

Abstract
Objective
This study was undertaken to determine whether a low residual quantity of dystrophin protein is associated with delayed clinical milestones in patients with DMD mutations.
Methods
We performed a retrospective multicentric cohort study by using molecular and clinical data from patients with DMD mutations registered in the Universal Mutation Database–DMD France database. Patients with intronic, splice site, or nonsense DMD mutations, with available muscle biopsy Western blot data, were included irrespective of whether they presented with severe Duchenne muscular dystrophy (DMD) or milder Becker muscular dystrophy (BMD). Patients were separated into 3 groups based on dystrophin protein levels. Clinical outcomes were ages at appearance of first symptoms; loss of ambulation; fall in vital capacity and left ventricular ejection fraction; interventions such as spinal fusion, tracheostomy, and noninvasive ventilation; and death.
Results
Of 3,880 patients with DMD mutations, 90 with mutations of interest were included. Forty‐two patients expressed no dystrophin (group A), and 31 of 42 (74%) developed DMD. Thirty‐four patients had dystrophin quantities < 5% (group B), and 21 of 34 (61%) developed BMD. Fourteen patients had dystrophin quantities ≥ 5% (group C), and all but 4 who lost ambulation beyond 24 years of age were ambulant. Dystrophin quantities of <5%, as low as <0.5%, were associated with milder phenotype for most of the evaluated clinical outcomes, including age at loss of ambulation (p < 0.001).
Interpretation
Very low residual dystrophin protein quantity can cause a shift in disease phenotype from DMD toward BMD. ANN NEUROL 2021;89:280–292
Mutations in the DMD gene that encodes dystrophin cause a spectrum of disease phenotypes including severe Duchenne muscular dystrophy (DMD) and the milder variant Becker muscular dystrophy (BMD). Dystrophin stabilizes striated muscle cells by linking cytoskeletal actin to the sarcolemmal dystrophin‐associated glycoprotein complex (DAGC). 1 DMD mutation cause quantitative and qualitative changes in dystrophin protein synthesis. 2 , 3 Decreases in dystrophin protein cause a group of X‐linked muscle diseases called dystrophinopathies, which are characterized by progressive muscle wasting and variable degrees of muscle weakness. The most severe form, DMD, when untreated, results in loss of ambulation (LoA) by 12 years of age, 4 near‐complete muscle paralysis before the age of 20 years, and markedly reduced life expectancy because of progressive respiratory insufficiency and cardiomyopathy. BMD is characterized by a milder course; ambulation is preserved beyond 16 years of age—with many patients never losing the ability to walk—and delayed involvement of respiratory and cardiac muscles. LoA between 13 and 16 years of age is defined as intermediate muscular dystrophy (IMD). 4 In >90% of patients with DMD/BMD, 5 , 6 phenotype can be predicted based on whether the transcriptional reading‐frame is maintained (rule of Monaco). 7
Phenotype severity in dystrophinopathy mainly depends on the amount of residual dystrophin in muscle: the higher the amount of dystrophin protein, the less severe the phenotype. 8 , 9 Western blot of skeletal muscle biopsies is the most reliable method to quantify dystrophin protein. Patients with residual dystrophin protein levels as high as 17% may develop BMD 10 ; however, a dystrophin protein level of 30% could protect against skeletal muscle symptoms. 11 Novel therapeutic agents such as eteplirsen (an exon 51–skipping agent), golodirsen (an exon 53–skipping agent), and ataluren (PTC128; a read‐through agent of premature stop codons) 12 can increase dystrophin protein levels. Eteplirsen can increase dystrophin protein levels by 0.44 to 0.93%, 13 , 14 and initial studies indicate slower disease progression in DMD patients treated with eteplirsen. 15 , 16 , 17 , 18 Golodirsen can increase dystrophin protein levels by 1.019%. 19 However, the minimum amount of dystrophin protein required for a shift toward milder disease phenotypes has not been reported. We hypothesized that studying the natural history of the disease in patients with very low residual dystrophin could yield this information. Alternative splicing can cause milder phenotypes by expression of truncated in‐frame or full‐length DMD transcripts to varying extents via spontaneous skipping of “in‐frame” exons that carry nonsense mutations, 5 , 20 , 21 , 22 residual expression of full‐length or exon skipped transcripts in splice site mutations, 5 , 21 , 22 , 23 or certain mid‐intronic mutations. 5 , 24 , 25 , 26 Many patients with DMD mutations prone to alternative splicing likely produce very low residual dystrophin or no dystrophin at all; however, the natural history of such patients has never been systematically studied.
The aim of this study was to determine the effects of very low dystrophin levels and of rare DMD mutations on muscular dystrophy disease phenotype, by comparing disease milestones in patients with no dystrophin and those in patients with dystrophin levels </≥ 5%.
Patients and Methods
Study Design
The patients included in this study were selected from individuals registered in the Universal Mutation Database (UMD)‐DMD France database irrespective of whether they clinically presented with severe DMD or milder IMD or BMD. Patients were eligible for inclusion in this study if (1) molecular diagnoses were performed at the Laboratory for Biochemistry and Molecular Genetics, University Hospital Cochin, Paris; (2) they had DMD pseudoexon mutations, splice site mutations, or nonsense mutations in exons whose deletion could lead to the production of an in‐frame transcript (“in‐frame” exons); and (3) Western blot of muscle biopsy included antidystrophin staining (using NCL‐DYS2 antibody) and antidysferlin staining on the same blot. Patients were excluded if (1) antidystrophin staining was positive with NCL‐DYS1 antibody but negative with NCL‐DYS2 antibody and (2) age was <7 years at last clinical follow‐up. The dysferlin band was chosen to normalize for dystrophin protein quantity, because proteins conventionally used for normalization, such as vimentin, were not included in the blots (see below). Patients younger than 7 years were excluded because the assessed disease milestones are unlikely to appear in the very young. 27
The study was performed in compliance with the ethical principles of the Declaration of Helsinki and was approved by the Commission Nationale de l'Informatique et des Libertés and the Comité d'Expertise pour les Recherches, les Etudes et les Evaluations dans le domaine de la Santé.
Western Blot Analysis
The photographic films of diagnostic muscle biopsy Western blots were retrospectively analyzed. Western blots for all patients were performed at the Laboratory for Biochemistry and Molecular Genetics, University Hospital Cochin, Paris, France, using the same methodology. Validated multiplex Western blot methods were used to assess dystrophin levels in the biopsy samples. 28 , 29 Each patient sample was run on 2 separate gels, each containing control samples. The membrane from gel 1 was developed using an antibody against the rod domain of dystrophin (clone Dys4/6D3, NCL‐DYS1), and the membrane from gel 2 using an antibody against the C‐terminal part of dystrophin (clone Dys8/6C5, NCL‐DYS2). Detection of dysferlin was performed on the membrane from gel 2 (clone Ham1/7B6, NCL‐Hamlet). Rabbit polyclonal horseradish peroxidase–conjugated antimouse IgG (P‐0260) was used as secondary antibody. Bands were visualized by chemiluminescence. Photographic films were exposed for 1, 5, and 60 minutes (T1, T5, and T60, respectively). We quantified the bands of NCL‐DYS2 and normalized it with the quantity of dysferlin protein, whose expression was considered unrelated to dystrophin levels. We first visually assessed the presence or absence of the NCL‐DYS2 band at T1, T5, and T60. Absence of NCL‐DYS2 bands was considered to indicate complete absence of dystrophin in the biopsy (0%), and was confirmed by absence of NCL‐DYS1 staining on gel 1. For blots on which dystrophin was detected, we quantified the bands only if the NCL‐DYS2 band from the patient was present on the T1 film, because bands from control patients were saturated at T5 and T60. Protein bands were quantified by measuring their integrated density within a rectangle that covered the entire individual band, subtracting the integrated density of an empty rectangle of exactly the same size in the vicinity, by using ImageJ software 1.51 (NIH, Bethesda, MD), and then normalizing it to the control values. We determined 0.5% dystrophin to be the lowest expression level at T1, and therefore considered the presence of a residual dystrophin band at T5 or T60, but absence at T1, to indicate dystrophin expression < 0.5% but too low for quantification. We included some patients who expressed no dystrophin and for whom dysferlin staining was not available, because densitometry was not required for these patients. Patients were classified into three groups according to the level of dystrophin protein quantity: group A, no dystrophin; group B, 0–5% dystrophin (>0 and < 5%); and group C, ≥5% dystrophin. In addition, we subdivided group B into subgroups B′ (0–0.5% dystrophin [>0 and <0.5%]) and B″ (0.5–5% dystrophin [≥0.5% and <5%]).
Disease Milestones
Anonymized patient data were collected from medical records maintained at the respective medical care services (French reference centers for neuromuscular disorders). The following disease milestones were recorded: age at loss of ambulation, age at appearance of first symptoms, age at start of noninvasive ventilation, age at tracheostomy, age at spinal fusion surgery, age at drop in vital capacity (VC) to <50% and to <20%, age at drop in left ventricular ejection fraction (LVEF) to <55% and to <30%, and age at death. LVEF was used as an index of left ventricular dysfunction. LVEF < 55% was considered to be abnormal, and LVEF < 30% to be an indication of severe cardiac failure. Data on medication were also collected.
Statistical Analysis
The Shapiro–Wilk test was used to assess the normality of variable distribution. Normally distributed continuous data were summarized as means ± standard deviation, and non‐normally distributed continuous data as medians with interquartile range (IQR). Values were compared between groups by using 1‐way analysis of variance or the Kruskal–Wallis test, as appropriate. Categorical data were summarized as frequencies and percentages. Kaplan–Meier plots and log‐rank tests were used to compare the differences in age at occurrence of landmark events between the three groups. Subsequent pairwise comparisons were used with Bonferroni correction for multiple comparisons. Univariate and multivariate Cox proportional hazard models were used to identify the factors associated with patient survival, and the hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated. Multivariate models were used to evaluate the influence of covariates such as treatment with corticosteroids, ataluren, and angiotensin‐converting enzyme (ACE) inhibitors. Prism 7 (GraphPad Software, San Diego, CA) and StatPlus 6.5.1.0 (AnalystSoft, Walnut, CA) were used for data analysis. A 2‐tailed p value < 0.05 was considered statistically significant.
Results
Patients
A total of 3,880 patients with DMD mutations were registered in the UMD‐DMD France database until 2018. Of 263 patients who met our inclusion criteria, 9 were excluded because they were <7 years old; 153 were excluded because Western blot data were unavailable or because of insufficient quality for quantification; and 11 were excluded because NCL‐DYS2 and NCL‐DYS1 staining was discordant. We therefore analyzed the data from 90 patients; 42 had no discernable dystrophin (group A), 34 had 0 to 5% dystrophin (group B), and 14 had ≥5% dystrophin. These patients were followed up (Fig 1) at 26 different centers across metropolitan France (84 patients), Martinique island (3 patients), and Réunion island (3 patients). Table 1 shows the distribution of mutations, clinical phenotypes, and relevant medications in the 3 groups. Figure 2 shows the detailed distribution of mutations based on type of mutation and level of dystrophin quantity. Thirty‐seven of 90 (41%) patients had canonical splice site mutations (mutations at the −2, −1 splice acceptor and +1, +2 splice donor positions), 27 of 90 (30%) had pseudoexon mutations and noncanonical splice site mutations (splice site mutations outside of the invariant −2, −1, +1, +2 intronic positions), and 26 of 90 (29%) had nonsense mutations in in‐frame exons. Mutation types were nonrandomly distributed in relation to presence or absence of dystrophin protein. Of the patients with 0% dystrophin (group A), 40% had canonical splice site mutations, 17% had pseudoexon/noncanonical splice site mutations, and 43% had nonsense mutations in in‐frame exons. Among patients with >0% dystrophin (groups B and C), 42% had canonical splice site mutations, 42% had pseudoexon/noncanonical splice site mutations, and 16% had nonsense mutation in in‐frame exons. Pseudoexon mutations and noncanonical splice site mutations are expected to preserve higher levels of normal splicing, which explains the higher proportion of patients with >0% dystrophin carrying these mutations; patients with 0% dystrophin mostly had canonical splice site and nonsense mutations. Tables S1 and S2 show the complete clinical data and Western blot results for each patient.
FIGURE 1.

Flow chart showing the process of patient selection for this study. UMD = Universal Mutation Database.
TABLE 1.
Biochemical, Genetic, and Treatment Characteristics of Patients
| Characteristic | Group A | Group B | Group C | p |
|---|---|---|---|---|
| n | 42 | 34 | 14 | |
| Dystrophin protein quantity, % | 0 | >0 and <5 | ≥5 | |
| Canonical splice sites mutations, n (%) | 17 (40) | 12 (35) | 8 (57) | 0.374 |
| Pseudoexon and noncanonical splice site mutations, n (%) | 7 (17) | 16 (47) | 4 (29) | 0.009 |
| Nonsense mutations in “in‐frame” exon, n (%) | 18 (43) | 6 (18) | 2 (14) | 0.023 |
| DMD [LoA at <13 yr of age], n (%) | 31 (74) | 6 (18) | 0 (0) | <0.001 |
| IMD [LoA at ≥13 and <16 yr of age], n (%) | 4 (10) | 1 (3) | 0 (0) | 0.283 |
| BMD [LoA at ≥16 yr of age], n (%) | 1 (2) | 21 (61) | 8 (57) | <0.001 |
| Undefined disease phenotype [age at LoA milestone not reached or unknown], n (%) | 6 (14) | 6 (18) | 6 (43) | 0.062 |
| Corticosteroid treatment, n (%) | 17 (40) | 9 (26) | 1 (1) | 0.053 |
| Ataluren treatment, n (%) | 4 (10) | 2 (6) | 0 (0) | 0.453 |
| ACE inhibitor treatment, n (%) | 37 (88) | 19 (56) | 8 (57) | 0.004 |
ACE = angiotensin‐converting enzyme; BMD = Becker muscular dystrophy; DMD = Duchenne muscular dystrophy; IMD = intermediate muscular dystrophy; LoA = loss of ambulation.
FIGURE 2.
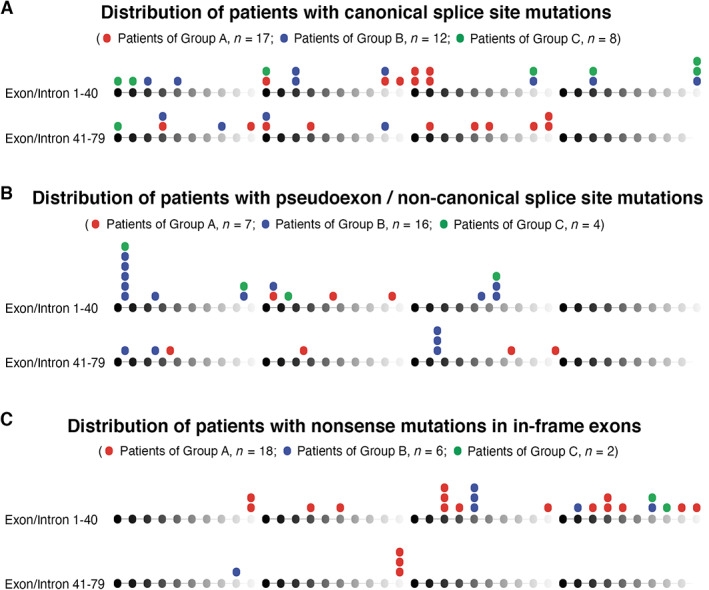
Genomic distribution of the analyzed mutations based on the quantity of dystrophin expressed. (A) Canonical splice site mutations. (B) Pseudoexon and noncanonical splice site mutations. (C) Nonsense mutations within “in‐frame” exons. DMD exons and introns 1 to 79 are symbolized by spheres and connecting lines from black to gray in repeats of 10. Mutations of the patients of group A, group B, and group C appear on the top of the respective exons/introns. Each sphere represents 1 patient.
Association between Residual Dystrophin Expression and Disease Milestones
Data on age at LoA were available for 88 of 90 (98%) patients. The age at LoA ranged from 7.5 to 20.0 years in group A, 10.6 to 65.2 years in group B, and 24.0 to 38.8 years in group C. Thirty‐one of 42 (74%) of group A patients developed DMD (LoA < 13 years of age), 21 of 34 (61%) of group B patients developed BMD (LoA ≥16 years of age), and 8 of 14 (57%) of group C patients developed BMD (see Table 1 for details). Residual dystrophin of 0 to 5% in group B was associated with delayed LoA compared to group A, and ambulation was almost completely conserved in group C (Fig 3A; p < 0.001). Median age at LoA was 10.5 years in group A, but could not be defined in groups B and C, because 21 of 34 patients and 9 of 13 patients, respectively, were still ambulant at the last clinical follow‐up.
FIGURE 3.
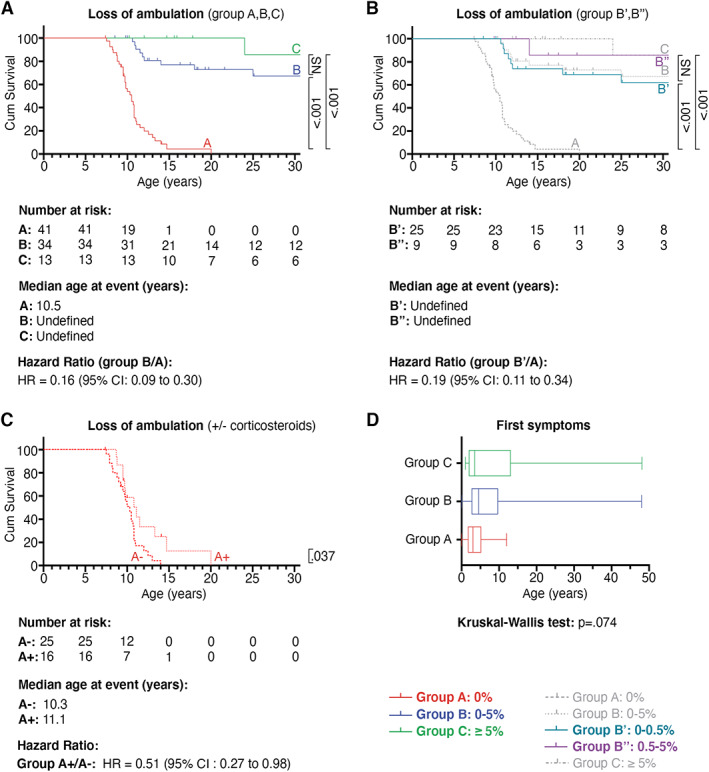
Time to loss of ambulation and age at appearance of first symptoms. Kaplan–Meier analysis shows the relationship for the age at loss of ambulation between (A) groups A, B, and C, (B) subgroups B′ and B″, and (C) patients of group A who were non–corticosteroid treated (subgroup A−) or corticosteroid treated (subgroup A+). Subsequent pairwise comparisons were used with Bonferroni correction for multiple comparisons. Probability values are shown on the right side of the diagram. (D) Box plot with median and interquartile range differences between groups A, B, and C. Kruskal–Wallis test was used and p calculated. CI = confidence interval; NS = non‐significant.
Subanalysis of group B revealed delayed LoA for subgroup B′ (expressing 0–0.5% dystrophin) and for subgroup B″ (expressing 0.5–5% dystrophin) compared to group A (see Fig 3B; p < .001). Comparison of groups A, B′, B″, and C revealed an inverse relationship between dystrophin quantity and LoA; higher dystrophin proteins levels were associated with lower proportions of patients who lost ambulation. Interestingly, not all group A patients had a DMD‐like disease course; 4 patients with no dystrophin lost ambulation between 13 and 16 years of age, and 1 lost ambulation at 20 years. The other disease milestones were also delayed in these 5 patients. Importantly, only 3 of 5 of the dystrophin‐negative patients who ambulated beyond the age of 12 years received corticosteroids and/or ataluren. Cox regression analysis with corticosteroid treatment, ataluren treatment, and dystrophin expression as covariates revealed that only dystrophin quantity was independently associated with age at LoA (HR = 0.05, 95% CI = 0.02–0.12, p < 0.001; Table 2).
TABLE 2.
Multivariate Cox Regression Analysis of Variables Respectively Associated with Loss of Ambulation and LVEF < 30%
| Clinical Endpoint | Cox Regression Factor | Number at Risk (%) | HR (95% CI) | p |
|---|---|---|---|---|
| Loss of ambulation | Corticosteroids | 26 (30) | 0.69 (0.35–1.34) | 0.270 |
| Ataluren | 6 (7) | 0.66 (0.18–2.36) | 0.520 | |
| Residual dystrophin | 47 (53) | 0.05 (0.02–0.12) | <0.001 | |
| LVEF < 30% | Corticosteroids | 19 (26) | 0.92 (0.11–7.53) | 0.938 |
| ACE inhibitors | 56 (77) | 0.57 (0.10–3.37) | 0.535 | |
| Residual dystrophin | 34 (47) | 0.29 (0.06–1.43) | 0.127 |
ACE = angiotensin‐converting enzyme; CI = confidence interval; HR = hazard ratio; LVEF = left ventricular ejection fraction.
Subanalysis of group A and not of group B showed a delayed LoA with the use of corticosteroid treatment. Within group A, median age of LoA shifted from 10.3 years to 11.1 years for patients treated with corticosteroids (see Fig 3C; p = 0.037). No effect of corticosteroids was detected in patients of group B; however, only a small number of patients received corticosteroid treatment (9/34, data not shown).
We determined how dystrophin quantity was associated with age at first appearance of clinical symptoms or signs (Table 3). Data were available for 61 of 90 (68%) patients. The median age at appearance of first symptoms was 3 years (IQR = 3.4 years) in group A, 4.6 years (IQR = 6.9 years) in group B, and 3.5 years (IQR = 11.0 years) in group C. No significant difference in appearance of first symptoms was detected (see Fig 3D; Kruskal–Wallis test, p = 0.074). We then determined the impact of dystrophin expression on the age at spinal fusion surgery and survival (Fig 4A, B). Patients of group B with 0 to 5% residual dystrophin had significantly delayed appearance of these milestones compared to patients of group A. Kaplan–Meier curves were similar for patients of groups B and C. Subanalysis for patients of group B′ (residual dystrophin quantity of 0–0.5%) compared to those of group A also showed a significantly higher age at spinal fusion surgery and survival (data not shown).
TABLE 3.
First Symptoms in the 3 Groups
| First Symptoms as Reported by Patients/Caretakers | Group A | Group B | Group C | Total |
|---|---|---|---|---|
| n | 42 | 34 | 14 | 90 |
| Exercise difficulties [eg, during running, cycling, climbing], n (%) | 5 (12) | 8 (24) | 2 (15) | 15 (17) |
| Walking difficulties [eg, toe walking, waddling gait], n (%) | 14 (33) | 12 (35) | 0 (0) | 26 (29) |
| Axial hypotonia, n (%) | 4 (9) | 1 (3) | 0 (0) | 5 (6) |
| Calf hypertrophy, n (%) | 9 (21) | 12 (35) | 9 (69) | 30 (33) |
| Abnormal psychomotor development [eg, speech delay], n (%) | 9 (21) | 4 (12) | 2 (15) | 15 (17) |
| Frequent falling, n (%) | 12 (28) | 5 (15) | 2 (15) | 19 (21) |
| Difficulties climbing stairs, n (%) | 7 (16) | 3 (9) | 1 (8) | 11 (12) |
| Rapid muscle fatigability, n (%) | 6 (14) | 4 (12) | 5 (38) | 15 (17) |
| Muscle weakness, n (%) | 2 (5) | 5 (15) | 1 (8) | 8 (9) |
| Myalgia, n (%) | 0 (0) | 5 (15) | 4 (31) | 9 (10) |
| Difficulties rising from the floor, n (%) | 4 (9) | 1 (3) | 1 (8) | 6 (7) |
| Non documented, n (%) | 10 (23) | 7 (21) | 0 (0) | 17 (19) |
FIGURE 4.
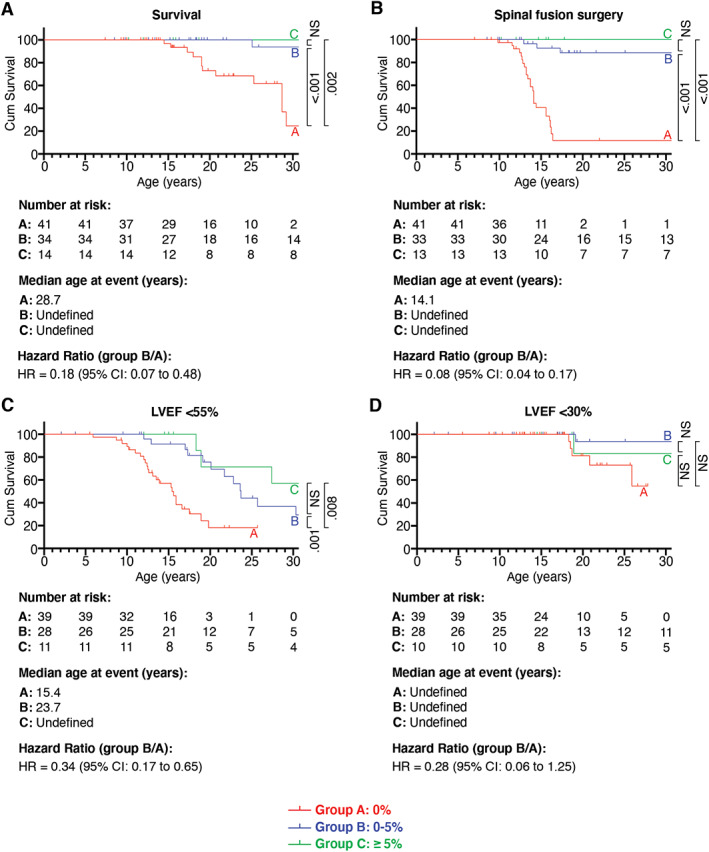
Effect of dystrophin on survival, heart function, and age at spinal fusion surgery. Kaplan–Meier analysis compares survival (A), and ages at spinal fusion surgery (B), left ventricular ejection fraction (LVEF) < 55% (C), and LVEF < 30% (D) between groups A, B, and C. Subsequent pairwise comparisons were used with Bonferroni correction for multiple comparisons. Probability values are shown on the right side of the diagram. CI = confidence interval; NS = non‐significant.
The impact of dystrophin quantity on heart function was examined by comparing echocardiogram‐derived LVEF between groups A, B, and C (see Fig 4C, D). Data on LVEF were available for 78 of 90 (87%) patients. Residual dystrophin quantity of 0 to 5% of group B was associated with delayed decline of LVEF compared to group A (HR = 0.34, 95% CI = 0.17–0.65, p = 0.001; Fig 5). The median age at which LVEF dropped to <55% was 15.4 years in group A and 23.7 years in group B. The age at which LVEF dropped to <30% was not significantly different between the two groups (HR = 0.28, 95% CI = 0.06–1.25, p = 0.125), probably because the number of patients with LVEF < 30% was too small in both groups (5/39 in group A and 3/28 in group B) and the observation period was too short. Cox regression analysis revealed that medication with ACE inhibitors and corticosteroids had no influence on the effects of residual dystrophin (HR = 0.29, 95% CI = 0.06–1.43, p = 0.127; see Table 2). Kaplan–Meier curves were similar for patients of groups B and C. Subanalysis for patients of group B′ (residual dystrophin quantity of 0–0.5%) compared to group A also showed a significant increase in age at which LVEF dropped to <55% (data not shown). Residual dystrophin quantity was also associated with delayed age at noninvasive ventilation, tracheostomy, and decline in VC to <50% and <20% (see Fig 5). Kaplan–Meier curves were similar for patients of groups B and C. Subanalysis for patients of group B′ (residual dystrophin quantity of 0–0.5%) compared to group A also revealed a significantly higher age at these disease milestones and delayed decline in VC (data not shown).
FIGURE 5.
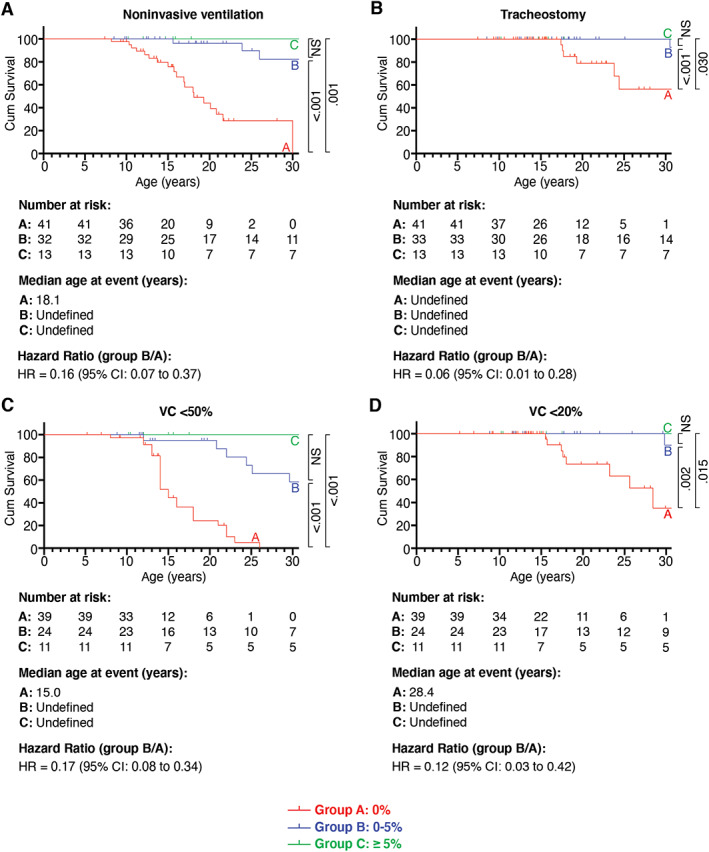
Effect of dystrophin on ages at start of noninvasive ventilation and tracheotomy and on vital capacity (VC). Kaplan–Meier analysis compares ages at start of noninvasive ventilation (A), tracheotomy (B), VC < 50% (C), and VC < 20% (D) between groups A, B, and C. Subsequent pairwise comparisons were used with Bonferroni correction for multiple comparisons. Probability values are shown on the right side of the diagram. CI = confidence interval; NS = non‐significant.
On Western blot, dystrophin bands of lower molecular weight were detected for 3 of 34 (9%) group B patients (data not shown). One of these patients lost ambulation at 46 years of age, and the remaining 2 were ambulant at last follow‐up, at ages 18 years and 40 years, respectively. Thus, residual dystrophin protein, even in minimal quantities, appears to be associated with milder dystrophinopathy phenotypes.
Discussion
The aim of this study was to determine the effect of very low dystrophin levels on the natural history of dystrophinopathy in patients with rare DMD mutations and disease severity ranging from severe DMD to mild BMD. The results showed that dystrophin protein quantities as low as <0.5% were associated with a shift from DMD toward milder phenotypes; median age at LoA shifted toward undefined later ages in these patients, and most were still ambulant at the time of the last follow‐up, with no patient having LoA before the age of 10 years. All the other disease milestones studied were also significantly delayed, except the age at first symptoms and age at which LVEF dropped to <30%, which was also delayed; however, the difference from dystrophin‐negative patients was not statistically significant, likely because most patients in both groups were still too young to have reached this disease milestone. Patients showing no dystrophin protein in their Western blots mostly had a disease course typical for DMD, with 74% suffering LoA before the age of 12 years; the median age at LoA was 10.5 years in this group. Our data showed that disease severity correlates with dystrophin quantities even at very low levels, as observed in patient groups with 0 to 0.5% and 0.5 to 5% dystrophin when compared to 0% dystrophin (group A) and ≥5% dystrophin (group C).
Our findings are supported by recently published studies on the mdx;utrn −/− ;Xist(Δhs) mouse model for DMD, which showed that dystrophin levels < 4% are sufficient to improve survival and motor function. 30 Waldrop et al reported a 10‐year‐old boy with a nonsense mutation in exon 42 and dystrophin quantity of 3.2% on quantitative Western blot; he showed an unusually good performance on the 6‐minute walk test for his age (157% compared to age‐matched DMD controls) and was therefore classified as having prospective IMD. 31
A recent genotype–phenotype correlation study revealed a milder phenotype in patients with DMD mutations amenable to exon 44 and exon 8 skipping, but not in patients amenable to exon 51 skipping. 32 The milder phenotype in these patients correlated with the presence of endogenous skipping of exons 44 and 8 in cultured fibroblasts, whereas no endogenous exon skipping was observed in fibroblasts from patients amenable to exon 51 skipping. These data together with our findings suggest that therapeutic restoration of low dystrophin quantities could be beneficial to DMD patients. Accumulating data from long‐term clinical studies suggest slower disease progression following therapeutic exon 51 skipping in DMD patients treated with eteplirsen, 15 , 16 , 17 , 18 although dystrophin restoration levels remained <1%. 13 , 14 However, the question remains as to whether therapeutic restoration of dystrophin, even if started later in life, would be equally beneficial to the congenital presence of low residual dystrophin in patients with endogenous skipping.
We showed that the effect of very low dystrophin quantity on disease progression was independent of medication with corticosteroids and ataluren. However, within‐group analysis of patients with no dystrophin showed that treatment with corticosteroids shifted the age at LoA from 10.3 years to 11.1 years. Thus, our retrospective data are in line with recently published prospective data. 33 Surprisingly, corticosteroids had minimal effects in the group with <5% dystrophin. We could not draw conclusions from this observation, because the effective number was very small and details about dosing and duration of treatment were unavailable.
Interestingly, 3 unrelated patients in our cohort shared the same nucleotide substitution (c.31 + 36947G > A) in intron 1, which resulted in <0.5% dystrophin levels. One patient was ambulant until death at the age of 73 years from a disease unrelated to myopathy; the other 2 patients were ambulant until the ages of 22 years and 66 years. RNA sequencing revealed expression of 2 types of transcripts: a full‐length transcript and a transcript that included a pseudoexon leading to a premature stop codon (data not shown). This suggests that translation of full‐length dystrophin at very low levels is sufficient for the mild phenotype observed in these patients.
Two previously described French patients with BMD with c.31 + 36947G > A and c.3432 + 2036A > G mutations were different from patients in this study with the same mutations. 24 , 25 One patient had relatively high dystrophin levels, and the other a relatively large real‐time polymerase chain reaction (RT‐PCR) band for normal transcripts. Although dystrophin quantification data were not available for these patients, we could not exclude the possibility that patients with the same genotype express different quantities of residual dystrophin. Furthermore, we could not account for whether different muscles express different quantities of residual dystrophin, and whether the quantity changes with time, because repetitive biopsies were not available for the patients in our cohort. The complexity in the medical conditions of the patient cohort was illustrated by Subject 55 of our study, who had no detectable dystrophin at the age of diagnosis (7 years). He never received corticosteroids and lost ambulation at the age of 12 years. However, he then presented with an unusually mild disease progression. At the age of 24 years, he could still move between his bed and wheelchair, had good autonomy of the upper limbs, and a normal VC. His similarly afflicted younger brother, for whom a muscle biopsy was not performed, was still ambulant at the age of 20 years. A future study with multiple‐point Western blot measurement of dystrophin, possibly including siblings, could be of great clinical value. A previously described French patient had the same mutation (c.3432 + 2036A > G) as Subject 42 of our study, and also presented a BMD phenotype. 25 RT‐PCR revealed similarly sized bands of normal and pseudoexon spliced fragments. Western blot data, however, were not available. Subject 42 had low quantities of normal and high quantity of pseudoexon spliced PCR products (data not shown), and 1.93% dystrophin protein in the biopsy. This suggests that the proportion of normally spliced RT‐PCR products in comparison to mutated products or controls has a low predictive value for protein quantity, likely due to variables such as noncontrolled PCR cycles, nonrandom decay of mutated transcripts, and unknown transcription/translation rates.
Our study had some unavoidable limitations. First, this was a retrospective study, and some bias is therefore inevitable. The patients included were born between 1940 and 2009, which is a long period over which there were many changes in diagnosis and standards of care. Patients were followed up at multiple clinical centers, and many were lost to follow‐up. Furthermore, we studied the natural history of disease by using clinical milestones and interventions. The interventions could reflect differences in treatment protocols at the different centers rather than differences in natural histories. However, in our opinion, interventions are also a reflection of clinical events occurring during disease, for example, vertebral fusion surgery performed for scoliosis or noninvasive ventilation started for nocturnal hypoventilation. We had to use these data for assessing disease progression, as approved methods for determining motor function, such as Motor Function Measure scale or 6‐minute walk test, were not performed for all patients. Moreover, although all Western blots were performed in the same laboratory, using the same protocol, and under the supervision of the same geneticist, minimizing interlaboratory variability, 34 exact quantification was not performed using a series of diluted control samples. 35 Control samples were obtained from different individuals, and healthy individuals express different levels of dystrophin. 36 The estimate of dystrophin level therefore depends on the control chosen. Furthermore, in the absence of conventional proteins such as vinculin or alpha‐actinin for normalization, we normalized dystrophin to dysferlin. Dysferlin is not part of the DAGC and, theoretically, the quantity should not vary. However, 2 previous studies showed an upregulation of dysferlin in DMD. 37 , 38 Dysferlin band intensity of the Western blots analyzed in our study did not vary systematically between patients and controls, and therefore we do not believe that we systematically underestimated dystrophin quantity. In our series, 0.51% dystrophin was the lowest quantity that we measured on films after 1‐minute exposure. Dystrophin bands that were only visualized on films after 5 minutes or 60 minutes of exposure were considered indicative of smaller quantities (although >0%). However, precise quantification was not possible for these patients. We also quantified dystrophin by using an antibody against C‐terminal dystrophin to avoid missing dystrophin isoforms with truncations in the core region. Variations in dystrophin quantification have been observed by other researchers using antibodies against the core region or N‐terminus. 8 , 9 , 39 Finally, although we ruled out confounding of the results by the medications received by the patients, we did not analyze the influence of genetic modifiers such as ACTN3 and LTBP4 variants, which are known to modify disease course. 40 , 41
In conclusion, our results demonstrate that residual dystrophin, even in very low quantities, could ameliorate the disease course and produce milder phenotypes.
Author Contributions
Y.d.F. and H.A. contributed to the conception and design of the study; Y.d.F., H.A., R.B.Y., F.L., K.W., I.D., L.S., C.S., and the FILNEMUS Network contributed to the acquisition and analysis of the data, Y.d.F., H.A., R.B.Y., F.L., K.W., A.S., I.D., L.S., and V.L. contributed to drafting the text and preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
TABLE S1 Detailed Milestones of Patients Included
TABLE S2: Detailed Mutations and Western Blot Data of Patients Included
APPENDIX S1 Supporting Information
Acknowledgments
This work was supported by the Duchenne Parents Project France, the Association Française Contre les Myopathies, the Association Monegasque Contre les Myopathies, and the Université Franco‐Allemande (CDFA‐06‐11).
We thank the FILNEMUS network and its collaborators who contributed to the UMD‐DMD database.
References
- 1. Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol 2006;7:762–773. [DOI] [PubMed] [Google Scholar]
- 2. Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod‐shaped cytoskeletal protein. Cell 1988;53:219–228. [DOI] [PubMed] [Google Scholar]
- 3. Hoffman EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in muscle‐biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med 1988;318:1363–1368. [DOI] [PubMed] [Google Scholar]
- 4. Flanigan KM. Duchenne and Becker muscular dystrophies. Neurol Clin 2014;32:671–688. [DOI] [PubMed] [Google Scholar]
- 5. Tuffery‐Giraud S, Béroud C, Leturcq F, et al. Genotype‐phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD‐DMD database: a model of nationwide knowledgebase. Hum Mutat 2009;30:934–945. [DOI] [PubMed] [Google Scholar]
- 6. Koenig M, Beggs AH, Moyer M, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 7. Monaco AP, Bertelson CJ, Liechti‐Gallati S, et al. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2:90–95. [DOI] [PubMed] [Google Scholar]
- 8. Nicholson LV, Johnson MA, Bushby KM, et al. Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data. Part 1. Trends across the clinical groups. J Med Genet 1993;30:728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bulman DE, Murphy EG, Zubrzycka‐Gaarn EE, et al. Differentiation of Duchenne and Becker muscular dystrophy phenotypes with amino‐ and carboxy‐terminal antisera specific for dystrophin. Am J Hum Genet 1991;48:295. [PMC free article] [PubMed] [Google Scholar]
- 10. Anthony K, Arechavala‐Gomeza V, Ricotti V, et al. Biochemical characterization of patients with in‐frame or out‐of‐frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol 2014;71:32–40. [DOI] [PubMed] [Google Scholar]
- 11. Neri M, Torelli S, Brown S, et al. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscul Disord 2007;17:913–918. [DOI] [PubMed] [Google Scholar]
- 12. Finkel RS, Flanigan KM, Wong B, et al. Phase 2a study of ataluren‐mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One 2013;8:e81302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology 2018;90:e2146–e2154. [DOI] [PubMed] [Google Scholar]
- 14. Center for Drug Evaluation and Research, US Food and Drug Administration , Department of Health and Human Services. Application number: 206488Orig1s000. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_summary%20review_Redacted.pdf. Accessed 17 November, 2020.
- 15. Mendell JR, Rodino‐Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–647. [DOI] [PubMed] [Google Scholar]
- 16. Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016;79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khan N, Eliopoulos H, Han L, et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and non‐ambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis 2019;6:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alfano LN, Charleston JS, Connolly AM, et al. Long‐term treatment with eteplirsen in nonambulatory patients with Duchenne muscular dystrophy. Medicine (Baltimore) 2019;98:e15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Frank DE, Schnell FJ, Akana C, et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020;94:e2270–e2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Disset A, Bourgeois CF, Benmalek N, et al. An exon skipping‐associated nonsense mutation in the dystrophin gene uncovers a complex interplay between multiple antagonistic splicing elements. Hum Mol Genet 2006;15:999–1013. [DOI] [PubMed] [Google Scholar]
- 21. Aartsma‐Rus A, Van Deutekom JCT, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading‐frame rule. Muscle Nerve 2006;34:135–144. [DOI] [PubMed] [Google Scholar]
- 22. Juan‐Mateu J, González‐Quereda L, Rodríguez MJ, et al. Interplay between DMD point mutations and splicing signals in dystrophinopathy phenotypes. PLoS One 2013;8:e59916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Todeschini A, Gualandi F, Trabanelli C, et al. Becker muscular dystrophy due to an intronic splicing mutation inducing a dual dystrophin transcript. Neuromuscul Disord 2016;26:662–665. [DOI] [PubMed] [Google Scholar]
- 24. Béroud C, Carrié A, Beldjord C, et al. Dystrophinopathy caused by mid‐intronic substitutions activating cryptic exons in the DMD gene. Neuromuscul Disord 2004;14:10–18. [DOI] [PubMed] [Google Scholar]
- 25. Tuffery‐Giraud S, Saquet C, Chambert S, Claustres M. Pseudoexon activation in the DMD gene as a novel mechanism for Becker muscular dystrophy. Hum Mutat 2003;21:608–614. [DOI] [PubMed] [Google Scholar]
- 26. Trabelsi M, Beugnet C, Deburgrave N, et al. When a mid‐intronic variation of DMD gene creates an ESE site. Neuromuscul Disord 2014;24:1111–1117. [DOI] [PubMed] [Google Scholar]
- 27. Arora H, Willcocks RJ, Lott DJ, et al. Longitudinal timed function tests in Duchenne muscular dystrophy: ImagingDMD cohort natural history. Muscle Nerve 2018;58:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Anderson LV, Davison K. Multiplex Western blotting system for the analysis of muscular dystrophy proteins. Am J Pathol 1999;154:1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deburgrave N, Daoud F, Llense S, et al. Protein‐ and mRNA‐based phenotype‐genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat 2007;28:183–195. [DOI] [PubMed] [Google Scholar]
- 30. van Putten M, Hulsker M, Young C, et al. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double‐knockout mice. FASEB J 2013;27:2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waldrop MA, Gumienny F, El Husayni S, et al. Low‐level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul Disord 2018;28:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang RT, Barthelemy F, Martin AS, et al. DMD genotype correlations from the Duchenne registry: endogenous exon skipping is a factor in prolonged ambulation for individuals with a defined mutation subtype. Hum Mutat 2018;39:1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDonald CM, Henricson EK, Abresch RT, et al. Long‐term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 2017;391:451–461. [DOI] [PubMed] [Google Scholar]
- 34. Anthony K, Arechavala‐Gomeza V, Taylor LE, et al. Dystrophin quantification: biological and translational research implications. Neurology 2014;83:2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schnell FJ, Donoghue C, Dworzak J, et al. Development of a validated Western blot method for quantification of human dystrophin protein used in phase 2 and 3 clinical trials of eteplirsen for the treatment of Duchenne muscular dystrophy. Neuromuscul Disord 2017;27:S16. [Google Scholar]
- 36. Beekman C, Janson AA, Baghat A, et al. Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS One 2018;13:e0195850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vontzalidis A, Terzis G, Manta P. Increased dysferlin expression in Duchenne muscular dystrophy. Anal Quant Cytopathol Histopathol 2014;36:15–22. [PubMed] [Google Scholar]
- 38. Waddell LB, Lemckert FA, Zheng XF, et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T‐system with stretch. J Neuropathol Exp Neurol 2011;70:302–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Na S‐J, Kim W‐J, Kim SM, et al. Clinical, immunohistochemical, Western blot, and genetic analysis in dystrophinopathy. J Clin Neurosci 2013;20:1099–1105. [DOI] [PubMed] [Google Scholar]
- 40. van den Bergen JC, Hiller M, Böhringer S, et al. Validation of genetic modifiers for Duchenne muscular dystrophy: a multicentre study assessing SPP1 and LTBP4 variants. J Neurol Neurosurg Psychiatry 2015;86:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hogarth MW, Houweling PJ, Thomas KC, et al. Evidence for ACTN3 as a genetic modifier of Duchenne muscular dystrophy. Nat Commun 2017;8:14143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Detailed Milestones of Patients Included
TABLE S2: Detailed Mutations and Western Blot Data of Patients Included
APPENDIX S1 Supporting Information