-Aldrich) substituents. Peptides were synthesized on 0.1–0.3 mmol of Fmoc-ProNovaSyn TGT resin (Novabiochem) in N-methylpyrrolidone, using 0.4 M Fmoc-protected amino acids, 0.44 M N-hydroxybenzotriazole, and 10% (v/v) N,N′-diisopropylcarbodiimide; 20% 4-methylpiperidine in dimethylformamide was used for Fmoc deprotections. Linear peptides were cleaved from resin and deprotected with trifluoracetic acid (TFA):triisopropylsilane:1,2-ethanedithiol:thioanisole:H2O (85:2.5:2.5:5:5 by volume) for ~3 h at room temperature, with removal of deprotecting groups monitored by MALDI-TOF mass spectrometry (Applied Biosystems). Cleaved peptide was filtered from resin, precipitated with cold diethyl ether, washed once with ether, and residual ether evaporated under a gentle stream of N2. The peptide pellet was then dissolved in H2O with 50% CH3CN or 50% AcOH, injected onto a preparatory C18 column (Vydac), and eluted with a linear gradient of CH3CN with 0.1% TFA. Single Pra GxTX mutants eluted at ~35% CH3CN and were lyophilized, redissolved in 50% CH3CN, and diluted to 50 μM, in oxidation buffer at 4° C (1 M guanidinium HCl, 0.1 M NH4OAc, 2.5 mM glutathione, 0.25 mM oxidized glutathione, and 1% CH3CN, adjusted to pH 8.0 with NH4OH).23 Oxidation was monitored by mass spectrometry and upon completion (~3 d), the solution was filtered, TFA was added to 0.1% and the product purified by HPLC as above. Refolded GxTX mutants were lyophilized and stored as powders at −20° C.
Toxin-fluorophore conjugation.
GxTX mutants with Pra substitutions at Lys10, Ser13, Lys27 or Phe33 were conjugated to JP azide using Cu(I)-catalyzed azide-alkyne click cycloaddition in the presence of BTTAA (bis-[(tertbutyltriazoyl)methyl]-[(2-carboxymethyltriazoyl)-methyl]-amine).54 Reagents were added sequentially to a polypropylene tube: 24 μL of a premixed aqueous solution containing 2.5 mM CuSO4 (60 nmol) with 15 mM BTTAA (360 nmol); followed by 30 μL of 1 mM JP-N3 (30 nmol) in DMSO, 42 μL of DMSO, and 8 μL of 1.9 mM GxTX (15 nmol); 10 μL of 150 mM freshly mixed 10:1 sodium ascorbate:ascorbic acid (1.5 μmol). The reaction mixture was briefly vortexed after each addition. The reaction was shaken in the dark at 25 °C for 4 h before quenching with 10 μL of 50 mM EDTA, pH 5, and purified by C18 HPLC as described above. GxTX-JP fractions were lyophilized and stored as powders at −20° C. GxTX-JP conjugates were resuspended into stock solutions containing arginine as an excipient to improve GxTX-JP solubility (1 M Arg-HCl, 50 mM Glu, pH 5.0 with NaOH), stored at −80 °C and diluted to working concentrations on the day of experiment.
Structural modeling of GxTx – Kv2.1 VSD complex.
We used a Rosetta-based model of Kv2.1 VSD20 and an X-ray structure of the ProTx-II-hNav1.7-NavAb chimera32 to generate GxTx-Kv2.1 VSD models in an activated state. We first superimposed a Rosetta model of Kv2.1 VSD20 onto the X-ray structure of hNav1.7-NavAb VSD chimera32 using the S3b segment of Kv2.1 VSD and the S3b segment of the hNav1.7-NavAb VSD chimera. We then superimposed an NMR structure of GxTX55 onto an X-ray structure of ProTx-II in the ProTx-II-hNav1.7-NavAb chimera complex.32 We used the RosettaMembrane energy function18, 33 to calculate membrane dimensions and embed the GxTx-Kv2.1 VSD complex in an implicit membrane environment (Figure 1B).
Cell culture.
All CHO-K1 cell lines were maintained in polystyrene dishes (BioLite, Thermo) at 37 °C in a 5% CO2 atmosphere in Ham’s F-12 media (Corning) containing 10% v/v FBS (Gibco), and penicillin–streptomycin (Sigma). A CHO-K1 cell line stably transfected with rat Kv2.1 (Kv2.1-CHO cells)56 was cultured with 1 μg/mL blasticidin and 25 μg/mL zeocin to retain stably transfected vectors. To induce expression of Kv2.1, minocycline (Enzo Life Sciences) from a stock of 2 mg/ml in 70% EtOH was added to cell culture media to a final concentration 1 μg/mL. Time of incubation with minocycline was chosen to induce a desirable amount of channel expression: 1.5 – 2 h for K+ conductance experiments, ~48 h for imaging and voltage clamp spectroscopy experiments. The CHO-K1 cell line stably transfected with BFP (BFP-CHO cells)23 expresses the blue fluorescent protein variant EBFP2 with a nuclear localization sequence.57 BFP-CHO cells were maintained in media containing 100 μg/mL of G418.
Live cell imaging.
For CHO cell imaging without patch-clamp, Kv2.1-CHO and BFP-CHO cells were co-plated in chambered cover glass (Nunc) and imaged in external neuronal solution (NE) (3.5 mM KCl, 155 mM NaCl, 1.5 CaCl2, 1 MgCl2, 10 mM HEPES, pH adjusted to 7.4 using NaOH, measured osmolarity 316 mOsm using a VAPRO Osmometer). When noted, solution was supplemented with either 10 mM glucose (NEG) or 10 mM glucose and 1% BSA (NEGB) (bovine serum albumin fraction V, Roche). BSA was included to prevent GxTX depletion by non-specific adsorption to chamber surfaces. For photostability measurements, cells were incubated in 100 nM of JP-GxTX conjugate for 5 min and then rinsed with NE. Epifluorescent images were obtained using an inverted Zeiss Axioobserver microscope with a 1.4 NA 40x oil objective (Plan-Apochromat) using low-fluorescence immersion oil (Immersol, Zeiss). AP or JP conjugates were excited at 530 nm with a 605/70 nm emission filter (Zeiss), exposure times were set to 70 msec, and images acquired with an EMCCD camera (Photometrics QuantEM 512SC). Emission was measured from manually selected regions of interest (ROIs), with background fluorescence measured from Kv2- cells with BFP fluorescence. To measure emission spectra, cells were incubated in 100 nM GxTX-JP conjugate, and confocal images were acquired on Zeiss 710 or Zeiss 880 laser scanning confocal microscopes excited at 543 nm, 561 nm, or 594 nm. Full spectra were collected with 32 PMTs, and imaged through a 63× 1.4NA oil objective (Plan-Apochromat) using low-fluorescence immersion oil (Immersol, Zeiss). Emission spectra were collected from 571 – 721 nm with 3.2- or 9.7-nm bandwidth PMT intervals. Images were acquired with ZEN Black software and exported to ImageJ to extract spectra corresponding to different regions of interest, including cell membranes and background. These spectra were then exported for further analysis including curve fitting, as described below.
Hippocampal neuron imaging.
All animal procedures were approved by the University of California at Davis Animal Care and Use Committee following NIH guidelines. Dissociated hippocampal neurons were prepared from embryonic day 18 rats of both sexes (Sprague-Dawley, Charles River Laboratories) as described previously58. Surface localization of Kv2.1 protein in similar cultures has been found to be maximal after two weeks in vitro36, and neurons were imaged after 17 days. Neurons were rinsed with NEG solution and imaged during incubation at room temperature in NEGB with 100 nM Lys27Pra(JP) GxTX (from 73 μM stock) and 200 nM tetrodotoxin (diluted from a 10 mM stock in 50 mM HEPES free acid, stored at −20°C). Spectral images were acquired on a Zeiss 880 laser scanning confocal microscope run by ZEN black v2.1. Images were collected using a 63× 1.4 NA Plan-Apochromat DIC oil objective. Lys27Pra(JP) GxTX was excited using a 561 nm laser set to 4.0% power. Emission spectra were collected using 34 different PMT detectors. Two detectors binned emission signals from 371 – 410 nm and 697 – 758 nm. The remaining detectors used 8.9-nm wide bins to detect emission signals from 410 – 694 nm. A final detector collected TPMT/brightfield images. Images were analyzed with Zen Blue Desk. The JP signal was isolated from broadband auto-fluorescence signals using Zeiss linear unmixing methods, with the auto-fluorescence ROI taken from a region outside of the cell boundary. Unmixed images were further processed in ImageJ as max intensity stack projections.
Voltage clamp spectroscopy.
Voltage clamp spectroscopy (VCS) was performed on Kv2.1-CHO cells bathed in a solution containing a GxTX-JP conjugate. Kv2.1-CHO cells were plated in glass-bottomed 35 mm dishes (Mat-Tek) with 1 μg/mL minocycline 40–48 hours before VCS and washed once with NEGB immediately prior to experiments. This solution was removed and replaced with 100 μL of NE containing 100 nM of a GxTX-JP conjugate. After 5 minutes, solution was diluted with 1 mL NEG for a working concentration of 9 nM GxTX-JP, or imaged as is. We observed no significant spectral shift or loss of JP fluorescence from cell membranes after this dilution step. Cells with visible JP surface fluorescence were voltage-clamped in whole cell mode using a patch clamp amplifier (HEKA EPC-10) run by Patchmaster software (HEKA). The patch pipette contained a K+-deficient Cs+ internal pipette solution (70 mM CsCl, 50 mM CsF, 35mM NaCl, 1 mM EGTA, 10 mM HEPES, adjusted to pH 7.4 with CsOH; measured osmolarity was 310 mOsm) to limit channel conductance and reduce current-induced series resistance voltage error. Borosilicate glass pipettes (1.5 mm OD, 1.1 mm ID, with filament; Sutter Instruments) were pulled with blunt tips to resistances between 2 and 7 MΩ in these solutions. Cells were held at −80 mV and stepped to indicated voltages for the duration of confocal imaging protocols which typically lasted several seconds. All VCS was conducted at room temperature. Confocal images were acquired on a Zeiss 880 laser scanning confocal microscope excited with 34 PMTs for spectral imaging, run by ZEN black v2.1. Images were collected using a 63× 1.15 NA water-immersion DIC objective (Zeiss). GxTX-JP was excited using a 543-nm laser set to 60% power or a 594-nm laser set to 15% power. For 543 nm laser emission spectra were collected from 557 nm to 691 nm with 8.9 nm bandwidth PMT intervals and the emission spectra were collected from 539 nm to 691 nm with 8.9 nm bandwidth PMT in 594 nm laser. Images were analyzed with ImageJ to extract spectra corresponding to different regions of interest, including cell membranes and background. Voltage clamp spectra are mean values of 4 independent cells. These spectra were then exported for further analysis including curve fitting, as described below. The calculated liquid junction potential of 5.3 mV between bath and pipette was corrected post hoc for Figure 5.
Association/dissociation rate measurements.
Whole cell voltage clamp was used to measure currents from Kv2.1-CHO cells before, during, and after application of Lys27Pra(JP) GxTX. Cells in 35-mm culture dishes were washed in divalent-free PBS (Gibco), dissociated in 0.5 mL of 0.48 mM EDTA solution (Gibco), transferred to a 1.5 mL polypropylene tube, and pelleted at 1000 × g for 2 min. The supernatant was removed, and the cells were then resuspended in 1 mL of NEG solution. The pelleting and resuspension were repeated, and resuspended cells rotated slowly at room temperature until use. In all experiments, aliquots of the cell solution were added to a solution exchange chamber (Warner Instruments), at room temperature (22 – 24 °C), with the recording chamber prefilled with approximately 50 μL NE solution. Prior to recording, cells were allowed to settle for 5 or more minutes and then were rinsed with NE solution using a gravity-driven perfusion system. Recording pipettes were pulled from thin wall borosilicate glass (1.5 mm OD, 1.1 mm ID, with filament; Sutter Instruments) on a horizontal micropipette puller as described previously22. Pipette tips were coated with a silicone elastomer (Sylgard 184) and fire polished. Pipettes were filled with a solution containing 35 mM KOH, 70 mM KCl, 50 mM KF, 50 mM HEPES, 5 mM EGTA adjusted to pH 7.2 with KOH, with measured osmolarity of 303 mOsm. Pipette-tip resistance with the above solutions were 1.5 to 3 MΩ. A calculated liquid junction potential of 8.5 mV was not corrected. Cells were voltage clamped in whole cell mode with a patch clamp amplifier (Axon 200B) run by Patchmaster software (HEKA). Currents were low-pass filtered at 10 kHz and digitized at 100 kHz. Holding potential was −100 mV. Remaining capacitance and ohmic leak were subtracted offline using traces recorded during P/5 voltage protocols from holding potential. In sequences of voltage steps, at least 2 s elapsed between the start times of each recording. To minimize voltage errors at the cell membrane due to series resistance, channel expression was induced with minocycline incubation for a minimal time (1.5 – 2 h) to limit Kv2.1 conductance magnitude. Series resistance of 2 – 16 MΩ (before compensation) were estimated from the value arrived at during whole-cell capacitive transient subtraction by manual adjustment of the whole-cell parameters circuit of the amplifier. Series-resistance compensation lag was set to 10 μs. To constrain voltage error, the series resistance compensation circuit was set between 60% and 80%. The series resistance compensation prediction (supercharging) circuit was not used. The product of current amplitude and estimated series resistance remaining after compensation indicated that voltage error at the cell membrane due to series resistance was ≤10 mV in all data used for analysis. After whole cell configuration was established, bath solution was replaced by a vehicle control solution with 5 μM tetrodotoxin and 0.1% BSA added. Tetrodotoxin was included to block current from endogenous voltage-gated sodium currents apparent in some CHO cells22. Solution was exchanged by manually flushing a volume of at least 100 μL through the recording chamber.
A microscopic association rate, first-order with respect to concentration, kon was determined by application of 100 nM Lys27Pra(JP) GxTX (73 μM stock) and measuring the change in amplitude of Kv2.1 currents during 100 ms steps to 0 mV repeated at 2 second intervals. A time constant (τ) was determined from a mono-exponential decay function fit to the amplitude of the first 25 steps to 0 mV in Lys27Pra(JP) GxTX. As Kv2.1 current during these voltage steps is expected to be22 eliminated after GxTX binds to even one of the four voltage sensors of a Kv2.1 homotetramer, kon was calculated from:
where [GxTX] is concentration of unlabeled GxTX or a JP-GxTX conjugate.
The microscopic dissociation rate (koff) was difficult to quantitate with this protocol, as less than 10% of current recovery was observed after 10 min of washing out Lys27Pra(JP) GxTX by continuous flushing of NE through the solution exchange chamber, using a gravity-driven perfusion system. Thus koff << 4kon[GxTX] and koff was set to zero for calculation of kon. To accelerate Lys27Pra(JP) GxTX dissociation, cells were stepped to 0 mV for 5 s every 10 s.23 τ was determined by fitting with:
koff was calculated from using [GxTX] = 0, assuming all dissociation occurred during the 5 s 0 mV steps. In Table 1, values for GxTX are from23. Values for Lys27Pra(JP) GxTX kon at −100 mV (n = 8) and koff at 0 mV (n = 8) were from experiments, other values were calculated with the assumption that rates for GxTX and Lys27Pra(JP) GxTX remain proportionate at all voltages.
Gating current measurements.
Channel expression was achieved by 48 hours incubation with minocycline during culture. The external gating current recording solution contained 150 mM TEA-Cl, 41 mM HEPES, 1 mM MgCl2 · 6H2O, 1.5 mM CaCl2, adjusted to pH 7.3 with NMDG; measured osmolarity was 311 mOsm. The internal (pipette) solution contained 90 mM NMDG, 1 mM NMDG-Cl, 50 mM HEPES, 5 mM EGTA, 50 mM NMDG-F, 0.01 mM CsCl, adjusted to pH 7.4 with methanesulfonic acid; with a measured osmolarity of 303 mOsm. To avoid monovalent cation contamination of the recording solution, the pH was determined from small aliquots that were discarded. Osmolarity was measured with a VAPRO vapor pressure osmometer 5520. Recording pipettes were pulled from thin walled borosilicate glass (1.5 mm outer diameter, 1.1 mm inner diameter, with filament; Sutter Instruments). Pipette resistances with gating current solutions were 6 – 14 MΩ. Cultured cells were dissociated with 0.48 mM EDTA in PBS for 30 sec and then harvested by scraping. Cells were pelleted by centrifugation (2 min × 1500g) and washed three times in NE. Following the final pelleting, cells were resuspended in the gating current external solution and left to sit in a 1.5 mL polypropylene tube. Prior to patching, 100 μL of cell suspension was added to the <100 μL recording chamber (Warner R-24N). Cells were given 4 min to adhere before gently washing with 10 mL of the gating current recording solution to dilute any monovalent cation contamination. Lys27Pra(JP) GxTX (100 μL of 100 nM) with 1% BSA was pipetted into the bath with vacuum line suctioning of the fluid surface to maintain constant bath volume. Seals with GΩ resistances were formed either before (n = 3) or after (n = 2) the addition of toxin. In all cases, the cell was incubated in Lys27Pra(JP) for at least 4 minutes before the gating current measurements were recorded. Patched cells were stepped from a −100 mV holding potential to the indicated potential for 100 ms and then to −140 mV to record OFF gating currents. Cell series resistances were 14 – 30 MΩ (before compensation) and 50% compensation used when series resistance exceeded 10 MΩ. Cell capacitances were 6 – 10 pF. Remaining capacitance and Ohmic leak were subtracted offline using the average of five traces recorded during P/5.9 voltage protocols from −133 mV holding potential. In sequences of voltage steps, at least 2 s elapsed between the start times of each recording. Qoff gating charge movement was quantified by integrating the area under the OFF gating currents in a 10.5 ms window following the end of any obvious fast capacitative artifacts created from the voltage step to −140 mV. Currents were baseline-subtracted with the average current elicited during a 10 ms-long period after current decay had ceased. This window began 10 ms following the −140 mV voltage step stimuli. Each Qoff curve was normalized using the average Qoff value elicited with 100–120 mV steps. The mean of the normalized data set is plotted with the standard error of the mean, which was computed following normalization. Fitting with a Boltzmann distribution was as described previously, where Vmid is the voltage where the function reaches half maximal conductance, and z is valence in units of elementary charge (e0).59 The calculated liquid junction potential of 13.5 mV between bath and pipette was corrected post hoc for Figure 5.
Emission spectra curve fitting.
Fluorescence emission spectra were decomposed into one or more 6-parameter split pseudo-Voigt curves39, 40, 60, 61 expressed as an inequality with the form:
where is a0 is the amplitude, a1 is the midpoint, and a2-a5 are shape definition parameters. The 4-parameter pseudo-Voigt function in each range is in the form:
In unconstrained fittings, all parameters (a0-a5) were optimized during fitting. Otherwise, only a0 and a1 were optimized, while a2-a5 were constrained to the reference spectrum, the split pseudo-Voigt fit of Lys27Pra(JP) GxTX on Kv2.1-expressing cells at resting membrane potential (Table S1). As a measure of the agreement this spectrum with a single-species spectrum, the R² of this spectrum relative to spectrum of JP in toluene is calculated to be 0.98.
The parameters were optimized by the Levenberg-Marquardt algorithm, with some modifications, using the curve-fitting software Fityk 0.9.8 (https://fityk.nieto.pl/). This algorithm optimized fits for lowest weighted sums of squared residuals (WSSR), iterating until the change in WSSR < 0.0001. Modifications include the usage of reference spectra constructed by fitting a single curve to a well-defined fluorescence spectrum from a single species of fluorophore in solvent. For some spectra, as noted, we replaced one or more component curves with reference curves, holding shape parameters constant and allowing only amplitude and peak position parameters to vary. Goodness of fit was determined by root-mean-squared deviation (R2) values, which are listed in Table S1 along with the parameters of each component function.
Reference spectra matching the spectral line shape of JP were constructed by fitting single six-component split pseudo-Voigt curves to the fluorescence emission spectra obtained from pure samples of JP in solvent. Reference spectra were collected for JP-N3 in toluene, acetone, MeOH, and H2O, as well as Lys27Pra(JP) GxTX on Kv2.1-expressing CHO cells in polarizing buffer, which has a similar line shape as JP azide in toluene. When fitting spectra with these reference curves, all 4 shape-defining parameters were held constant, and the amplitude and position wavelength of each reference curve allowed to vary. Spectra corresponding to the lineshape of a single reference curve with R2 > 0.98 were considered to be well defined by a single species (e.g., JP in solvent, Lys27Pra(JP) GxTX on Kv2.1 at −100 mV). Spectra with R2 < 0.98 for a single reference curve were fitted to 2 contributory species.
Fluorescence emission spectra suspected to contain multiple species centered at different wavelengths were decomposed into a sum of multiple reference curves using the Levenberg-Marquardt algorithm. The optimized amplitude and position components of each curve function were used to estimate the wavelengths and relative contributions of the predominant fluorescent species. The function amplitudes were adjusted according to the relative fluorescent intensity of JP species emitting at the corresponding wavelengths and the resulting values were used as a proxy for the relative concentrations of the species.
Supplementary Material
ACKNOWLEDGMENT
We thank Victor Mann and Robert Stewart for helpful discussions; Kenneth Eum, Mark Lillya, and Michael Kirmiz for contributions to the early phases of this project; and Olivia Buonarati in the laboratory of Mary Horne and Johannes Hell for preparation of dissociated hippocampal neurons. This work was supported by National Institutes of Health award R01NS096317 (B.E.C. and J.T.S.), R21EY026449 (J.T.S), T32GM007377 (R.J.S.), F31NS108614 (R.J.S.), the UC Davis Senate (J.T.S), and American Heart Association award 17POST33670698 (P.T.). Work at the Molecular Foundry was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Funding Sources
National Institutes of Health
American Heart Association
U.S. Department of Energy
University of California
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. This includes additional cell microscopy with JP-GxTX mutants, curve fitting parameters, voltage dependent fluorescence spectra, and structures of the JP click adducts (PDF)
chemical structures, confocal images, curve-fitting parameters, live-cell spectra, and channel-toxin models
REFERENCES
- 1.Wulff H; Christophersen P; Colussi P; Chandy KG; Yarov-Yarovoy V, Antibodies and venom peptides: new modalities for ion channels. Nat. Rev. Drug Discov 2019, 18, 339–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal SK; MacKinnon R, Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 1996, 16, 1169–1177. [DOI] [PubMed] [Google Scholar]
- 3.Seoh SA; Sigg D; Papazian DM; Bezanilla F, Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 1996, 16, 1159–1167. [DOI] [PubMed] [Google Scholar]
- 4.Tao X; Lee A; Limapichat W; Dougherty DA; MacKinnon R, A gating charge transfer center in voltage sensors. Science 2010, 328, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zagotta WN; Hoshi T; Dittman J; Aldrich RW, Shaker potassium channel gating. II: Transitions in the activation pathway. J. Gen. Physiol 1994, 103 (2), 279–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole KS; Moore JW, Potassium ion current in the squid giant axon: dynamic characteristic. Biophys J 1960, 1, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoppa NE; Sigworth FJ, Activation of shaker potassium channels. I. Characterization of voltage-dependent transitions. J. Gen. Physiol 1998, 111, 271–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoshi T; Zagotta WN; Aldrich RW, Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 1990, 250, 533–538. [DOI] [PubMed] [Google Scholar]
- 9.Ledwell JL; Aldrich RW, Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol 1999, 113, 389–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pathak MM; Yarov-Yarovoy V; Agarwal G; Roux B; Barth P; Kohout S; Tombola F; Isacoff EY, Closing in on the resting state of the Shaker K(+) channel. Neuron 2007, 56, 124–140. [DOI] [PubMed] [Google Scholar]
- 11.Larsson HP; Baker OS; Dhillon DS; Isacoff EY, Transmembrane movement of the shaker K+ channel S4. Neuron 1996, 16, 387–397. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y; Holmgren M; Jurman ME; Yellen G, Gated access to the pore of a voltage-dependent K+ channel. Neuron 1997, 19, 175–184. [DOI] [PubMed] [Google Scholar]
- 13.Villalba-Galea CA; Sandtner W; Starace DM; Bezanilla F, S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 17600–17607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabe M; Lai HC; Jain M; Jan YN; Jan LY, Structure prediction for the down state of a potassium channel voltage sensor. Nature 2007, 445, 550–553. [DOI] [PubMed] [Google Scholar]
- 15.Long SB; Tao X; Campbell EB; MacKinnon R, Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [DOI] [PubMed] [Google Scholar]
- 16.Sun J; MacKinnon R, Structural Basis of Human KCNQ1 Modulation and Gating. Cell 2020, 180 (2), 340–347.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hite RK; MacKinnon R, Structural Titration of Slo2.2, a Na(+)-Dependent K(+) Channel. Cell 2017, 168, 390–399.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Catterall WA; Cestele S; Yarov-Yarovoy V; Yu FH; Konoki K; Scheuer T, Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [DOI] [PubMed] [Google Scholar]
- 19.Herrington J; Zhou YP; Bugianesi RM; Dulski PM; Feng Y; Warren VA; Smith MM; Kohler MG; Garsky VM; Sanchez M; Wagner M; Raphaelli K; Banerjee P; Ahaghotu C; Wunderler D; Priest BT; Mehl JT; Garcia ML; McManus OB; Kaczorowski GJ; Slaughter RS, Blockers of the Delayed-Rectifier Potassium Current in Pancreatic -Cells Enhance Glucose-Dependent Insulin Secretion. Diabetes 2006, 55, 1034–1042. [DOI] [PubMed] [Google Scholar]
- 20.Gupta K; Zamanian M; Bae C; Milescu M; Krepkiy D; Tilley DC; Sack JT; Yarov-Yarovoy V; Kim JI; Swartz KJ, Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. elife 2015, 4, e06774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu PW; Bean BP, Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J. Neurosci 2014, 34, 4991–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tilley DC; Angueyra JM; Eum KS; Kim H; Chao LH; Peng AW; Sack JT, The tarantula toxin GxTx detains K(+) channel gating charges in their resting conformation. J. Gen. Physiol 2019, 151, 292–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tilley DC; Eum KS; Fletcher-Taylor S; Austin DC; Dupre C; Patron LA; Garcia RL; Lam K; Yarov-Yarovoy V; Cohen BE; Sack JT, Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc. Natl. Acad. Sci. U. S. A 2014, 111, e4789–e4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thapa P; Stewart R; Sepela RJ; Vivas O; Parajuli LK; Lillya M; Fletcher-Taylor S; Cohen BE; Zito K; Sack JT, Optical measurement of endogenous ion channel activation in live tissue. bioRxiv 2019, 541805. [Google Scholar]
- 25.Kubota T; Durek T; Dang B; Finol-Urdaneta RK; Craik DJ; Kent SBH; French RJ; Bezanilla F; Correa AM, Mapping of voltage sensor positions in resting and inactivated mammalian sodium channels by LRET. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E1857–E1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon SE; Munari M; Zagotta WN, Visualizing conformational dynamics of proteins in solution and at the cell membrane. eLife 2018, 7, e37248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macgregor RB; Weber G, Estimation of the polarity of the protein interior by optical spectroscopy. Nature 1986, 319, 70–73. [DOI] [PubMed] [Google Scholar]
- 28.Sackett DL; Wolff J, Nile red as a polarity-sensitive fluorescent probe of hydrophobic protein surfaces. Analytical Biochemistry 1987, 167, 228–234. [DOI] [PubMed] [Google Scholar]
- 29.Cohen BE; McAnaney TB; Park ES; Jan YN; Boxer SG; Jan LY, Probing protein electrostatics with a synthetic fluorescent amino acid. Science 2002, 296, 1700–1703. [DOI] [PubMed] [Google Scholar]
- 30.Pierce DW; Boxer SG, Dielectric relaxation in a protein matrix. J. Phys. Chem 1992, 96, 5560–5566. [Google Scholar]
- 31.Cohen BE; Pralle A; Yao X; Swaminath G; Gandhi CS; Jan YN; Kobilka BK; Isacoff EY; Jan LY, A fluorescent probe designed for studying protein conformational change. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu H; Li T; Rohou A; Arthur CP; Tzakoniati F; Wong E; Estevez A; Kugel C; Franke Y; Chen J; Ciferri C; Hackos DH; Koth CM; Payandeh J, Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 2019, 176, 702–715.e14. [DOI] [PubMed] [Google Scholar]
- 33.Yarov-Yarovoy V; Schonbrun J; Baker D, Multipass membrane protein structure prediction using Rosetta. Proteins 2006, 62, 1010–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mann VR; Powers AS; Tilley DC; Sack JT; Cohen BE, Azide-Alkyne Click Conjugation on Quantum Dots by Selective Copper Coordination. ACS Nano 2018, 12, 4469–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Titus JA; Haugland R; Sharrow SO; Segal DM, Texas Red, a hydrophilic, red-emitting fluorophore for use with fluorescein in dual parameter flow microfluorometric and fluorescence microscopic studies. J. Immunol. Methods 1982, 50, 193–204. [DOI] [PubMed] [Google Scholar]
- 36.Antonucci DE; Lim ST; Vassanelli S; Trimmer JS, Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 2001, 108, 69–81. [DOI] [PubMed] [Google Scholar]
- 37.Johnson B; Leek AN; Sole L; Maverick EE; Levine TP; Tamkun MM, Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl. Acad. Sci. U. S. A 2018, 115, e7331–e7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirmiz M; Palacio S; Thapa P; King AN; Sack JT; Trimmer JS, Remodeling neuronal ER-PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol. Biol. Cell 2018, 29, 2410–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jha SK; Ji M; Gaffney KJ; Boxer SG, Site-specific measurement of water dynamics in the substrate pocket of ketosteroid isomerase using time-resolved vibrational spectroscopy. J. Phys. Chem. B 2012, 116, 11414–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greathouse JA; Cygan RT; Bradshaw RW; Majzoub EH; Simmons BA, Computational and Spectroscopic Studies of Dichlorofluoroethane Hydrate Structure and Stability. J. Phys. Chem. C 2007, 111, 16787–16795. [Google Scholar]
- 41.Hodgkin AL; Huxley AF, A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol 1952, 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao X; Hite RK; MacKinnon R, Cryo-EM structure of the open high-conductance Ca2+-activated K+ channel. Nature 2016, 541, 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kintzer AF; Green EM; Dominik PK; Bridges M; Armache JP; Deneka D; Kim SS; Hubbell W; Kossiakoff AA; Cheng Y; Stroud RM, Structural basis for activation of voltage sensor domains in an ion channel TPC1. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E9095–E9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wisedchaisri G; Tonggu L; McCord E; Gamal El-Din TM; Wang L; Zheng N; Catterall WA, Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 2019, 178, 993–1003.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen H; Liu D; Wu K; Lei J; Yan N, Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [DOI] [PubMed] [Google Scholar]
- 46.Cohen BE; Grabe M; Jan LY, Answers and questions from the KvAP structures. Neuron 2003, 39, 395–400. [DOI] [PubMed] [Google Scholar]
- 47.Lee SY; MacKinnon R, A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [DOI] [PubMed] [Google Scholar]
- 48.Zagotta WN; Hoshi T; Aldrich RW, Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol 1994, 103, 321–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee CH; MacKinnon R, Voltage Sensor Movements during Hyperpolarization in the HCN Channel. Cell 2019, 179, 1582–1589.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koide Y; Urano Y; Hanaoka K; Piao W; Kusakabe M; Saito N; Terai T; Okabe T; Nagano T, Development of NIR fluorescent dyes based on Si-rhodamine for in vivo imaging. J. Am. Chem. Soc 2012, 134, 5029–5031. [DOI] [PubMed] [Google Scholar]
- 51.Grimm JB; Muthusamy AK; Liang Y; Brown TA; Lemon WC; Patel R; Lu R; Macklin JJ; Keller PJ; Ji N; Lavis LD, A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 2017, 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milescu M; Bosmans F; Lee S; Alabi AA; Kim JI; Swartz KJ, Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol 2009, 16, 1080–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Revell Phillips L; Milescu M; Li-Smerin Y; Mindell JA; Kim JI; Swartz KJ, Voltage-sensor activation with a tarantula toxin as cargo. Nature 2005, 436, 857–860. [DOI] [PubMed] [Google Scholar]
- 54.Besanceney-Webler C; Jiang H; Zheng T; Feng L; Soriano del Amo D; Wang W; Klivansky LM; Marlow FL; Liu Y; Wu P, Increasing the efficacy of bioorthogonal click reactions for bioconjugation: a comparative study. Angew. Chem. Int. Ed. Engl 2011, 50, 8051–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee S; Milescu M; Jung HH; Lee JY; Bae CH; Lee CW; Kim HH; Swartz KJ; Kim JI, Solution structure of GxTX-1E, a high-affinity tarantula toxin interacting with voltage sensors in Kv2.1 potassium channels. Biochemistry 2010, 49, 5134–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapani JG; Korn SJ, Control of ion channel expression for patch clamp recordings using an inducible expression system in mammalian cell lines. BMC Neurosci 2003, 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Subach OM; Cranfill PJ; Davidson MW; Verkhusha VV, An enhanced monomeric blue fluorescent protein with the high chemical stability of the chromophore. PLoS One 2011, 6, e28674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matt L; Kim K; Hergarden AC; Patriarchi T; Malik ZA; Park DK; Chowdhury D; Buonarati OR; Henderson PB; Gokcek Sarac C; Zhang Y; Mohapatra D; Horne MC; Ames JB; Hell JW, alpha-Actinin Anchors PSD-95 at Postsynaptic Sites. Neuron 2018, 97, 1094–1109 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sack JT; Aldrich RW; Gilly WF, A gastropod toxin selectively slows early transitions in the Shaker K channel’s activation pathway. J. Gen. Physiol 2004, 123, 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aschaffenburg DJ; Moog RS, Probing hydrogen bonding environments: solvatochromic effects on the CN vibration of benzonitrile. J. Phys. Chem. B 2009, 113, 12736–12743. [DOI] [PubMed] [Google Scholar]
- 61.Adhikary R; Zimmermann J; Dawson PE; Romesberg FE, IR probes of protein microenvironments: utility and potential for perturbation. Chemphyschem 2014, 15, 849–853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.