

ABSTRACT
Pulmonary complications in CTD are common and can involve the interstitium, airways, pleura and pulmonary vasculature. ILD can occur in all CTD (CTD‐ILD), and may vary from limited, non‐progressive lung involvement, to fulminant, life‐threatening disease. Given the potential for major adverse outcomes in CTD‐ILD, accurate diagnosis, assessment and careful consideration of therapeutic intervention are a priority. Limited data are available to guide management decisions in CTD‐ILD. Autoimmune‐mediated pulmonary inflammation is considered a key pathobiological pathway in these disorders, and immunosuppressive therapy is generally regarded the cornerstone of treatment for severe and/or progressive CTD‐ILD. However, the natural history of CTD‐ILD in individual patients can be difficult to predict, and deciding who to treat, when and with what agent can be challenging. Establishing realistic therapeutic goals from both the patient and clinician perspective requires considerable expertise. The document aims to provide a framework for clinicians to aid in the assessment and management of ILD in the major CTD. A suggested approach to diagnosis and monitoring of CTD‐ILD and, where available, evidence‐based, disease‐specific approaches to treatment have been provided.
Keywords: clinical diagnosis and management, collagen vascular disease, connective tissue disease, interstitial lung disease
Abbreviations
- AAV
ANCA‐associated vasculitis
- Ab
antibody
- AE
adverse event
- AIP
acute interstitial pneumonia
- ANA
anti‐nuclear antibody
- ANCA
anti‐neutrophil cytoplasmic antibody
- ASSIST
American Scleroderma Stem Cell versus Immune Suppression Trial
- ASTIS
autologous stem cell transplantation
- ATS
American Thoracic Society
- AZA
azathioprine
- BAL
bronchoalveolar lavage
- BP
blood pressure
- CADM
clinically amyopathic DM
- CCP
cyclic citrullinated peptide
- COPD
chronic obstructive pulmonary disease
- CPFE
combined pulmonary fibrosis and emphysema
- CT
computed tomography
- CTD
connective tissue disease
- CTD‐ILD
CTD‐associated ILD
- CYC
cyclophosphamide
- DAD
diffuse alveolar damage
- DLCO
diffusing capacity for carbon monoxide
- DM
dermatomyositis
- dsDNA
double‐stranded DNA
- ENA
extractable nuclear antigen
- ERS
European Respiratory Society
- ESC
European Society of Cardiology
- EUC
electrolyte and urea concentration
- FB
follicular bronchiolitis
- FBC
full blood count
- f/u
follow‐up
- FVC
forced vital capacity
- GAP
gender, age and pulmonary function tests
- GI
gastrointestinal
- GORD
gastro‐oesophageal reflux disease
- GPA
granulomatosis with polyangiitis
- HBV
hepatitis B virus
- HRQoL
health‐related quality of life
- HSCT
haematopoietic stem cell transplant
- IBM
inclusion body myositis
- IIM
idiopathic inflammatory myopathy
- IIM‐ILD
IIM‐associated ILD
- IL
interleukin
- ILD
interstitial lung disease
- IMNM
immune‐mediated necrotizing myopathy
- INBUILD
Nintedanib in Progressive Fibrosing Lung Disease
- IPAF
interstitial pneumonia with autoimmune feature
- IPF
idiopathic pulmonary fibrosis
- IVIG
IV immunoglobulin
- LFT
liver function test
- LIP
lymphocytic interstitial pneumonia
- LTBI
latent TB infection
- LTOT
long‐term oxygen therapy
- MALT
mucosa‐associated lymphoid tissue
- MCTD
mixed CTD
- MCTD‐ILD
MCTD‐associated ILD
- MDM
multidisciplinary meeting
- MMF
mycophenolate mofetil
- MPA
microscopic polyangiitis
- MPO
myeloperoxidase
- mRSS
modified Rodnan skin score
- MSA
myositis‐specific autoantibody
- MTX
methotrexate
- NSIP
non‐specific interstitial pneumonia
- NXP
nuclear matrix protein
- OP
organizing pneumonia
- PAH
pulmonary arterial hypertension
- PFT
pulmonary function test
- PH
pulmonary hypertension
- PJP
Pneumocystis jirovecii pneumonia
- PM
polymyositis
- PR
pulmonary rehabilitation
- PR3
proteinase 3
- RA
rheumatoid arthritis
- RA‐ILD
RA‐associated ILD
- RCT
randomized controlled trial
- RF
rheumatoid factor
- RNP
ribonucleoprotein
- SAE
serious AE
- SCOT
Scleroderma: Cyclophosphamide or Transplant trial
- SENSCIS
Safety and Efficacy of Nintedanib in Systemic Sclerosis
- SjS
Sjögren's syndrome
- SjS‐ILD
SjS‐associated ILD
- SLE
systemic lupus erythematosus
- SLS
Scleroderma Lung Study
- SSc
systemic sclerosis
- SSc‐ILD
SSc‐associated ILD
- TB
tuberculosis
- TPMT
test for thiopurine methyltransferase
- TSANZ
Thoracic Society of Australia and New Zealand
- UCTD
undifferentiated CTD
- UIP
usual interstitial pneumonia
- VATS
video‐assisted thoracoscopic surgery
- WHO
World Health Organization
CONTENTS
Introduction
Methods
Approach to diagnosing CTD‐ILD
Clinical diagnosis of CTD
Serology
Imaging
Histopathology
Lung biopsy selection and techniques
Bronchoscopy and lavage
Multidisciplinary meeting
Approach to management of CTD‐ILD
General principles of monitoring and treatment
Staging and prognostication
Deciding when to treat
Pharmacological/immunosuppressive treatment
Special considerations
Latent tuberculosis infection
Hepatitis B
Pneumocystis jirovecii
Specific CTD‐ILD
Systemic sclerosis
Stem cell treatment for scleroderma‐associated ILD
Rheumatoid arthritis
Idiopathic inflammatory myopathy
Other CTD
Systemic lupus erythematosus
Sjögren's syndrome
Undifferentiated and mixed connective tissue disease
Treatment of SLE, SjS, UCTD and MCTD‐ILD
Interstitial pneumonia with autoimmune features
Vasculitis and ILD
Psoriasis and ILD
Special considerations in CTD‐ILD management
CTD‐ILD with pulmonary hypertension
Lung transplantation for CTD
Non‐pharmacological management in CTD‐ILD
Pulmonary rehabilitation
Supplemental oxygen
Symptom management and palliative care
Future directions
Summary and conclusions
INTRODUCTION
The connective tissue diseases (CTD) include a variety of disease entities characterized by end‐organ damage mediated by immune system overactivity. Pulmonary complications are common and can involve the interstitium, airways, pleura, pulmonary vasculature and chest wall. 1 Interstitial lung disease (ILD) can occur in all CTD (CTD‐associated ILD (CTD‐ILD)), and may vary from limited, non‐progressive lung involvement, to fulminant, life‐threatening disease. Given the potential for major adverse outcomes in CTD‐ILD, accurate diagnosis, assessment and careful consideration of therapeutic intervention are a priority.
In contrast to well‐defined diagnostic criteria and evidence‐based treatment recommendations for idiopathic pulmonary fibrosis (IPF), limited data are available to guide management decisions in CTD‐ILD. 2 , 3 Autoimmune‐mediated pulmonary inflammation is considered a key pathobiological pathway in these disorders, with immunosuppressive therapy generally accepted as the cornerstone of treatment for severe and/or progressive CTD‐ILD. However, the natural history of CTD‐ILD in individual patients can be difficult to predict, and deciding who to treat, when and with what agent can be challenging. Establishing realistic therapeutic goals from both the patient and clinician perspective requires considerable expertise.
This document aims to provide a framework for clinicians to aid in the assessment and management of ILD in the major CTD. Non‐ILD and extra‐pulmonary manifestations of CTD will not be discussed in detail. A suggested approach to diagnosis and monitoring of CTD‐ILD and, where available, evidence‐based, disease‐specific approaches to treatment have been provided.
Methods
This position paper highlights important aspects in the diagnosis and treatment of CTD‐ILD, with particular reference to Australia and New Zealand. The document is not intended as a treatment guideline. G.J.K. was appointed as the chair of a 26‐member panel with expertise in CTD‐ILD, including 17 respiratory physicians, two rheumatologists and an immunologist, pathologist, radiologist, physiotherapist, exercise physiologist, respiratory nurse and patient representative.
After initial face‐to‐face discussion, specific sections were assigned to individual authors for comprehensive literature review. All authors had the opportunity to review all content and referenced articles, and contribute to all sections in subsequent meetings (conducted as both face‐to‐face and telephone discussions). Summary statements have been provided at the end of each major section. These statements are based on published data where available (with specific references cited) or expert opinion by consensus agreement where evidence is lacking. Two authors (A.S.J. and G.J.K.) compiled and edited the final manuscript that was reviewed and approved by all authors. The committee did not receive any commercial sponsorship and all panel members worked on an honorary basis.
APPROACH TO DIAGNOSING CTD‐ILD
Clinical diagnosis of CTD
All CTD can be associated with ILD and, when present, accounts for substantial morbidity and mortality. 4 The estimated prevalence of ILD in the major CTD is shown in Table 1. In most cases of CTD‐ILD, the underlying CTD is established at the time of diagnosis. However, ILD can be the presenting (forme fruste) or only manifestation of an underlying CTD. For example, ILD is the presenting disease manifestation in up to 10% of rheumatoid arthritis (RA) patients, while in clinically amyopathic dermatomyositis (CADM), anti‐synthetase syndrome and systemic sclerosis (SSc) sine scleroderma, ILD may persist as the only manifestation of the CTD. 18 , 19 , 20 , 21 , 22 Therefore, in all patients with ILD it is vital to assess for subtle and non‐volunteered features of CTD based on history and clinical examination.
Table 1.
Summary of estimated prevalence, clinical signs and symptoms, manifestations and non‐ILD respiratory manifestation in the major CTD
| CTD | Prevalence of ILD | Clinical signs and symptoms | ILD manifestations | Non‐ILD respiratory manifestations 5 |
|---|---|---|---|---|
| SSc 6 |
Detectable: up to 75% |
Raynaud's phenomenon Sclerodactyly Digital ulcers/pits Telangiectasia Skin thickening GI involvement |
Most common: NSIP Other: UIP, OP |
PAH Pulmonary lymphoma Aspiration pneumonia |
| RA 9 |
Detectable: 30–60% Clinically evident: 10–30% 10 |
Polyarticular synovitis Rheumatoid skin nodules |
Most common: UIP Other: NSIP, OP |
Bronchiectasis, bronchiolitis Rheumatoid lung nodules Pleural disease ± effusion |
| IIM 11 |
Up to 80% in anti‐synthetase syndrome |
Proximal symmetrical muscle weakness Gottron's sign Gottron's papules Heliotrope rash Shawl sign Mechanic's hands |
Most common: NSIP Other: OP, UIP, AIP/DAD |
Respiratory muscle weakness |
| SLE 14 |
Detectable: up to 30% Clinically evident: 3–11% 15 |
Photosensitive and/or malar rash Oral ulcers Alopecia Serositis |
Most common: NSIP Other: LIP, OP, UIP, FB, AIP/DAD |
Pleuritis Diffuse alveolar haemorrhage Respiratory muscle weakness ‘Shrinking lung syndrome’ |
| SjS 16 | 10–30% 8 |
Sicca symptoms Enlarged parotid glands |
Most common: NSIP Other: OP,LIP, UIP, FB |
MALT lymphoma Amyloidosis Bronchiectasis |
| MCTD 17 | 20–85% 8 |
Swollen hands Synovitis Myositis Acrosclerosis Raynaud's phenomenon |
Most common: NSIP Other: UIP, OP |
Pleuritis Diffuse alveolar haemorrhage Respiratory muscle weakness Aspiration pneumonia |
AIP, acute interstitial pneumonia; CTD, connective tissue disease; DAD diffuse alveolar damage; FB, follicular bronchiolitis; GI, gastrointestinal; IIM, idiopathic inflammatory myopathy; LIP, lymphocytic interstitial pneumonia; MALT, mucosa‐associated lymphoid tissue; MCTD, mixed CTD; NSIP, non‐specific interstitial pneumonia; OP, organizing pneumonia; PAH, pulmonary arterial hypertension; RA, rheumatoid arthritis; SjS, Sjögren's syndrome; SLE, systemic lupus erythematosus; SSc, systemic sclerosis; UIP, usual interstitial pneumonia.
A non‐exhaustive list of the clinical signs and symptoms associated with the major CTD is presented in Table 1. In practice, a reasonable screening approach would include enquiry for the presence of inflammatory joint pain or stiffness, Raynaud's phenomenon (especially recent onset), rashes, sicca symptoms (dry eyes and/or dry mouth) or skeletal muscle symptoms (e.g. proximal weakness and myalgia), combined with examination for synovitis, cutaneous signs (e.g. mechanic's hands and malar rash) and proximal muscle strength. A rheumatology review may be helpful if symptoms or signs are difficult to interpret or if further investigations are being considered.
International consensus classification criteria for specific CTD are referenced (Table 1) and are a helpful guide for diagnosis. In addition, some patients have ‘overlap’ features across several diagnoses, and some patients do not fulfil criteria for specific CTD. In the context of ILD, these patients may be considered as meeting the research criteria for interstitial pneumonia with autoimmune feature (IPAF).
Serology
Testing for the presence of autoantibodies is an integral step in the diagnostic work up for a suspected CTD and closely interacts with the clinical assessment summarized above. Certain autoantibody profiles are characteristic or strongly suggestive of a particular CTD, can focus clinical assessment for other organ involvement and assist with determining prognosis and treatment. The development of tests for extended autoantibody profiles, such as the myositis‐specific autoantibody (MSA) panel, has helped more accurately identify and characterize patients with CTD‐ILD, especially when disease manifestations are subtle. Typical radiological findings (such as rapidly progressive organizing pneumonia (OP), characteristic of anti‐synthetase syndrome) may also prompt more detailed serological testing. Major treatment decisions can hinge upon serology, for example, in amyopathic dermatomyositis (DM) where the combination of a positive autoantibody and ILD may be the only evidence of a CTD. Autoantibodies and their disease associations are shown in Table 2 and discussed in more detail in disease‐specific sections.
Table 2.
Most common auto‐Ab associations in the major CTD
| CTD | Auto‐Ab | Clinical associations |
|---|---|---|
| Systemic sclerosis | ANA | |
| Anti‐centromere | Strong association with PAH; ‘limited’ disease | |
| Anti‐Scl 70 (topoisomerase I) | Strong association with ILD; ‘diffuse’ disease | |
| Anti‐RNA polymerase I, III |
Increased risk of renal crisis with concomitant corticosteroid use Not included on most ENA panels |
|
| RA | RF | |
| Anti‐CCP | Strong association with RA‐ILD | |
| Idiopathic inflammatory myositis |
ANA Myositis‐specific Ab: Anti‐tRNA synthetase (Jo‐1, PL7, PL12, EJ, OJ) |
Associated with high prevalence of ILD |
| MDA5 | May be associated with rapidly progressive ILD | |
| Mi2, TIF1y, NXP2, SAE | TIF1y, NXP2 increased risk of malignancy | |
|
SRP, HMGCR |
IMNM; severe muscle disease | |
|
Myositis‐associated Ab: PM‐Scl, Ro52, Ku |
Also detected in other CTD | |
| Systemic lupus erythematosus | ANA | Present in >95% of cases |
| Anti‐dsDNA | Titre often correlates with disease activity | |
| Anti‐Sm, ‐RNP, ‐ribosomal P | ||
| Anti‐Ro60 (SSA), ‐Ro52, ‐La (SSB) | ||
| Sjögren's syndrome | ANA | |
| Anti‐Ro60 (SSA), ‐Ro52, ‐La (SSB) | Some patients may have a positive RF | |
| Mixed connective tissue disease | ANA | |
| Anti‐RNP |
Ab, antibody; ANA, anti‐nuclear antibody; CCP, cyclic citrullinated peptide; CTD, connective tissue disease; dsDNA, double‐stranded DNA; ENA, extractable nuclear antigen; ILD, interstitial lung disease; IMNM, immune‐mediated necrotizing myopathy; NXP, nuclear matrix protein; PAH, pulmonary arterial hypertension; PM, polymyositis; RA, rheumatoid arthritis; RA‐ILD, RA‐associated ILD; RF, rheumatoid factor; RNP, ribonucleoprotein; SAE, serious adverse event.
The most common methods for detecting autoantibodies are indirect immunofluorescence (e.g. anti‐nuclear antibody (ANA) and double‐stranded DNA (dsDNA) autoantibodies), enzyme‐linked immunosorbent assay (ELISA; e.g. extractable nuclear antigen (ENA) screen, dsDNA autoantibodies and anti‐cyclic citrullinated peptide (CCP) antibody) and line immunoblot (e.g. MSA panel). Individual centres may have access to immunoprecipitation or other bespoke immunological techniques. Each technique has unique strengths and weaknesses, balancing sensitivity, specificity, reproducibility, cost and convenience. Different laboratories employ various techniques and may modify the thresholds used to report ‘positive/negative’ or ‘borderline’ results. Thus, serology results must always be interpreted within the clinical context of each patient and not simply taken at ‘face value’. The input of a rheumatologist or immunologist on the relative importance of positive results can be helpful in the clinical context.
Imaging
The principal roles of high‐resolution computed tomography (HRCT) in the evaluation of suspected CTD‐ILD are to: (i) confirm the presence of ILD and (ii) provide a radiological classification of disease pattern. 23 Almost all radiological patterns can occur in CTD‐ILD, with a non‐specific interstitial pneumonia (NSIP) pattern the most frequently observed (Table 1, Fig. 1). The exception is RA where the usual interstitial pneumonia (UIP) pattern predominates and is predictive of poorer outcome. 24 , 25 Other common radiological patterns include OP, lymphocytic interstitial pneumonia (LIP) and some patients may demonstrate a variety of overlapping HRCT patterns. 23 , 26 , 27 , 28
Figure 1.
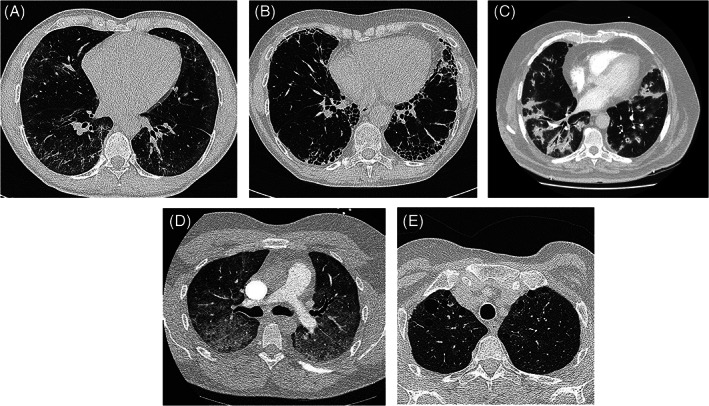
Common radiological patterns of interstitial lung disease (ILD) in connective tissue disease (CTD). (A) Non‐specific interstitial pneumonia (NSIP): ground‐glass change and limited reticulation; predominantly basal, subpleural distribution. (B) Usual interstitial pneumonia (UIP): subpleural, basal predominant reticulation with honeycomb change. No ground‐glass change, nodules or air‐trapping (features ‘inconsistent’ with radiological UIP). (C) Organizing pneumonia (OP): dense, patchy predominantly peribronchial consolidation affecting the lower lobes bilaterally and symmetrically. (D) Acute interstitial pneumonia (AIP): widespread ground‐glass infiltrates in a patient with previously undiagnosed systemic lupus erythematosus (SLE) presenting with acute respiratory failure. (E) Lymphocytic interstitial pneumonia (LIP): variably sized thin‐walled cysts. Ground‐glass change and centrilobular nodules may also occur (not seen here).
Thin‐section volumetric computed tomography (CT, 0.5–1.5 mm) is essential for optimal spatial resolution and appreciation of parenchymal detail. 29 Prone sequences help establish whether abnormalities reflect true disease or gravity‐dependent change, particularly relevant in the radiological diagnosis of early (or mild) NSIP. Sequences obtained during full expiration highlight features of air‐trapping, relevant in the CTD associated with small airways disease (including bronchiectasis, obliterative bronchiolitis and follicular bronchiolitis). Indeed, airways disease can be an early manifestation of RA‐associated ILD (RA‐ILD), and inspiratory sequences may appear normal or near‐normal. 29 , 30 HRCT also aids the assessment of complications such as infection, drug‐induced pneumonitis, acute exacerbations and malignancy, with increased incidence among certain CTD. 23 , 27 , 29 , 30 , 31 , 32 HRCT also allows rapid evaluation of ILD extent, largely independent of confounding factors (e.g. concurrent emphysema or pulmonary hypertension (PH)) which may impact pulmonary function tests (PFT).
The syndrome of combined pulmonary fibrosis with emphysema (CPFE) has been described in RA and SSc, sometimes without a significant smoking history. 33 As with idiopathic CPFE, this may represent a distinct clinical phenotype in genetically susceptible individuals with appropriate tobacco exposure. Pleuroparenchymal fibroelastosis (PPFE) is a rare entity that has recently been described in a mixed cohort of CTD patients and may be associated with increased risk of respiratory‐related death. 34
Histopathology
Similar to radiological patterns, NSIP is the predominant histopathological sub‐type found in CTD‐ILD (except for RA) and often overlaps with other patterns such as OP and UIP (Table 1). 35 , 36 , 37 Even where radiology may initially demonstrate more inflammatory changes such as ground‐glass opacification, CTD‐ILD can evolve to a pattern more in‐keeping with UIP, with ‘honeycomb’ cystic lung destruction and irreversible fibrosis. Extent of traction bronchiectasis and extent of honeycombing are independent predictors of mortality and are more likely to develop if the inflammatory ILD is left untreated or under‐treated in its earlier stages. 38
Lung biopsy selection and techniques
Lung biopsy is generally not required for the diagnosis of CTD‐ILD, with HRCT highly specific for discriminating disease pattern in this population. 37 , 39 Histopathology can have prognostic implications but this information seldom influences treatment decisions. Lung biopsy may be necessary to exclude non‐autoimmune causes for pulmonary infiltrates or nodules (e.g. malignancy) and a reasonable strategy would be to discuss the need for biopsy at a multidisciplinary meeting (MDM) prior to proceeding.
In the rare situations where lung biopsy is considered in CTD‐ILD, options include transbronchial lung biopsy using forceps, transbronchial lung cryobiopsy and video‐assisted thoracoscopic surgery (VATS) surgical lung biopsy. For investigation of nodules, radiologic‐guided core biopsy may be reasonable, but has limited utility outside of this indication. VATS lung biopsy is the gold standard for histopathological sampling of lung parenchyma and provides a diagnostic yield of over 88% in ILD. 2 With an overall morbidity rate of 10–30% and mortality risk of 1–2%, this modality should be reserved for younger patients with well‐preserved physiology and fewer comorbidities. 40 , 41 In general, the small biopsy size and sampling error of traditional transbronchial biopsy limit its role in the diagnosis of ILD, but may be considered in airway‐centred processes such as OP. Cryobiopsy has been shown to have superior diagnostic yield compared with conventional forceps biopsy in studies of mixed ILD populations, with larger tissue sample size and absence of crush artefact. 42 The recently published Cryobiopsy versus Open Lung Biopsy in the Diagnosis of Interstitial Lung Disease (COLDICE) study reported high levels of agreement between cryobiopsy and surgical lung biopsy for both histopathological interpretation and MDM diagnoses in patients with difficult to classify ILD who required histopathological evaluation to aid diagnosis. 43 Current American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Society (ATS/ERS/JRS/ALAT) IPF diagnostic guidelines do not make recommendations for or against the use of cryobiopsy for ILD diagnosis outside of expert centres. 2 A recent expert consensus statement from the American College of Chest Physicians provides guidance for the procedural aspects of cryobiopsy, without specific recommendations on cryobiopsy application in CTD‐ILD. 44
Bronchoscopy and lavage
Bronchoscopy with bronchoalveolar lavage (BAL) may be indicated in the CTD‐ILD patients with unexplained radiological lung infiltrates. Analysis of BAL cellular composition and microbial cultures can help distinguish possible differential diagnoses including infection, haemorrhage and drug‐induced eosinophilic infiltrate. BAL leucocyte differential counts in CTD‐ILD have been described in the research setting but seldom influence treatment decisions. 45
Multidisciplinary meeting
Comprehensive description of the core features, governance and constitution of the ILD‐MDM have recently been detailed in a joint Thoracic Society of Australia and New Zealand (TSANZ)/Lung Foundation Australia position statement. 46 In brief, the ILD‐MDM consists of the face‐to‐face discussion of clinical and diagnostic material by clinicians caring for the patient with respiratory physicians, a radiologist and, if relevant, a histopathologist with expertise in ILD (Fig. 2). Rheumatologists may also play an important role in the ILD‐MDM, with CTD‐ILD among the most common presentations to the ILD‐MDM after IPF. 47
Figure 2.
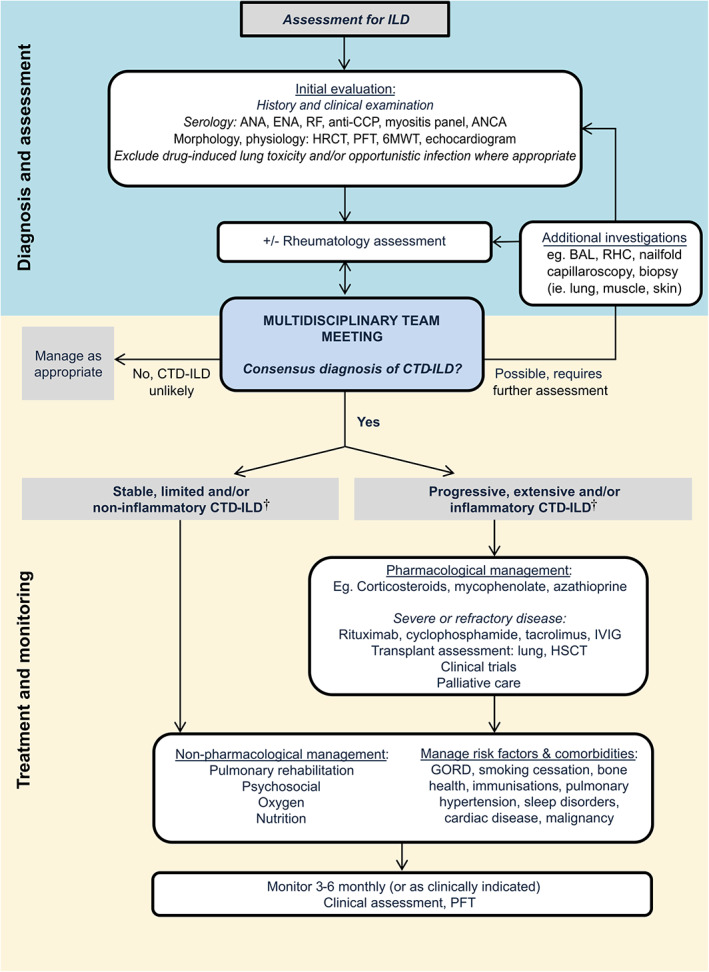
Suggested algorithm for the diagnosis and management of connective tissue disease‐associated interstitial lung disease (CTD‐ILD). †For detailed discussion, see the section ‘Deciding when to treat’.
Specific studies on the role of MDM in CTD‐ILD are limited. Walsh et al. investigated the agreement between seven ILD‐MDM for the diagnosis of 70 ILD cases. 48 Agreement for a diagnosis of CTD‐ILD was the highest for any ILD (weighted kappa: 0.73). 48 However, independent clinician agreement before MDM discussion was already high (weighted kappa: 0.76), suggesting that MDM discussion may not be essential if CTD‐ILD diagnosis is clear cut. However, MDM also aid the re‐classification of previously unrecognized CTD‐ILD, mainly from unclassifiable and ‘IPF’ referrals, with important implications for prognosis and management. The frequency of CTD‐ILD diagnosis increased from 10% to 21% after MDM discussion in a study of 90 consecutive ILD‐MDM cases. 49
With the evolution of ILD nomenclature (including the Fleischner criteria), and the nuances of designating likely histology from HRCT appearances, the ILD‐MDM is an increasingly important forum for integrating ‘clinical context’ with HRCT patterns, informing prediction of disease behaviour and treatment decisions. 50 In addition, in complex CTD‐ILD cases, the ILD‐MDM may be useful in staging severity and assessing serial change. 51
Summary.
Integration of clinical, serological and HRCT findings, coupled with ILD‐MDM discussion, is key to confirming an accurate diagnosis of CTD‐ILD. 46 , 51
NSIP is the most frequent radiological and histological ILD pattern seen in association with CTD. 23 , 35 , 36 , 37
Lung biopsy generally does not contribute significant additional diagnostic or prognostic information and should be reserved for cases of major diagnostic uncertainty following ILD‐MDM discussion. 37 , 39 , 40 , 41
APPROACH TO MANAGEMENT OF CTD‐ILD
General principles of monitoring and treatment
Given the heterogeneous disease course of CTD‐ILD, accurate prognostication involving staging of ILD severity and assessment of disease progression is important when making treatment decisions. An understanding of the natural history of the specific CTD‐ILD in question also provides important information. For example, whilst SSc‐associated ILD (SSc‐ILD) may follow a relatively indolent course and require only careful monitoring, myositis‐associated ILD (idiopathic inflammatory myopathy‐associated ILD (IIM‐ILD)) can progress rapidly, necessitating intensive immunosuppressive therapy. Importantly, drug‐induced ILD and infection (including opportunistic infection) should always be considered in the setting of new‐onset interstitial changes in CTD, particularly in those receiving immunosuppressive therapy.
Staging and prognostication
Staging CTD‐ILD severity at diagnosis allows survival estimation and establishment of a baseline from which to gauge future change. In addition to clinical examination, baseline investigations typically include PFT, HRCT and echocardiography in certain circumstances (e.g. SSc‐ILD).
There is no universally accepted staging system for CTD‐ILD, with several different models proposed. The ILD‐GAP index (ILD subtype, gender, age and pulmonary function tests) has been validated across a range of ILD, including CTD‐ILD. 52 An important exception appears to be IIM‐ILD, where the ILD‐GAP model is a poor predictor of mortality risk. 53 In SSc‐ILD, an algorithm based on HRCT appearances and PFT defines ‘limited’ or ‘extensive’ disease (Fig. 3). 54 Both staging systems demonstrate similar survival estimates for patients with more severe or extensive CTD‐ILD, identifying a population that may benefit from treatment intervention.
Figure 3.
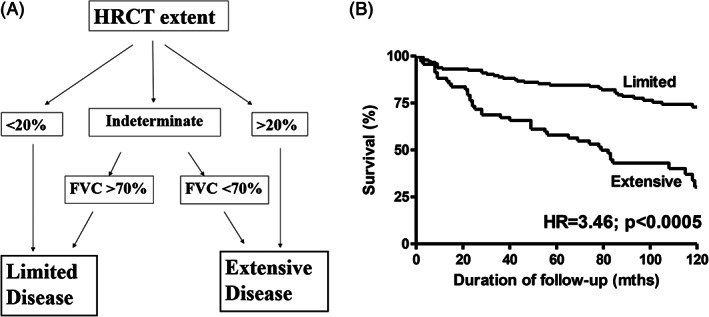
(A) A simple staging system for systemic sclerosis‐associated interstitial lung disease (SSc‐ILD). (B) Survival curves for SSc‐ILD patients with ‘limited’ versus ‘extensive’ disease (hazard ratio (HR) = 3.46; P < 0.0005) (Adapted from Goh et al., 54 with permission).
Disease trajectory is a powerful prognostic determinant and longitudinal measurement of PFT is vital to monitoring ILD progression. 55 , 56 A decline in forced vital capacity (FVC) ≥10% and/or diffusing capacity for carbon monoxide (DLCO) ≥15% on serial measures indicates clinically significant disease progression and is associated with increased mortality risk. 38 , 56 , 57 , 58 However, even ‘marginal’ declines (e.g. <10% FVC) can represent progressive ILD in the appropriate context, particularly if associated with deterioration in symptoms and other measures (e.g. oxygen saturation and 6‐min walk distance). 59 A number of unique confounders should be kept in mind when interpreting PFT in CTD‐ILD, including concomitant PH contributing to a reduced DLCO, and the presence of muscular weakness, pleural disease and/or thoracic skin thickening (particularly in diffuse cutaneous SSc) that can reduce lung volumes independent of the ILD.
HRCT provides important diagnostic and staging information, but serial HRCT is not recommended to routinely monitor disease progression. Repeat HRCT imaging may be useful to corroborate a decline in PFT and exclude other causes of deterioration (e.g. infection, pulmonary embolism and malignancy). Computer‐based quantitative CT analysis and molecular and genetic biomarkers as objective measures of disease prognosis are exciting and evolving research areas, not yet validated for clinical use. 60 , 61
Deciding when to treat
Accurately distinguishing patients with inherently stable CTD‐ILD who can be carefully observed without treatment, from those at risk of progression requiring therapy is one of the most challenging aspects of CTD‐ILD management. 62 , 63 Whilst the goal of treatment in most cases of CTD‐ILD is to achieve stability and prevent further progression, a number of important exceptions to this rule exist and are discussed in more detail below. The decision to treat and the intensity of treatment are influenced by a variety of factors including the CTD subtype, ILD severity, observed disease trajectory and mitigating patient factors. Progressive ILD, particularly in the context of recent onset systemic CTD manifestations, is another important factor influencing treatment decisions. The presence of extra‐pulmonary CTD manifestations (e.g. active synovitis in RA for which mycophenolate and azathioprine are largely ineffective) may also impact treatment strategies, highlighting the importance of multidisciplinary involvement between respiratory physicians, rheumatologists and immunologists. Referral to an ILD expert centre may be useful when deciding who, when and how to treat. Treatment approaches based on estimated disease behaviour, coupled with treatment goals and monitoring strategies, have been adapted from the idiopathic interstitial pneumonias (IIP) and summarized in Table 3.
Table 3.
Disease behaviour, treatment goals and intervention in CTD‐ILD
| Disease behaviour | Treatment goal | Intervention and monitoring |
|---|---|---|
| Reversible, self‐limited | Achieve regression | Short‐term (3–6 months) observation to confirm disease regression |
| Reversible, with risk of progression | Achieve regression and then stabilize | Intensive/higher dose treatment with short‐term monitoring to ensure response. Then, rationalize to maintenance treatment and long‐term observation to ensure stability |
| Stable, risk of progression | Maintain status | Maintenance treatment. Monitor for disease progression |
| Progressive, irreversible with potential for stabilization | Stabilize | Intensive/higher dose treatment and then rationalize to maintenance treatment. Short‐term monitoring to ensure response and then long‐term observation to ensure stability |
| Progressive, despite treatment | Slow disease progression |
Rescue therapy Trials Transplantation Palliative/best supportive case |
Adapted from Travis et al., 64 with permission.
CTD‐ILD, connective tissue disease‐associated interstitial lung disease.
Treatment strategies are largely drawn from studies in SSc‐ILD and extrapolated to the other CTD‐ILD. Whilst reasonable in the absence of specific evidence, caution is required, especially in more progressive CTD‐ILD, such as IIM‐ILD. CTD‐ILD that is limited (HRCT extent <20% and/or FVC >70%) with stable lung function over time can generally be closely monitored with serial PFT and clinical assessment (Fig. 3A). In contrast, extensive and/or progressive ILD usually warrants treatment. 56 , 58 , 65 However, extensive ILD alone is not an indication for therapy with established fibrosis with stable lung function and symptoms less likely to respond to immunosuppression and careful monitoring without pharmacotherapy may be appropriate. A suggested algorithm for the broad management of CTD‐ILD is shown in Figure 2.
Pharmacological/immunosuppressive treatment
Immunosuppression is the mainstay of pharmacotherapy in CTD‐ILD, aiming to reduce inflammation, minimize progression to established fibrosis and permanent loss of lung function. 62 The general mechanism of action is to interfere with critical pathways in the inflammatory cascade including B‐ and T‐cell function, and inhibit pro‐inflammatory cytokines, although precise mechanisms are poorly understood. 66 The major classes of immunosuppressive medications, their mechanism of action and potential adverse effects are summarized in Table 4. Prior to commencement of immunosuppressive therapy, screening for active/latent microorganisms and organ dysfunction is recommended due to the increased risk of new or reactivated infection (e.g. tuberculosis (TB) and hepatitis) and side effects. 67
Table 4.
Common immunosuppressive therapies utilized in CTD‐ILD
| Medication and mechanism | Dosage | Adverse effects | Screening and monitoring |
|---|---|---|---|
|
Corticosteroids Bind to the intracellular glucocorticoid receptor → inhibits cytokine transcription → (1) ↓ T cells (IL2 inhibited) (2) Eosinophil apoptosis (directly or by inhibition of IL5) (3) Macrophage inhibition (blocking IL1 and TNF‐α) (4) Leucocytosis B cells are not significantly inhibited |
Start 0.5 mg/kg/day. Aim to taper down to 10–20 mg maintenance |
Diabetes Weight gain Hypertension Myopathy Osteoporosis Accelerated atherosclerosis Sleep disturbance Cataracts Glaucoma Dyspepsia Pregnancy risk category A |
BP Serum glucose Lipid profile Eye examination Bone densitometry |
|
AZA Inhibits DNA and RNA synthesis in mainly T cells but also B cells |
2.0–2.5 mg/kg/day if TPMT within normal limits |
Bone marrow suppression GI intolerance Hepatotoxicity Increased malignancy risk (skin and lymphoproliferative) Avoid allopurinol Pregnancy risk category D |
TPMT FBC and LFT |
|
MMF and enteric‐coated mycophenolate sodium Inhibits DNA synthesis in T and B cells |
MMF: start 500 mg bd, titrating to 2–3 g daily (in two divided doses) MMF 500 mg = enteric‐coated mycophenolate sodium 360 mg |
Diarrhoea Bone marrow suppression Hepatotoxicity Increased malignancy risk (skin and lymphoproliferative) Progressive multifocal leucoencephalopathy Pregnancy risk category D contraindicated in pregnancy (category D) |
FBC, LFT, renal function |
|
Cyclophosphamide Alkylating agent toxic to all human cells to differing degrees with haematopoietic cells forming a sensitive target |
600 mg/m2, maximum dose 1000 mg Monthly for maximum 6 months |
Toxicity to bladder and gonads
Contraindicated in pregnancy (category D) |
Maintain adequate fluid intake to avoid bladder toxicity Monthly urinalysis FBC and LFT |
|
Tacrolimus Prevents calcineurin‐dependent gene transcription in T cells |
Start 1 mg bd titrating by 1–2 mg daily with at least 7 days between adjustments. Aim for 12 h trough level 5–8 ng/mL |
Increased vascular constriction → ↑BP, ↓ renal perfusion Contraindicated in pregnancy (category D) |
Trough levels 10–14 days after initiating and at regular intervals Monitor BP, BGL, FBC, EUC, LFT, lipids |
|
Rituximab B‐cell depletion by targeting CD20 lasting 6–9 months |
Initiation: two 1 g infusions, 2 weeks apart Maintenance: 1 g every 6–12 months |
Hepatitis B reactivation Should not be taken in pregnancy (category C) Hypogammaglobulinaemia |
Screen for hepatitis B (surface antigen and core antibody) |
|
Methotrexate Inhibits dihydrofolate reductase |
Start 5–15 mg/week, escalating by 5 mg/month to maximum 25–30 mg/week |
Pulmonary toxicity Hepatotoxicity Bone marrow suppression Alopecia Mouth ulcers Contraindicated in pregnancy (category D) |
FBC, LFT, EUC |
AZA, azathioprine; bd, twice daily; BGL, blood glucose level; BP, blood pressure; CTD‐ILD, connective tissue disease‐associated interstitial lung disease; EUC, electrolyte and urea concentration; FBC, full blood count; GI, gastrointestinal; LFT, liver function test; MMF, mycophenolate mofetil; TNF, tumour necrosis factor; TPMT, test for thiopurine methyltransferase.
The intensity of immunosuppressive therapy is tailored to the individual CTD, acuity of ILD presentation, therapeutic expectations and patient factors. For example, rapidly progressive ILD as seen in IIM and systemic lupus erythematosus (SLE) often presents with a radiological pattern of OP or diffuse alveolar damage (DAD), suggesting a predominantly inflammatory process. In such situations, intensive immunosuppression with pulse intravenous (IV) methylprednisolone, IV cyclophosphamide or rituximab may be required with the expectation of achieving reversal of inflammation and minimizing permanent lung damage. 62 , 63 In contrast, SSc‐ILD may be more slowly progressive and lower dose maintenance therapy to achieve stabilization rather than major improvement is a realistic therapeutic goal. Specific treatment approaches are discussed in more detail in relevant disease sections and currently available evidence from treatment trials is summarized in Table 5.
Table 5.
Important treatment trials in CTD‐ILD
| Study | Study design | Intervention | Outcome |
|---|---|---|---|
| Systemic sclerosis | |||
| Scleroderma Lung Study I 68 |
Prospective RCT n = 158 Duration: 12 months |
Oral CYC (up to 2 mg/kg daily) vs placebo | Mean absolute improvement in FVC at 12 months of 2.53%, favouring CYC, greater frequency of AE in the CYC arm |
| FAST study 69 |
Prospective RCT n = 45 Duration: 12 months |
IV CYC (600 mg/m2, monthly for six doses) + low‐dose prednisolone (20 mg alternate days) followed by AZA (2.5 mg/kg/day; max 200 mg) vs placebo | Trend to improvement FVC (4.19%; P = 0.08) in the active treatment arm, although statistically non‐significant |
| BUILD 2 (Bosentan) 70 |
Prospective RCT n = 163 Duration: 12 months |
Bosentan 125 mg bd vs placebo |
Negative primary end point with no difference in 6MWD between the groups No increase in SAE in the active treatment group |
| Scleroderma Lung Study II 71 |
Prospective RCT n = 142 Duration: 24 months |
MMF 1.5 g bd vs oral CYC (up to 2.0 mg/kg/day) for 12 months followed by placebo for 12 months |
Significant improvement in FVC from baseline in both arms (2.2% and 2.9%: MMF vs CYC), with no difference in FVC change between the groups Greater frequency of AE in the CYC group |
| LOTUSS (pirfenidone) 72 |
Randomized, open label n = 63 Duration: 16 weeks |
Comparison of two pirfenidone up‐titration regimens: 2‐week titration vs 4‐week titration, to a maintenance dose of 801 mg tds | ‘Acceptable’ tolerability profile, although a longer up‐titration may be associated with better tolerability |
|
Pomalidomide (CC‐4047) 73 |
Prospective RCT n = 23 Duration: 12 months |
Pomalidomide 1 mg/day vs placebo |
Negative co‐primary end points with no difference in FVC, mRSS or GI symptoms at 52 weeks Enrolment discontinued early due to difficult recruitment |
| faSScinate 74 |
Prospective RCT n = 87 Duration: 48 weeks |
Tocilizumab 162 mg subcutaneous weekly vs placebo | Negative secondary end point with no change in FVC or DLCO from baseline to 48 weeks |
| SENSCIS (nintedanib) 75 |
Prospective RCT n = 576 Duration: 12 months |
Nintedanib 150 mg bd vs placebo | Positive primary end point with reduced annual rate of change in FVC in subjects receiving nintedanib compared to placebo (−52.4 mL vs −93.3 mL per year; P = 0.04) |
| Rituximab 76 |
Open‐label, comparative study n = 51 Duration: 4 years (range: 1–7 years) |
Rituximab (n = 33) vs conventional immunosuppression (MMF, AZA or MTX) (n = 18) | At 2 years, improvement in FVC compared to baseline in rituximab group, with no change in FVC in conventional immunosuppression group |
| SCOT study 77 |
Randomized, open‐label n = 75 Duration: 54 months |
Myeloablative stem cell transplantation (n = 36) vs IV CYC (n = 39) | Positive composite primary end point (incorporating death, event‐free survival, FVC and mRSS) favouring stem cell transplantation |
|
ASTIS study 78 |
Randomized, open‐label n = 156 Duration: 5.8 years |
Autologous stem cell transplantation (n = 79) vs IV CYC (n = 77) |
Positive primary end point (death or persistent major organ failure) favouring stem cell transplantation Increased stem cell treatment‐associated mortality in the first 12 months |
| ASSIST study 79 |
Randomized, open‐label n = 19 Duration: 12 months |
Non‐myeloablative stem cell transplantation (n = 10) vs IV CYC (n = 9) | Positive primary end point (improvement in mRSS or FVC) favouring stem cell transplantation |
|
RA | |||
| Song et al. 80 |
Retrospective review n = 84 Median f/u: 33 months |
All had RA with UIP. Forty‐one percent received glucocorticoids ± immunosuppression due to poor baseline lung function or progressive RA with UIP | Fifty percent of treated group improved or had stable lung function |
|
Fischer et al. 81 |
Retrospective review n = 18 Median f/u: 2.5 years |
MMF (1–1.5 g bd) |
Trend to improvement in FVC following MMF commencement, with reduction in prednisolone requirement |
|
IIM | |||
| Schnabel et al. 82 |
Prospective open label n = 20 Median f/u 35 months |
Rapidly progressive IIM‐ILD (n = 10): IV CYC IIM‐ILD but less rapidly progressive (n = 10) |
Stabilization in IIM‐ILD following IV CYC |
| Kameda et al. 83 |
Prospective open label n = 27 |
Rapidly progressive IIM‐ILD (n = 10): IV CYC (10–30 mg/kg every 3–4 weeks), prednisolone (>0.5 mg/kg/day) and cyclosporine A (2–4 mg/kg/day) | Fifty percent mortality rate at 3 months |
|
Huapaya JA et al. 148 |
Retrospective review n = 110 f/u: 24–60 months |
AZA (n = 66) MMF (n = 44) |
Improved FVC% and DLCO% predicted in AZA group Improved FVC% predicted in MMF group Lower prednisolone dose in both groups More frequent AE in AZA group |
6MWD, 6‐min walk distance; AE, adverse event; ASSIST, American Scleroderma Stem Cell versus Immune Suppression Trial; ASTIS, Autologous Stem cell transplantation; AZA, azathioprine; bd, twice daily; CTD‐ILD, connective tissue disease‐associated ILD; CYC, cyclophosphamide; DLCO, diffusing capacity for carbon monoxide; f/u, follow‐up; FVC, forced vital capacity; GI, gastrointestinal; IIM, idiopathic inflammatory myopathy; IIM‐ILD, IIM‐associated ILD; ILD, interstitial lung disease; IV, intravenous; MMF, mycophenolate mofetil; mRSS, modified Rodnan skin score; MTX, methotrexate; n, number; RA, rheumatoid arthritis; RCT, randomized controlled trial; SAE, serious AE; SCOT, Scleroderma: Cyclophosphamide or Transplantation trial; SENSCIS, Safety and Efficacy of Nintedanib in Systemic Sclerosis; tds, three times daily; UIP, usual interstitial pneumonia.
Anti‐fibrotic therapy has recently been found to be of benefit in CTD‐ILD. Annual rate of decline in FVC was lower in patients with SSc‐ILD and progressive fibrosing‐ILD who received nintedanib compared with placebo in the Safety and Efficacy of Nintedanib in Systemic Sclerosis (SENSCIS) and Nintedanib in Progressive Fibrosing Lung Disease (INBUILD) trials, respectively. 85 , 86 INBUILD included a range of fibrosing ILD, incorporating 170 patients with ‘autoimmune’ ILD, including RA‐ILD, SSc‐ILD and mixed CTD (MCTD)‐associated ILD (MCTD‐ILD). Although not powered to provide evidence for a benefit of nintedanib in specific diagnostic subgroups, results suggest that nintedanib reduces the rate of ILD progression (measured by FVC decline) in patients with chronic, progressive fibrosing ILD. 87 Pirfenidone has also shown potential benefit in patients with progressive unclassifiable ILD, including a subgroup with IPAF. Slower disease progression was observed in study participants receiving pirfenidone compared with placebo over 24 weeks. 88 Whilst encouraging, neither nintedanib nor pirfenidone has regulatory approval for the treatment of CTD‐ILD in Australia or New Zealand at this time and the exact role of these agents (including patient selection, timing and combination with immunosuppressive therapy) for CTD‐ILD remains to be determined.
Special considerations
Identification of patients at risk of infectious complications is prudent prior to commencement of immunosuppressive therapy. Screening for latent TB infection (LTBI) and hepatitis B is the most well recognized, while hepatitis C, human immunodeficiency virus (HIV) and strongyloides infection should be considered on an individual patient basis. 67 , 89 , 90
Latent TB infection
The lifetime risk of reactivation in LTBI is approximately 5–10% and increases with immunosuppressive therapy. The risk of TB reactivation increases by up to 25‐fold, highest in the first 12 weeks, after commencement of tumour necrosis factor (TNF) inhibitors (e.g. infliximab and adalimumab). Thus, LTBI screening with an interferon‐gamma release assay (IGRA) or Mantoux tuberculin skin test (TST) is recommended in all patients where TNF inhibitors are being considered. 91 Patients with LTBI should be treated prior to commencement of TNF inhibitor therapy, which reduces the incidence of TB reactivation by more than 80%. 68 , 89
LTBI screening/treatment for other immunosuppressive medications is less clear. Studies have reported a seven‐fold increased risk of TB reactivation in patients on corticosteroids. 69 Evidence to recommend LTBI treatment for all patients is currently insufficient, but it is reasonable to evaluate overall risk (steroid dose, concomitant immunosuppressive medications, exposures and background incidence), and consider LTBI testing and treatment on an individual basis. 90
Hepatitis B
Hepatitis B virus (HBV) reactivation associated with immunosuppressive therapy is associated with significant morbidity and mortality. The American Gastroenterological Association guidelines on the prevention and treatment of HBV reactivation during immunosuppressive drug therapy stratify patients into high‐, moderate‐ or low‐risk groups based on the serological pattern (e.g. HBV surface antigen positivity) and immunosuppressive agent. 92 Expert hepatology opinion is strongly recommended if active or previous HBV is detected. 89 , 90
Pneumocystis jirovecii
Pneumocystis jirovecii pneumonia (PJP) is a potentially life‐threatening infection, most prevalent in patients receiving high‐dose glucocorticoids combined with other immunosuppressive agents. There are published guidelines for the use of PJP prophylaxis in HIV, cancer and solid organ transplant recipients. 71 No guidelines exist for CTD patients on immunosuppression, but is an important consideration given the generally long‐term nature of immunosuppressive therapy in CTD. 75 , 93 It is reasonable to recommend prophylaxis (e.g. sulfamethoxazole and trimethoprim) in patients receiving sustained daily corticosteroids of 20 mg/day or greater. Prophylaxis in the context of other immunosuppressants is less clear but should be considered in patients on combinations of immunosuppressants despite lower doses of prednisolone.
Summary.
Treatment decisions in CTD‐ILD are influenced by CTD diagnosis and duration since diagnosis, ILD severity, disease trajectory, extra‐pulmonary CTD manifestations and individual patient factors.
Progressive CTD‐ILD with a predominant inflammatory component (determined following clinical and radiological evaluation) may be amenable to significant reversal with appropriate intensive immunosuppression. 62 , 94
Progressive and/or severe CTD‐ILD with a predominant fibrotic component typically do not regress significantly with intensive immunosuppression, although stabilization may be achieved with low‐dose corticosteroids, with or without a steroid‐sparing immunosuppressive agent. New data support the potential role for anti‐fibrotic therapy in such cases, although this is not yet approved for use in Australia or New Zealand. 85 , 86 , 87
Serial PFT is the cornerstone of CTD‐ILD monitoring, including response to treatment. 38 , 55 , 56 , 57
Evaluation for latent infection (e.g. TB and hepatitis) and appropriate opportunistic infection prophylaxis should be considered on an individual patient basis prior to initiation of immunosuppressive therapy.
SPECIFIC CTD‐ILD
Systemic sclerosis
SSc (also known as scleroderma) is characterized by autoimmunity, fibrosis of the skin and internal organs, and vasculopathy. Pulmonary complications, including ILD and pulmonary arterial hypertension (PAH), are the leading causes of morbidity and mortality in SSc. 77 , 95 Whilst ILD may occur in up to 80% of SSc patients, only 25–30% have progressive disease and the key challenge is identifying patients at risk of disease progression and likely to benefit from treatment, whilst avoiding unnecessary drug toxicities in indolent disease. 79
Risk factors for developing severe, progressive SSc‐ILD include: (i) SSc disease duration of less than 3 years 78 , 96 ; (ii) diffuse cutaneous SSc 96 ; (iii) presence of anti Scl‐70/DNA topoisomerase 1 autoantibodies 96 ; (iv) severe gastro‐oesophageal reflux disease (GORD) 97 , 98 ; (v) extensive ILD 54 ; and (vi) declining lung function. 55 , 56 , 58 Extent of ILD severity, traditionally quantified by FVC and DLCO, is the most important factor, especially in the early stages of disease. The risk of developing ILD is greatest in the first few years after SSc symptom onset, but patients can still progress after this ‘at‐risk’ period. 99 Vigilance for disease progression must always be maintained, with declining lung function associated with increased mortality. 78 , 100 , 101
A simple staging system, incorporating semi‐quantitative assessment of disease extent on HRCT and FVC, classifies patients into limited and extensive disease (Fig. 3A). 54 Patients who have <20% or ≥20% disease on HRCT are classified as limited or extensive disease, respectively. If HRCT extent is ‘indeterminate’, an FVC threshold of above or below 70% predicted is used to distinguish limited versus extensive disease, respectively. This approach has been validated in other SSc‐ILD cohorts and provides greater prognostic insight than either HRCT extent or FVC measures alone (Fig. 3B). 65
The goal of treatment in most cases of severe and/or progressive SSc‐ILD is to achieve disease stabilization, rather than major reversal. The use of corticosteroids remains debatable, but when used, dosage should be minimized (<10–15 mg/day), to reduce the risk of side effects and SSc renal crisis. The Scleroderma Lung Study I (SLS I) demonstrated modest improvements in lung function, dyspnoea and health‐related quality of life (HRQoL) with cyclophosphamide, administered as either a monthly infusion or daily oral dose for 12 months. 102 , 103 Although the latest European League Against Rheumatism (EULAR) recommendations advocate for the use of cyclophosphamide in SSc‐ILD, this is now outdated. Instead, mycophenolate mofetil (MMF) has increasingly been the first drug of choice following the findings from the Scleroderma Lung Study II (SLS II). 104 Whilst the beneficial effect on lung function was similar between oral MMF and oral cyclophosphamide given over 24 months, MMF was associated with better tolerability and less adverse effects. 105 However, in patients with rapidly progressive disease, IV cyclophosphamide is still considered the first drug of choice with a quicker onset of action compared to MMF. In these cases, IV cyclophosphamide could be given monthly for 6 months as the ‘induction phase’ of treatment, followed by MMF in the ‘consolidation phase’ of treatment.
The exact duration of ongoing maintenance therapy remains unclear. In the SLS I study, the benefits of cyclophosphamide were lost 12 months after the cessation of treatment. 102 Long‐term follow‐up of SLS I and SLS II patients (median follow‐up of 8 years) demonstrated no difference in survival between cyclophosphamide or MMF, and patients on placebo. 55 It is likely that more than 2 years of immunosuppression is required to sustain improvements in lung function and symptoms with longer term studies required. An observational study of 30 SSc‐ILD patients receiving 24 months of IV cyclophosphamide followed by ongoing low‐dose immunosuppression (azathioprine, methotrexate (MTX) or MMF) demonstrated sustained improvement in lung function in the ensuing 5 years and no significant adverse events. 106
The SENSCIS trial demonstrated a reduced decline in FVC in SSc‐ILD patients on nintendanib compared with placebo (−52.4 vs −93.3 mL/year, respectively, P = 0.04), but does not support nintedanib as a substitution for immunotherapy in SSc‐ILD. 85 The magnitude of benefit depended on MMF use, suggesting the potential benefit of MMF on lung function. 85 Furthermore, there were no benefits reported with HRQoL measures or modified Rodnan skin score (mRSS) and nintedanib should not be considered a disease‐modifying agent for SSc. 85 It remains unclear if nintedanib should be used following or together with immunosuppressive therapy. The U.S. Food and Drug Administration recently approved nintedanib for the treatment of SSc‐ILD, but is not yet approved for this indication in Australia or New Zealand.
Several additional therapeutic options including pirfenidone, tocilizumab and rituximab are currently being evaluated for SSc‐ILD. 107 Tocilizumab, an anti‐IL6 monoclonal antibody, has recently been investigated in a phase III study of SSc patients, including 31% with ILD (NCT02453256). 108 Preliminary analysis demonstrated preservation of FVC over 48 weeks, less decline in FVC compared with placebo and improvements of fibrosis as assessed by HRCT with final analysis awaited. 109
Stem cell transplantation for SSc‐ILD
There are no stem cell trials in SSc‐ILD per se, but secondary lung function end points in broader SSc studies are interesting. Two randomized trials of non‐myeloablative autologous haematopoietic stem cell transplant (HSCT)—the American Scleroderma Stem Cell versus Immune Suppression Trial (ASSIST) and Autologous Stem cell transplantation (ASTIS) trial—reported less SSc disease progression with HSCT compared with cyclophosphamide. 110 , 111 ASTIS also demonstrated superior event‐free survival, overall survival and improvement in FVC and total lung capacity (TLC) over a median follow‐up time of 5.8 years. 111 The Scleroderma: Cyclophosphamide or Transplantation (SCOT) trial compared myeloablative autologous HSCT to cyclophosphamide in severe SSc, including 73 (97%) participants with pulmonary involvement. HSCT was superior to cyclophosphamide in terms of Global Rank Composite Score (comprising in hierarchical order: death, event‐free survival, FVC, Health Assessment Questionnaire‐Disability Index (HAQ‐DI) and mRSS) and change in FVC. 112 Notably, patients with severe lung disease (FVC <45% or DLCO <40% predicted) were excluded, as were those with left ventricular ejection fraction <50%, severe renal impairment, PAH or more than 6 months of previous treatment with cyclophosphamide. 112
Summary.
Risk factors for severe and/or progressive SSc‐ILD include greater extent of ILD on HRCT, declining PFT, Scl‐70 autoantibody positivity and development of ILD early in the SSc disease course. 54 , 55 , 78 , 79 , 96
MMF has been demonstrated to stabilize SSc‐ILD and is generally regarded as the initial therapy of choice when treatment is required. Azathioprine or cyclophosphamide (typically IV, given the lower cumulative toxicity) may be an alternative to MMF. 102 , 103 , 104
The use of concomitant corticosteroids is controversial and if prescribed, the dose should be minimized (<10–15 mg/day) to reduce side effects including the risk of SSc renal crisis.
Nintedanib has been shown to slow lung function decline in SSc‐ILD, although it is not yet approved for this indication in Australia or New Zealand. 85
HSCT may improve SSc‐ILD and survival in highly selected SSc patients, although its role is likely to be limited to specialist centres. 111 , 112
Rheumatoid arthritis
Interstitial changes on HRCT occur in up to 40–50% of newly diagnosed RA patients, with up to 10% developing clinically significant ILD during their lifetime. 18 , 113 ILD may precede articular symptoms or appear many years after RA diagnosis. Male gender, older age at RA onset, smoking history, positive rheumatoid factor (RF) and anti‐CCP antibodies are associated with increased risk of ILD. 19 , 114 , 115 Both RA‐ILD and IPF share a number of genetic risk factors, including the MUC5B promoter variant, and genes associated with telomere maintenance (TERT, TERC and TINF‐2). 116 , 117 Given the high prevalence of subclinical ILD in RA, a low threshold for respiratory investigation in patients with seropositive RA is suggested. 10 ILD has been reported in patients with anti‐CCP antibodies without a clinical diagnosis of RA, but it is unknown what proportion progress and develop clinically apparent joint disease. 81
UIP is the most common radiological and histopathological pattern in RA, followed by NSIP, OP and, rarely, acute interstitial pneumonia (AIP) or LIP. 37 , 113 RA‐UIP is associated with poorer survival, with most but not all studies suggesting a prognosis similar to IPF. 24 , 25 , 38 Other negative prognostic factors include lower baseline PFT and declining PFT in the first 6 months. 24 , 30 , 118
There are no randomized controlled trials (RCT) assessing immunosuppressive therapy specifically in RA‐ILD. Case series and retrospective series report stabilization or improvements in lung function with corticosteroids, conventional immunosuppressive agents (e.g. cyclophosphamide, azathioprine and MMF) and rituximab. 119 , 120 , 121 RA‐ILD with UIP may be less responsive to immunosuppressive therapy, although retrospective series have reported stabilization of pulmonary function with MMF. 122 Given the increased morbidity and mortality associated with immunosuppression in IPF, the decision to immunosuppress in RA‐ILD requires caution. 123 The recently reported INBUILD study demonstrated reduced rate of decline in FVC in a mixed cohort of patients with progressive fibrosing ILD, including 89 patients with RA‐ILD. 86 , 87 As of July 2020, a multicentre trial of pirfenidone in RA‐ILD (NCT02808871) is ongoing.
In patients with established RA‐ILD, the safety of MTX and other potentially pneumotoxic disease‐modifying antirheumatic drugs remains under debate. 124 , 125 While MTX pneumonitis is a recognized rare adverse event (estimated prevalence of 0.43% in RA patients), the impact of MTX on established RA‐ILD remains uncertain. 126 MTX treatment was not associated with an increased risk of RA‐ILD in a cohort of 2701 patients with newly diagnosed RA. 127 Furthermore, a case series of 78 patients with RA‐ILD (two‐thirds receiving MTX) reported improved survival in the MTX subgroup. 128 Thus, pre‐existing ILD is not a specific contraindication to MTX therapy, but caution is advised in patients with limited respiratory reserve. 90
Leflunomide and other biologic agents (e.g. TNF inhibitors, rituximab and tocilizumab) have all been associated with pulmonary toxicity, but it is unknown if pre‐existing ILD is an independent risk factor. 129 , 130 , 131 , 132 , 133 , 134 Given these uncertainties, caution and vigilance for a decline in lung function is required if considering these agents in patients with RA‐ILD. 120 , 135
Summary.
In RA‐ILD, a UIP pattern, lower baseline PFT and declining PFT are associated with a poorer prognosis. 24 , 25 , 30 , 118
Controlled treatment trial data for managing RA‐ILD are lacking. A variety of immunosuppressive agents (including cyclophosphamide, azathioprine, MMF or rituximab) may be effective in ameliorating decline in RA‐ILD.
In the setting of progressive RA‐ILD, consideration should be given to cessation of disease‐modifying anti‐rheumatic drugs (leflunomide and TNF inhibitors), given the potential for these agents to precipitate worsening ILD.
Clinical trials evaluating nintedanib and pirfenidone for RA‐ILD are ongoing.
Idiopathic inflammatory myopathy
The IIM spectrum of disorders includes polymyositis (PM), DM, inclusion body myositis (IBM) and immune‐mediated necrotizing myopathy (IMNM). 136 ILD is a major cause of morbidity and mortality in PM/DM, with an estimated prevalence of 20–65%, including rapidly progressive ILD. 137 , 138 IBM and IMNM are not typically associated with pulmonary involvement and will not be discussed. 139 , 140
The recently revised IIM diagnostic criteria incorporate demographic, clinical, serological and muscle biopsy features to determine the likelihood of disease (Table 1). 136 , 140 , 141 Pragmatically, myositis in the presence of cutaneous features (including mechanic's hands, Gottron's papules and a heliotrope rash) supports a diagnosis of DM, while positive muscle biopsy findings and elevated serum creatinine kinase (CK) in the absence of cutaneous features suggests PM. CADM is a subgroup of IIM with cutaneous findings in the absence of myositis. 140 , 142 , 143
Autoantibodies are a key diagnostic feature in IIM and are broadly classified into MSA and myositis‐associated autoantibodies (MAA) (Table 2). In most pathology laboratories in Australia and New Zealand, these antibodies are not included in standard ENA testing, and need to be specifically requested. Of the MSA, the presence of anti‐tRNA synthetase antibody (most commonly anti‐Jo1) is a defining feature of the anti‐synthetase syndrome and highly associated with ILD (prevalence up to 71%). 137 , 140 While the anti‐MDA5 antibody may be associated with CADM and rapidly progressive ILD (particularly in individuals of Asian ethnicity), it may also be associated with a more benign clinical course. 84 , 141 , 142 , 143 Specific IIM phenotypes are associated with an increased risk of malignancy, especially lung, breast, colorectal and pancreas and targeted screening may be appropriate. Positive anti‐TIF1γ and anti‐nuclear matrix protein (NXP)‐2 antibodies in particular have a reported 27‐ and 3.68‐fold higher risk of malignancy, respectively, among DM patients. 144
Subclinical ILD appears to be less common in IIM compared with other CTD, with the majority of IIM‐ILD patients tending to have more severe, progressive lung involvement requiring treatment. In the absence of randomized controlled data, several treatment recommendations have been proposed by expert groups. Oral prednisolone (0.25–1 mg/kg) for 3–8 weeks followed by slow wean has been reported to result in clinical improvement in 90–100% of patients. 11 , 140 , 145 Treatment of rapidly progressive IIM‐ILD includes pulse IV methylprednisolone or IV cyclophosphamide, followed by steroid‐sparing oral agents. 145 , 146 Azathioprine, MMF, calcineurin inhibitors (cyclosporine and tacrolimus) and rituximab have all demonstrated benefit in case series with small patient numbers. 145 , 147 , 148 , 149 , 150 A recent retrospective study of 110 IIM‐ILD patients treated with azathioprine (n = 66) or MMF reported similar improvements in PFT and reduced prednisolone requirements, although a higher frequency of adverse events were observed in the azathioprine group. 148 IV immunoglobulin (IVIG) is beneficial in active muscle disease, although efficacy for ILD specifically remains unclear. 151 Plasmapheresis is generally reserved for severe multiorgan disease, although evidence for its role specifically in IIM‐ILD is limited. 152 , 153 , 154 Due to the risk of worsening or relapsed ILD, immunosuppression is generally maintained for a relatively long duration (often >2–5 years), although controlled treatment data are lacking. Despite treatment, morbidity and mortality related to IIM‐ILD are significant, with 25% of patients deteriorating despite treatment and an overall ILD‐associated mortality rate of 13.5% at 5 years in one case series. 146
Summary.
ILD is a major contributor to IIM mortality and warrants expedited evaluation and treatment. 137 , 138
Initial intensive immunosuppression with high‐dose corticosteroids (oral or IV), IV cyclophosphamide or rituximab is often required to achieve control in progressive IIM‐ILD. IVIG and plasmapheresis may play a role in the acute setting. 145 , 147 , 151 , 152 , 154
Maintenance immunosuppression with low‐dose corticosteroids and steroid‐sparing agents (including MMF, azathioprine and calcineurin inhibitors) is generally required following initial high‐dose therapy, without evidence of an obviously superior agent. 146 , 147 , 148 , 149
Other CTD
Systemic lupus erythematosus
Clinically significant pulmonary complications involving the interstitium, pulmonary vasculature and/or pleura occur in 3–8% of SLE patients. 155 ILD is uncommon, but in rare cases, heralded by life‐threatening acute lupus pneumonitis and diffuse alveolar haemorrhage. Whilst elevated erythrocyte sedimentation rate (ESR) and/or hypocomplementaemia are indicators of systemic SLE disease activity, the presence of autoantibodies (e.g. ANA and anti‐dsDNA) do not correlate with the risk of ILD. 156 , 157 , 158
Pleural disease is the most common pulmonary manifestation, with pleurisy and/or pleural effusion occurring in over 60% of patients. 159 Shrinking lung syndrome is a rare, poorly understood complication of SLE, characterized by pleuritic chest pain, restrictive physiology and no interstitial changes on imaging. 5 Other pulmonary manifestations of SLE include PH and thromboembolic disease, especially in association with antiphospholipid syndrome. 160 , 161
Sjögren's syndrome
Clinically significant ILD is rare in Sjögren's syndrome (SjS), and most commonly follows a mild, self‐limited disease course. 155 , 162 SjS‐associated ILD (SjS‐ILD) is typically more female predominant, with a median age of onset of 60 years and occurring 5–10 years after SjS diagnosis. 134 Autoantibodies do not predict risk of SjS‐ILD, although presence of anti‐Ro/SS‐A autoantibodies correlated with a lower DLCO in one case series. 163 LIP was previously considered the most common radiological pattern in SjS, but recent reports suggest NSIP predominates. 39 It is important to distinguish dry mucous membranes (xerotrachea) as a cause or contributor to symptoms, especially dry cough, independent of ILD. 155
SjS patients are at increased risk for lymphoma, especially mucosa‐associated lymphoid tissue (MALT) lymphoma. 155 , 164 Pulmonary lymphoma occurs in 1–2% of patients with primary SjS, and accounts for approximately 20% of all SjS‐associated lymphoma. 165 It is important to distinguish pulmonary lymphoma from LIP. Both can manifest with ground‐glass opacification, cysts, parenchymal nodules and lymphadenopathy. 158 Other rare pulmonary complications in SjS include pleuritis, vascular disease (PAH and pulmonary embolism), amyloidosis, ‘shrinking lung’ and ‘middle lobe’ syndromes. 158 , 166 , 167 , 168 , 169
Undifferentiated and mixed connective tissue disease
Undifferentiated CTD (UCTD) is a term used to describe the early, ‘undifferentiated’ phase of a CTD with clinical symptoms and signs of a CTD and non‐specific laboratory abnormalities (e.g. ANA and ENA), not satisfying diagnostic criteria for a defined CTD. UCTD does not have a well‐accepted definition and is broadly encompassed within the IPAF definition (discussed later). 170 Approximately one‐third of UCTD patients progress to a definable CTD over 5‐year follow‐up. 171
MCTD has a distinct clinical phenotype and human leukocyte antigen (HLA) profile, although controversy remains with four available diagnostic criteria. 17 , 172 , 173 The central concept of MCTD is an overlap syndrome associated with anti‐U1 ribonucleoprotein (anti‐RNP) antibodies. 174 Peak incidence is around the age of 40 years. 174 Clinical features always include Raynaud's phenomenon and at least two of: myositis, arthritis, oesophageal dysmotility, leucopaenia, serositis and ILD or PAH. 174 Diagnosis is challenging due to the overlap of clinical features with other CTD and requires continual reassessment.
HRCT abnormalities occur in approximately half of MCTD patients, with one‐third demonstrating ILD. 175 Anti‐Ro52 antibodies are a potential marker of lung involvement. 175 MCTD‐ILD is severe in approximately one‐fifth of patients, and associated with a 21% mortality compared with 3% in patients with a normal HRCT. 175
Treatment of SLE, SjS, UCTD and MCTD‐ILD
The optimal therapy for ILD associated with SLE, SS, UCTD and MCTD is unknown due to a lack of controlled treatment trials. In clinical practice, broad CTD‐ILD treatment principles remain pertinent with therapeutic intervention based on ILD severity and rate of progression, other organ involvement and comorbidities. Extrapolating from treatment trials in other CTD, mild, non‐progressive ILD may be monitored, whilst treatment with immunosuppression (e.g. corticosteroids, azathioprine and MMF) is reasonable where there is evidence of progression. Treatment trials on the utility of rituximab in these CTD are ongoing. 176
Summary.
ILD is a recognized complication in SLE, SjS, UCTD and MCTD but is typically less frequent than the other CTD.
Limited data exist to guide treatment decisions in this group with therapeutic approaches generally extrapolated from other CTD.
In severe and/or progressive ILD, high‐dose corticosteroids in combination with a steroid‐sparing agent (MMF or azathioprine) is a reasonable initial treatment approach.
Several unique pulmonary complications may occur in SjS (lymphoma and amyloidosis) and SLE (lupus pneumonitis and pulmonary haemorrhage), requiring a high degree of diagnostic suspicion to enable timely diagnosis.
Interstitial pneumonia with autoimmune features
While ILD can develop in the context of a known CTD, it is increasingly recognized that some patients with idiopathic ILD have features of autoimmune disease, but do not fulfil criteria for a defined CTD. Historically, various terms including ‘UCTD‐associated ILD’, ‘lung‐dominant CTD’ or ‘autoimmune‐featured ILD’ have been used to describe such patients, impeding uniform study of this important cohort. 177 , 178 , 179 In 2015, a ERS/ATS Taskforce defined this group as ‘IPAF’. 180 The proposed classification criteria remain a research framework rather than clinical classification, requiring revision and validation in future studies.
The full criteria consist of three domains—clinical, serological and morphological (Table 6). Qualifying individuals must demonstrate characteristics from at least two of the three domains. Serological evaluation includes a broad array of autoantibodies including an extended ENA and myositis panel, high titre ANA and RF level. 180 Multi‐compartment involvement in the morphological domain includes ILD with unexplained pleuro‐pericardial involvement, pulmonary vasculopathy or airways disease.
Table 6.
ERS/ATS criteria for the diagnosis of IPAF
| Domain | Characteristics |
|---|---|
| (A) Clinical |
Distal digital fissuring (i.e. ‘mechanic's hands’) Distal digital tip ulceration Inflammatory arthritis or polyarticular morning joint stiffness ≥60 min Palmar telangiectasia Raynaud's phenomenon Unexplained digital oedema Unexplained fixed rash on the digital extensor surfaces (Gottron's sign) |
| (B) Serological |
ANA ≥1:320 titre, diffuse, speckled, homogeneous patterns or ANA nucleolar pattern (any titre) orANA centromere pattern (any titre) RF ≥2 × ULN Anti‐CCP Anti‐dsDNA Anti‐Ro (SS‐A) Anti‐La (SS‐B) Anti‐ribonucleoprotein Anti‐Smith Anti‐topoisomerase (Scl‐70) Anti‐tRNA synthetase (e.g. Jo‐1, PL‐7, PL‐12, others are: EJ, OJ, KS, Zo, YRS) Anti‐PM‐Scl Anti‐CADM (MDA5) |
| (C) Morphological |
(1) Suggestive radiology patterns by HRCT NSIP OP NSIP with OP overlap LIP (2) Histopathology patterns or features by surgical lung biopsy NSIP OP NSIP with OP overlap LIP Interstitial lymphoid aggregates with germinal centres Diffuse lymphoplasmacytic infiltration (with or without lymphoid follicles) (3) Multi‐compartment involvement (not otherwise explained) Pleural effusion or thickening Pericardial effusion or thickening Small airways disease (by PFT, imaging or pathology) Pulmonary vasculopathy |
Adapted from Fischer et al., 180 with permission.
ANA, anti‐nuclear antibody; ATS, American Thoracic Society; CADM, clinically amyopathic dermatomyositis; CCP, cyclic citrullinated peptide; dsDNA, double‐stranded DNA; ERS, European Respiratory Society; HRCT, high‐resolution computed tomography; IPAF, interstitial pneumonia with autoimmune feature; LIP, lymphocytic interstitial pneumonia; NSIP, non‐specific interstitial pneumonia; OP, organizing pneumonia; PFT, pulmonary function test; PM, polymyositis; RF, rheumatoid factor; ULN, upper limit of normal.
Since publication of the IPAF classification criteria, several retrospective single‐centre studies have described IPAF populations with heterogeneous demographics, clinical features and natural history. 181 One landmark study demonstrated no difference in survival between IPAF and IPF patients, although there was a clear demarcation in survival between those with and without a UIP pattern on HRCT. 182 However, other studies have not replicated this finding and larger multicentre, prospective studies are needed. 183
Little is known about the best clinical approach to managing patients who meet the IPAF research criteria. Indeed, IPAF is likely to be comprised of a heterogenous group of patients, in which different therapies are targeted at different patient phenotypes. At present, a pragmatic approach would be to consider the underlying ILD diagnosis by ILD‐MDM and treat the patient accordingly. In patients who are considered to have CTD‐ILD, treatment will usually follow an approach similar to SSc‐ILD, whereas, in patients considered to have IPF, anti‐fibrotic therapy may be appropriate. The recently published pirfenidone in unclassifiable ILD study included 33 (13%) patients with IPAF, too small a number to make meaningful conclusions. 88 Ongoing studies in this patient population are required.
Summary.
IPAF identifies a heterogeneous patient group with ILD and features suggestive of a CTD, not meeting established classification criteria for a specific CTD. 180
It remains a research framework rather than clinical diagnosis.
There is no current consensus for the management of patients meeting IPAF criteria, with several ongoing studies evaluating the role of immunosuppressive and antifibrotic therapies
Vasculitis and ILD
Anti‐neutrophil cytoplasmic antibody (ANCA)‐associated vasculitis (AAV), incorporating granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA) and eosinophilic GPA (EGPA) may affect the lung parenchyma, blood vessels and airways. While diffuse alveolar haemorrhage and parenchymal mass lesions (with or without cavitation) are the most frequent pulmonary manifestations of AAV, ILD is an increasingly recognized complication, particularly in MPA where there is an estimated prevalence of 7–36% and significant associated morbidity. 184 , 185 , 186
Positive ANCA serology has been reported in 4–10% of patients with a confirmed diagnosis of IPF. 184 , 187 , 188 However, the significance of myeloperoxidase (MPO) or proteinase 3 (PR3) antibodies remains uncertain, with inconsistent clinical and radiological findings reported in several small single‐centre studies. 184 , 186 , 187 , 188 , 189 , 190 Recently, a large retrospective cohort of 745 IPF patients from two North American expert ILD centres identified ANCA in 34 (4.6%) patients at the time of IPF diagnosis. 191 Of the ANCA‐positive patients, 18 (53%) were MPO‐antibody positive, 10 PR3‐antibody positive (29%) and 6 (18%) with non‐specific ANCA positivity 191 ANCA positivity was not associated with baseline IPF severity or transplant‐free survival. During follow‐up (median: <18 months), 28% of MPO‐positive patients developed clinically significant vasculitis, most commonly renal disease and mononeuritis multiplex. No PR3‐positive or non‐specific ANCA patients developed clinical vasculitis. 191
Treatment for systemic vasculitis usually involves guideline‐recommended immunosuppressive regimens, but data specific to AAV‐associated ILD (AAV‐ILD) are limited. 187 , 192 Some case series suggest clinical or radiological improvement with immunosuppressive therapy, but impact on overall survival and progression of lung disease remains controversial and further research is required. 184 , 186 , 187 , 188 , 193 Generally speaking, ILD occurring in the context of positive ANCA serology without clinical evidence of vasculitis should be treated on its merits, based on the principles discussed in section ‘Approach to management of CTD‐ILD’. 194 , 195
Summary.
The association between AAV and ILD, particularly IPF, is increasingly recognized, although variable clinical manifestations have been reported in published series. 184 , 185 , 186
Positive MPO serology appears to be more strongly linked to the development of systemic vasculitis. 191
Optimal treatment, including the use of immunosuppressive therapy, remains the focus of ongoing research.
Psoriasis and ILD
There are small retrospective case reports and case series of ILD in patients with psoriasis and psoriatic arthritis, although a clear pathogenic association is yet to be determined. Observed radiological patterns on HRCT include UIP, NSIP and OP with descriptions of non‐specific bilateral lower zone predominant ground‐glass opacities with irregular reticular changes. 196 , 197 , 198 , 199 , 200 Most reported cases are mild with minimal or no respiratory symptoms. 196 , 197 Correlation between dermatological and pulmonary disease activity, and response of pulmonary disease to immunosuppressive treatment for the skin, is also variable. 196 , 197 , 198 Small case numbers limit meaningful conclusions and further prospective studies are required to determine if a clear association with ILD exists, and to clarify prevalence and pathogenic mechanisms. Careful observation for pulmonary complications in patients with psoriasis, as with any systemic inflammatory disease, is reasonable.
SPECIAL CONSIDERATIONS IN CTD‐ILD MANAGEMENT
CTD‐ILD with pulmonary hypertension
PH is defined as a mean pulmonary artery pressure of ≥25 mm Hg at rest on right heart catheterization (RHC), although a revised threshold of >20 mm Hg in combination with a pulmonary vascular resistance of ≥3 Wood Units has recently been proposed. 201 Clinical conditions associated with PH are classified into five groups based on similar pathophysiological mechanisms, characteristics and management, as outlined in the updated World Health Organization (WHO) World Symposium on Pulmonary Hypertension classification. 201 In CTD‐ILD, it is important to ascertain whether PH is a primary vasculopathy (i.e. PAH ‘CTD‐PAH’; WHO Group 1 PAH) or secondary to the underlying CTD‐associated ILD (‘CTD‐ILD‐PH’; WHO Group 3 PH). Furthermore, PH secondary to left heart disease (WHO Group 2 PH) may also be present, further complicating classification of these patients. The presence of PH regardless of cause is a poor prognostic marker. The ASPIRE Registry estimated a 5‐year survival of 25% for SSc‐associated PAH and 30% for PH secondary to lung disease. 202 A recent study demonstrated similarly dismal prognosis in patients with severe ILD and mild–moderate PH, and severe PAH with minor ILD, with 3‐year survival rates of 49.9% and 61.9%, respectively. 203
Clues suggest that PH may include breathlessness out of proportion to lung function or radiological severity, features of right heart failure (e.g. syncope, pre‐syncope or fluid retention) or it may be detected on screening echocardiogram (as recommended in patients with SSc). Differentiating Group 1 PAH from Group 3 PH in CTD‐ILD patients is often complex and requires careful evaluation at an experienced PH centre. Ancillary investigations suggesting Group 3 PH include an FVC <70% predicted, extensive interstitial changes on HRCT and exhausted ventilatory reserve on cardiopulmonary exercise testing (e.g. reduced breathing reserve, normal O2 pulse, normal CO/VO2 slope and an increase in PaCO2 during exercise). 204 Ultimately, expert clinical judgement is often required for accurate diagnosis. The presence of mild ILD does not preclude the presence of concomitant Group 1 PAH, and major clinical trials evaluating PAH‐specific therapies permitted mild impairments in lung function.
In Group 1 PAH, pulmonary vasodilator therapies have proven therapeutic benefit. 205 , 206 , 207 , 208 SSc is the dominant CTD associated with PAH (estimated prevalence of 8–12%), but PAH can also occur in RA, SLE and MCTD. 209 PAH‐specific therapies include endothelin receptor antagonists, phosphodiesterase (PDE5) inhibitors, the cyclic GMP stimulator riociguat and prostacyclin analogues. The European Society of Cardiology(ESC)/ERS guidelines advocate a ‘treat to target’ approach using combination PAH–drug therapy, with commencement of therapy in those with WHO Functional Class II symptoms. 210 Current regulatory restrictions in Australia and New Zealand only fund one agent at a time for patients with WHO Functional Class II or above symptoms.
Conversely, PAH‐specific therapies are generally not beneficial in Group 3 PH, with potential for worsening ventilation/perfusion (V/Q) mismatch and consequent hypoxaemia. Management should focus on treating the ILD and hypoxaemia. Evidence‐based guidelines for the management of PH coexisting with moderate‐to‐severe CTD‐ILD do not currently exist.
General management of all CTD patients with PH includes diuresis, supervised exercised programmes and domiciliary oxygen as required. As for all PH, there is no clear evidence for anticoagulation (in the absence of another indication), pregnancy should be avoided and anaesthesia carries significant additional risks.
Summary.
Elevated pulmonary pressure in the setting of CTD carries a significant negative prognostic impact and warrants evaluation at an experienced PH centre.
In CTD‐PAH (WHO Group 1), pulmonary vasodilator therapies are beneficial and should be instituted once a PAH diagnosis is confirmed, ideally at WHO functional class II level. 206 , 207 , 208 , 210
Treatment escalation of CTD‐PAH with combination therapy as per the ESC/ERS guidelines is recommended. 210
PH complicating CTD‐ILD (WHO Group 3) should prompt optimization of ILD therapy and correction of hypoxaemia.
Lung transplantation for CTD
Approximately 9–12% of CTD‐ILD patients progress to end‐stage respiratory failure, despite appropriate immunosuppressive therapy and lung transplantation are potential treatment options. CTD is not a relative or absolute contraindication to transplantation, yet less than 2% of all lung transplants worldwide between 1995 and 2010 were for patients with CTD‐associated lung disease. 211 , 212 , 213 Factors complicating transplant management and outcomes in CTD include immune dysregulation, GORD, renal disease and active myositis. Oesophageal dysfunction is common in CTD patients, especially in those with SSc, and GORD is potentially associated with the development of chronic lung allograft dysfunction and post‐transplant morbidity and mortality. 214 The presence of untreatable significant dysfunction of another major organ system (e.g. heart, liver, kidney or brain) is an absolute contraindication unless combined organ transplantation can be performed. 213
Recent studies suggest that carefully selected CTD patients with pulmonary disease warrant transplantation assessment, with similar post‐transplant outcomes to other cohorts. Evaluation of 284 lung transplantations in CTD patients from 1991 to 2009 included in the United States Organ Procurement and Transplantation Network database reported that there were no post‐transplant recurrences of CTD lung disease, extra‐pulmonary flares of the CTD were rare (one possible flare per 20.3 patient years) and cumulative survival was similar to IPF patients. 215
This was echoed by a single‐centre cohort study of 892 lung transplant recipients, including 72 with SSc‐ILD and 311 with pulmonary fibrosis from other causes (other ILD). 216 SSc‐ILD recipients demonstrated satisfactory short‐term survival (100% 30‐day and 80.6% 1‐year survival) and no difference in 5‐year survival (conditional on 1‐year survival) compared to other ILD (66% vs 58%, respectively, P = 0.249). 216 Importantly, SSc‐ILD recipients also had superior bronchiolitis obliterans syndrome (BOS)‐free survival. 216 Careful evaluation with 24 h pH monitoring and oesophageal manometry (before and/or early after transplant) may aid reflux management, including planning laparoscopic fundoplication post‐transplant as a strategy for long‐term allograft protection.
Case series of successful transplant outcomes are emerging for myositis patients, but the presence of active myositis requiring immunosuppression pre‐transplant, especially steroids, may complicate post‐transplant management, rehabilitation and outcomes. 217
Summary.
In selected CTD‐ILD patients, survival following lung transplantation is comparable to non‐CTD patients with similar restrictive lung disease. 215
Given the unique challenges in CTD‐ILD, early assessment by an experienced transplant physician should be undertaken in those patients with severe and/or progressive lung disease who may be suitable transplant recipients.
Non‐pharmacological management in CTD‐ILD
People with CTD‐ILD often experience dyspnoea, fatigue, anxiety, depression, cough and reduced exercise tolerance. 155 , 218 These symptoms are debilitating and substantially affect HRQoL. 219 , 220 The CTD are further complicated by other systemic manifestations including arthropathy, vasculopathy, exercise‐induced hypoxaemia and corticosteroid‐induced myopathies which may further impair exercise tolerance. Non‐pharmacological strategies such as pulmonary rehabilitation (PR), supplemental oxygen and the early integration of palliative care can positively impact symptoms and HRQoL in this population.
Pulmonary rehabilitation
There is increasing evidence that PR is effective at improving exercise tolerance, dyspnoea and HRQoL in ILD. 221 , 222 , 223 Whilst there are no RCT of PR in CTD‐ILD specifically, one study including patients with CTD‐ILD found clinically meaningful benefits in HRQoL in subgroup analysis, despite limited improvements in exercise capacity. PR is strongly recommended in the British Thoracic Society PR guidelines for all patients functionally limited by dyspnoea. 222 , 224 It is possible that musculoskeletal limitations (joint pain, swelling and stiffness) may limit the gains in exercise capacity that can be achieved with traditional PR. Alternative strategies could include water‐based training, which is effective in COPD patients with physical comorbidities. 225 PR also incorporates education, nutrition counselling and psychological support, which may assist to improve emotional and physical well‐being in this population. 226 , 227
Supplemental oxygen
Clinical practice guidelines provide strong recommendations for the use of long‐term oxygen therapy (LTOT ≥18 h per day) for people with ILD who have resting hypoxaemia. 228 , 229 This is based on studies in COPD where it is known to improve survival. Supplemental oxygen may provide symptomatic relief in patient with advanced ILD. 230 , 231 The role of oxygen therapy in people with ILD who have exertional desaturation without resting hypoxaemia is less clear. Two small studies in IPF found that oxygen significantly increased endurance time and reduced exertional dyspnoea during constant load cycling. 232 , 233 This supports the ATS/ERS PR guidelines recommendation for supplemental oxygen during exercise training in patients who desaturate on exertion. 226 A recent crossover trial of short‐term (2 weeks) use of ambulatory oxygen in fibrotic ILD demonstrated a significant improvement in HRQoL. 234 Longer term outcomes remain unclear. There are two current RCT including a comparison of the effects of ambulatory oxygen with room air over 3 months (NCT02286063 and NCT02551068) and the benefits of oxygen during an 8‐week exercise programme (NCT02551068).
Symptom management and palliative care
Palliative care aims to alleviate symptom burden, especially dyspnoea and cough, address advance care planning and optimize HRQoL throughout the disease course. 231 , 235 Palliative care should occur in parallel with all other treatments and not be delayed until the disease is advanced. 231 , 236 As discussed, supplemental oxygen and PR play an important role in self‐management and reducing dyspnoea. 221 Opioids, up to a dose of 30 mg morphine daily, might safely relieve dyspnoea. 3 , 230 , 231 , 237 , 238 Evidence for the use of benzodiazepines is limited, but may be an alternative in those with dyspnoea associated with anxiety or not controlled by opioids. 231 , 235 , 238 , 239 Caution is required with both opioids and low‐dose benzodiazepines associated with increased hospital admissions and mortality in patients with oxygen‐dependent ILD. 240
Treatment of cough is difficult and requires investigation for, and treatment of, GORD given its high prevalence in CTD‐ILD. 236 , 241 Thalidomide has been shown to reduce cough in IPF, although there is no evidence of its effect in CTD‐ILD. 231 , 241 Gabapentin appears effective for refractory chronic cough and mucolytic agents may be useful in assisting with sputum clearance, but CTD‐ILD specific data are lacking. 242 , 243 Low‐dose opiates may be considered to palliate cough when alternative treatments have been unsuccessful. 238 , 241 , 244
Non‐pharmacological interventions such as handheld fans, rollator devices, breathing exercises and speech pathology may also prove beneficial in relieving symptoms. 231 , 238 , 244 , 245 Symptom management should also address depression, anxiety, fatigue and deconditioning, for which PR and psychotherapy may be of assistance. 227 , 231 , 238 , 246 , 247 , 248 , 249 Education, support and assistance for self‐management for both patients and caregivers is important in optimizing quality of life and in advanced care planning. Timely patient‐centred end‐of‐life care should be offered to all patients where progressive disease is evident or expected. 231
Summary.
In CTD‐ILD, PR can help improve exercise tolerance, dyspnoea and HRQoL.
LTOT should be offered in those patients with resting hypoxaemia. Supplemental oxygen during exercise training in those with exertional desaturation may facilitate PR outcomes. 221 , 229 , 233 , 234 , 236
Palliative care should be considered in parallel with other treatment interventions to optimize symptom management. 235
FUTURE DIRECTIONS
The next 5 years will see the most significant changes in the available therapies and the evidence base supporting treatment choice for CTD‐ILD. Some of the most important treatment trials in CTD‐ILD are listed in Table 5. Anti‐fibrotics additional to immunosuppression are an active area of current research. As discussed, the SENSCIS study demonstrated efficacy of nintedanib in SSc‐ILD, and long‐term safety is being evaluated in an open‐label extension study (NCT03313180). 176 The utility of pirfenidone in combination with MMF for SSc‐ILD is being assessed in the Scleroderma Lung Study III (NCT03221257) and the TRAIL1 study is evaluating pirfenidone in RA‐ILD (NCT02808871).
Rituximab is generally reserved for severe CTD‐ILD progressing despite conventional therapy. However, rituximab as ‘induction’ therapy (1 g at baseline and 2 weeks) is being evaluated in two ongoing clinical trials—one comparing rituximab with placebo and background MMF therapy in both arms in a broad range of CTD‐ILD (NCT02990286), and a second comparing rituximab with IV cyclophosphamide (600 mg/m2 4 weekly for six doses) in SSc‐ILD, IIM and MCTD (NCT01862926). 176 As discussed, tocilizumab was recently found to be associated with stabilization of FVC in SSc with final analysis awaited. 109
Genetic studies in CTD‐ILD are only just being reported, but it seems likely that there will be increasing recognition of gene–environment interactions and their impact on disease pathogenesis and prognosis, similar to IPF and chronic hypersensitivity pneumonitis. 116 , 250 The incorporation of genomic markers to aid in diagnosis, risk stratification and clinical decision‐making together with the identification of other molecular biomarkers may be of particular importance in CTD‐ILD, given the characteristically variable disease course. 251
The rarity of CTD‐ILD in general will continue to present difficulties obtaining evidence and reimbursement for therapeutic agents, especially for orphan CTD‐ILD (e.g. SjS‐ILD and anti‐synthetase syndrome). However, advances in diagnostic and therapeutic modalities, and the evidence base that underpins their place in clinical practice, will significantly improve both the safety and efficacy of care for all patients with CTD‐ILD.
SUMMARY AND CONCLUSIONS
Management of CTD‐ILD is a challenging endeavour, requiring a multidisciplinary approach in order to achieve the best possible outcomes for patients and their caregivers. Accurate diagnosis, staging of severity and monitoring progression of CTD‐ILD require the at times complicated integration of clinical, radiological, physiological and serologic data, often with substantial input from non‐respiratory specialists. While immunosuppression remains the mainstay of treatment for severe and/or progressive CTD‐ILD, deciding when to start therapy, which agent to use and duration of therapy is a nuanced decision impacted by many clinical and patient specific factors. An ever‐expanding array of treatment options, including monoclonal antibodies targeting specific molecular pathways and the anti‐fibrotic agents, are being evaluated with promising early results and are providing new insights into disease pathophysiology.
In this position statement, we have endeavoured to provide both an overview of general principles of CTD‐ILD monitoring and treatment in addition to disease‐specific guidance. However, it is important to note that given the heterogeneous clinical course of many CTD‐ILD, no single management strategy will capture every clinical scenario, and continued assessment and calibration of treatment approach is vital. Given the rarity of some of the CTD‐ILD and absence of specific treatment evidence, by necessity therapeutic approaches are often extrapolated from other CTD‐ILD with assumed similar disease mechanisms. This paucity of treatment evidence represents a major unmet clinical need across many CTD‐ILD, and should remain a priority for future research. Finally, non‐pharmacological therapies are a key and often overlooked aspect in the holistic care of patients with these multi‐system disorders.
Disclosure statement
M.J.S.P. has received speaker's fees from Boehringer Ingelheim; J.P.W. has received speaking honorarium and adboard honorarium from Roche and Boehringer Ingelheim; T.J.C. has received grants (Boehringer Ingelheim, Roche, Gilead, Bayer, Intermune and BMS) and personal fees (Boehringer Ingelheim, Roche, BMS, Astra Zeneca, Ad Alta and Promedior) during the conduct of the study; I.N.G. has received speaker's fees from Boehringer Ingelheim and Roche and acted as a consultant to Ad Alta, Avalyn Pharma, Boehringer Ingelheim and Roche; D.C.C. acted as a paid consultant to Boehringer Ingelheim; C.G. conducts contracted fundamental science research on behalf of Boehringer Ingelheim on the subject of fibrotic lung disease and has been a paid advisory board membership for Boehringer Ingelheim and Roche; L.K.T. has received speaker's fees from Boehringer Ingelheim and Roche, and works in a unit that has received unrestricted educational grants from Boehringer Ingelheim and Roche. G.J.K., A.S.J., P.H., L.M.S., L.D., S.C., K.S., P.N.R., S.d.B., T.R., A.E.H., H.E.J., J.F.B., M.L.W., N.S.L.G., R.S. and Y.M. have no conflicts of interest to declare in relation to this document.
Acknowledgements
This work was supported by the NHMRC Centre of Research Excellence in Pulmonary Fibrosis, Australia, which is funded by the NHMRC and supported by Foundation partner Boehringer Ingelheim and Program Partners Roche and Galapagos.
Jee AS, Sheehy R, Hopkins P, et al. Diagnosis and management of connective tissue disease‐associated interstitial lung disease in Australia and New Zealand: A position statement from the Thoracic Society of Australia and New Zealand*. Respirology. 2021;26:23–51. 10.1111/resp.13977
*This document was endorsed by the Thoracic Society of Australia and New Zealand (TSANZ) Board of Directors on 14 February 2020 after review by the TSANZ Clinical Care and Resources Subcommittee.
Received 11 May 2020; invited to revise 29 June and 6 September 2020; revised 10 August and 26 September 2020; accepted 22 October 2020
Associate Editor: Elisabetta Renzoni; Senior Editor: Philip Bardin
REFERENCES
- 1. Wells A, Denton CP. Pulmonary complications of connective tissue disease. Semin. Respir. Crit. Care Med. 2019; 40: 145–6. [DOI] [PubMed] [Google Scholar]
- 2. Raghu G, Remy‐Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F et al Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018; 198: e44–68. [DOI] [PubMed] [Google Scholar]
- 3. Jo HE, Troy LK, Keir G, Chambers DC, Holland A, Goh N, Wilsher M, de Boer S, Moodley Y, Grainge C et al Treatment of idiopathic pulmonary fibrosis in Australia and New Zealand: a position statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia. Respirology 2017; 22: 1436–58. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki A, Kondoh Y, Fischer A. Recent advances in connective tissue disease related interstitial lung disease. Expert Rev. Respir. Med. 2017; 11: 591–603. [DOI] [PubMed] [Google Scholar]
- 5. Mira‐Avendano I, Abril A, Burger CD, Dellaripa PF, Fischer A, Gotway MB, Lee AS, Lee JS, Matteson EL, Yi ES et al Interstitial lung disease and other pulmonary manifestations in connective tissue diseases. Mayo Clin. Proc. 2019; 94: 309–25. [DOI] [PubMed] [Google Scholar]
- 6. van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Matucci‐Cerinic M, Naden RP, Medsger TA Jr, Carreira PE et al 2013 Classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Ann. Rheum. Dis. 2013; 72: 1747–55. [DOI] [PubMed] [Google Scholar]
- 7. Ha Y, Lee Y, Kang E. Lung involvements in rheumatic diseases: update on the epidemiology, pathogenesis, clinical features, and treatment. Biomed. Res. Int. 2018; 2018: 6930297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wallace B, Vummidi D, Khanna D. Management of connective tissue diseases associated interstitial lung disease: a review of the published literature. Curr. Opin. Rheumatol. 2016; 28: 236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD et al 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum. 2010; 62: 2569–81. [DOI] [PubMed] [Google Scholar]
- 10. Doyle TJ, Patel AS, Hatabu H, Nishino M, Wu G, Osorio JC, Golzarri MF, Traslosheros A, Chu SG, Frits ML et al Detection of rheumatoid arthritis‐interstitial lung disease is enhanced by serum biomarkers. Am. J. Respir. Crit. Care Med. 2015; 191: 1403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, Alfredsson L, Amato AA, Barohn RJ, Liang MH et al 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann. Rheum. Dis. 2017; 76: 1955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fathi M, Vikgren J, Boijsen M, Tylen U, Jorfeldt L, Tornling G, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis: longitudinal evaluation by pulmonary function and radiology. Arthritis Rheum. 2008; 59: 677–85. [DOI] [PubMed] [Google Scholar]
- 13. Connors GR, Christopher‐Stine L, Oddis CV, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies: what progress has been made in the past 35 years? Chest 2010; 138: 1464–74. [DOI] [PubMed] [Google Scholar]
- 14. Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey‐Goldman R, Smolen JS, Wofsy D, Boumpas DT, Kamen DL et al 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann. Rheum. Dis. 2019; 78: 1151–9. [DOI] [PubMed] [Google Scholar]
- 15. Roca F, Dominique S, Schmidt J, Smail A, Duhaut P, Levesque H, Marie I. Interstitial lung disease in primary Sjogren's syndrome. Autoimmun. Rev. 2017; 16: 48–54. [DOI] [PubMed] [Google Scholar]
- 16. Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ et al 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren's syndrome: a consensus and data‐driven methodology involving three international patient cohorts. Arthritis Rheumatol. 2017; 69: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alarcon‐Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J. Rheumatol. 1989; 16: 328–34. [PubMed] [Google Scholar]
- 18. Bongartz T, Nannini C, Medina‐Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, Vassallo R, Gabriel SE, Matteson EL. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population‐based study. Arthritis Rheum. 2010; 62: 1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kelly CA, Saravanan V, Nisar M, Arthanari S, Woodhead FA, Price‐Forbes AN, Dawson J, Sathi N, Ahmad Y, Koduri G et al Rheumatoid arthritis‐related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics – a large multicentre UK study. Rheumatology (Oxford) 2014; 53: 1676–82. [DOI] [PubMed] [Google Scholar]
- 20. Satoh M, Tanaka S, Ceribelli A, Calise SJ, Chan EK. A comprehensive overview on myositis‐specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol 2017; 52: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Y, Yang CS, Li YJ, Liu XD, Wang JN, Zhao Q, Xiao WG, Yang PT. Predictive factors of rapidly progressive‐interstitial lung disease in patients with clinically amyopathic dermatomyositis. Clin. Rheumatol. 2016; 35: 113–6. [DOI] [PubMed] [Google Scholar]
- 22. Fischer A, Meehan RT, Feghali‐Bostwick CA, West SG, Brown KK. Unique characteristics of systemic sclerosis sine scleroderma‐associated interstitial lung disease. Chest 2006; 130: 976–81. [DOI] [PubMed] [Google Scholar]
- 23. Nair A, Walsh SL, Desai SR. Imaging of pulmonary involvement in rheumatic disease. Rheum. Dis. Clin. North Am. 2015; 41: 167–96. [DOI] [PubMed] [Google Scholar]
- 24. Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez‐Perez ER, Fischer A, Frankel SK, Hobbs SB, Huie TJ, Ketzer J et al Predictors of mortality in rheumatoid arthritis‐associated interstitial lung disease. Eur. Respir. J. 2016; 47: 588–96. [DOI] [PubMed] [Google Scholar]
- 25. Kim EJ, Elicker BM, Maldonado F, Webb WR, Ryu JH, Van Uden JH, Lee JS, King TE, Collard HR. Usual interstitial pneumonia in rheumatoid arthritis‐associated interstitial lung disease. Eur. Respir. J. 2010; 35: 1322–8. [DOI] [PubMed] [Google Scholar]
- 26. Aparicio IJ, Lee JS. Connective tissue disease‐associated interstitial lung diseases: unresolved issues. Semin. Respir. Crit. Care Med. 2016; 37: 468–76. [DOI] [PubMed] [Google Scholar]
- 27. Bryson T, Sundaram B, Khanna D, Kazerooni EA. Connective tissue disease‐associated interstitial pneumonia and idiopathic interstitial pneumonia: similarity and difference. Semin. Ultrasound CT MR 2014; 35: 29–38. [DOI] [PubMed] [Google Scholar]
- 28. Tanaka N, Kunihiro Y, Kubo M, Kawano R, Oishi K, Ueda K, Gondo T. HRCT findings of collagen vascular disease‐related interstitial pneumonia (CVD‐IP): a comparative study among individual underlying diseases. Clin. Radiol. 2018; 73: 833.e1–e10. [DOI] [PubMed] [Google Scholar]
- 29. Lynch DA. Lung disease related to collagen vascular disease. J. Thorac. Imaging 2009; 24: 299–309. [DOI] [PubMed] [Google Scholar]
- 30. Tsuchiya Y, Takayanagi N, Sugiura H, Miyahara Y, Tokunaga D, Kawabata Y, Sugita Y. Lung diseases directly associated with rheumatoid arthritis and their relationship to outcome. Eur. Respir. J. 2011; 37: 1411–7. [DOI] [PubMed] [Google Scholar]
- 31. Franquet T. High‐resolution CT of lung disease related to collagen vascular disease. Radiol. Clin. North Am. 2001; 39: 1171–87. [DOI] [PubMed] [Google Scholar]
- 32. Ruano CA, Lucas RN, Leal CI, Lourenco J, Pinheiro S, Fernandes O, Figueiredo L. Thoracic manifestations of connective tissue diseases. Curr. Probl. Diagn. Radiol. 2015; 44: 47–59. [DOI] [PubMed] [Google Scholar]
- 33. Cottin V, Reix P, Khouatra C, Thivolet‐Bejui F, Feldmann D, Cordier JF. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 2011; 66: 918–9. [DOI] [PubMed] [Google Scholar]
- 34. Enomoto Y, Nakamura Y, Colby TV, Johkoh T, Sumikawa H, Nishimoto K, Yoshimura K, Matsushima S, Oyama Y, Hozumi H et al Radiologic pleuroparenchymal fibroelastosis‐like lesion in connective tissue disease‐related interstitial lung disease. PLoS One. 2017; 12: e0180283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakamura Y, Chida K, Suda T, Hayakawa H, Iwata M, Imokawa S, Tsuchiya T, Ida M, Gemma H, Yasuda K et al Nonspecific interstitial pneumonia in collagen vascular diseases: comparison of the clinical characteristics and prognostic significance with usual interstitial pneumonia. Sarcoidosis Vasc. Diffuse Lung Dis. 2003; 20: 235–41. [PubMed] [Google Scholar]
- 36. Kim EJ, Collard HR, King TE Jr. Rheumatoid arthritis‐associated interstitial lung disease: the relevance of histopathologic and radiographic pattern. Chest 2009; 136: 1397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee HK, Kim DS, Yoo B, Seo JB, Rho JY, Colby TV, Kitaichi M. Histopathologic pattern and clinical features of rheumatoid arthritis‐associated interstitial lung disease. Chest 2005; 127: 2019–27. [DOI] [PubMed] [Google Scholar]
- 38. Walsh SL, Sverzellati N, Devaraj A, Keir GJ, Wells AU, Hansell DM. Connective tissue disease related fibrotic lung disease: high resolution computed tomographic and pulmonary function indices as prognostic determinants. Thorax 2014; 69: 216–22. [DOI] [PubMed] [Google Scholar]
- 39. Parambil JG, Myers JL, Lindell RM, Matteson EL, Ryu JH. Interstitial lung disease in primary Sjogren syndrome. Chest 2006; 130: 1489–95. [DOI] [PubMed] [Google Scholar]
- 40. Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In‐hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am. J. Respir. Crit. Care Med. 2016; 193: 1161–7. [DOI] [PubMed] [Google Scholar]
- 41. Hunninghake GW, Zimmerman MB, Schwartz DA, King TE Jr, Lynch J, Hegele R, Waldron J, Colby T, Muller N, Lynch D et al Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2001; 164: 193–6. [DOI] [PubMed] [Google Scholar]
- 42. Lentz RJ, Argento AC, Colby TV, Rickman OB, Maldonado F. Transbronchial cryobiopsy for diffuse parenchymal lung disease: a state‐of‐the‐art review of procedural techniques, current evidence, and future challenges. J. Thorac. Dis. 2017; 9: 2186–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Troy LK, Grainge C, Corte TJ, Williamson JP, Vallely MP, Cooper WA, Mahar A, Myers JL, Lai S, Mulyadi E et al Diagnostic accuracy of transbronchial lung cryobiopsy for interstitial lung disease diagnosis (COLDICE): a prospective, comparative study. Lancet Respir. Med. 2020; 8: 171–181. [DOI] [PubMed] [Google Scholar]
- 44. Maldonado F, Danoff SK, Wells AU, Colby TV, Ryu JH, Liberman M, Wahidi MM, Frazer L, Hetzel J, Rickman OB et al Transbronchial cryobiopsy for the diagnosis of interstitial lung diseases: CHEST guideline and expert panel report. Chest 2020; 157: 1030–42. [DOI] [PubMed] [Google Scholar]
- 45. Goh NSL, Veeraraghavan S, Desai SR, Cramer D, Hansell DM, Denton CP, Black CM, Du Bois RM, Wells AU. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis–associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007; 56: 2005–12. [DOI] [PubMed] [Google Scholar]
- 46. Prasad JD, Mahar A, Bleasel J, Ellis SJ, Chambers DC, Lake F, Hopkins PMA, Corte TJ, Allan H, Glaspole IN. The interstitial lung disease multidisciplinary meeting: a position statement from the Thoracic Society of Australia and New Zealand and the Lung Foundation Australia. Respirology 2017; 22: 1459–72. [DOI] [PubMed] [Google Scholar]
- 47. Jo HE, Corte TJ, Moodley Y, Levin K, Westall G, Hopkins P, Chambers D, Glaspole I. Evaluating the interstitial lung disease multidisciplinary meeting: a survey of expert centres. BMC Pulm. Med. 2016; 16: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walsh SLF, Wells AU, Desai SR, Poletti V, Piciucchi S, Dubini A, Nunes H, Valeyre D, Brillet PY, Kambouchner M et al Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case‐cohort study. Lancet Respir. Med. 2016; 4: 557–65. [DOI] [PubMed] [Google Scholar]
- 49. Jo HE, Glaspole IN, Levin KC, McCormack SR, Mahar AM, Cooper WA, Cameron R, Ellis SJ, Cottee AM, Webster SE et al Clinical impact of the interstitial lung disease multidisciplinary service. Respirology 2016; 21: 1438–44. [DOI] [PubMed] [Google Scholar]
- 50. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, Goldin JG, Hansell DM, Inoue Y, Johkoh T et al Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir. Med. 2018; 6: 138–53. [DOI] [PubMed] [Google Scholar]
- 51. Wells A, Devaraj A, Renzoni EA, Denton CP. Multidisciplinary evaluation in patients with lung disease associated with connective tissue disease. Semin. Respir. Crit. Care Med. 2019; 40: 184–93. [DOI] [PubMed] [Google Scholar]
- 52. Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD, Elicker BM, Wolters PJ, Koth LL, King TE Jr et al Predicting survival across chronic interstitial lung disease: the ILD‐GAP model. Chest 2014; 145: 723–8. [DOI] [PubMed] [Google Scholar]
- 53. Brusca RM, Pinal‐Fernandez I, Psoter K, Paik JJ, Albayda J, Mecoli C, Tiniakou E, Mammen AL, Christopher‐Stine L, Danoff S et al The ILD‐GAP risk prediction model performs poorly in myositis‐associated interstitial lung disease. Respir. Med. 2019; 150: 63–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, Corte TJ, Sander CR, Ratoff J, Devaraj A et al Interstitial lung disease in systemic sclerosis: a simple staging system. Am. J. Respir. Crit. Care Med. 2008; 177: 1248–54. [DOI] [PubMed] [Google Scholar]
- 55. Volkmann ER, Tashkin DP, Sim M, Li N, Goldmuntz E, Keyes‐Elstein L, Pinckney A, Furst DE, Clements PJ, Khanna D et al Short‐term progression of interstitial lung disease in systemic sclerosis predicts long‐term survival in two independent clinical trial cohorts. Ann. Rheum. Dis. 2019; 78: 122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Goh NS, Hoyles RK, Denton CP, Hansell DM, Renzoni EA, Maher TM, Nicholson AG, Wells AU. Short‐term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. 2017; 69: 1670–8. [DOI] [PubMed] [Google Scholar]
- 57. ATS/ERS . American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 58. Moore OA, Proudman SM, Goh N, Corte TJ, Rouse H, Hennessy O, Morrisroe K, Thakkar V, Sahhar J, Roddy J et al Quantifying change in pulmonary function as a prognostic marker in systemic sclerosis‐related interstitial lung disease. Clin. Exp. Rheumatol. 2015; 33(4 Suppl. 91): S111–6. [PubMed] [Google Scholar]
- 59. Zappala CJ, Latsi PI, Nicholson AG, Colby TV, Cramer D, Renzoni EA, Hansell DM, du Bois RM, Wells AU. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010; 35: 830–6. [DOI] [PubMed] [Google Scholar]
- 60. Jacob J, Bartholmai BJ, Rajagopalan S, Brun AL, Egashira R, Karwoski R, Kokosi M, Wells AU, Hansell DM. Evaluation of computer‐based computer tomography stratification against outcome models in connective tissue disease‐related interstitial lung disease: a patient outcome study. BMC Med. 2016; 14: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jee AS, Sahhar J, Youssef P, Bleasel J, Adelstein S, Nguyen M, Corte TJ. Review: serum biomarkers in idiopathic pulmonary fibrosis and systemic sclerosis associated interstitial lung disease – frontiers and horizons. Pharmacol. Ther. 2019; 202: 40–52. [DOI] [PubMed] [Google Scholar]
- 62. Wells AU, Denton CP. Interstitial lung disease in connective tissue disease – mechanisms and management. Nat. Rev. Rheumatol. 2014; 10: 728–39. [DOI] [PubMed] [Google Scholar]
- 63. Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ 2016; 352: h6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU et al An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013; 188: 733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moore OA, Goh N, Corte T, Rouse H, Hennessy O, Thakkar V, Byron J, Sahhar J, Roddy J, Gabbay E et al Extent of disease on high‐resolution computed tomography lung is a predictor of decline and mortality in systemic sclerosis‐related interstitial lung disease. Rheumatology (Oxford) 2013; 52: 155–60. [DOI] [PubMed] [Google Scholar]
- 66. Wiseman AC. Immunosuppressive medications. Clin. J. Am. Soc. Nephrol. 2016; 11: 332–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Meyer KC. Immunosuppressive agents and interstitial lung disease: what are the risks? Expert Rev. Respir. Med. 2014; 8: 263–6. [DOI] [PubMed] [Google Scholar]
- 68. Carmona L, Gómez‐Reino JJ, Rodríguez‐Valverde V, Montero D, Pascual‐Gómez E, Mola EM, Carreño L, Figueroa M; BIOBADASER Group . Effectiveness of recommendations to prevent reactivation of latent tuberculosis infection in patients treated with tumor necrosis factor antagonists. Arthritis Rheum. 2005; 52: 1766–72. [DOI] [PubMed] [Google Scholar]
- 69. Jick SS, Lieberman ES, Rahman MU, Choi HK. Glucocorticoid use, other associated factors, and the risk of tuberculosis. Arthritis Rheum. 2006; 55: 19–26. [DOI] [PubMed] [Google Scholar]
- 70. Seibold JR, Denton CP, Furst DE, Guillevin L, Rubin LJ, Wells A, Matucci Cerinic M, Riemekasten G, Emery P, Chadha‐Boreham H et al Randomized, prospective, placebo‐controlled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010; 62: 2101–8. [DOI] [PubMed] [Google Scholar]
- 71. Cooley L, Dendle C, Wolf J, Teh BW, Chen SC, Boutlis C, Thursky KA. Consensus guidelines for diagnosis, prophylaxis and management of Pneumocystis jirovecii pneumonia in patients with haematological and solid malignancies, 2014. Intern. Med. J. 2014; 44: 1350–63. [DOI] [PubMed] [Google Scholar]
- 72. Khanna D, Albera C, Fischer A, Khalidi N, Raghu G, Chung L, Chen D, Schiopu E, Tagliaferri M, Seibold JR et al An open‐label, phase II study of the safety and tolerability of Pirfenidone in patients with scleroderma‐associated interstitial lung disease: the LOTUSS trial. J. Rheumatol. 2016; 43: 1672–9. [DOI] [PubMed] [Google Scholar]
- 73. Hsu V, Denton C, Domsic R, Furst D, Rischmueller M, Stanislav M, Steen V, Distler JW, Korish S, Cooper A et al Pomalidomide in patients with interstitial lung disease due to systemic sclerosis: a phase II, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study. J. Rheumatol. 2018; 45: 405–10. [DOI] [PubMed] [Google Scholar]
- 74. Khanna D, Denton CP, Lin CJ, van Laar JM, Frech TM, Anderson ME, Baron M, Chung L, Fierlbeck G, Lakshminarayanan S et al Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open‐label period of a phase II randomised controlled trial (faSScinate). Ann. Rheum. Dis. 2018; 77: 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reid AB, Chen SC, Worth LJ. Pneumocystis jirovecii pneumonia in non‐HIV‐infected patients: new risks and diagnostic tools. Curr. Opin. Infect. Dis. 2011; 24: 534–44. [DOI] [PubMed] [Google Scholar]
- 76. Daoussis D, Melissaropoulos K, Sakellaropoulos G, Antonopoulos I, Markatseli TE, Simopoulou T, Georgiou P, Andonopoulos AP, Drosos AA, Sakkas L et al A multicenter, open‐label, comparative study of B‐cell depletion therapy with rituximab for systemic sclerosis‐associated interstitial lung disease. Semin. Arthritis Rheum. 2017; 46: 625–31. [DOI] [PubMed] [Google Scholar]
- 77. Hao Y, Hudson M, Baron M, Carreira P, Stevens W, Rabusa C, Tatibouet S, Carmona L, Joven BE, Huq M et al Early mortality in a multinational systemic sclerosis inception cohort. Arthritis Rheumatol. 2017; 69: 1067–77. [DOI] [PubMed] [Google Scholar]
- 78. Steen VD, Conte C, Owens GR, Medsger TA Jr. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994; 37: 1283–9. [DOI] [PubMed] [Google Scholar]
- 79. Khanna D, Nagaraja V, Tseng CH, Abtin F, Suh R, Kim G, Wells A, Furst DE, Clements PJ, Roth MD et al Predictors of lung function decline in scleroderma‐related interstitial lung disease based on high‐resolution computed tomography: implications for cohort enrichment in systemic sclerosis‐associated interstitial lung disease trials. Arthritis Res. Ther. 2015; 17: 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Song JW, Lee CK, Chae EJ, Jang SJ, Colby TV, Kim DS. Clinical course and outcome of rheumatoid arthritis‐related usual interstitial pneumonia. Sarcoidosis Vasc. Diffuse Lung Dis. 2013; 30: 103–12. [PubMed] [Google Scholar]
- 81. Fischer A, Solomon JJ, du Bois RM, Deane KD, Olson AL, Fernandez‐Perez ER, Huie TJ, Stevens AD, Gill MB, Rabinovitch AM et al Lung disease with anti‐CCP antibodies but not rheumatoid arthritis or connective tissue disease. Respir. Med. 2012; 106: 1040–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schnabel A, Reuter M, Biederer J, Richter C, Gross WL. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin. Arthritis Rheum. 2003; 32: 273–84. [DOI] [PubMed] [Google Scholar]
- 83. Kameda H, Nagasawa H, Ogawa H, Sekiguchi N, Takei H, Tokuhira M, Amano K, Takeuchi T. Combination therapy with corticosteroids, cyclosporin A, and intravenous pulse cyclophosphamide for acute/subacute interstitial pneumonia in patients with dermatomyositis. J. Rheumatol. 2005; 32: 1719. [PubMed] [Google Scholar]
- 84. Ceribelli A, Fredi M, Taraborelli M, Cavazzana I, Tincani A, Selmi C, Chan JY, Chan EK, Satoh M, Franceschini F. Prevalence and clinical significance of anti‐MDA5 antibodies in European patients with polymyositis/dermatomyositis. Clin. Exp. Rheumatol. 2014; 32: 891–7. [PubMed] [Google Scholar]
- 85. Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M et al Nintedanib for systemic sclerosis–associated interstitial lung disease. N. Engl. J. Med. 2019; 380: 2518–28. [DOI] [PubMed] [Google Scholar]
- 86. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S et al Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019; 381: 1718–27. [DOI] [PubMed] [Google Scholar]
- 87. Wells AU, Flaherty KR, Brown KK, Inoue Y, Devaraj A, Richeldi L, Moua T, Crestani B, Wuyts WA, Stowasser S et al Nintedanib in patients with progressive fibrosing interstitial lung diseases‐subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double‐blind, placebo‐controlled, parallel‐group trial. Lancet Respir. Med. 2020; 8: 453–60. [DOI] [PubMed] [Google Scholar]
- 88. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina‐Molina M, Axmann J, Kirchgaessler K‐U, Samara K, Gilberg F et al Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double‐blind, randomised, placebo‐controlled, phase 2 trial. Lancet Respir. Med. 2020; 8: 147–157. [DOI] [PubMed] [Google Scholar]
- 89. Holroyd CR, Seth R, Bukhari M, Malaviya A, Holmes C, Curtis E, Chan C, Yusuf MA, Litwic A, Smolen S et al The British Society for Rheumatology biologic DMARD safety guidelines in inflammatory arthritis. Rheumatology (Oxford) 2019; 58: e3–e42. [DOI] [PubMed] [Google Scholar]
- 90. Ledingham J, Gullick N, Irving K, Gorodkin R, Aris M, Burke J, Gordon P, Christidis D, Galloway S, Hayes E et al BSR and BHPR guideline for the prescription and monitoring of non‐biologic disease‐modifying anti‐rheumatic drugs. Rheumatology (Oxford) 2017; 56: 865–8. [DOI] [PubMed] [Google Scholar]
- 91. Ai JW, Ruan QL, Liu QH, Zhang WH. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg. Microbes Infect. 2016; 5: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reddy K, Beavers K, Hammond S. American Gastroenterological Association Institute guideline on the prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology 2015; 148: 215–219. [DOI] [PubMed] [Google Scholar]
- 93. Mori S, Sugimoto M. Pneumocystis jirovecii infection: an emerging threat to patients with rheumatoid arthritis. Rheumatology (Oxford) 2012; 51: 2120–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jee AS, Corte TJ. Current and emerging drug therapies for connective tissue disease‐interstitial lung disease (CTD‐ILD). Drugs 2019; 79: 1511–28. [DOI] [PubMed] [Google Scholar]
- 95. Poudel DR, Jayakumar D, Danve A, Sehra ST, Derk CT. Determinants of mortality in systemic sclerosis: a focused review. Rheumatol. Int. 2018; 38: 1847–58. [DOI] [PubMed] [Google Scholar]
- 96. Nihtyanova SI, Denton CP. Autoantibodies as predictive tools in systemic sclerosis. Nat. Rev. Rheumatol. 2010; 6: 112–6. [DOI] [PubMed] [Google Scholar]
- 97. Richardson C, Agrawal R, Lee J, Almagor O, Nelson R, Varga J, Cuttica MJ, Dematte JD, Chang RW, Hinchcliff ME. Esophageal dilatation and interstitial lung disease in systemic sclerosis: a cross‐sectional study. Semin. Arthritis Rheum. 2016; 46: 109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Christmann RB, Wells AU, Capelozzi VL, Silver RM. Gastroesophageal reflux incites interstitial lung disease in systemic sclerosis: clinical, radiologic, histopathologic, and treatment evidence. Semin. Arthritis Rheum. 2010; 40: 241–9. [DOI] [PubMed] [Google Scholar]
- 99. Guler SA, Winstone TA, Murphy D, Hague C, Soon J, Sulaiman N, Li KH, Dunne J, Wilcox PG, Ryerson CJ. Does systemic sclerosis‐associated interstitial lung disease burn out? Specific phenotypes of disease progression. Ann. Am. Thorac. Soc. 2018; 15: 1427–33. [DOI] [PubMed] [Google Scholar]
- 100. Morgan C, Knight C, Lunt M, Black CM, Silman AJ. Predictors of end stage lung disease in a cohort of patients with scleroderma. Ann. Rheum. Dis. 2003; 62: 146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Steen VD, Medsger TA. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000; 43: 2437–44. [DOI] [PubMed] [Google Scholar]
- 102. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, Arriola E, Silver R, Strange C, Bolster M et al Cyclophosphamide versus placebo in scleroderma lung disease. N. Engl. J. Med. 2006; 354: 2655–66. [DOI] [PubMed] [Google Scholar]
- 103. Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, Roberts C, Desai S, Herrick AL, McHugh NJ et al A multicenter, prospective, randomized, double‐blind, placebo‐controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006; 54: 3962–70. [DOI] [PubMed] [Google Scholar]
- 104. Kowal‐Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore Y, Distler O, Clements P, Cutolo M, Czirjak L et al Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017; 76: 1327–39. [DOI] [PubMed] [Google Scholar]
- 105. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, Goldin J, Arriola E, Volkmann ER, Kafaja S et al Mycophenolate mofetil versus oral cyclophosphamide in scleroderma‐related interstitial lung disease (SLS II): a randomised controlled, double‐blind, parallel group trial. Lancet Respir. Med. 2016; 4: 708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Pavlov‐Dolijanovic S, Vujasinovic Stupar N, Zugic V, Ostojic P, Zekovic A, Zivanovic Radnic T, Jeremic I, Tadic I. Long‐term effects of immunosuppressive therapy on lung function in scleroderma patients. Clin. Rheumatol. 2018; 37: 3043–50. [DOI] [PubMed] [Google Scholar]
- 107. Khanna D, Tashkin DP, Denton CP, Lubell MW, Vazquez‐Mateo C, Wax S. Ongoing clinical trials and treatment options for patients with systemic sclerosis–associated interstitial lung disease. Rheumatology 2018; 58: 567–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Khanna D, Lin CJF, Kuwana M, Allanore Y, Batalov A, Butrimiene I, Carreira P, Cerinic MM, Distler O, Kaliterna DM et al Efficacy and safety of tocilizumab for the treatment of systemic sclerosis: results from a phase 3 randomized controlled trial. 2018 ACR/ARHP Annual Meeting. Chicago, IL: Arthritis and Rheumatology, 2018.
- 109. Khanna D, Lin CJF, Goldin J, Kim G, Kuwana M, Allanore Y, Batalov A, Butrimiene I, Carreira P, Matucci‐Cerinic M et al Preservation of lung function observed in a phase 3 randomized controlled trial of tocilizumab for the treatment of early systemic sclerosis. American Thoracic Society International Congress [Abstract]. Am. J. Respir. Crit. Care Med. 2020; 201: A2627–A2627. https://www.atsjournals.org/doi/10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A2627. [Google Scholar]
- 110. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, Craig R, Hirano I, Marshall K, Ruderman E et al Autologous non‐myeloablative haemopoietic stem‐cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open‐label, randomised phase 2 trial. Lancet 2011; 378: 498–506. [DOI] [PubMed] [Google Scholar]
- 111. van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, Schuerwegh AJ, Marijt EW, Vonk MC, Schattenberg AV et al Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014; 311: 2490–8. [DOI] [PubMed] [Google Scholar]
- 112. Sullivan KM, Goldmuntz EA, Keyes‐Elstein L, McSweeney PA, Pinckney A, Welch B, Mayes MD, Nash RA, Crofford LJ, Eggleston B et al Myeloablative autologous stem‐cell transplantation for severe scleroderma. N. Engl. J. Med. 2018; 378: 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zamora‐Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Patterns of interstitial lung disease and mortality in rheumatoid arthritis. Rheumatology (Oxford) 2017; 56: 344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wilsher M, Voight L, Milne D, Teh M, Good N, Kolbe J, Williams M, Pui K, Merriman T, Sidhu K et al Prevalence of airway and parenchymal abnormalities in newly diagnosed rheumatoid arthritis. Respir. Med. 2012; 106: 1441–6. [DOI] [PubMed] [Google Scholar]
- 115. Gabbay E, Tarala R, Will R, Carroll G, Adler B, Cameron D, Lake FR. Interstitial lung disease in recent onset rheumatoid arthritis. Am. J. Respir. Crit. Care Med. 1997; 156(2 Pt 1): 528–35. [DOI] [PubMed] [Google Scholar]
- 116. Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, Kannengiesser C, Ottaviani S, Oka S, Tohma S et al MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N. Engl. J. Med. 2018; 379: 2209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Juge PA, Borie R, Kannengiesser C, Gazal S, Revy P, Wemeau‐Stervinou L, Debray MP, Ottaviani S, Marchand‐Adam S, Nathan N et al Shared genetic predisposition in rheumatoid arthritis‐interstitial lung disease and familial pulmonary fibrosis. Eur. Respir. J. 2017; 49: 1602314. [DOI] [PubMed] [Google Scholar]
- 118. Zamora‐Legoff JA, Krause ML, Crowson CS, Ryu JH, Matteson EL. Progressive decline of lung function in rheumatoid arthritis‐associated interstitial lung disease. Arthritis Rheumatol. 2017; 69: 542–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Keir GJ, Maher TM, Ming D, Abdullah R, Lauretis A, Wickremasinghe M, Nicholson AG, Hansell DM, Wells AU, Renzoni EA. Rituximab in severe, treatment‐refractory interstitial lung disease. Respirology 2014; 19: 353–9. [DOI] [PubMed] [Google Scholar]
- 120. Iqbal K, Kelly C. Treatment of rheumatoid arthritis‐associated interstitial lung disease: a perspective review. Ther. Adv. Musculoskelet. Dis. 2015; 7: 247–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Md Yusof MY, Kabia A, Darby M, Lettieri G, Beirne P, Vital EM, Dass S, Emery P. Effect of rituximab on the progression of rheumatoid arthritis‐related interstitial lung disease: 10 years' experience at a single centre. Rheumatology (Oxford) 2017; 56: 1348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fischer A, Brown KK, Du Bois RM, Frankel SK, Cosgrove GP, Fernandez‐Perez ER, Huie TJ, Krishnamoorthy M, Meehan RT, Olson AL et al Mycophenolate mofetil improves lung function in connective tissue disease‐associated interstitial lung disease. J. Rheumatol. 2013; 40: 640–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Idiopathic Pulmonary Fibrosis Clinical Research Network . Prednisone, azathioprine, and N‐acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012; 366: 1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Roubille C, Haraoui B. Interstitial lung diseases induced or exacerbated by DMARDS and biologic agents in rheumatoid arthritis: a systematic literature review. Semin. Arthritis Rheum. 2014; 43: 613–26. [DOI] [PubMed] [Google Scholar]
- 125. Alarcon GS, Kremer JM, Macaluso M, Weinblatt ME, Cannon GW, Palmer WR, St Clair EW, Sundy JS, Alexander RW, Smith GJ et al Risk factors for methotrexate‐induced lung injury in patients with rheumatoid arthritis. A multicenter, case‐control study. Methotrexate‐Lung Study Group. Ann. Intern. Med. 1997; 127: 356–64. [DOI] [PubMed] [Google Scholar]
- 126. Salliot C, van der Heijde D. Long‐term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann. Rheum. Dis. 2009; 68: 1100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kiely P, Busby AD, Nikiphorou E, Sullivan K, Walsh DA, Creamer P, Dixey J, Young A. Is incident rheumatoid arthritis interstitial lung disease associated with methotrexate treatment? Results from a multivariate analysis in the ERAS and ERAN inception cohorts. BMJ Open 2019; 9: e028466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rojas‐Serrano J, Herrera‐Bringas D, Pérez‐Román D, Pérez‐Dorame R, Mateos‐Toledo H, Mejía M. Rheumatoid arthritis‐related interstitial lung disease (RA‐ILD): methotrexate and the severity of lung disease are associated to prognosis. Clin. Rheumatol. 2017; 36: 1493–500. [DOI] [PubMed] [Google Scholar]
- 129. Suissa S, Hudson M, Ernst P. Leflunomide use and the risk of interstitial lung disease in rheumatoid arthritis. Arthritis Rheum. 2006; 54: 1435–9. [DOI] [PubMed] [Google Scholar]
- 130. Curtis JR, Sarsour K, Napalkov P, Costa LA, Schulman KL. Incidence and complications of interstitial lung disease in users of tocilizumab, rituximab, abatacept and anti‐tumor necrosis factor [alpha] agents, a retrospective cohort study (Report). Arthritis Res. Ther. 2015; 17: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Hallowell R, Horton M. Interstitial lung disease in patients with rheumatoid arthritis: spontaneous and drug induced. Drugs 2014; 74: 443–50. [DOI] [PubMed] [Google Scholar]
- 132. Dixon WG, Hyrich KL, Watson KD, Lunt M, Symmons DP. Influence of anti‐TNF therapy on mortality in patients with rheumatoid arthritis‐associated interstitial lung disease: results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2010; 69: 1086–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Koo BS, Hong S, Kim YJ, Kim Y‐G, Lee C‐K, Yoo B. Mortality in patients with rheumatoid arthritis‐associated interstitial lung disease treated with an anti‐tumor necrosis factor agent. Korean J. Intern. Med. 2015; 30: 104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hagiwara K, Sato T, Takagi‐Kobayashi S, Hasegawa S, Shigihara N, Akiyama O. Acute exacerbation of preexisting interstitial lung disease after administration of etanercept for rheumatoid arthritis. J. Rheumatol. 2007; 34: 1151–4. [PubMed] [Google Scholar]
- 135. Saravanan V, Kelly C. Drug‐related pulmonary problems in patients with rheumatoid arthritis. Rheumatology (Oxford) 2006; 45: 787–9. [DOI] [PubMed] [Google Scholar]
- 136. Lundberg IE, de Visser M, Werth VP. Classification of myositis. Nat. Rev. Rheumatol. 2018; 14: 269–78. [DOI] [PubMed] [Google Scholar]
- 137. Lilleker JB, Vencovsky J, Wang G, Wedderburn LR, Diederichsen LP, Schmidt J, Oakley P, Benveniste O, Danieli MG, Danko K et al The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann. Rheum. Dis. 2018; 77: 30–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Lega JC, Reynaud Q, Belot A, Fabien N, Durieu I, Cottin V. Idiopathic inflammatory myopathies and the lung. Eur. Respir. Rev. 2015; 24: 216–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lundberg IE, Miller FW, Tjärnlund A, Bottai M. Diagnosis and classification of idiopathic inflammatory myopathies. J. Intern. Med. 2016; 280: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hallowell RW, Danoff SK. Interstitial lung disease associated with the idiopathic inflammatory myopathies and the antisynthetase syndrome: recent advances. Curr. Opin. Rheumatol. 2014; 26: 684–9. [DOI] [PubMed] [Google Scholar]
- 141. Hall JC, Casciola‐Rosen L, Samedy LA, Werner J, Owoyemi K, Danoff SK, Christopher‐Stine L. Anti‐melanoma differentiation‐associated protein 5‐associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res. 2013; 65: 1307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Cao H, Pan M, Kang Y, Xia Q, Li X, Zhao X, Shi R, Lou J, Zhou M, Kuwana M et al Clinical manifestations of dermatomyositis and clinically amyopathic dermatomyositis patients with positive expression of anti‐melanoma differentiation‐associated gene 5 antibody. Arthritis Care Res. 2012; 64: 1602–10. [DOI] [PubMed] [Google Scholar]
- 143. Labrador‐Horrillo M, Martinez MA, Selva‐O'Callaghan A, Trallero‐Araguas E, Balada E, Vilardell‐Tarres M, Juarez C. Anti‐MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J. Immunol. Res. 2014; 2014: 290797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Selva‐O'Callaghan A, Martinez‐Gomez X, Trallero‐Araguas E, Pinal‐Fernandez I. The diagnostic work‐up of cancer‐associated myositis. Curr. Opin. Rheumatol. 2018; 30: 630–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Morisset J, Johnson C, Rich E, Collard HR, Lee JS. Management of myositis‐related interstitial lung disease. Chest 2016; 150: 1118–28. [DOI] [PubMed] [Google Scholar]
- 146. Marie I, Hachulla E, Chérin P, Dominique S, Hatron PY, Hellot MF, Devulder B, Herson S, Levesque H, Courtois H. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Care Res. 2002; 47: 614–22. [DOI] [PubMed] [Google Scholar]
- 147. Cottin V, Thivolet‐Bejui F, Reynaud‐Gaubert M, Cadranel J, Delaval P, Ternamian PJ, Cordier JF. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur. Respir. J. 2003; 22: 245–50. [DOI] [PubMed] [Google Scholar]
- 148. Huapaya JA, Silhan L, Pinal‐Fernandez I, Casal‐Dominguez M, Johnson C, Albayda J, Paik JJ, Abanti S, Mammen AL, Christopher‐Stine L et al Long‐term treatment with azathioprine and mycophenolate mofetil for myositis‐related interstitial lung disease. Chest 2019; 156: 896–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003; 362: 971–82. [DOI] [PubMed] [Google Scholar]
- 150. Tsuji T, Arai T, Hirose M, Sugimoto C, Tachibana K, Akira M, Kitaichi M, Hayashi S, Inoue Y. The role of detection of myositis specific and associated antibodies in Japanese patient with interstitial lung disease. Eur. Respir. J. 2014; 44: P714. [Google Scholar]
- 151. Suzuki Y, Hayakawa H, Miwa S, Shirai M, Fujii M, Gemma H, Suda T, Chida K. Intravenous immunoglobulin therapy for refractory interstitial lung disease associated with polymyositis/dermatomyositis. Lung 2009; 187: 201–6. [DOI] [PubMed] [Google Scholar]
- 152. Yagishita M, Kondo Y, Terasaki T, Terasaki M, Shimizu M, Honda F, Oyama A, Takahashi H, Yokosawa M, Asashima H et al Clinically amyopathic dermatomyositis with interstitial pneumonia that was successfully treated with plasma exchange. Intern. Med. 2018; 57: 1935–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Fujita Y, Fukui S, Suzuki T, Ishida M, Endo Y, Tsuji S, Takatani A, Igawa T, Shimizu T, Umeda M et al Anti‐MDA5 antibody‐positive dermatomyositis complicated by autoimmune‐associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern. Med. 2018; 57: 3473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Endo Y, Koga T, Suzuki T, Hara K, Ishida M, Fujita Y, Tsuji S, Takatani A, Shimizu T, Sumiyoshi R et al Successful treatment of plasma exchange for rapidly progressive interstitial lung disease with anti‐MDA5 antibody‐positive dermatomyositis: a case report. Medicine 2018; 97: e0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gutsche M, Rosen GD, Swigris JJ. Connective tissue disease‐associated interstitial lung disease: a review. Curr. Respir. Care Rep. 2012; 1: 224–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. King TEJ, Kim EJ, Kinder BW. Connective tissue diseases In: Schwarz MI, King TE Jr (ed), Interstitial Lung Disease, 5th edn Shelton, CT, People's Medical Publishing House‐USA, 2011; 689–742. [Google Scholar]
- 157. Weinrib L, Sharma OP, Quismorio FP Jr. A long‐term study of interstitial lung disease in systemic lupus erythematosus. Semin. Arthritis Rheum. 1990; 20: 48–56. [DOI] [PubMed] [Google Scholar]
- 158. Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand‐Adam S. Pulmonary manifestations of Sjogren's syndrome. Eur. Respir. Rev. 2016; 25: 110–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tselios K, Urowitz MB. Cardiovascular and pulmonary manifestations of systemic lupus erythematosus. Curr. Rheumatol. Rev. 2017; 13: 206–18. [DOI] [PubMed] [Google Scholar]
- 160. Kanakis MA, Kapsimali V, Vaiopoulos AG, Vaiopoulos GA, Samarkos M. The lung in the spectrum of antiphospholipid syndrome. Clin. Exp. Rheumatol. 2013; 31: 452–7. [PubMed] [Google Scholar]
- 161. Martinez‐Martinez MU, Abud‐Mendoza C. Predictors of mortality in diffuse alveolar haemorrhage associated with systemic lupus erythematosus. Lupus 2011; 20: 568–74. [DOI] [PubMed] [Google Scholar]
- 162. Nannini C, Jebakumar AJ, Crowson CS, Ryu JH, Matteson EL. Primary Sjogren's syndrome 1976‐2005 and associated interstitial lung disease: a population‐based study of incidence and mortality. BMJ Open 2013; 3: e003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Davidson BK, Kelly CA, Griffiths ID. Ten year follow up of pulmonary function in patients with primary Sjogren's syndrome. Ann. Rheum. Dis. 2000; 59: 709–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Fox RI. Sjogren's syndrome. Lancet 2005; 366: 321–31. [DOI] [PubMed] [Google Scholar]
- 165. Hansen LA, Prakash UB, Colby TV. Pulmonary lymphoma in Sjogren's syndrome. Mayo Clin. Proc. 1989; 64: 920–31. [DOI] [PubMed] [Google Scholar]
- 166. Tavoni A, Vitali C, Cirigliano G, Frigelli S, Stampacchia G, Bombardieri S. Shrinking lung in primary Sjogren's syndrome. Arthritis Rheum. 1999; 42: 2249–50. [DOI] [PubMed] [Google Scholar]
- 167. Chen HA, Lai SL, Kwang WK, Liu JC, Chen CH, Huang DF. Middle lobe syndrome as the pulmonary manifestation of primary Sjogren's syndrome. Med. J. Aust. 2006; 184: 294–5. [DOI] [PubMed] [Google Scholar]
- 168. Launay D, Hachulla E, Hatron PY, Jais X, Simonneau G, Humbert M. Pulmonary arterial hypertension: a rare complication of primary Sjogren syndrome: report of 9 new cases and review of the literature. Medicine (Baltimore). 2007; 86: 299–315. [DOI] [PubMed] [Google Scholar]
- 169. Cain HC, Noble PW, Matthay RA. Pulmonary manifestations of Sjogren's syndrome. Clin. Chest Med. 1998; 19: 687–99, viii. [DOI] [PubMed] [Google Scholar]
- 170. Kelly BT, Moua T. Overlap of interstitial pneumonia with autoimmune features with undifferentiated connective tissue disease and contribution of UIP to mortality. Respirology 2018; 23: 600–5. [DOI] [PubMed] [Google Scholar]
- 171. Bodolay E, Csiki Z, Szekanecz Z, Ben T, Kiss E, Zeher M, Szucs G, Danko K, Szegedi G. Five‐year follow‐up of 665 Hungarian patients with undifferentiated connective tissue disease (UCTD). Clin. Exp. Rheumatol. 2003; 21: 313–20. [PubMed] [Google Scholar]
- 172. Doria A, Ghirardello A, de Zambiasi P, Ruffatti A, Gambari PF. Japanese diagnostic criteria for mixed connective tissue disease in Caucasian patients. J. Rheumatol. 1992; 19: 259–64. [PubMed] [Google Scholar]
- 173. Kahn M, Appelboom T. Les maladies systemiques, 3rd edn Paris, Flammarion, 1991. [Google Scholar]
- 174. Gunnarsson R, Hetlevik SO, Lilleby V, Molberg O. Mixed connective tissue disease. Best Pract. Res. Clin. Rheumatol. 2016; 30: 95–111. [DOI] [PubMed] [Google Scholar]
- 175. Gunnarsson R, El‐Hage F, Aalokken TM, Reiseter S, Lund MB, Garen T, Molberg O. Associations between anti‐Ro52 antibodies and lung fibrosis in mixed connective tissue disease. Rheumatology (Oxford) 2016; 55: 103–8. [DOI] [PubMed] [Google Scholar]
- 176. Saunders P, Tsipouri V, Keir GJ, Ashby D, Flather MD, Parfrey H, Babalis D, Renzoni EA, Denton CP, Wells AU et al Rituximab versus cyclophosphamide for the treatment of connective tissue disease‐associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials 2017; 18: 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Corte TJ, Copley SJ, Desai SR, Zappala CJ, Hansell DM, Nicholson AG, Colby TV, Renzoni E, Maher TM, Wells AU. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur. Respir. J. 2012; 39: 661–8. [DOI] [PubMed] [Google Scholar]
- 178. Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, Elicker B, Jones KD, King TE Jr. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am. J. Respir. Crit. Care Med. 2007; 176: 691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Fischer A, West SG, Swigris JJ, Brown KK, du Bois RM. Connective tissue disease‐associated interstitial lung disease: a call for clarification. Chest 2010; 138: 251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, du Bois RM, Lee JS, Leslie KO, Lynch DA, Matteson EL et al An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015; 46: 976–87. [DOI] [PubMed] [Google Scholar]
- 181. Ahmad K, Barba T, Gamondes D, Ginoux M, Khouatra C, Spagnolo P, Strek M, Thivolet‐Béjui F, Traclet J, Cottin V. Interstitial pneumonia with autoimmune features: clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir. Med. 2017; 123: 56–62. [DOI] [PubMed] [Google Scholar]
- 182. Oldham JM, Adegunsoye A, Valenzi E, Lee C, Witt L, Chen L, Husain AN, Montner S, Chung JH, Cottin V et al Characterisation of patients with interstitial pneumonia with autoimmune features. Eur. Respir. J. 2016; 47: 1767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Graney BA, Fischer A. Interstitial pneumonia with autoimmune features. Ann. Am. Thorac. Soc. 2019; 16: 525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Arulkumaran N, Periselneris N, Gaskin G, Strickland N, Ind PW, Pusey CD, Salama AD. Interstitial lung disease and ANCA‐associated vasculitis: a retrospective observational cohort study. Rheumatology (Oxford) 2011; 50: 2035–43. [DOI] [PubMed] [Google Scholar]
- 185. Tzelepis GE, Kokosi M, Tzioufas A, Toya SP, Boki KA, Zormpala A, Moutsopoulos HM. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur. Respir. J. 2010; 36: 116–21. [DOI] [PubMed] [Google Scholar]
- 186. Comarmond C, Crestani B, Tazi A, Hervier B, Adam‐Marchand S, Nunes H, Cohen‐Aubart F, Wislez M, Cadranel J, Housset B et al Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)‐associated vasculitis: a series of 49 patients and review of the literature. Medicine (Baltimore). 2014; 93: 340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Alba MA, Flores‐Suarez LF, Henderson AG, Xiao H, Hu P, Nachman PH, Falk RJ, Charles Jennette J. Interstitial lung disease in ANCA vasculitis. Autoimmun. Rev. 2017; 16: 722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Hervier B, Pagnoux C, Agard C, Haroche J, Amoura Z, Guillevin L, Hamidou MA. Pulmonary fibrosis associated with ANCA‐positive vasculitides. Retrospective study of 12 cases and review of the literature. Ann. Rheum. Dis. 2009; 68: 404–7. [DOI] [PubMed] [Google Scholar]
- 189. Fernandez Casares M, Gonzalez A, Fielli M, Caputo F, Bottinelli Y, Zamboni M. Microscopic polyangiitis associated with pulmonary fibrosis. Clin. Rheumatol. 2015; 34: 1273–7. [DOI] [PubMed] [Google Scholar]
- 190. Tanaka T, Otani K, Egashira R, Kashima Y, Taniguchi H, Kondoh Y, Kataoka K, Shiraki A, Kitasato Y, Leslie KO et al Interstitial pneumonia associated with MPO‐ANCA: clinicopathological features of nine patients. Respir. Med. 2012; 106: 1765–70. [DOI] [PubMed] [Google Scholar]
- 191. Liu GY, Ventura IB, Achtar‐Zadeh N, Elicker BM, Jones KD, Wolters PJ, Collard HR, Adegunsoye A, Strek ME, Ley B. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in North American patients with idiopathic pulmonary fibrosis. Chest 2019; 156: 715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Yates M, Watts RA, Bajema IM, Cid MC, Crestani B, Hauser T, Hellmich B, Holle JU, Laudien M, Little MA et al EULAR/ERA‐EDTA recommendations for the management of ANCA‐associated vasculitis. Ann. Rheum. Dis. 2016; 75: 1583–94. [DOI] [PubMed] [Google Scholar]
- 193. Homma S, Suzuki A, Sato K. Pulmonary involvement in ANCA‐associated vasculitis from the view of the pulmonologist. Clin. Exp. Nephrol. 2013; 17: 667–71. [DOI] [PubMed] [Google Scholar]
- 194. Thompson GE, Specks U. Update on the management of respiratory manifestations of the antineutrophil cytoplasmic antibodies‐associated vasculitides. Clin. Chest Med. 2019; 40: 573–82. [DOI] [PubMed] [Google Scholar]
- 195. Sebastiani M, Manfredi A, Vacchi C, Cassone G, Faverio P, Cavazza A, Sverzellati N, Salvarani C, Luppi F. Epidemiology and management of interstitial lung disease in ANCA‐associated vasculitis. Clin. Exp. Rheumatol. 2020; 38(Suppl. 124): 221–31. [PubMed] [Google Scholar]
- 196. Kawamoto H, Hara H, Minagawa S, Numata T, Araya J, Kaneko Y, Umezawa Y, Asahina A, Nakagawa H, Kuwano K. Interstitial pneumonia in psoriasis. Mayo Clin. Proc. Innov. Qual. Outcomes 2018; 2: 370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Sato R, Ohshima N, Masuda K, Suzuki J, Higaki N, Inoue E, Suzuki J, Matsui H, Nagai H, Akagawa S et al Investigation of interstitial pneumonia cases with psoriasis vulgaris. American Thoracic Society International Congress [Abstract]. Am. J. Respir. Crit. Care Med. 2020; 201: A1589 https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2012.185.1_MeetingAbstracts.A1589. [Google Scholar]
- 198. Hara H, Kuwano K, Kawamoto H, Nakagawa H. Psoriasis‐associated interstitial pneumonia. Eur. J. Dermatol. 2018; 28: 395–6. [DOI] [PubMed] [Google Scholar]
- 199. Ishikawa G, Acquah S, Salvatore M, Padilla ML. Interstitial lung disease with psoriasis [Abstract]. American Journal of Respiratory and Critical Care Medicine 2017; 195: A7122 https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2017.195.1_MeetingAbstracts.A7122 [Google Scholar]
- 200. Peluso R, Iervolino S, Vitiello M, Bruner V, Lupoli G, Di Minno MN. Extra‐articular manifestations in psoriatic arthritis patients. Clin. Rheumatol. 2015; 34: 745–53. [DOI] [PubMed] [Google Scholar]
- 201. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019; 53: 1801913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Hurdman J, Condliffe R, Elliot CA, Davies C, Hill C, Wild JM, Capener D, Sephton P, Hamilton N, Armstrong IJ et al ASPIRE registry: assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur. Respir. J. 2012; 39: 945–55. [DOI] [PubMed] [Google Scholar]
- 203. Launay D, Montani D, Hassoun PM, Cottin V, Le Pavec J, Clerson P, Sitbon O, Jais X, Savale L, Weatherald J et al Clinical phenotypes and survival of pre‐capillary pulmonary hypertension in systemic sclerosis. PLoS One 2018; 13: e0197112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Seeger W, Adir Y, Barbera JA, Champion H, Coghlan JG, Cottin V, De Marco T, Galie N, Ghio S, Gibbs S et al Pulmonary hypertension in chronic lung diseases. J. Am. Coll. Cardiol. 2013; 62(25 Suppl.): D109–16. [DOI] [PubMed] [Google Scholar]
- 205. Galie N, Barbera JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, Peacock AJ, Simonneau G, Vachiery JL, Grunig E et al Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N. Engl. J. Med. 2015; 373: 834–44. [DOI] [PubMed] [Google Scholar]
- 206. Pulido T, Adzerikho I, Channick RN, Delcroix M, Galie N, Ghofrani HA, Jansa P, Jing ZC, Le Brun FO, Mehta S et al Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013; 369: 809–18. [DOI] [PubMed] [Google Scholar]
- 207. Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF et al Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013; 62(25 Suppl.): D34–41. [DOI] [PubMed] [Google Scholar]
- 208. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galie N, Ghofrani HA, Hoeper MM, Lang IM, Preiss R et al Selexipag for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2015; 373: 2522–33. [DOI] [PubMed] [Google Scholar]
- 209. Chung L, Liu J, Parsons L, Hassoun PM, McGoon M, Badesch DB, Miller DP, Nicolls MR, Zamanian RT. Characterization of connective tissue disease‐associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest 2010; 138: 1383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Galiè N, Humbert M, Vachiery J‐L, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M et al 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2015; 46: 903–75. [DOI] [PubMed] [Google Scholar]
- 211. Hadley R, Chan KM. Lung transplantation for connective tissue disease‐associated lung disease In: Dellaripa P, Fischer A, Flaherty K (eds) Pulmonary Manifestations of Rheumatic Disease. New York, Springer, 2014; 179–91. [Google Scholar]
- 212. Christie JD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Dobbels F, Kirk R, Rahmel AO, Stehlik J, Hertz MI. The Registry of the International Society for Heart and Lung Transplantation: 29th adult lung and heart‐lung transplant report‐2012. J. Heart Lung Transplant. 2012; 31: 1073–86. [DOI] [PubMed] [Google Scholar]
- 213. Weill D, Benden C, Corris PA, Dark JH, Davis RD, Keshavjee S, Lederer DJ, Mulligan MJ, Patterson GA, Singer LG et al A consensus document for the selection of lung transplant candidates: 2014 – an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015; 34: 1–15. [DOI] [PubMed] [Google Scholar]
- 214. Gasper WJ, Sweet MP, Golden JA, Hoopes C, Leard LE, Kleinhenz ME, Hays SR, Patti MG. Lung transplantation in patients with connective tissue disorders and esophageal dysmotility. Dis. Esophagus 2008; 21: 650–5. [DOI] [PubMed] [Google Scholar]
- 215. Takagishi T, Ostrowski R, Alex C, Rychlik K, Pelletiere K, Tehrani R. Survival and extrapulmonary course of connective tissue disease after lung transplantation. J. Clin. Rheumatol. 2012; 18: 283–9. [DOI] [PubMed] [Google Scholar]
- 216. Crespo MM, Bermudez CA, Dew MA, Johnson BA, George MP, Bhama J, Morrell M, D'Cunha J, Shigemura N, Richards TJ et al Lung transplant in patients with scleroderma compared with pulmonary fibrosis. Short‐ and long‐term outcomes. Ann. Am. Thorac. Soc. 2016; 13: 784–92. [DOI] [PubMed] [Google Scholar]
- 217. Delplanque M, Gatfosse M, Ait‐Oufella H, Mercier O, Savale L, Fain O, Mekinian A. Bi‐lung transplantation in anti‐synthetase syndrome with life‐threatening interstitial lung disease. Rheumatology (Oxford) 2018; 57: 1688–9. [DOI] [PubMed] [Google Scholar]
- 218. Holland AE, Fiore JF, Bell EC, Goh N, Westall G, Symons K, Dowman L, Glaspole I. Dyspnoea and comorbidity contribute to anxiety and depression in interstitial lung disease. Respirology 2014; 19: 1215–21. [DOI] [PubMed] [Google Scholar]
- 219. Madison JM, Irwin RS. Chronic cough in adults with interstitial lung disease. Curr. Opin. Pulm. Med. 2005; 11: 412–6. [DOI] [PubMed] [Google Scholar]
- 220. Chang JA, Curtis JR, Patrick DL, Raghu G. Assessment of health‐related quality of life in patients with interstitial lung disease. Chest 1999; 116: 1175–82. [DOI] [PubMed] [Google Scholar]
- 221. Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst. Rev. 2014; (10): CD006322. [DOI] [PubMed] [Google Scholar]
- 222. Dowman LM, McDonald CF, Hill CJ, Lee AL, Barker K, Boote C, Glaspole I, Goh NSL, Southcott AM, Burge AT et al The evidence of benefits of exercise training in interstitial lung disease: a randomised controlled trial. Thorax 2017; 72: 610–9. [DOI] [PubMed] [Google Scholar]
- 223. Perez‐Bogerd S, Wuyts W, Barbier V, Demeyer H, Van Muylem A, Janssens W, Troosters T. Short and long‐term effects of pulmonary rehabilitation in interstitial lung diseases: a randomised controlled trial. Respir. Res. 2018; 19: 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Bolton CE, Bevan‐Smith EF, Blakey JD, Crowe P, Elkin SL, Garrod R, Greening NJ, Heslop K, Hull JH, Man WD‐C et al British Thoracic Society guideline on pulmonary rehabilitation in adults: accredited by NICE. Thorax 2013; 68(Suppl. 2): ii1–ii30. [DOI] [PubMed] [Google Scholar]
- 225. McNamara RJ, McKeough ZJ, McKenzie DK, Alison JA. Water‐based exercise in COPD with physical comorbidities: a randomised controlled trial. Eur. Respir. J. 2013; 41: 1284–91. [DOI] [PubMed] [Google Scholar]
- 226. Spruit MA, Singh SJ, Garvey C, ZuWallack R, Nici L, Rochester C, Hill K, Holland AE, Lareau SC, Man WDC et al An official American Thoracic Society/European Respiratory Society statement: key concepts and advances in pulmonary rehabilitation. Am. J. Respir. Crit. Care Med. 2013; 188: e13–64. [DOI] [PubMed] [Google Scholar]
- 227. Nakazawa A, Cox NS, Holland AE. Current best practice in rehabilitation in interstitial lung disease. Ther. Adv. Respir. Dis. 2017; 11: 115–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA et al An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Hardinge M, Annandale J, Bourne S, Cooper B, Evans A, Freeman D, Green A, Hippolyte S, Knowles V, MacNee W et al British Thoracic Society guidelines for home oxygen use in adults. Thorax 2015; 70(Suppl. 1): i1–43. [DOI] [PubMed] [Google Scholar]
- 230. Ferrara G, Luppi F, Birring SS, Cerri S, Caminati A, Sköld M, Kreuter M. Best supportive care for idiopathic pulmonary fibrosis: current gaps and future directions. Eur. Respir. Rev. 2018; 27: 170076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Kreuter M, Bendstrup E, Russell AM, Bajwah S, Lindell K, Adir Y, Brown CE, Calligaro G, Cassidy N, Corte TJ et al Palliative care in interstitial lung disease: living well. Lancet Respir. Med. 2017; 5: 968–80. [DOI] [PubMed] [Google Scholar]
- 232. Dowman LM, McDonald CF, Bozinovski S, Vlahos R, Gillies R, Pouniotis D, Hill CJ, Goh NSL, Holland AE. Greater endurance capacity and improved dyspnoea with acute oxygen supplementation in idiopathic pulmonary fibrosis patients without resting hypoxaemia. Respirology 2017; 22: 957–64. [DOI] [PubMed] [Google Scholar]
- 233. Schaeffer MR, Ryerson CJ, Ramsook AH, Molgat‐Seon Y, Wilkie SS, Dhillon SS, Mitchell RA, Sheel AW, Khalil N, Camp PG et al Effects of hyperoxia on dyspnoea and exercise endurance in fibrotic interstitial lung disease. Eur. Respir. J. 2017; 49: 1602494. [DOI] [PubMed] [Google Scholar]
- 234. Visca D, Mori L, Tsipouri V, Fleming S, Firouzi A, Bonini M, Pavitt MJ, Alfieri V, Canu S, Bonifazi M et al Effect of ambulatory oxygen on quality of life for patients with fibrotic lung disease (AmbOx): a prospective, open‐label, mixed‐method, crossover randomised controlled trial. Lancet Respir. Med. 2018; 6: 759–70. [DOI] [PubMed] [Google Scholar]
- 235. Lindell K, Raghu G. Palliative care for patients with pulmonary fibrosis: symptom relief is essential. Eur. Respir. J. 2018; 52: 1802086. [DOI] [PubMed] [Google Scholar]
- 236. Lindell KO. Nonpharmacological therapies for interstitial lung disease. Curr. Pulmonol. Rep. 2018; 7: 126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Ekström MP, Bornefalk‐Hermansson A, Abernethy AP, Currow DC. Safety of benzodiazepines and opioids in very severe respiratory disease: national prospective study. BMJ 2014; 348: g445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Gillon S, Clifton IJ. Breathlessness in palliative care: a practical guide. Br. J. Hosp. Med. 2019; 80: 72–7. [DOI] [PubMed] [Google Scholar]
- 239. Simon ST, Higginson IJ, Booth S, Harding R, Weingärtner V, Bausewein C. Benzodiazepines for the relief of breathlessness in advanced malignant and non‐malignant diseases in adults. Cochrane Database Syst. Rev. 2016; 10: CD007354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Bajwah S, Davies JM, Tanash H, Currow DC, Oluyase AO, Ekström M. Safety of benzodiazepines and opioids in interstitial lung disease: a national prospective study. Eur. Respir. J. 2018; 52: 1801278. [DOI] [PubMed] [Google Scholar]
- 241. Birring SS, Kavanagh JE, Irwin RS, Keogh KA, Lim KG, Ryu JH. Treatment of interstitial lung disease associated cough: CHEST guideline and expert panel report. Chest 2018; 154: 904–17. [DOI] [PubMed] [Google Scholar]
- 242. Ryan NM, Birring SS, Gibson PG. Gabapentin for refractory chronic cough: a randomised, double‐blind, placebo‐controlled trial. Lancet 2012; 380: 1583–9. [DOI] [PubMed] [Google Scholar]
- 243. Tarrant BJ, Le Maitre C, Romero L, Steward R, Button BM, Thompson BR, Holland AE. Mucoactive agents for chronic, non‐cystic fibrosis lung disease: a systematic review and meta‐analysis. Respirology 2017; 22: 1084–92. [DOI] [PubMed] [Google Scholar]
- 244. Assayag D, Camp PG, Fisher J, Johannson KA, Kolb M, Lohmann T, Manganas H, Morisset J, Ryerson CJ, Shapera S et al Comprehensive management of fibrotic interstitial lung diseases: a Canadian Thoracic Society position statement. Can. J. Respir. Crit. Care Sleep Med. 2018; 2: 234–43. [Google Scholar]
- 245. Bourke SJ, Peel ET. Palliative care of chronic progressive lung disease. Clin. Med. (Lond.) 2014; 14: 79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Swigris JJ, Fairclough DL, Morrison M, Make B, Kozora E, Brown KK, Wamboldt FS. Benefits of pulmonary rehabilitation in idiopathic pulmonary fibrosis. Respir. Care 2011; 56: 783–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Ryerson CJ, Cayou C, Topp F, Hilling L, Camp PG, Wilcox PG, Khalil N, Collard HR, Garvey C. Pulmonary rehabilitation improves long‐term outcomes in interstitial lung disease: a prospective cohort study. Respir. Med. 2014; 108: 203–10. [DOI] [PubMed] [Google Scholar]
- 248. van Manen MJG, Geelhoed JJM, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2017; 11: 157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Wallaert B, Duthoit L, Drumez E, Behal H, Wemeau L, Chenivesse C, Grosbois J‐M. Long‐term evaluation of home‐based pulmonary rehabilitation in patients with fibrotic idiopathic interstitial pneumonias. ERJ Open Res. 2019; 5: 00045‐2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Ley B, Torgerson DG, Oldham JM, Adegunsoye A, Liu S, Li J, Elicker BM, Henry TS, Golden JA, Jones KD et al Rare protein‐altering telomere‐related gene variants in patients with chronic hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2019; 200: 1154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, Torres F, Kaza V, Girod CE, Jones KD et al Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir. Med. 2014; 2: 557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]