

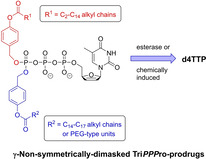
Abstract
Nucleoside analogue reverse transcriptase inhibitors (NRTI) and nucleoside analogue monophosphate prodrugs are used in combination antiretroviral therapy (cART). The design of antivirally active nucleoside triphosphate prodrugs is a recent and an important advancement in the field of nucleoside analogue drug development. Here, we report on TriPPPro‐derivatives of nucleoside analogue triphosphates (NTPs) that comprised two different acyloxybenzyl‐masks at the γ‐phosphate of the NTP aiming to achieve the metabolic bypass. Thus, γ‐non‐symmetrically dimasked TriPPPro‐compounds (γ‐(AB,ab)‐d4TTPs) were synthesized and they proved to be active against HIV‐1 and HIV‐2 in cultures of infected wild‐type human CD4+ T‐lymphocyte (CEM/0) cells and more importantly also in thymidine kinase‐deficient CD4+ T‐cells (CEM/TK‐). From hydrolysis studies both in phosphate buffer (PB, pH 7.3) and CEM cell extracts, there was surprisingly no differentiation in the cleavage of the two acyloxybenzyl prodrug‐masks. However, if within one of the two acyloxybenzyl groups a short PEG‐type methoxytriglycol group was introduced, the “standard” acyloxybenzyl‐mask was cleaved with high preference.
Keywords: antiviral agents, nucleoside analogue, nucleoside triphosphate prodrugs, bioreversible protection, nucleotides
Masked and ready: Lipophilic nucleoside triphosphate prodrugs bearing two different bioreversible masking groups attached to the γ‐position of the triphosphate moiety were studied. The aim to use two different masking groups was to enhance the selectivity of their removal during the cleavage process in order to deliver nucleoside triphosphates intracellularly. The compounds proved to be active against HIV even in cells lacking a phosphorylating enzyme.
Introduction
In the past a number of nucleoside analogues was discovered and applied in antitumor and antiviral chemotherapy. These compounds still play an important role to combat viral infections in the clinic, such as HIV, hepatitis B and C, influenza or most recently SARS‐CoV‐2 infections.[ 1 , 2 ] Generally, the targets of these nucleoside analogues are the viral DNA‐ or RNA polymerases which are involved in the virus replication, such as HIV's reverse transcriptase (HIV‐RT).[ 3 , 4 ] Till now, several nucleoside analogues have been approved as HIV‐RT inhibitors (NRTIs) [6] and they are nowadays part of the highly effective combination antiretroviral therapy (cART). However, the antiviral activity of nucleoside analogues such as 3'‐deoxy‐2',3'‐didehydrothymidine 1 (d4T), is strongly dependent on an in‐vivo phosphorylation into the nucleoside triphosphate forms (NTPs) mainly by host cell kinases. D4TTP 1 t is formed via a stepwise phosphorylation from the nucleoside analogue into the nucleoside mono‐ (1 m, d4TMP), the diphosphate (1 d, d4TDP) and finally to d4TTP (1 t) (Scheme 1).[ 7 , 8 ] However, the stepwise transformation into the triphosphates often occurs insufficiently due to the substrate specificity of the involved kinases. Often the first metabolic step, the monophosphorylation, is rate limiting but examples for bottlenecks in the following second and third phosphorylation steps are known as well. Further limitations such as poor biological half‐lives due to catabolic elimination, mutations of nucleoside transporters, variable bioavailability after oral administration or selection of drug resistance have been observed for nucleoside analogues. [9] To overcome some of these hurdles, nucleoside monophosphate (NMP) prodrugs have been explored in the past and resulted in orally administrable forms of some antiviral NMPs and others are currently under continuing development.[ 10 , 11 , 12 , 13 ] Amongst others, two examples of efficient NMP‐prodrugs are the nucleoside phosphoramidates and cycloSal‐nucleoside phosphate triesters.[ 14 , 15 , 16 , 17 , 18 , 19 ]
Scheme 1.
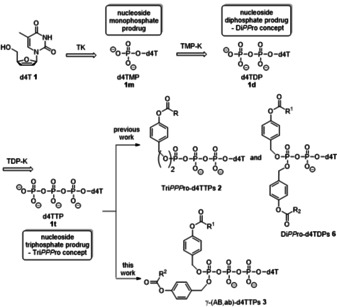
Metabolism of nucleoside analogues such as d4T 1 and the corresponding nucleotide prodrugs.
The aim of the above‐mentioned nucleotide prodrugs was the delivery of NMPs into cells, to bypass the first phosphorylation step and finally improve antiviral activity. Next, an approach towards nucleoside diphosphate prodrugs was developed by attaching two lipophilic, bioreversible masks (R1=R2 or R1≠R2) at the β‐phosphate,[ 20 , 21 , 22 , 23 ] such as DiPPro‐d4TDPs 6 (Scheme 1). [23] Moreover, we also reported on a first delivery system of NTPs. These first generation TriPPPro‐compounds 2 comprised two identical acyloxybenzyl‐masking moieties at the γ‐phosphate moiety to achieve membrane permeability (Scheme 1).[ 24 , 25 , 26 , 27 , 28 ] It was shown that these TriPPPro‐d4TTPs 2 with long‐chain acyloxybenzyl masks (AB‐mask) exhibited also higher antiviral activity and longer half‐life in phosphate buffer (PB) and CEM cell extracts than those containing two short acyloxybenzyl‐masks. Such TriPPPro‐compounds not only proved to be antivirally active against HIV‐1 and HIV‐2 in wild‐type CEM/0 cells, but they even retained high anti‐HIV activity in HIV‐2‐infected mutant CEM/TK–cell cultures whereas the parent nucleoside d4T 1 was virtually inactive due to the lack of phosphorylation. The stepwise enzyme‐driven cleavage of the two AB‐masks of the TriPPPro‐compounds 2 was achieved by an ester bond cleavage within the AB‐moiety forming an intermediate of type 4 and a subsequent spontaneous cleavage of the remaining part of the mask leading to d4TTP 1 t and two equivalents of 4‐hydroxybenzyl alcohol (Scheme 2). [26] The cellular uptake of these compounds was proven by using a fluorescent nucleoside analogue. [27] However, although this approach worked satisfactory and NTPs were formed in chemical and PLE‐catalyzed studies predominately, we also observed some concomitant formation of the corresponding NDPs and also (very) small amounts of the NMPs. Particularly in cell extracts, very small amounts of d4TTP and large amounts of d4TDP were detected due to the fast dephosphorylation of d4TTP by cellular phosphorylases or kinases. [26] During our development of the previously described DiPPro‐compounds 6, we observed the exclusive formation of NDPs when we introduced two different acyloxybenzyl masking groups. [23] Thus, a combination of a short‐chain acyloxybenzyl moiety and a long‐chain, lipophilic acyloxybenzyl moiety led for DiPPro‐compounds 6 to a selective first fast cleavage of the short‐chain residue and formation of the intermediate comprising the long lipophilic AB‐moiety. Moreover, we have shown that some concomitant cleavage of the pyrophosphate linkage of the DiPPro‐compounds to yield NMPs only happened at the level of the doubly‐masked starting DiPPro‐compound. From the intermediate no NMP formation was detected and only the second bioreversible group was cleaved leading to NDPs.[ 20 , 21 , 22 , 23 ] Recently, we also reported on bis‐alkoxycarbonyloxybenzyl (ACB)‐TriPPPro‐compounds as well as on non‐symmetric‐TriPPPro compounds which are active against HIV‐2 in mutant CEM/TK–cell cultures. The prodrugs comprised two different ACB‐moieties or a combination of an ACB and an AB mask at γ‐phosphate. For the latter compounds it has proven that the acyloxybenzyl‐mask was faster cleaved to give almost selectively the γ‐ACB‐NTP‐intermediates in chemical hydrolysis studies and also in cell extracts. [29]
Scheme 2.
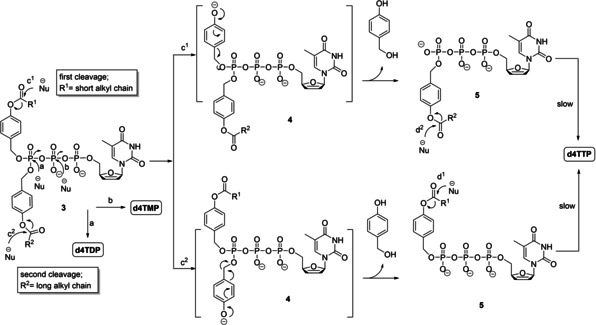
Proposed hydrolysis mechanism of non‐symmetric TriPPPro‐compounds 3.
In this report, we transferred the promising results from the DiPPro‐compounds to the TriPPPro‐approach and studied a series of non‐symmetrically esterified TriPPPro‐compounds 3 (Scheme 1). We disclose here the synthesis and the properties of such non‐symmetrically modified TriPPPro‐derivatives of the nucleoside d4T 1 comprising an ab‐moiety with an alkyl/polyether chain and a mask with different alkyl AB‐moiety. The aim was to achieve a highly selective delivery of d4TTP, as in the case of the DiPPro‐counterparts. It was expected that the short mask should be cleaved fast in cells forming the monomasked intermediate which subsequently would be converted into the target d4TTP (Scheme 2).
In addition to acyl groups bearing simple alkyl chains we also studied PEG‐units linked by an ester group as the acyl‐moiety (ab‐PEG mask) in order to increase the hydrophilicity due to the low solubility of some TriPPPro‐compounds 2 comprising two long lipophilic ab‐groups in aqueous media. [26]
Results and Discussion
Synthesis
D4TMP 1 m was prepared applying the method described by Sowa and Ouchi. [30] For the synthesis of γ‐(AB,ab)‐d4TTPs 3, the H‐phosphonate route was used as disclosed previously by us.[ 27 , 29 ] Briefly, diphenyl hydrogen phosphonate (DPP) was selectively reacted with two different acyloxybenzyl alcohols 7 which led to H‐phosphonates 8 in moderate yields ranging from 44 % to 52 %. Next, H‐phosphonates 8 were converted into the corresponding phosphorochloridates by an oxidative chlorination using N‐chlorosuccinimide (NCS) which were then reacted with tetra‐n‐butylammonium phosphate to yield pyrophosphates 9. The conversion was almost quantitative and after extraction in CH2Cl2/water, the crude products were immediately used for the next step without further purification. Compounds 9 were stepwise activated with trifluoroacetic acid anhydride (TFAA) and N‐methylimidazole and coupled with d4TMP 1 m to yield γ‐(AB,ab)‐d4TTPs 3 (Bu4N+ form). After a reverse‐phase column chromatography of the crude products, ion‐exchange using Dowex 50WX8 (NH4 +), a second reverse‐phase column chromatography and freeze‐drying, γ‐(AB,ab)‐d4TTPs 3 in their NH4 +‐form were isolated. The total yields obtained in these conversions of d4TMP 1 m to give the TriPPPro‐compounds 3 varied between 28 %–64 % (Scheme 3).
Scheme 3.
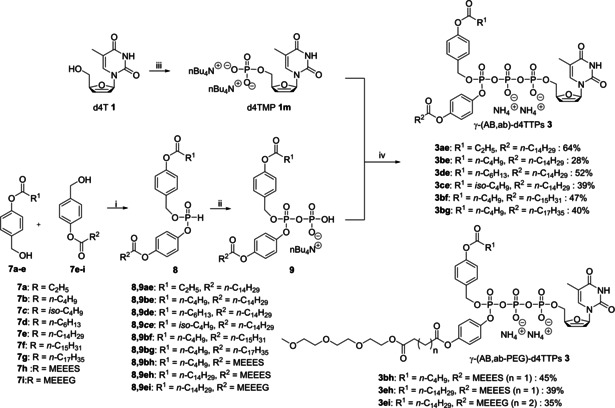
Reagents and conditions: i) diphenyl hydrogen phosphonate (DPP), pyridine, 0 °C–38 °C, 3.5 h; ii) a. NCS, CH3CN, RT, 2 h, b) (H2PO4)Bu4N, CH3CN, RT, 1 h; iii) d4T 1, POCl3, pyridine, H2O, CH3CN, 0 °C‐rt, 5 h; iv) a. TFAA, Et3N, CH3CN, 0 °C, 10 min, b. 1‐methylimidazol, Et3N, CH3CN, 0 °C‐rt, 10 min, c. d4TMP 1 m, RT, 3–5 h, RP‐chromatography, Dowex 50WX8 (NH4 + form) ion exchange, RP‐chromatography.
Compounds 3 bh, 3 eh and 3 ei are TriPPPro‐derivatives with one PEG‐comprising ab‐moiety. The hydrophilic PEG‐bearing benzyl alcohols 7 h and 7 i were synthesized starting from 2‐(2‐(2‐methoxyethoxy)ethoxy)ethan‐1‐ol 10 (MEEE). Succinic anhydride 11 h and glutaric anhydride 11 i were used to form a diester linker between the PEG moiety and 4‐hydroxybenzyl alcohol (Scheme 4). MEEE‐succinate 7 h (MEEES) and MEEE‐glutarate 7 i (MEEEG) are hygroscopic and should be carefully handled when exposed to air.
Scheme 4.
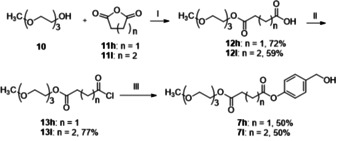
Reagents and conditions: i) DMAP, CH2Cl2, RT; ii) oxalyl chloride, cat. DMF, CH2Cl2, 0 °C‐rt; iii) 4‐hydroxybenzyl alcohol, Et3N, CH2Cl2, 0 °C‐rt.
Stability studies
To study the hydrolytic stability of the prodrugs and the products distribution, TriPPPro‐compounds 3 were incubated in phosphate buffer (PB, 25 mM, pH 7.3), human CD4 + T‐lymphocyte cell extracts or pig liver esterase (PLE) in PB, pH 7.3. Samples of the hydrolysis mixtures at different time points were analyzed by analytical RP18‐HPLC. The calculated half‐lives (t 1/2) of compounds 3 reflect the removal of the first bioreversible AB‐ or ab‐group to yield the corresponding monomasked intermediates 5. The half‐lives (t 1/2) of prodrugs 3 in phosphate buffer (PB, 25 mM, pH 7.3) and human CD4 + T‐lymphocyte cell extracts are summarized in Table 1.
Table 1.
Hydrolysis of TriPPPro‐compounds 3 in various media.
|
Prodrug |
R1 |
R2 |
PB pH=7.3 t 1/2 [h] |
CEM/0 cell extracts t 1/2 [h] |
|---|---|---|---|---|
|
3 ae |
C2H5 |
n‐C14H29 |
59 |
0.8 |
|
3 be |
n‐C4H9 |
n‐C14H29 |
45 |
2.5 |
|
3 de |
n‐C6H13 |
n‐C14H29 |
61 |
2.3 |
|
3 ce |
iso‐C4H9 |
n‐C14H29 |
64 |
3.8 |
|
3 bf |
n‐C4H9 |
n‐C15H31 |
49 |
3.1 |
|
3 bg |
n‐C4H9 |
n‐C17H35 |
50 |
3.3 |
|
3 bh |
n‐C4H9 |
MEEES |
22 |
0.8 |
|
3 eh |
n‐C14H29 |
MEEES |
65 |
1.1 |
|
3 ei |
n‐C14H29 |
MEEEG |
86 |
2.4 |
|
D4T |
/ |
/ |
>40 |
n.a.[a] |
|
D4TTP31 |
/ |
/ |
>40 |
0.6 |
The hydrolysis experiments of TriPPPro‐compounds 3 were performed in aqueous 25 mM phosphate buffer (PB, pH=7.3) and in CEM/0 cell extracts. The hydrolysis products were detected by analytical RP18 HPLC. [a] n.a.: not available.
Chemical stability in phosphate buffer (PB, pH 7.3)
In PB, the chemical stability of the TriPPPro‐compounds 3 ae‐3 bg bearing two different acyloxybenzyl masking groups hydrolyzed with half‐lives between 45 h and 64 h without showing a clear trend following the cleavage mechanism depicted in Scheme 2. A stability difference was observed for TriPPPro‐derivatives 3 be (45 h) and 3 ce (64 h) bearing a n‐butyl moiety (3 be) and a branched i‐butyl group (3 ce), respectively. Generally, d4TTP 1 t and d4TDP 1 d were detected as main hydrolysis products in addition to d4TMP 1 m which was detected as well although in very low concentrations (<4 %). After complete consumption of the TriPPPro‐prodrugs 3, an increase in the d4TTP 1 t concentration was formed in the case of TriPPPro‐compounds 3 in PB (pH 7.3). As an example, when TriPPPro‐compounds 3 bf was totally consumed after incubation of 480 h, the ratio of 1 t:1 d:1 m was 14 : 11 : 1. Both intermediates γ‐(ab‐C15)‐d4TTP 5 f and γ‐(AB−C4)‐d4TTP 5 b were formed in almost identical concentrations and thus, the hydrolysis proceeded without marked selectivity, which was in agreement with the results obtained from the studies of γ‐(ACB−C4,ACB−C12)‐d4TTPs in PB, but in contrast with results as γ‐(AB−C2,ACB−C16)‐d4TTPs. [29] Compared to the half‐lives in PB of symmetric γ‐(AB−C4,AB−C4)‐d4TTP (t1/2=22 h) and γ‐(AB−C9,AB−C9)‐d4TTP (t1/2=44 h), the half‐life of all non‐symmetric TriPPPro‐compounds studied here showed half‐lives close to that of γ‐(AB−C9,AB−C9)‐d4TTP, e. g. γ‐(AB−C4,AB−C14)‐d4TTP 3 be; t1/2=45 h, although most of them comprise also one short acyloxybenzyl moieties. [26] As reported previously for the TriPPPro‐compounds 2, the half‐lives of the mono‐masked intermediates 5 were also much higher than those of the TriPPPro‐prodrugs 3. A side reaction occurred and thymine was formed due to the cleavage of the glycosidic bond.
Interestingly, for the PEG‐containing MEEES‐ and MEEEG‐TriPPPro‐derivatives 3 bh (t 1/2=2 h), 3 eh (t 1/2=65 h) and 3 ei (t 1/2=86 h) a broad range of stability was determined. Just by changing the alkyl residue in one of the acyloxybenzyl‐groups from n‐butyl‐ (3 bh) to a n‐C14‐fragment (3 eh) and keeping the PEG‐bearing masking group unchanged, the stability increased by a 3‐fold. For prodrugs 3 eh and 3 ei, in which the linker structure changed from succinic diester (3 eh) to glutaric diester (3 ei), the t 1/2 of the prodrug increased by 1.3‐fold. The distribution of the hydrolysis products was identical to the results of the non‐PEG‐containing TriPPPro‐compounds 3 showing almost no selectivity. Both intermediates γ‐(AB−C14)‐d4TTP 5 e and γ‐(ab‐MEEES)‐d4TTP 5 h were formed to a highest concentration after 200 h incubation and decreased when the hydrolysis proceeded. TriPPPro‐compounds 3 eh was totally consumed after incubation of 500 h, and at that time point the ratio of 5 e:5 h:1 t:1 d:1 m was 3 : 5 : 15 : 8 : 1. During the hydrolysis, almost no d4TMP 1 m was formed.
Hydrolysis in CEM cell extracts
The stability of TriPPPro‐prodrugs 3 was further investigated in human CD4 + T‐lymphocyte CEM cell extracts. Here, an enzymatic cleavage reaction took place, because the half‐lives of the prodrugs 3 in CEM/0 cell extracts ranging from 0.8 h to 3.8 h were significantly lower than the half‐lives in PB buffer. Compounds 3 ae and 3 bh, comprising a propanoyl‐ester and pentanoyl‐ester moiety, respectively, were found to be the most labile compounds. This is in accordance to our previous results of the DiPPro‐compounds [23] or the TriPPPro‐compounds 2. [26] In all those cases the predominate formation of d4TDP 1 d was observed. Mono‐masked Intermediates 5 and d4TTP 1 t were only observed in very low concentration. This result was in accordance to our previous studies. [26] In CEM/0 cell extracts, the half‐life of d4TDP 1 d was 59 h and that of d4TTP 1 t was only 38 min, [26] which means that the observed low levels of d4TTP 1 t in these hydrolysis studies were the result of a fast enzymatic dephosphorylation of d4TTP 1 t. It can not fully exclude that the d4TDP found in these studies is a result of a direct cleavage of the anhydride bond between the γ‐ and β‐phosphate. However, it seems to be unlikely because here in the cell extracts a considerable amount of esterases is present which are responsible for the ester cleavage within the masking group. This pathway might be more relevant in the phosphate buffer hydrolysis studies due to the lack of the esterase. But even in those studies the main product, e. g. for compound 3 bf was still d4TTP 1 t. And in contrast to the phosphate buffer, pH 7.3, the cell extracts contain phosphatases. No thymine was detected during the incubation period within 10 h for all prodrugs. It was not possible to calculate the exact peak area for d4TMP 1 m using this HPLC method because of the overlapped peaks between d4TMP 1 m and cell extracts.
In addition, both intermediates 5 f and 5 b were observed in low concentration. Thus, as above and in contrast to our previous work on the DiPPro‐compounds 6 [23] and γ‐(AB,ACB)‐d4TTPs [29] no selective cleavage of the different masks was observed in the hydrolysis in CEM cell extracts. In our previous work, the half‐lives of symmetric γ‐(AB−C4,AB−C4)‐d4TTP and γ‐(AB−C9,AB−C9)‐d4TTP in CEM/0 cell extracts were t1/2=0.43 h and t1/2=2.8 h, respectively. Thus, similar to the results in PB, the determined half‐life of non‐symmetric γ‐(AB−C4,AB−C14)‐d4TTP 3 be (t1/2=2.5 h) was almost identical to that of symmetric γ‐(AB−C9,AB−C9)‐d4TTP but comprising a short AB‐group. [26] This points to a compensation of the lability of short chain comprising TriPPPro‐derivatives if a lipophilic alkyl masking group is also present in the molecule.
The situation was different in the case of the PEG‐containing derivatives 3 bh, 3 eh, and 3 ei. After 3 h incubation in the CEM/0 cell extracts, low concentrations d4TTP 1 t (3 %) and more importantly only small amounts of the intermediate γ‐(AB−C14)‐d4TTP 5 e (9 %) were detected. Here, the predominant product was intermediate γ‐(AB−MEEES)‐d4TTP 5 h (52 %). Intermediate PEG‐d4TTP 5 h was detected and found to be much more stable than γ‐(AB−C14)‐d4TTPs 5 e (Figure 2, B). D4TDP 1 d increased constantly to an amount of 15 % after 3 h incubation and 32 % after 9 h incubation. Thus, by introducing the hydrophilic PEG‐bearing acyloxybenzyl‐mask, the “normal” lipophilic acyloxybenzyl‐mask was cleaved preferably in these hydrolysis studies, which is in agreement with the results obtained from the studies as reported before. [29]
Figure 2.
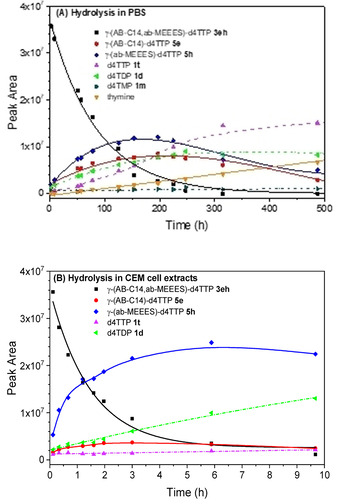
Hydrolysis of γ‐(AB−C14,AB−MEEES)‐d4TTP 3 eh in PB (A) and in CEM cell extracts (B).
Hydrolysis in pig liver esterase (PLE)
TriPPPro‐ d4TTP 3 bf was treated with pig liver esterase (PLE) to prove a possible selectivity in the removal of the masking groups (Figure 1, C). The determined half‐life here was 0.75 h. The cleavage of the masking units in 3 bf occurred much faster than that in PB (t 1/2=49 h) and also faster as compared to the study in CEM/0 cell extracts (t 1/2=3.1 h). Again, both 5 f and 5 b were formed in identical concentrations. Both, d4TTP 1 t and d4TDP 1 d were observed in low levels during PLE hydrolysis. The half‐lives for intermediates 5 f, 5 b were also found to be higher than that for γ‐(AB−C4,ab‐C15)‐d4TTP 3 bf, probably due to the additional negative charge at the γ‐phosphate.
Figure 1.
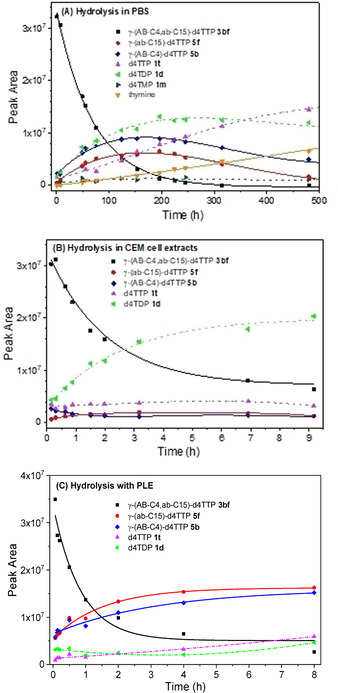
Hydrolysis of γ‐(AB−C4,AB−C15)‐d4TTP 3 bf in PB (A), CEM cell extracts (B) and pig liver esterase (PLE; C).
Anti‐HIV activities in CEM/0 and CEM/TK− cells
All TriPPPro‐compounds 3 were evaluated for their antiviral activity. For this purpose, HIV‐1‐ or HIV‐2‐infected wild‐type CEM/0 as well as mutant thymidine kinase‐deficient CEM cell cultures (CEM/TK−) were treated with the TriPPPro‐d4TTPs 3.
In wild‐type CEM/0 cell cultures, all γ‐(AB,ab)‐d4TTPs 3 showed higher activities against HIV‐2 and similar or slightly better activities against HIV‐1 as compared to the parent nucleoside d4T 1 or d4TTP 1 t (Table 2). More importantly, except compound 3 bh (C4/PEG) all prodrugs 3 were highly potent in thymidine kinase‐deficient CEM/TK− cells whereas d4T 1 and d4TTP 1 t lacked any relevant anti‐HIV activity in this assay (EC50: >50 μM for d4T 1 and >100 μM for d4TTP 1 t). Correlated with a substantial increase of the lipophilicity of the mask, TriPPPro‐compound 3 eh was 12‐fold more active than 3 bh against HIV‐2 in CEM/TK− cells. This point to sufficient lipophilicity of compound 3 eh combined with a relatively slow cleavage of the bioreversible AB‐moiety which led to the formation of γ‐(ab‐MEEES)‐d4TTP 5 h (Figure 2, B). To enable membrane passage of the non‐symmetrically‐masked TriPPPro‐compounds 3, one of the two masking groups should comprise a long lipophilic alkyl chain which then ensures that the prodrug is active against HIV, which in agreement with the results as γ‐(AB,ACB)‐d4TTPs. [29]
Table 2.
Anti‐HIV activity and cytotoxicity of TriPPPro compounds 3.
|
|
EC50 [μM][a] |
CC50 [μM][b] |
||
|---|---|---|---|---|
|
Prodrug |
CEM/0 |
CEM/TK− |
CEM/0 |
|
|
|
HIV‐1 |
HIV‐2 |
HIV‐1 |
|
|
3 ae |
0.24±0.17 |
0.39±0.22 |
1.1±0.82 |
20±0 |
|
3 be |
0.14±0.11 |
0.32±0.26 |
0.69±0.52 |
18±0 |
|
3 de |
0.15±0.087 |
0.29±0.24 |
0.74±0.46 |
22±3 |
|
3 ce |
0.17±0.085 |
0.23±0.18 |
0.39±0.21 |
24±3 |
|
3 bf |
0.15±0.12 |
0.27±0.25 |
1.1±0.81 |
24±15 |
|
3 bg |
0.12±0.05 |
0.10±0.03 |
0.54±0.41 |
33±7 |
|
3 bh |
0.34±0.23 |
0.76±0.58 |
16±4.0 |
23±9 |
|
3 eh |
0.26±0.14 |
0.43±0.30 |
1.3±0.64 |
28±3 |
|
3 ei |
0.31±0.16 |
0.65±0.59 |
2.0±1.1 |
34±14 |
|
d4T 1 |
0.33±0.13 |
0.97±0.50 |
>50 |
>50 |
|
d4TTP31 |
1.05±0.35 |
0.40±0.26 |
>100 |
>100 |
[a] Antiviral activity of the compounds in CD4+ T‐lymphocyte CEM cell cultures: 50 % effective concentration; values are the mean ±SD of n=2–3 independent experiments. [b] Cytotoxicity: 50 % cytostatic concentration or compound concentration required to inhibit CD4+ T‐cell (CEM) proliferation by 50 %; values are the mean ±SD of n=2–3 independent experiments.
Interestingly, the antiviral activity and the chemical or biological stability of prodrug 3 ce are 1.5‐fold higher than that of prodrug 3 be. Due to this favorable hydrolysis data, the antiviral activity observed in the wild‐type CEM/0 cell cultures was nearly retained in the case of γ‐(AB−C4,AB−C14)‐d4TTP 3 ce (EC50: 0.39 μM) in mutant thymidine kinase‐deficient CEM cell cultures (CEM/TK−). TriPPPro‐compound 3 ce is the most potent compound of all the listed derivatives (>128‐fold more active in the TK‐def. cell assay than the parent d4T).
Conclusion
In summary, a series of TriPPPro‐compounds 3 bearing two different AB‐masks attached to the γ‐phosphate was synthesized by using H‐phosphonate route. Hydrolysis in CEM cell extracts and pig liver esterase proved the delivery mechanism that the hydrolysis is mainly triggered by enzymes. However, compared to γ‐(AB,ACB)‐d4TTPs, [29] hydrolysis studies showed that no obvious selective cleavage occurred between short acetyl ester and long acetyl ester. Antiviral evaluation showed that most of the compounds exhibited higher activity against HIV‐1 and HIV‐2 in cultures of infected wild‐type human CD4+ T‐lymphocyte (CEM/0) cells and more importantly in thymidine kinase‐deficient CD4+ T‐cells (CEM/TK–). Interestingly, if one AB‐mask was modified with methoxytriglycol group with a diester linker, the other AB‐mask was cleaved predominately. However, these compounds with hydrophilic groups are less potent than the TriPPPro‐compounds with two alkylacyl AB‐masks.
Experimental Section
Chemicals and instrumentation. General: Without further noticed, all manipulations were carried out under an atmosphere of nitrogen using standard Schlenk techniques and all solvents were dried by using standard procedures. Triethylamine (Et3N) were dried by being heated under reflux over calcium hydride for 5 days and followed by distillation. Trifluoroacetic anhydride (TFAA) was dried over phosphorus pentoxide for one hour and distilled under nitrogen. Anhydrous CH3CN (CH3CN), tetrahydrofuran (THF), dichloromethane (CH2Cl2),) and pyridine were obtained by the MBraun solvent purification system (MB SPS‐800). Other dry solvents were purchased from Acros Organics (extra dry over molecular sieves). For eluent of column chromatography, technique grade ethyl acetate (EA), petroleum ether (PE) 50–70, CH2Cl2, and CH3OH were distilled before use. Tetra‐n‐butylammonium phosphate monobasic solution (0.4 M in CH3CN) was purchased from Acros Organics directly. Column chromatography: Normal phase column chromatography were performed with Macherey‐Nagel silica gel 60 M (0.04–0.063 mm). For automatic reversed‐phase chromatography an Interchim Puriflash 430 in combination with Chromabond® Flash C [18] ec was used. Analytical thin‐layer chromatography (TLC): For thin layer chromatography Macherey‐Nagel pre‐coated TLC sheets Alugram® Xtra SIL G/UV254 were used. Phosphomolybdic acid (PMA) stain was used and prepared by dissolving 10 g of phosphomolybdic acid in 100 mL of ethanol. Potassium permanganate stain was used and prepared by 1.5 g of KMnO4, 10 g K2CO3, and 1.25 mL 10 % NaOH in 200 mL water. High performance liquid chromatography (HPLC): A VWR‐Hitachi LaChromElite HPLC system (L‐2130, L‐2200, L‐2455) with an EzChromElite software and a Nucleodur 100–5 C18ec column (Macherey‐Nagel) was used. HPLC grade CH3CN was obtained from VWR and ultrapure water (Mili‐Q water) from a Sartorius Aurium® pro apparatus (Sartopore 0.2 μm, UV). 2 mM tetra‐n‐butylammonium acetate solution (TBAA, pH 6.3) was used as buffer. Method: Nucleodur 100–5 C18ec; 0–20 min: TBAA buffer/CH3CN gradient (5–80 %); 20–30 min: buffer/CH3CN (80 %); 30–33 min: buffer/CH3CN (80–5 %); 33–38 min: buffer/CH3CN (5 %); flow: 1 mL/min. TBAA buffer (2 mM): 4.05 mL tetra‐n‐butylammonium hydroxide in water (ca. 40 %) was diluted with 3000 mL ultrapure water. Then glacial acetic acid was added to adjust the buffer to pH 6.3. Nuclear Magnetic Resonance spectroscopy (NMR): NMR spectra were recorded at room temperature in automation mode on either of a Varian Gemini 2000BB, Bruker AMX 400, Bruker DRX 500 or Bruker AVIII 600 spectrometer. Chloroform‐d1 (CDCl3), methanol‐d4 (MeOD), dimethyl sulfoxide‐d6 (DMSO) and deuterium oxide (D2O) were purchased form Euriso‐Top. All 1H, 31P, and 13C NMR chemical shifts are quoted in parts per million (ppm). Reference peaks for chloroform‐d in 1H NMR and 13C NMR spectra were set at 7.26 ppm and 77.0 ppm, respectively. For methanol‐d4 reference peaks in 1H NMR and 13C NMR spectra were set at 3.31 ppm and 49.0 ppm respectively. All final compounds were isolated analytically pure, ≥95 % purity by HPLC and NMR spectroscopy. Mass Spectrometry (MS): HRMS (ESI) mass spectra were recorded on an Agilent 6224 ESI‐TOF spectrometer. MALDI measurements (matrix: 9‐aminoacridine [9AA] or 2,5‐dihydroxybenzoic acid [DHB]) were performed with a Bruker UltrafelXtreme spectrometer. Infrared spectroscopy (IR): IR spectra were recorded on a Bruker Alpha P FT‐IR at room temperature in the range of 400–4000 cm−1. Freeze dryer: Alpha 2–4 LDPlus freeze dryer from Christ Co. was used. Ultrapure water: Arium®pro ultrapure water system was used. pH meter: pH value was tested with ProLab 300 from Schott Co. Thermomixer: Eppendorf Thermomixer 5436 and CellMedia Thermoschüttler basic were used. Ultrasonic cleaning device: Sonorex RK512H from Bandelin Co. was used. Centrifuges: Heraeus Biofuge Pico (13000 u/min) and Biofuge Primo R (8000 u/min) was used.
General synthesis procedures
General procedure A: preparation of 4‐(hydroxymethyl)phenylalkanoates 7. 4‐Hydroxybenzyl alcohol and triethylamine in CH2Cl2 were cooled down to 0 °C or at room temperature. The corresponding acyl chloride in CH2Cl2 was added dropwise and the mixture was stirred at room temperature (rt). The precipitate was filtered, and the solvent was removed in vacuum. The residue was diluted with CH2Cl2 and washed once with water. The organic layer was dried with Na2SO4 and the solvent was removed. The crude was purified by SiO2 column chromatography to give compounds 7.
General procedure B: preparation of non‐symmetric (AB,ab)‐H‐phosphonates 8. Under dry conditions, diphenyl phosphonate (1.2 equiv.) was dissolved in 3 mL pyridine and cooled to 0 °C. Compounds 7 e–h (1.0 equiv.) were added and stirred at 0 °C for 30 min and then heated up to 38 °C. Following, compounds 7 a–e (1.4 equiv.) were added and the mixture was stirred for 3 h. Then the solvent was removed in vacuo. The crude products were purified by flash column chromatography (silica) with EtOAc/petroleum ether/0.5 % acetic acid as eluent.
General procedure C: preparation of non‐symmetric TriPPPro‐compounds 3. The reactions were performed under dry conditions. a) H‐Phosphonates 8 (1.0 eq.) were dissolved in 6 mL CH3CN and N‐chlorosuccinimide (2.0 eq.) was added. After stirring for 2 h at RT, tetra‐n‐butylammonium phosphate monobasic solution (0.4 M in CH3CN) (3.0 eq.) was added dropwise. The mixture was stirred for 1 h and the solvent was removed in vacuum. The residue was extracted with CH2Cl2/H2O. The organic phase was dried over sodium sulfate and the solvent was removed by evaporation to afford corresponding pyrophosphates in nearly quantitative yield. B) The corresponding pyrophosphates were dissolved in CH3CN and cooled down to 0 °C. A mixture of trifluoroacetic anhydride (TFAA, 5.0 eq.) and trimethylamine (Et3N, 8.0 eq.) in 3 mL CH3CN was cooled to 0 °C and added to the mixture. After stirring for 10 min, all volatile components were removed in vacuum. The residue was once again co‐evaporated with 3 mL CH3CN and subsequently dissolved in 3 mL CH3CN at 0 °C. 1‐methylimidazole (3.0 eq.) and trimethylamine (Et3N, 8.0 eq.) was added. The suspension was warmed up to RT and stirred for 10 min. The resulting activated imidazolidate formed and the corresponding d4TMP 1 m (0.7–1.0 eq.) in 3 mL CH3CN was added. The reaction was stirred at room temperature for 3–5 h and dried in vacuum. The crude product was purified by automatic RP18 flash chromatography, and then followed by ion‐exchange to the ammonium form with Dowex 50WX8 cation‐exchange resin and a second RP18 chromatography purification step. Product‐containing fractions were collected, and the organic solvent evaporated. The remaining aqueous solutions were freeze‐dried, and the desired product obtained as a white solid.
4‐(Hydroxymethyl)phenyl propionate 7 a: According to General Procedure A, under atmosphere, 3.41 g 4‐hydroxybenzylalcohol (27.5 mmol, 1.1 equiv.) and 3.47 mL triethylamine (25 mmol, 1.0 equiv.) was dissolved in 15 mL CH2Cl2 at 0 °C. 2.17 mL propionyl chloride (25 mmol, 1.0 equiv.) in 25 mL CH2Cl2 was added dropwise. Reaction time was 2.5 h at RT. Column chromatography (SiO2, PE/EE 6 : 4 v/v) Yield: 60 %, 2.70 g, as colorless oil. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.36 (d, 3 J=8.0 Hz, 2H), 7.06 (d, 3 J=8.0 Hz, 2H), 4.66 (s), 2.59 (q, J=7.5 Hz, 2H), 1.91 (s, 1H), 1.26 (t, J HH=7.5 Hz, 3H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=173.0, 150.1, 138.4, 128.0, 121.6, 64.7, 27.7, 9.0. HRMS (ESI‐TOF) m/z: calculated for C10H12NaO3 [M+Na]+ 203.0679, found 203.0618. IR: ν [cm−1]=3405, 2982, 2941, 2883, 1752, 1606, 1509, 1473, 1456, 1413, 1383, 1351, 1201, 1170, 1138, 1073, 1025, 999, 946, 891, 830, 804, 761, 727, 577, 559, 515, 423, 409.
4‐(Hydroxymethyl)phenyl pentanoate 7 b: According to General Procedure A, under atmosphere, 1.22 g 4‐hydroxybenzylalcohol (9.79 mmol, 1.2 equiv.) and 1.36 mL triethylamine (9.79 mmol, 1.2 equiv.) was dissolved in 20 mL CH2Cl2 at RT. 1 mL pentanoyl chloride (8.16 mmol, 1.0 equiv.) in 30 mL CH2Cl2 was added dropwise. Reaction time was 4 h at RT. Column chromatography (SiO2, PE/EE 7 : 3 v/v). Yield: 78 %, 1.33 g, as colorless oil. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.36–7.27 (d, J=8.1 Hz, 2H), 7.08–6.97 (d, J=8.4 Hz, 2H), 4.57 (s, 2H), 2.54 (t, J=7.5 Hz, 2H), 2.75–2.45 (br, s, 1H), 1.73 (tt, J=7.5, 7.5 Hz, 2H), 1.44 (tq, J=8.0, 7.2 Hz, 2H), 0.97 (t, J=7.4 Hz, 3H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.4, 149.9, 138.4, 127.9, 121.5, 64.4, 34.0, 26.9, 22.1, 13.6. HRMS (ESI‐TOF) m/z: calculated for C12H16NaO3 [M+Na]+ 231.0992, found 231.0972. IR: ν [cm−1]=3393, 2958, 2932, 2872, 1754, 1606, 1506, 1464, 1417, 1365, 1345, 1310, 1198, 1163, 1143, 1101, 1046, 1014, 940, 919, 848, 811, 753, 733, 643, 561, 505, 458, 405, 389.
4‐(Hydroxymethyl)phenyl 3‐methylbutanoate 7 c: According to General Procedure A, under atmosphere, 3.0 g 4‐hydroxybenzylalcohol (24.0 mmol, 1.0 equiv.) and 3.67 mL triethylamine (26.4 mmol, 1.1 equiv.) was dissolved in 15 mL CH2Cl2 at 0 °C. 3.21 mL 3‐methylbutanoyl chloride (25 mmol, 1.0 equiv.) in 25 mL CH2Cl2 was added dropwise. The mixture was stirred for 2.5 h at RT. Column chromatography (SiO2, PE/EE 7 : 3 v/v). Yield: 64 %, 3.21 g, as colorless oil. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.40–7.27 (m, 2H), 7.10–7.01 (m, 2H), 4.66 (s, 2H), 2.43 (d, J=7.2 Hz, 2H), 2.34–2.18 (m, 1H), 2.09–1.77 (m, 1H), 1.06 (d, J=6.6 Hz, 6H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.4, 149.9, 138.4, 127.9, 121.5, 64.4, 34.0, 26.9, 22.1, 13.6. HRMS (ESI‐TOF) m/z: calculated for C12H16NaO3 [M+Na]+ 231.0992, found 231.0989. IR: ν [cm−1]=3350, 2959, 2932, 2873, 1753, 1606, 1466, 1417, 1388, 1369, 1292, 1245, 1197, 1152, 1099, 1042, 1014, 964, 914, 890, 850, 832, 811, 628, 562, 500.
4‐(Hydroxymethyl)phenyl heptanoate 7 d: According to General Procedure A, under atmosphere, 3.0 g 4‐hydroxybenzylalcohol (24.0 mmol, 1.0 equiv.) and 3.67 mL triethylamine (26.4 mmol, 1.1 equiv.) was dissolved in 20 mL CH2Cl2 at 0 °C. 4.1 mL n‐heptanoyl chloride (26.4 mmol, 1.1 equiv.) in 30 mL CH2Cl2 was added dropwise. The mixture was stirred for 2.5 h at RT. Column chromatography (SiO2, PE/EE 7 : 3 v/v). Yield: 58 %, 3.31 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.45–7.32 (m, 2H), 7.13–7.02 (m, 2H), 4.66 (s, 2H), 2.55 (t, J=7.5 Hz, 2H), 1.75 (quint, J=7.0 Hz, 2H), 1.69 (br, s), 1.49–1.21 (m, 2H), 0.91 (t, J=7.0 Hz, 3H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.4, 150.0, 138.4, 128.0, 121.6, 64.6, 34.3, 31.4, 28.7, 22.4, 24.8, 14.0. HRMS (ESI‐TOF) m/z: calculated for C14H20NaO3 [M+Na]+ 259.1305, found 259.1230. IR: ν [cm−1]=3380, 2956, 2928, 2860, 1754, 1510, 1459, 1417, 1379, 1195, 1164, 1140, 1015, 848, 811, 503.
4‐(Hydroxymethyl)phenyl pentadecanoate 7 e: According to General Procedure A, under atmosphere, 1.22 g 4‐hydroxybenzylalcohol (9.9 mmol, 1.2 equiv.) and 1.36 mL triethylamine (9.9 mmol, 1.2 equiv.) was dissolved in 20 mL CH2Cl2 at room temperature. 2.14 g n‐pentadecanoyl chloride (8.25 mmol, 1.0 equiv.) in 30 mL CH2Cl2 was added dropwise. The mixture was stirred for overnight at RT. Column chromatography (SiO2, PE/EE 7 : 3 v/v). Yield: 63 %, 1.82 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.35 (dt, J=8.8, 2.6 Hz, 2H), 7.06 (dt, 3 J HH=8.4, 2.4 Hz, 2H), 4.65 (s, 2H), 2.55 (t, J=7.6 Hz, 2H), 1.75 (p, J=7.5 Hz, 2H), 1.46–1.19 (m, 22H), 0.89 (t, J=6.8 Hz, 3H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.4, 150.1, 138.4, 128.0, 121.6, 64.7, 34.4, 31.9, 29.66, 29.65, 29.57, 29.43, 29.33, 29.23, 29.08, 22.7, 24.9, 14.1. HRMS (ESI‐TOF) m/z: calculated for C22H40NO3 [M+NH4]+ 366.3003, found 366.3000. IR: ν [cm−1]=3321, 2955, 2914, 2847, 1748, 1605, 1509, 1463, 1411, 1386, 1318, 1294, 1270, 1246, 1218, 1166, 1150, 1094, 1037, 1014, 950, 925, 846, 817, 760, 745, 719, 580, 515.
4‐(Hydroxymethyl)phenyl hexadecanoate 7 f: According to General Procedure A, under atmosphere,1.22 g 4‐hydroxybenzylalcohol (9.8 mmol, 1.2 equiv.) and 1.36 mL triethylamine (9.8 mmol, 1.2 equiv.) was dissolved in 20 mL CH2Cl2 at RT. 2.48 mL n‐hexadecanoyl chloride (8.16 mmol, 1.0 equiv.) in 30 mL CH2Cl2 was added dropwise. The mixture was stirred for overnight at RT. Column chromatography (SiO2, PE/EE 7 : 3 v/v). Yield: 80 %, 2.36 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.37 (d, J=8.5 Hz, 2H), 7.06 (dt, J=8.4, 2.4 Hz, 2H), 4.68 (s, 2H), 2.55 (t, J=7.5 Hz, 2H), 1.75 (p, J=7.5 Hz, 2H), 1.48–1.18 (m, 24H), 0.88 (t, J=7.0 Hz, 3H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.4, 150.1, 138.3, 128.0, 121.7, 64.8, 34.4, 31.9, 29.68, 29,67, 29.64, 29.59, 29.45, 29.35, 29.25,29.10, 22.68, 24.9, 14.1. HRMS (ESI‐TOF) m/z: calculated for C23H38NaO3 [M+Na]+ 385.2719, found 385.2782. IR: ν [cm−1]=3322, 2955, 2914, 2847, 1748, 1606, 1509, 1471, 1463, 1410, 1387, 1348, 1330, 1308, 1286, 1263, 1241, 1218, 1166, 1150, 1097, 1039, 1014, 950, 925, 846, 817, 779, 960, 739, 727, 719, 581, 515.
4‐(Hydroxymethyl)phenyl octadecanoate 7 g: According to General Procedure A, under atmosphere, 2.46 g 4‐hydroxybenzylalcohol (19.8 mmol, 1.2 equiv.) and 2.75 mL triethylamine (19.8 mmol, 1.2 equiv.) was added in 100 mL CH2Cl2. 5.57 mL octadecanoyl chloride (16.5 mmol, 1.0 equiv.) in 150 mL CH2Cl2 was added dropwise and the mixture was stirred at RT overnight. The precipitate was filtered, and the solvent was removed in vacuum. The residue was diluted with CH2Cl2 and washed once with water. The organic layer was dried with Na2SO4 and the solvent was removed. Column chromatography (SiO2, petrol ether/ethyl acetate 6 : 4 v/v). Yield: 78 %, 5.0 g, as white solid. 1H NMR (500 MHz, CDCl3): δ [ppm]=7.37 (d, J=8.4 Hz, 2H), 7.07 (dt, J=8.4 Hz, 2H), 4.68 (s, 2H), 2.55 (t, J=7.5 Hz, 2H), 1.74 (p, J=7.5 Hz, 2H), 1.64 (br, s, 1H), 1.49–1.12 (m, 28H), 0.88 (t, J=6.8 Hz, 3H). 13C‐NMR (126 MHz, CDCl3): δ [ppm]=172.38, 150.16, 138.32, 128.03, 121.69, 64.79, 34.39, 31.92, 29.69, 29.67, 29.65, 29.64, 29.59, 29.45, 29.35, 29.25, 29.10, 24.94, 22.68, 14.11. HRMS (ESI‐TOF) m/z: calculated for C25H46NO3 [M+NH4]+ 408.3472, found 408.3478. IR: ν [cm−1]=3318, 2955, 2914, 2847, 1748, 1605, 1509, 1471, 1463, 1410, 1387, 1331, 1312, 1292, 1272, 1252, 1232, 1218, 1166, 1150, 1101, 1034, 1014, 950, 924, 846, 817, 760, 727, 719, 580, 511.
12‐Oxo‐2,5,8,11‐tetraoxapentadecan‐15‐oic acid 12 h: Under atmosphere, 10 g 2‐(2‐(2‐Methoxyethoxy)ethoxy)ethan‐1‐ol 10 (60.9 mmol, 1.0 equiv.) and 7.3 g succinic anhydride 11 h (73.1 mmol, 1.2 equiv.) was added in 40 mL CH2Cl2. Then 1.49 g DMAP (12.2 mmol, 0.2 equiv.) was added and the mixture was stirred at RT overnight. The reaction mixture was then quenched with 4 mL water, diluted with 20 mL CH2Cl2 and extract with (3×10 mL) 10 % NaHSO4 and (1×10 mL) brine. The organic phase was dried with MgSO4 and the solvent was removed in vacuum. The product was used directly without further purification. Yield: 72 %, 11.6 g, as colorless liquid. 1H NMR (400 MHz, DMSO‐d6): δ [ppm]=4.16–4.07 (m, 2H), 3.65–3.56 (m, 2H), 3.56–3.48 (m, 6H), 3.48–3.38 (m, 2H) 3.24 (s, 3H), 2.57 −2.42 (m, 4H). 13C‐NMR (101 MHz, DMSO‐d6): δ [ppm]=173.24, 172.07, 71.23, 69.76, 69.68, 69.56, 68.20, 63.33, 58.00, 28.60. HRMS (ESI‐TOF): calculated for C11H20NaO7 [M+Na]+ 287.1101, found 287.1072. IR: ν [cm−1]=2877, 1730, 1452, 1383, 1349, 1244, 1199, 1160, 1097, 1027, 988, 942, 849, 623, 564, 405.
4‐(Hydroxymethyl)phenyl (2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl) succinate 7 h: 5.0 g 12‐oxo‐2,5,8,11‐tetraoxapentadecan‐15‐oic acid 12 h (18.9 mmol, 1.0 equiv.) was dissolved in 100 mL CH2Cl2 and cooled to 0 °C. 0.7 mL oxalyl chloride (22.7 mmol, 1.2 equiv.) was added to the flask and 3 drops of DMF was then added. Afterwards, the mixture was warm up to RT and stirred until no more gas generated (around 2–4 h). Then oxalyl chloride was evaporated to afford target acyl chloride as colorless liquid. The yield was calculated as quantitative. The crude product 13 h was used directly without further purification. According to General Procedure A, under atmosphere, 2.25 g 4‐hydroxybenzylalcohol (18.1 mmol, 1.2 equiv.) and 2.52 mL triethylamine (18.1 mmol, 1.2 equiv.) was added in 20 mL CH2Cl2. 4.27 g 2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl 4‐chloro‐4‐oxobutanoate 13 h (15.1 mmol, 1.0 equiv.) in 30 mL CH2Cl2 was added dropwise and the mixture was stirred at room temperature overnight. The precipitate was filtered and the solvent was removed in vacuum. The residue was diluted with CH2Cl2 and washed once more with water. The organic layer was dried with Na2SO4 and the solvent was removed. Column chromatography (SiO2, petrol ether/ethyl acetate 2 : 8 v/v). Yield: 50 %, 3.0 g, as colorless oil. 1H NMR (400 MHz, CDCl3) δ [ppm]=7.36 (d, J=8.5 Hz, 2H), 7.07 (d, J=8.5 Hz, 2H), 4.66 (s, 2H), 4.32–4.22 (m, 2H), 3.74–3.67 (m, 2H), 3.67–3.58 (m, 6H), 3.56–3.50 (m, 2H) 3.37 (s, 3H), 2.87 (t, J=6.8 Hz, 2H), 2.76 (t, J=6.8 Hz, 2H), 1.97 (s, 1H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.07, 170.89, 149.97, 138.58, 128.00, 121.54, 71.88, 70.53, 70.50, 69.02, 64.66, 63.96, 58.98, 29.26, 29.04. HRMS (ESI‐TOF): calculated for C18H26NaO8 [M+Na]+ 393.1520, found 393.1514. IR: ν [cm−1]=3437, 2875, 1856, 1732, 1607, 1507, 1453, 1411, 1350, 1244, 1196, 1164, 1131, 1099, 1015, 997, 944, 888, 847, 811, 550, 506.
12‐Oxo‐2,5,8,11‐tetraoxahexadecan‐16‐oic acid 12 i: Under atmosphere, 10 g 2‐(2‐(2‐Methoxyethoxy)ethoxy)ethan‐1‐ol 10 (60.9 mmol, 1.0 equiv.) and 8.4 g glutaric anhydride 11 i (73.1 mmol, 1.2 equiv.) was added in 50 mL CH2Cl2. Then 1.49 g DMAP (12.2 mmol, 0.2 equiv.) was added and the mixture was stirred at RT overnight. The reaction mixture was then quenched with 4 mL water, diluted with 20 mL CH2Cl2 and extract with (3×10 mL) 10 % NaHSO4 and (1×10 mL) brine. The organic phase was dried with MgSO4 and the solvent was removed in vacuum. The product was used directly without further purification. Yield: 59 %, 9.8 g, as colorless liquid. 1H NMR (400 MHz, CDCl3) δ [ppm]=4.31–4.20 (m, 2H), 3.75–3.61 (m, 8H), 3.60–3.52 (m, 2H), 3.38 (s, 3H), 2.44 (td, J=7.1, 3.5 Hz, 4H), 1.98 (quint, J=7.2 Hz, 2H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.9, 71.9, 70.61, 70.49, 70.46, 69.10, 63.57, 58.95, 33.10, 32.74, 19.88. HRMS (ESI‐TOF): calculated for C12H22NaO7 [M+Na]+ 301.1258, found 301.1263. IR: ν [cm−1]=2880, 1731, 1451, 1385, 1344, 1240, 1189, 1155, 1098, 1024, 978, 951, 832, 611, 542, 410.
4‐(Hydroxymethyl)phenyl (2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl) glutarate 7 i: 4.3 g 12‐oxo‐2,5,8,11‐tetraoxahexadecan‐16‐oic acid 12 i (15.6 mmol, 1.0 equiv.) was dissolved in 50 mL CH2Cl2 and cooled to 0 °C. 1.58 mL oxalyl chloride (18.7 mmol, 1.2 equiv.) was added to the flask and 3 drops of DMF was then added. Afterwards, the mixture was warm up to RT and stirred until no more gas generated (around 2–4 h). Then oxalyl chloride was evaporated to afford 4.3 g target acyl chloride as colorless liquid. The yield was calculated as 77 %. The crude product 13 i was used directly without further purification. According to General Procedure A, under atmosphere, 2.32 g 4‐hydroxybenzylalcohol (18.7 mmol, 1.2 equiv.) and 2.59 mL triethylamine (18.7 mmol, 1.2 equiv.) was added in 20 mL CH2Cl2. 4.3 g 2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl 5‐chloro‐5‐oxopentanoate 13 i (15.6 mmol, 1.0 equiv.) in 30 mL CH2Cl2 was added dropwise and the mixture was stirred at RT overnight. The precipitate was filtered, and the solvent was removed in vacuum. The residue was diluted with CH2Cl2 and washed once more with water. The organic layer was dried with Na2SO4 and the solvent was removed. Column chromatography (SiO2, petrol ether/ethyl acetate 2 : 8 v/v). Yield: 50 %, 3.0 g, as colorless oil. 1H NMR (400 MHz, CDCl3) δ [ppm]=7.37 (d, J=8.5 Hz, 2H), 7.07 (d, J=9.2 Hz, 2H), 4.68 (d, J=4.3 Hz, 2H), 4.32–4.20 (m, 2H), 3.75–3.67 (m, 2H), 3.67–3.60 (m, 6H), 3.57–3.51 (m, 2H) 3.37 (s, 3H), 2.64 (t, J=7.4 Hz, 2H,), 2.49 (t, J=7.3 Hz, 2H), 2.07 (quint, J=7.3 Hz, 2H), 1.80 (s, 1H). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.8, 171.5, 150.0, 138.5, 128.0, 121.6, 71.93, 70.60, 70.57, 69.11, 64.73, 63.63, 59.0, 33.30, 33.06, 20.03. HRMS (ESI‐TOF) m/z: calculated for C19H28NaO8 [M+Na]+ 407.1676, found 407.1703. IR: ν [cm−1]=3439, 2874, 1731, 1601, 1507, 1452, 1417, 1381, 1352, 1195, 1163, 1127, 1016, 944, 849, 562, 507, 385.
(AB‐C2H5,ab‐C14H29)‐H‐phosphonate 8 ae: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.35 g 4‐(hydroxymethyl)phenyl pentadecanoate 7 e (1.0 mmol, 1.0 equiv.) was added and followed with 0.25 g 4‐(hydroxymethyl)phenyl propionate 7 a (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 7 : 3:0.005 v/v/v). Yield: 50 %, 0.29 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.39–7.30 (m, 4H, H‐2''), 7.17–7.02 (m, 4H, H‐3''), 6.94 (d, 1 J PH=708 Hz, 1H, P−H), 5.13–4.93 (m, 4H, Ph‐CH2), 2.59 (q, 3 J HH=8.0 Hz, 2H, H‐q), 2.55 (t, 3 J HH=6.3 Hz, 2H, H‐b), 1.75 (p, J=7.5 Hz, 2H, H‐c), 1.47–1.25 (m, 25H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐r), 0.88 (t, 3 J HH=6.7 Hz, 3H, H‐o). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.7 (C‐p),172.1 (C‐a), 151.0 (2×C‐4''), 132.98, 132.92 (2×C‐1''), 129.2 (4×C‐2''), 121.90, 121.87 (4×C‐3''), 66.65 (d, 3 J CP=6.1 Hz, Ph‐CH2), 34.4 (C‐b), 31.9, 29.65, 29,64, 29.61, 29.57, 29.43, 29.32, 29.22, 29.08 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 27.7 (C‐q), 24.9 (C‐c), 22.7 (C‐n), 14.1 (C‐o), 9.0 (C‐r). 31P‐NMR (162 MHz, CDCl3): δ [ppm]=7.71. HRMS (ESI‐TOF) m/z: calculated for C32H47NaO7P [M+Na]+ 597.2952, found 597.2952. IR: ν [cm−1]=2956, 2915, 2848, 1754, 1607, 1510, 1463, 1412, 1386, 1359, 1318, 1295, 1269, 1249, 1219, 1167, 1149, 1061, 996, 925, 896, 876, 827, 806, 769, 720, 806, 769, 720, 581, 538, 515, 455, 422, 410.
(AB‐C4H9,ab‐C14H29)‐H‐phosphonate 8 be: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.35 g 4‐(hydroxymethyl)phenyl pentadecanoate 7 e (1.0 mmol, 1.0 equiv.) was added and followed with 0.29 g 4‐(hydroxymethyl)phenyl pentanoate 7 b (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 7 : 3:0.005 v/v/v). Yield: 48 %, 0.29 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.44–7.31 (m, 4H, H‐2''), 7.17–7.01 (m, 4H, H‐3''), 6.94 (d, 1 J PH=708 Hz, 1H, P−H), 5.16–4.93 (m, 4H, Ph‐CH2), 2.56 (t, 3 J HH=7.5 Hz, 2H, H‐q), 2.55 (t, 3 J HH=7.5 Hz, 2H, H‐b), 1.85–1.65 (m, 4H, H‐c, H‐r), 1.53–1.17 (m, 24H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐s), 0.97 (t, 3 J HH=7.3 Hz, 3H, H‐t), 0.88 (t, 3 J HH=6.7 Hz, 3H, H‐o). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.08 (C‐p),172.06 (C‐a), 151.0 (2×C‐4''), 132.98, 132.92 (2×C‐1''), 129.2 (4×C‐2''), 121.90 (4×C‐3''), 66.69, 66.63 (2×Ph‐CH2), 34.4 (C‐b), 34.1 (C‐q), 31.9, 29.66, 29,65, 29.62, 29.57, 29.43, 29.33, 29.23, 29.08 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 26.9 (C‐r), 24.9 (C‐c), 22.7 (C‐n), 22.2 (C‐s), 14.1 (C‐o), 13.7 (C‐t). 31P‐NMR (162 MHz, CDCl3): δ [ppm]=7.70. HRMS (ESI‐TOF) m/z: calculated for C34H55NO7P [M+NH4]+ 620.3711, found 620.3709. IR: ν [cm−1]=2956, 2915, 2848, 1748, 1608, 1510, 1465, 1413, 1383, 1350, 1317, 1295, 1268, 1250, 1219, 1167, 1150, 1105, 1061, 1009, 996, 925, 897, 878, 834, 769, 720, 692, 580, 559, 538, 515, 456, 420, 408.
(AB‐C6H13,ab‐C14H29)‐H‐phosphonate 8 de: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.35 g 4‐(hydroxymethyl)phenyl pentadecanoate 7 d (1.0 mmol, 1.0 equiv.) was added and followed with 0.33 g 4‐(hydroxymethyl)phenyl heptanoate 7 e (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 7 : 3:0.005 v/v/v). Yield: 52 %, 0.33 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.51–7.32 (m, 4H, H‐2''), 7.18–6.97 (m, 4H, H‐3''), 6.93 (d, 1 J PH=708 Hz, 1H, P−H), 5.20–4.85 (m, 4H, Ph‐CH2), 2.55 (m, 4H, H‐q, H‐b), 1.75 (m, 4H, H‐c, H‐r), 1.55–1.18 (m, 28H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐s, H‐t, H‐u), 0.940.84 (m, 6H, H‐v, H‐o). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.05 (C‐p, C‐a), 151.0 (2×C‐4''), 132.97, 132.91 (2×C‐1''), 129.2 (4×C‐2''), 121.90 ( 4×C‐3''), 66.67, 66.61 (2×Ph‐CH2), 34.3 (C‐b, C‐q), 31.88, 31.38, 29.64, 29,63, 29.60, 29.56, 29.42, 29.31, 29.21, 29.07, 28.72 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐s, C‐t), 24.87,24,82 (C‐r, C‐c), 22.6 (C‐n), 22.4 (C‐u), 14.1 (C‐o), 14.0 (C‐v). 31P‐NMR (162 MHz, CDCl3): δ [ppm]=7.70. HRMS (ESI‐TOF) m/z: calculated for C36H59NO7P [M+NH4]+ 648.4024, found 648.4037. IR: ν [cm−1]=2956, 2915, 2848, 1748, 1608, 1510, 1465, 1411, 1384, 1293, 1270, 1250, 1236, 1218, 1167, 1149, 1116, 1061, 996, 925, 878, 834, 769, 739, 721, 581, 539, 515, 453, 427.
(AB‐iso‐C4H9,ab‐C14H29)‐H‐phosphonate 8 ce: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.35 g 4‐(hydroxymethyl)phenyl pentadecanoate 7 e (1.0 mmol, 1.0 equiv.) was added and followed with 0.29 g 4‐(hydroxymethyl)phenyl 3‐methylbutanoate 7 c (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 7 : 3:0.005 v/v/v). Yield: 48 %, 0.28 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.33–7.23 (m, 4H, H‐2''), 7.08–6.95 (m, 4H, H‐3''), 6.87 (d, 1 J PH=708 Hz, 1H, P−H), 5.16–4.89 (m, 4H, Ph‐CH2), 2.48 (t, 3 J HH=7.5 Hz, 2H, H‐b), 2.36 (d, 3 J HH=7.2 Hz, 2H, H‐q), 2.17 (tq, 3 J HH=6.8 Hz, 1H, H‐r), 1.67 (p, 3 J HH=7.5 Hz, 2H, H‐c), 1.40–1.11 (m, 22H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 0.99 (d, 3 J HH=6.7 Hz, 6H, H‐t), 0.81 (t, 3 J HH=6.8 Hz, 3H, H‐o). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.08 (C‐a), 171.31 (C‐p), 150.99, 150.94 (2×C‐4''), 133.01, 132.95 (2×C‐1''), 129.22, 129.21 (4×C‐2''), 121.94, 121.92 (4×C‐3''), 66.70, 66.65 (2×Ph‐CH2), 43.30 (C‐q), 34.36 (C‐b), 31.90, 29.66, 29.65, 29.62, 29.58, 29.44, 29.33, 29.23, 29.09 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 25.83 (C‐r), 24.89 (C‐c), 22.66 (C‐n), 22.37 (C‐s, C‐t), 14.09 (C‐o). 31P‐NMR (162 MHz, CDCl3): δ [ppm]=7.70. HRMS (ESI‐TOF) m/z: calculated for C34H51NaO7P [M+Na]+ 625.3265, found 625.3262. IR: ν [cm−1]=2956, 2915, 2848, 1748, 1608, 1510, 1464, 1413, 1385, 1317, 1295, 1250, 1218, 1166, 1150, 1106, 1060, 996, 924, 877, 832, 768, 719, 693, 580, 558, 538, 515, 456, 423.
(AB‐C4H9,ab‐C15H31)‐H‐phosphonate 8 bf: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.36 g 4‐(hydroxymethyl)phenyl hexadecanoate 7 f (1.0 mmol, 1.0 equiv.) was added and followed with 0.29 g 4‐(hydroxymethyl)phenyl pentanoate 7 b (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 7 : 3:0.005 v/v/v). Yield: 44 %, 0.27 g, as white solid. 1H NMR (600 MHz, CDCl3): δ [ppm]=7.39–7.34 (m, 4H, H‐2''), 7.10–7.04 (m, 4H, H‐3''), 6.93 (d, 1 J PH=708 Hz, 1H, P−H), 5.12–4.95 (m, 4H, Ph‐CH2), 2.56 (t, 3 J HH=7.6 Hz, 2H, H‐q), 2.55 (t, 3 J HH=7.5 Hz, 2H, H‐b), 1.74 (m, 4H, H‐c, H‐r), 1.54–1.19 (m, 26H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐o, H‐s), 0.97 (t, 3 J HH=7.4 Hz, 3H, H‐t), 0.88 (t, 3 J HH=7.0 Hz, 3H, H‐u). 13C‐NMR (151 MHz, CDCl3): δ [ppm]=172.09 (C‐p),172.08 (C‐a), 151.0 (2×C‐4''), 132.97, 132.93 (2×C‐1''), 129.2 (4×C‐2''), 121.90 (4×C‐3''), 66.68, 66.64 (2×Ph‐CH2), 34.4 (C‐b), 34.1 (C‐q), 31.9, 29.66, 29,65, 29.63, 29.62, 29.58, 29.44, 29.33, 29.23, 29.09 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐n), 26.9 (C‐r), 24.9 (C‐c), 22.7 (C‐o), 22.2 (C‐s), 14.1 (C‐u), 13.7 (C‐t). 31P‐NMR (243 MHz, CDCl3): δ [ppm]=7.70. HRMS (ESI‐TOF) m/z: calculated for C35H57NO7P [M+NH4]+ 634.3867, found 634.3861. IR: ν [cm−1]=2956, 2915, 2848, 1748, 1608, 1510, 1465, 1413, 1383, 1348, 1250, 1240, 1219, 1167, 1150, 1105, 1061, 1008, 996, 925, 897, 879, 833, 769, 720, 580, 538, 514, 451, 421, 403.
(AB‐C4H9,ab‐C17H35)‐H‐phosphonate 8 bg: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.39 g 4‐(hydroxymethyl)phenyl octadecanoate 7 g (1.0 mmol, 1.0 equiv.) was added and followed with 0.29 g 4‐(hydroxymethyl)phenyl pentanoate 7 b (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 8 : 2:0.005 v/v/v). Yield: 43 %, 0.28 g, as white solid. 1H NMR (600 MHz, CDCl3): δ [ppm]=7.39–7.33 (m, 4H, H‐2''), 7.11–7.05 (m, 4H, H‐3''), 6.93 (d, 1 J PH=708 Hz, 1H, P−H), 5.16–4.91 (m, 4H, Ph‐CH2), 2.56 (t, 3 J HH=7.5 Hz, 2H, H‐q), 2.55 (t, 3 J HH=7.5 Hz, 2H, H‐b), 1.74 (m, 4H, H‐c, H‐r), 1.49–1.20 (m, 30H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐o, H‐u, H‐v, H‐s), 0.97 (t, 3 J HH=7.3 Hz, 3H, H‐t), 0.88 (t, 3 J HH=7.0 Hz, 3H, H‐w). 13C‐NMR (151 MHz, CDCl3): δ [ppm]=172.08 (C‐p),172.06 (C‐a), 151.0 (2×C‐4''), 132.95, 132.91 (2×C‐1''), 129.2 (4×C‐2''), 121.90 (4×C‐3''), 66.67, 66.63 (2×Ph‐CH2), 34.3 (C‐b), 34.1 (C‐q), 31.9, 29.66, 29,64, 29.62, 29.61, 29.57, 29.43, 29.33, 29.22, 29.08 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐n, C‐o, C‐u), 26.9 (C‐r), 24.9 (C‐c), 22.7 (C‐v), 22.2 (C‐s), 14.1 (C‐w), 13.7 (C‐t). 31P‐NMR (243 MHz, CDCl3): δ [ppm]=7.71. HRMS (ESI‐TOF) m/z: calculated for C37H61NO7P [M+NH4]+ 62.4180, found 662.4200. IR: ν [cm−1]=2956, 2915, 2848, 1748, 1608, 1510, 1465, 1383, 1251, 1235, 1220, 1167, 1150, 1104, 1062, 1010, 997, 925, 878, 834, 769, 720, 510.
(AB‐C4H9,ab‐MEEES)‐H‐phosphonate 8 bh: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.28 g 4‐(hydroxymethyl)phenyl pentanoate 7 b (1.0 mmol, 1.0 equiv.) was added and followed with 0.52 g 4‐(hydroxymethyl)phenyl (2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl) succinate 7 h (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 2 : 8:0.005 v/v/v). Yield: 52 %, 0.33 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.40–7.32 (m, 4H, H‐2''), 7.09 (m, 4H, H‐3''), 6.93 (d, 1 J PH=708 Hz, 1H, P−H), 5.13–4.93 (m, 4H, Ph‐CH2), 4.27 (t, 3 J HH=4.8 Hz, 2H, H‐t), 3.74–3.68 (m, 2H, H‐u), 3.68–3.60 (m, 6H, H‐v, H‐w, H‐x), 3.57–3.51 (m, 2H, H‐y), 3.37 (s, 3H, H‐z), 2.88 (t, 3 J HH=6.9 Hz, 2H, H‐q), 2.77 (t, 3 J HH=6.9 Hz, 2H, H‐r), 2.56 (t, 3 J HH=7.6 Hz, 2H, H‐b), 1.73 (quint, 3 J HH=7.5 Hz, 2H, H‐c), 1.44 (tq, 3 J HH=7.6, 7.5 Hz, 2H, H‐d), 0.97 (t, 3 J HH=7.3 Hz, 3H, H‐e). 13C‐NMR (101 MHz, CDCl3): δ [ppm]=172.06, 171.99 (C‐p, C‐s), 170.67 (C‐a), 150.97, 150.81 (2×C‐4''), 133.13 (d, 3 J CP=6.1 Hz, C‐1''), 132.93 (d, 3 J CP=6.2 Hz, C‐1''), 129.20 (2×C‐2''), 121.86 (d, J CP=8.5 Hz, C‐3''), 71.90 (C‐z), 70.56, 70.54 (C‐v, C‐w, C‐x), 69.02 (C‐u), 66.70, 66.64, 66.58 (Ph‐CH2), 63.98 (C‐t), 58.99 (C‐z), 34.05 (C‐b), 29.25 (C‐q), 28.99 (C‐r), 26.92 (C‐c), 22.20 (C‐d), 13.67 (C‐e) 31P‐NMR (162 MHz, CDCl3): δ [ppm]=7.71. HRMS (ESI‐TOF) m/z: calculated for C30H45NO12P [M+NH4]+ 642.2674, found 642.2687. IR: ν [cm−1]=3435, 2874, 1756, 1733, 1607, 1508, 1456, 1417, 1350, 1248, 1198, 1165, 1131, 1101, 1029, 1016, 947, 888, 849, 811, 553, 504, 429.
(AB‐C14H29,ab‐MEEES)‐H‐phosphonate 8 eh: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.35 g 4‐(hydroxymethyl)phenyl pentadecanoate 7 e (1.0 mmol, 1.0 equiv.) was added and followed with 0.52 g 4‐(hydroxymethyl)phenyl (2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl) succinate 7 h (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/ CH3COOH 2 : 8:0.005 v/v/v). Yield: 52 %, 0.39 g, as white solid. 1H NMR (600 MHz, CDCl3): δ [ppm]=7.47–7.29 (m, 4H, H‐2''), 7.15–7.00 (m, 4H, H‐3''), 6.92 (d, 1 J PH=708 Hz, 1H, P−H), 5.11–4.93 (m, 4H, Ph‐CH2), 4.26 (t, 3 J HH=5.7 Hz, 2H, H‐t), 3.76–3.67 (m, 2H, H‐u), 3.68–3.57 (m, 6H, H‐v, H‐w, H‐x), 3.57–3.49 (m, 2H, H‐y), 3.36 (s, 3H, H‐z), 2.87 (t, 3 J HH=6.4 Hz, 2H, H‐q), 2.76 (t, 3 J HH=6.7 Hz, 2H, H‐r), 2.54 (t, 3 J HH=7.5 Hz, 2H, H‐b), 1.73 (quint, 3 J HH=7.5 Hz, 2H, H‐c), 1.43–1.36 (m, 2H, H‐d), 1.35–1.16 (m, 20H, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 0.87 (t, 3 J HH=7.0 Hz, 3H, H‐o). 13C‐NMR (151 MHz, CDCl3): δ [ppm]=172.07, 171.98 (C‐p, C‐s), 170.66 (C‐a), 150.95, 150.85 (2×C‐4''), 133.14 (d, 3 J CP=6.1 Hz, C‐1''), 132.96 (d, 3 J CP=6.2 Hz, C‐1''), 129.21 (2×C‐2''), 121.88 (d, J CP=8.5 Hz, C‐3''), 71.91 (C‐y), 70.55, 70.54 (C‐v, C‐w, C‐x), 69.03 (C‐u), 66.70, 66.66, 66.60 (Ph‐CH2), 63.98 (C‐t), 58.99 (C‐z), 34.40 (C‐b), 31.90, 29.64, 29.64, 29.62, 29.52, 29.44, 29.35, 29.27, 29.11 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐q), 28.99 (C‐r), 24.91 (C‐c), 22.68 (C‐n), 14.15 (C‐o). 31P‐NMR (243 MHz, CDCl3): δ [ppm]=7.69. HRMS (ESI‐TOF) m/z: calculated for C40H65NO12P [M+NH4]+ 782.4239, found 782.4233. IR: ν [cm−1]=2955, 2915, 2848, 1747, 1607, 1510, 1464, 1423, 1412, 1387, 1340, 1317, 1296, 1270, 1250, 1222, 1200, 1166, 1138, 1062, 1009, 995, 925, 835, 770, 727, 720, 657, 580, 554, 538, 516, 455, 441, 421.
(AB‐C14H29,ab‐MEEEG)‐H‐phosphonate 8 ei: According to General Procedure B, 0.23 mL diphenyl phosphonate (1.2 mmol, 1.2 equiv.) was added to 5 mL pyridine at 0 °C. Then 0.35 g 4‐(hydroxymethyl)phenyl pentadecanoate 7 e (1.0 mmol, 1.0 equiv.) was added and followed with 0.54 g 4‐(hydroxymethyl)phenyl (2‐(2‐(2‐methoxyethoxy)ethoxy)ethyl) glutarate 7 i (1.4 mmol, 1.4 equiv.). The mixture was stirred for 3 h at RT. Column chromatography (SiO2, petrol ether/ethyl acetate/CH3COOH 2 : 8:0.005 v/v/v). Yield: 46 %, 0.36 g, as white solid. 1H NMR (400 MHz, CDCl3): δ [ppm]=7.48–7.30 (m, 4H, H‐2''), 7.17–7.00 (m, 4H, H‐3''), 6.92 (d, 1 J PH=712 Hz, 1H, P−H), 5.15–4.90 (m, 4H, Ph‐CH2), 4.25 (t, 3 J HH=4.9 Hz, 2H, H‐u), 3.75–3.67 (m, 2H, H‐v), 3.67–3.60 (m, 6H, H‐w, H‐x, H‐y), 3.57–3.51 (m, 2H, H‐z), 3.36 (s, 3H, ‐OCH3), 2.64 (t, J=7.3 Hz, 2H, H‐q), 2.54 (t, 3 J HH=7.5 Hz, 2H, H‐b), 2.49 (t, J=7.2 Hz, 2H, H‐s), 2.06 (quint, 3 J HH=7.0 Hz, 2H, H‐r), 1.74 (quint, 3 J HH=7.4 Hz, 2H, H‐c), 1.46–1.19 (m, 22H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 0.87 (t, 3 J HH=6.5 Hz, 3H, H‐o). 13C‐NMR (151 MHz, CDCl3): δ [ppm]=172.7 (C‐t), 172.1 (C‐a), 171.2 (C‐p), 151.0, 150.8 (2×C‐4''), 133.07 (d, 4 J CP=6.0 Hz, C‐1''), 133.92 (d, 3 J CP=6.0 Hz, C‐1''), 129.2 (2×C‐2''), 121.92, 121.86 (2×C‐3''), 71.9 (C‐z), 70.60, 70.54 (C‐w, C‐x, C‐y), 69.1 (C‐v), 66.69, 66.66 (Ph‐CH2), 64.7 (Ph''‐CH2), 63.6 (C‐u), 59.0 (‐OCH3), 34.4 (C‐b), 33.3 (C‐q), 33.0 (C‐s), 31.89, 29.65, 29.64, 29.61, 29.56, 29.43, 29.32, 29.22, 29.07 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 24.9 (C‐c), 22.7 (C‐n), 20.0 (C‐r), 14.1 (C‐o). 31P‐NMR (162 MHz, CDCl3): δ [ppm]=7.71. HRMS (ESI‐TOF) m/z: calculated for C41H67NO12P [M+NH4]+ 796.4395, found 796.4416. IR: ν [cm−1]=2915, 2849, 1756, 1747, 1607, 1510, 1464, 1412, 1390, 1270, 1250, 1223, 1196, 1167, 1136, 1063, 1021, 1011, 997, 925, 877, 835, 770, 726, 582, 540, 516, 455.
γ‐(AB‐C2H5,ab‐C14H29)‐d4TTP (ammonium salt) 3 ae: According to General Procedure C, the reactions were performed under dry conditions using 100 mg H‐phosphonate 8 ae (0.174 mmol, 1.0 equiv.) and 137 mg d4TMP 2×nBu4N+ salt (0.174 mmol, 1.0 equiv.). Yield: 64 %, 111 mg, as white solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.69–7.62 (m, 1H, Hhet‐6), 7.40 (dt, 3 J HH=8.6, 2.5 Hz, 4H, H‐2''), 7.08–7.03 (m, 4H, H‐3''), 6.92 (m, 1H, H‐1'), 6.45 (dt, 3 J HH=5.9, 4 J HH=1.8 Hz, 1H, H‐3'), 5.80 (dt, 3 J HH=5.8 Hz, 4 J HH=1.8 Hz, 1H, H‐2'), 5.15 (d, 3 J HP=8.1 Hz, 4H, Ph‐CH2), 4.96–4.91 (m, 1H, H‐4'), 4.30–4.15 (m, 2H, H‐5'), 2.60 (q, 3 J HH=7.8 Hz, 2H, H‐q), 2.57 (t, 3 J HH=7.2 Hz, 2H, H‐b), 1.89 (s, 3H, Hhet‐7), 1.76–1.69 (m, 2H, H‐c), 1.47–1.25 (m, 22H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 1.23 (t, 3 J HH=7.6 Hz, 3H, H‐r), 0.90 (t, 3 J HH=6.9 Hz, 3H, H‐o). 13C‐NMR (151 MHz, MeOD): δ [ppm]=174.5 (C‐p), 173.8 (C‐a), 166.5.3 (Chet‐4), 152.8 (Chet‐2), 152.4 (2×C‐4''), 138.6 (Chet‐6), 135.7 (C‐3'), 134.9 (d, 3 J CP=7.6 Hz, 2×C‐1''), 130.5 (d, 4 J CP=2.9 Hz, 4×C‐2''), 127.2 (C‐2'), 122.89, 122.86, 122.82 (4×C‐3''), 112.1 (Chet‐5), 90.8 (C‐1'), 87.2 (d, 3 J CP=9.1 Hz, C‐4'), 70.39 (d, 3 J CP=5.7 Hz, Ph‐CH2), 67.9 (d, 2 J CP=5.8 Hz, C‐5'), 35.0 (C‐b), 33.0 (C‐q), 30.78, 30.77, 30.75, 30.71, 30.60, 30.46, 30.40, 30.16, 28.4 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 26.0 (C‐c), 23.7 (C‐n), 14.4 (C‐o), 12.5 (Chet‐7), 9.3 (C‐r). 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.81 (d, 2 J PP=19.6 Hz, P‐α), −13.23 (d, 2 J PP=17.2 Hz, P‐γ), −23.74 (br, s, P‐β). HRMS (ESI‐TOF) m/z: calculated for C42H58N2O17P3 [M−H]− 955.2954, found 955.2911. IR: ν [cm−1]=2922, 2852, 1757, 1689, 1509, 1460, 1422, 1248, 1218, 1168, 1128, 1079, 1008, 903, 837, 806, 784, 768, 721, 697, 645, 577, 488, 426, 401.
γ‐(AB‐C4H9,ab‐C14H29)‐d4TTP (ammonium salt) 3 be: According to General Procedure C, the reactions were performed under dry conditions using 276 mg H‐phosphonate 8 be (0.458 mmol, 1.0 equiv.) and 360 mg d4TMP 2×nBu4N+ salt (0.458 mmol, 1.0 equiv.). Yield: 28 %, 131 mg, as white solid. 1H‐NMR (400 MHz, MeOD): δ [ppm]=7.68 (d, 4 J HH=1.2 Hz, 1H, Hhet‐6), 7.55–7.25 (m, 4H, H‐2''), 7.12–6.99 (m, 4H, H‐3''), 6.92 (dt, 3 J HH=3.5 Hz, 4 J HH=1.6 Hz, 1H, H‐1'), 6.46 (dt, 3 J HH=6.0, 4 J HH=1.8 Hz, 1H, H‐3'), 5.83–5.76 (m, 1H, H‐2'), 5.16 (d, 3 J HP=8.0 Hz, 4H, Ph‐CH2), 4.99–4.93 (m, 1H, H‐4'), 4.36–4.16 (m, 2H, H‐5'), 2.58 (t, 3 J HH=7.2 Hz, 2H, H‐q), 2.57 (t, 3 J HH=7.2 Hz, 2H, H‐b), 1.90 (d, 4 J HH=1.2 Hz, 3H, Hhet‐7), 1.84–1.66 (m, 4H, H‐c, H‐r), 1.57–1.23 (m, 24H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐s), 0.99 (t, 3 J HH=7.4 Hz, 3H, H‐t), 0.90 (t, 3 J HH=6.8 Hz, 3H, H‐o). 13C‐NMR (101 MHz, MeOD): δ [ppm]=174.6 (C‐p, C‐a), 167.3 (Chet‐4), 153.6 (Chet‐2), 153.2 (2×C‐4''), 139.5 (Chet‐6), 136.6 (C‐3'), 135.8 (d, 3 J CP=7.5 Hz, 2×C‐1''), 131.4 (d, 4 J CP=2.9 Hz, 4×C‐2''), 128,0 (C‐2'), 123.73, 123.72 (4×C‐3''), 112.9 (Chet‐5), 91.7 (C‐1'), 88.1 (d, 3 J CP=9.1 Hz, C‐4'), 71.3 (d, J CP=6.1 Hz, Ph‐CH2), 71.2 (d, J CP=5.3 Hz, Ph‐CH2), 68.9 (d, 2 J CP=5.8 Hz, C‐5'), 35.9 (C‐b), 35.6 (C‐q), 33.9, 31.64, 31.63, 31.61, 31.57, 31.46, 31.32, 31.26, 31.03 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 28.9 (C‐r), 26.9 (C‐c), 24.6 (C‐n), 24.1 (C‐s), 15.3 (C‐o), 15.0 (C‐t), 12.5 (Chet‐7). 31P‐NMR (81 MHz, MeOD): δ [ppm]=‐11.81 (br, s, P‐α), −13.24 (d, J=17.2 Hz, P‐γ), −23.77 (br, s, P‐β). HRMS (ESI‐TOF) m/z: calculated for C44H62N2O17P3 [M−H]− 983.3267, found 983.3229. IR: ν [cm−1]=3183, 3042, 2923, 2853, 1756, 1689, 1509, 1462, 1380, 1248, 1218, 1202, 1167, 1128, 1112, 1082, 1008, 908, 837, 784, 723, 697, 644, 488, 421, 401.
γ‐(AB‐C6H13,ab‐C14H29)‐d4TTP (ammonium salt) 3 de: According to General Procedure C, the reactions were performed under dry conditions using 100 mg H‐phosphonate 8 de (0.159 mmol, 1.0 equiv.) and 125 mg d4TMP 2×nBu4N+ salt (0.159 mmol, 1.0 equiv.). Yield: 52 %, 87 mg, as white solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.70 −7.62 (m, 1H, Hhet‐6), 7.44–7.37 (m, 4H, H‐2''), 7.12–7.01 (m, 4H, H‐3''), 6.94 (dt, 3 J HH=3.5 Hz, 4 J HH=1.7 Hz, 1H, H‐1'), 6.47 (dt, 3 J HH=5.9 Hz, 4 J HH=1.8 Hz, 1H, H‐3'), 5.84–5.79 (m, 1H, H‐2'), 5.17 (d, 3 J HP=8.2 Hz, 4H, Ph‐CH2), 4.99–4.93 (m, 1H, H‐4'), 4.33–4.17 (m, 2H, H‐5'), 2.59 (t, 3 J HH=7.4 Hz, 4H, H‐q, H‐b), 1.91 (d, 4 J HH=1.3 Hz, 3H, Hhet‐7), 1.82–1.69 (m, 4H, H‐c, H‐r), 1.52–1.23 (m, 28H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐s, H‐t, H‐u), 0.95 (t, 3 J HH=7.2 Hz, 3H, H‐v), 0.92 (t, 3 J HH=7.0 Hz, 3H, H‐o). 13C‐NMR (151 MHz, MeOD): δ [ppm]=173.76 (C‐a, C‐p), 166.5 (Chet‐4), 152.8 (Chet‐2), 152.4 (2×C‐4''), 138.6 (Chet‐6), 135.7 (C‐3'), 134.9 (d, 3 J CP=7.6 Hz, 2×C‐1''), 130.49 (d, 4 J CP=4.5 Hz, 4×C‐2''), 127.2 (C‐2'), 122.88, 122.87 (4×C‐3''), 112.1 (Chet‐5), 90.9 (C‐1'), 87.12 (d, 3 J CP=9.1 Hz, C‐4'), 70.42 (d, J CP=3.8 Hz, Ph‐CH2), 70.39 (d, J CP=4.4 Hz, Ph‐CH2), 67.96 (d, 2 J CP=5.3 Hz, C‐5'), 35.0 (C‐b, C‐q), 33.07, 32.66, 30.79, 30.77, 30.75, 30.72, 30.60, 30.47, 30.41, 30.17, 29.8 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐s, C‐t), 25.96 (C‐r), 25.93 (C‐c), 23.73 (C‐n), 23.58 (C‐u), 14.44 (C‐o), 14.39 (C‐v), 12.5 (Chet‐7). 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.84 (d, 2 J PP=18.2 Hz, P‐α), −13.18 (d, 2 J PP=16.8 Hz, P‐γ), −23.82 (t, 2 J PP=16.3 Hz, P‐β). HRMS (ESI‐TOF) m/z: calculated for C46H66N2O17P3 [M−H]− 1011.3580, found .1011.3472. IR: ν [cm−1]=3184, 3045, 2955, 2922, 2852, 1754, 1690, 1509, 1464, 1380, 1249, 1220, 1168, 1128, 1113, 1084, 1007, 910, 838, 784, 768, 722, 696, 643, 577, 494, 421, 400.
γ‐(AB‐iso‐C4H9,ab‐C14H29)‐d4TTP (ammonium salt) 3 ce: According to General Procedure C, the reactions were performed under dry conditions using 90.8 mg H‐phosphonate 8 ce (0.151 mmol, 1.0 equiv.) and 106 mg d4TMP 2×nBu4N+ salt (0.151 mmol, 1.0 equiv.). Yield: 39 %, 60 mg, as white solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.68 (d, 4 J HH=1.2 Hz, 1H, Hhet‐6), 7.47–7.35 (m, 4H, H‐2''), 7.09–7.04 (m, 4H, H‐3''), 6.94 (ddd, 3 J HH=3.5 Hz, 4 J HH=1.6, 1.3 Hz, 1H, H‐1'), 6.48 (dt, 3 J HH=6.0 Hz, 4 J HH=1.7 Hz, 1H, H‐3'), 5.81 (ddd, 3 J HH=6.1 Hz, 3 J HH=2.4 Hz, 4 J HH=1.4 Hz, 1H, H‐2'), 5.17 (d, 3 J HP=8.1 Hz, 4H, Ph‐CH2), 4.98–4.93 (m, 1H, H‐4'), 4.29 (ddd, 2 J HH=11.6 Hz, 3 J HH=6.8 Hz, 4 J HH=3.3 Hz, 1H, H‐5'), 4.21 (ddd, 2 J HH=11.6 Hz, 3 J HH=5.4 Hz, 4 J HH=3.1 Hz, 1H, H‐5'), 2.59 (td, 3 J HH=7.4 Hz, 4 J HH=1.3 Hz 2H, H‐b), 2.47 (dd, 2 J HH=7.2 Hz, 4 J HH=1.3 Hz, 2H, H‐q), 2.28–2.17 (m, 1H, H‐r), 1.91 (d, 4 J HH=1.2 Hz, 3H, Hhet‐7), 1.80–1.69 (m, 2H, H‐c), 1.51–1.22 (m, 22H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 1.08 (dd, 3 J HH=6.7 Hz, 4 J HH=0.8 Hz, 6H, H‐t, H‐s), 0.92 (t, 3 J HH=7.0 Hz, 3H, H‐o). 13C‐NMR (151 MHz, MeOD): δ [ppm]=173.8 (C‐p), 173.0 (C‐a), 166.5 (Chet‐4), 152.8 (Chet‐2), 152.4, 152.3 (2×C‐4''), 138.7 (Chet‐6), 135.8 (C‐3'), 135.8 (2×C‐1''), 130.5 (d, 4 J CP=4.5 Hz, 4×C‐2''), 127.2 (C‐2'), 122.9 (4×C‐3''), 112.1 (Chet‐5), 90.9 (C‐1'), 87.2 (d, 3 J CP=9.6 Hz, C‐4'), 70.4 (2×Ph‐CH2), 67.88 (d, 2 J CP=5.6 Hz, C‐5'), 44.0 (C‐q), 35.0 (C‐b), 33.1, 30.79, 30.78, 30.76, 30.72, 30.61, 30.48, 30.41, 30.17 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 27.0 (C‐r), 26.0 (C‐c), 23.7 (C‐n), 22.7 (C‐s, C‐t), 14.4 (C‐o), 12.5 (Chet‐7) 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.80 (d, J=19.6 Hz, P‐α), −13.21 (d, J=17.3 Hz, P‐γ), −23.72 (t, J=19.3 Hz, P‐β). HRMS (ESI‐TOF) m/z: calculated for C44H62N2O17P3 [M−H]− 983.3267, found 983.3160. IR: ν [cm−1]=3046, 2957, 2923, 2853, 1756, 1690, 1509, 1464, 1369, 1247, 1218, 1202, 1167, 1128, 1112, 1083, 1009, 909, 837, 784, 769, 722, 696, 644, 573, 490, 425.
γ‐(AB‐C4H9,ab‐C15H31)‐d4TTP (ammonium salt) 3 bf: According to General Procedure C, the reactions were performed under dry conditions using 100 mg H‐phosphonate 8 bf (0.162 mmol, 1.0 equiv.) and 128 mg d4TMP 2×nBu4N+ salt (0.162 mmol, 1.0 equiv.). Yield: 47 %, 78 mg, as white solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.68 (d, 4 J HH=1.2 Hz„ 1H, Hhet‐6), 7.44–7.38 (m, 4H, H‐2''), 7.14–7.01 (m, 4H, H‐3''), 6.94 (dt, 3 J HH=3.5, 4 J HH=1.6 Hz, 1H, H‐1'), 6.48 (dt, 3 J HH=6.1 Hz, 4 J HH=1.7 Hz, 1H, H‐3'), 5.81 (ddd, 3 J HH=6.1, 3 J HH=2.4 Hz, 4 J HH=1.3 Hz, 1H, H‐2'), 5.17 (d, 3 J HP=8.4 Hz, 4H, Ph‐CH2), 4.99–4.92 (m, 1H, H‐4'), 4.32–4.16 (m, 2H, H‐5'), 2.63–2.56 (m, 4H, H‐b, H‐q), 1.91 (d, 4 J HH=1.2 Hz, 3H, Hhet‐7), 1.79–1.69 (m, 4H, H‐c, H‐r), 1.52–1.23 (m, 26H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐o, H‐s), 1.01 (t, 3 J HH=7.4 Hz, 3H, H‐t), 0.92 (t, 3 J HH=7.0 Hz, 3H, H‐u). 13C‐NMR (151 MHz, MeOD): δ [ppm]=173.78 (C‐p, C‐a), 166.52 (Chet‐4), 152.76 (Chet‐2), 152.35 (2×C‐4''), 138.66 (Chet‐6), 135.76 (C‐3'), 134.93 (d, 3 J CP=7.4 Hz, 2×C‐1''), 130.48 (d, 4 J CP=2.9 Hz, 4×C‐2''), 127.16 (C‐2'), 122.87 (d, 3 J CP=2.1 Hz, 4×C‐3''), 112.06 (Chet‐5), 90.84 (C‐1'), 87.20 (d, 3 J CP=9.1 Hz, C‐4'), 70.41, 70.38, 70.36 (2×Ph‐CH2), 67.88 (d, 2 J CP=5.6 Hz, C‐5'), 35.03, 34.76 (C‐q, C‐b), 33.07, 30.78, 30.77, 30.75, 30.71, 30.60, 30.46, 30.40, 30.17(C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐n), 28.07 (C‐r), 25.96 (C‐c), 23.73 (C‐o), 23.25 (C‐s), 14.44 (C‐u), 14.10 (C‐t), 12.48 (Chet‐7). 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.73 (br, s, P‐α), −13.18 (d, J=17.2 Hz, P‐γ), −23.68 (br, s, P‐β). HRMS (ESI‐TOF) m/z: calculated for C45H64N2O17P3 [M−H]− 997.3423, found 997.3389. IR: ν [cm−1]=3191, 3045, 2956, 2921, 2852, 1755, 1689, 1509, 1464, 1380, 1249, 1219, 1168, 1128, 1082, 1008, 908, 838, 784, 769, 721, 696, 644, 492, 422, 401.
γ‐(AB‐C4H9,ab‐C17H35)‐d4TTP (ammonium salt) 3 bg: According to General Procedure C, the reactions were performed under dry conditions using 97 mg H‐phosphonate 8 bg (0.150 mmol, 1.0 equiv.) and 118 mg d4TMP 2×nBu4N+ salt (0.150 mmol, 1.0 equiv.). Yield: 40 %, 64 mg, as white solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.71–7.66 (m, 1H, Hhet‐6), 7.44–7.38 (m, 4H, H‐2''), 7.09–7.04 (m, 4H, H‐3''), 6.94 (dt, 3 J HH=3.5 Hz, 4 J HH=1.6 Hz, 1H, H‐1'), 6.48 (dt, 3 J HH=6.0 Hz, 4 J HH=1.8 Hz, 1H, H‐3'), 5.81 (ddd, 3 J HH=6.1 Hz, 3 J HH=2.4 Hz, 4 J HH=1.3 Hz, 1H, H‐2'), 5.17 (d, 3 J HP=8.2 Hz, 4H, Ph‐CH2), 4.98–4.93 (m, 1H, H‐4'), 4.33–4.15 (m, 2H, H‐5'), 2.63–2.56 (m, 4H, H‐b, H‐q), 1.91 (d, 4 J HH=1.2 Hz, 3H, Hhet‐7), 1.79–1.69 (m, 4H, H‐c, H‐r), 1.52–1.23 (m, 30H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n, H‐o, H‐u, H‐v, H‐s), 1.01 (t, 3 J HH=7.4 Hz, 3H, H‐t), 0.92 (t, 3 J HH=7.0 Hz, 3H, H‐w). 13C‐NMR (151 MHz, MeOD): δ [ppm]=173.77 (C‐p, C‐a), 166.53 (Chet‐4), 152.76 (Chet‐2), 152.35 (2×C‐4''), 138.67 (Chet‐6), 135.76 (C‐3'), 134.93 (d, 3 J CP=7.5 Hz, 2×C‐1''), 130.48 (d, 4 J CP=2.9 Hz, 4×C‐2''), 127.16 (C‐2'), 122.88 (4×C‐3''), 112.05 (Chet‐5), 90.83 (C‐1'), 87.20 (d, 3 J CP=9.1 Hz, C‐4'), 70.38 (2×Ph‐CH2), 67.87 (d, 2 J CP=5.6 Hz, C‐5'), 35.03, 34.76 (C‐q, C‐b), 33.07, 30.79, 30.77, 30.75, 30.72, 30.61, 30.47, 30.42, 30.18 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐n, C‐o, C‐u), 28.07 (C‐r), 25.96 (C‐c), 23.74 (C‐v), 23.26 (C‐s), 14.45 (C‐w), 14.11 (C‐t), 12.49 (Chet‐7). 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.77 (d, J=19.6 Hz, P‐α), −13.20 (d, J=17.3 Hz, P‐γ), −23.70 (t, J=18.1 Hz, P‐β). HRMS (ESI‐TOF) m/z: calculated for C47H68N2O17P3 [M−H]− 1025.3736, found 1025.3703. IR: ν [cm−1]=3040, 2920, 2851, 1755, 1690, 1509, 1465, 1380, 1249, 1219, 1168, 1128, 1082, 1007, 910, 838, 784, 768, 721, 644, 491, 420.
γ‐(AB‐C4H9,ab‐MEEES)‐d4TTP (ammonium salt) 3 bh: According to General Procedure C, the reactions were performed under dry conditions using 144 mg H‐phosphonate 8 bh (0.231 mmol, 1.0 equiv.) and 187 mg d4TMP 2×nBu4N+ salt (0.231 mmol, 1.0 equiv.). Yield: 45 %, 101 mg, as colorless solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.69 (d, 4 J HH=1.3 Hz, 1H, Hhet‐6), 7.46–7.39 (m, 4H, H‐2''), 7.12–7.04 (m, 4H, H‐3''), 6.94 (dt, 3 J HH=3.5 Hz, 4 J HH=1.7 Hz, 1H, H‐1'), 6.48 (dt, 3 J HH=6.0 Hz, 4 J HH=1.8 Hz, 1H, H‐3'), 5.83–5.76 (m, 1H, H‐2'), 5.17 (dd, 3 J HP=8.1, 4.8 Hz, 4H, Ph‐CH2), 4.98–4.93 (m, 1H, H‐4'), 4.32–4.17 (m, 4H, H‐5', H‐t), 3.74–3.51 (m, 10H, H‐u, H‐v, H‐w, H‐x, H‐y), 3.26 (s, 3H, H‐z), 2.93–2.87 (m, 2H, H‐q), 2.78 (t, 3 J HH=6.6 Hz, 2H, H‐r) 2.61 (td, 3 J HH=7.4, 1.3 Hz, 2H, H‐b), 1.91 (d, 4 J HH=1.2 Hz, 3H, Hhet‐7), 1.78–1.70 (m, 2H, H‐c), 1.52–1.44 (m, 2H, H‐d), 1.01 (t, 3 J HH=7.4 Hz, 3H, H‐e). 13C‐NMR (151 MHz, MeOD): δ [ppm]=173.9,173.8 (C‐p, C‐s), 172.6 (C‐a), 166.53 (Chet‐4), 152.8 (Chet‐2), 152.4, 152.3 (2×C‐4''), 138.7 (Chet‐6), 135.8 (C‐3'), 135.8 (2×C‐1''), 130.50,130.46 (4×C‐2''), 127.1 (C‐2'), 122.88, 122.86, 122.84, 122.83 (4×C‐3''), 112.1 (Chet‐5), 90.8 (C‐1'), 87.2 (d, 3 J CP=9.5 Hz, C‐4'), 72.9, 71,54, 71.50, 71,3, 70.07 (C‐u, C‐v, C‐w, C‐x, C‐y), 70.38, 70.35, 70.35, 70.32 (2×Ph‐CH2), 67.85 (d, 2 J CP=6.0 Hz, C‐5'), 65.02 (C‐t), 59.08 (C‐z), 34.8 (C‐b), 30.1, 29.9 (C‐q, C‐r), 28.1 (H‐c), 23.3 (C‐d), 14.1 (C‐e), 12.5 (Chet‐7) 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.77 (d, J=19.8 Hz, P‐α), −13.24 (d, J=17.0 Hz, P‐γ), −23.71 (t, J=19.0 Hz, P‐β). HRMS (ESI‐TOF) m/z: calculated for C40H52N2O22P3 [M−H]− 1005.2230, found 1005.2268. IR: ν [cm−1]=3187, 2932, 2874, 1755, 1736, 1688, 1509, 1453, 1422, 1247, 1218, 1200, 1167, 1127, 1082, 1007, 903, 837, 806, 783, 731, 696, 644, 480, 422, 401.
γ‐(ab‐C14H29,ab‐MEEES)‐d4TTP (ammonium salt) 3 eh: According to General Procedure C, the reactions were performed under dry conditions using 100 mg H‐phosphonate 8 eh (0.162 mmol, 1.0 equiv.) and 127 mg d4TMP 2×nBu4N+ salt (0.162 mmol, 1.0 equiv.). Yield: 39 %, 75 mg, as colorless solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.69 (m, 1H, Hhet‐6), 7.46–7.38 (m, 4H, H‐2''), 7.13–7.03 (m, 4H, H‐3''), 6.94 (dt, 3 J HH=3.5 Hz, 4 J HH=1.6 Hz, 1H, H‐1'), 6.49 (dt, 3 J HH=5.6 Hz, 4 J HH=1.7 Hz, 1H, H‐3'), 5.81 (ddd, 3 J HH=5.6 Hz, 3 J HH=1.9 Hz, 4 J HH=1.8 Hz, 1H, H‐2'), 5.17 (d, 3 J HP=7.3 Hz, 4H, Ph‐CH2), 4.98–4.93 (m, 1H, H‐4'), 4.33–4.17 (m, 4H, H‐5', H‐t), 3.75–3.69 (m, 2H, H‐u), 3.67–3.59 (m, 6H, H‐v, H‐w, H‐x), 3.56–3.51 (m, 2H, H‐y), 3.36 (m, 3H, H‐z), 2.91 (t, 3 J HH=6.6 Hz, 2H, H‐q), 2.78 (t, 3 J HH=6.3 Hz, 2H, H‐r), 2.60 (t, 3 J HH=7.5 Hz, 2H, H‐b), 1.91 (d, 4 J HH=2.9 Hz, 3H, Hhet‐7), 1.75 (quint, 3 J HH=7.5 Hz, 2H, H‐c), 1.49–1.24 (m, 22H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 0.92 (t, 3 J HH=5.7 Hz, 3H, H‐o). 13C‐NMR (151 MHz, MeOD): δ [ppm]=173.88, 173.79, 172.59 (C‐p, C‐s, C‐a), 166.53 (Chet‐4), 152.77 (Chet‐2), 152.36, 152.29 (C‐4''), 138.70 (Chet‐6), 135.83 (C‐3'), 135.10 (d, 4 J CP=7.4 Hz, C‐1''), 134.97 (d, 3 J CP=7.7 Hz, C‐1''), 130.50, 130.47 (4×C‐2''), 127.12 (C‐2'), 122.88, 122.86, 122.84, 122.82 (4×C‐3''), 112.08 (Chet‐5), 90.83 (C‐1'), 87.25 (d, 3 J CP=8.9 Hz, C‐4'), 72.95 (C‐y), 71.55, 71.51, 71.38 (C‐w, C‐x, C‐y), 70.39, 70.36, 70.33 (2×Ph‐CH2), 70.08 (C‐u), 67.84 (d, 2 J CP=5.5 Hz, C‐5'), 65.03 (C‐t), 59.09 (C‐z), 35.03 (C‐b), 33.07, 30.79, 30.78, 30.76, 30.72, 30.61, 30.47, 30.42, 30.18, 30.12, 29.92 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m, C‐q, C‐r), 25.97 (C‐c), 23.74 (C‐n), 14.44 (C‐o), 12.49 (Chet‐7). 31P‐NMR (243 MHz, MeOD): δ [ppm]=‐11.71 (d, J=19.6 Hz, P‐α), −13.16 (d, J=17.1 Hz, P‐γ), −23.60 (t, J=17.1 Hz, P‐β). HRMS (ESI‐TOF) m/z: calculated for C50H72N2O22P3 [M−H]− 1145.3795, found 1145.3786. IR: ν [cm−1]=3184, 2923, 2853,1756, 1738, 1689, 1509, 1456, 1367, 1248, 1219, 1200, 1167, 1128, 1084, 1009, 906, 838, 807, 784, 722, 645, 486, 423, 399.
γ‐(AB‐C14H29,ab‐MEEEG)‐d4TTP (ammonium salt) 3 ei: According to General Procedure C, the reactions were performed under dry conditions using 117 mg H‐phosphonate 8 ei (0.150 mmol, 1.0 equiv.) and 118 mg d4TMP 2×nBu4N+ salt (0.150 mmol, 1.0 equiv.). Yield: 35 %, 62 mg, as colorless solid. 1H‐NMR (600 MHz, MeOD): δ [ppm]=7.69 (d, 4 J HH=1.4 Hz, 1H, Hhet‐6), 7.47–7.38 (m, 4H, H‐2''), 7.13–7.03 (m, 4H, H‐3''), 6.94 (dt, 3 J HH=3.5 Hz, 4 J HH=1.6 Hz, 1H, H‐1'), 6.48 (dt, 3 J HH=6.0 Hz, 4 J HH=1.7 Hz, 1H, H‐3'), 5.81 (ddd, 3 J HH=5.9 Hz, 3 J HH=2.4 Hz, 4 J HH=1.9 Hz, 1H, H‐2'), 5.17 (d, 3 J HP=8.1 Hz, 4H, Ph‐CH2), 4.98–4.93 (m, 1H, H‐4'), 4.32–4.17 (m, 4H, H‐5', H‐u), 3.74–3.70 (m, 2H, H‐v), 3.67–3.61 (m, 6H, H‐w, H‐x, H‐y), 3.56–3.52 (m, 2H, H‐z), 3.36 (s, 3H, ‐OCH3), 2.69 (td, 3 J HH=7.4 Hz, 4 J HH=1.3 Hz, 2H, H‐q), 2.60 (td, 3 J HH=7.4 Hz, 4 J HH=1.3 Hz, 2H, H‐b), 2.52 (t, 3 J HH=7.2 Hz, 2H, H‐s), 2.04 (quint, 3 J HH=7.2 Hz, 2H, H‐r), 1.91 (d, 4 J HH=1.3 Hz, 3H, Hhet‐7), 1.75 (quint, 3 J HH=7.4 Hz, 2H, H‐c), 1.49–1.25 (m, 22H, H‐d, H‐e, H‐f, H‐g, H‐h, H‐i, H‐j, H‐k, H‐l, H‐m, H‐n), 0.92 (t, 3 J HH=7.0 Hz, 3H, H‐o). 13C‐NMR (151 MHz, MeOD): δ [ppm]=174.6 (C‐t), 173.8 (C‐a), 173.1 (C‐p), 166.5 (Chet‐4), 152.8 (Chet‐2), 152.36, 152.27 (C‐4''), 138.7 (Chet‐6), 135.80 (C‐3'), 135.03 (d, 4 J CP=7.6 Hz, C‐1''), 134.96 (d, 3 J CP=7.7 Hz, C‐1''), 130.50, 130.47 (4×C‐2''), 127.2 (C‐2'), 122.89, 122.87 (4×C‐3''), 112.1 (Chet‐5), 90.8 (C‐1'), 87.23 (d, 3 J CP=9.0 Hz, C‐4'), 73.0 (C‐z), 71.53, 71.39 (C‐w, C‐x, C‐y), 70.39, 70.36, 70.33 (2×Ph‐CH2), 70.12 (C‐v), 67.86 (d, 2 J CP=6.0 Hz, C‐5'), 64.7 (C‐u), 59.1 (‐OCH3), 35.03 (C‐b), 33.98, 33.91 (C‐q, C‐s), 33.08, 30.80, 30.79, 30.76, 30.73, 30.62, 30.48, 30.42, 30.18 (C‐d, C‐e, C‐f, C‐g, C‐h, C‐i, C‐j, C‐k, C‐l, C‐m), 25.97 (C‐c), 23.74 (C‐n), 21.19 (C‐r), 14.45 (C‐o), 12.5 (Chet‐7). 31P‐NMR (243 MHz, MeOD): δ [ppm] −11.80 (d, J=19.9 Hz, P‐α), −13.23 (d, J=17.2 Hz, P‐γ), −23.75 (t, J=18.6 Hz, P‐β).HRMS (ESI‐TOF) m/z: calculated for C51H74N2O22P3 [M−H]− 1159.3952, found 1159.3885.IR: ν [cm−1]=3184, 2922, 2853, 1755, 1736, 1689, 1509, 1455, 1247, 1219, 1200, 1167, 1127, 1083, 1009, 905, 838, 784, 722, 643, 489, 420, 399.
Preparation of phosphate buffer (PB, pH 7.3)
5.47 g disodium hydrogen and phosphate 1.55 g Potassium dihydrogen phosphate were dissolved in 1 L ultrapure water. Then titrated with diluted phosphoric acid to pH 7.3. All prodrugs were incubated in this buffer to study their chemical stability.
Hydrolysis studies
Chemical hydrolysis in PB
Stock solutions (50 mM in DMSO) of TriPPPro‐NTPs were prepared. After dilution of 11 μL stock solution with 189 μL ultrapure water and 100 μL DMSO to 1.83 mM hydrolysis solutions the reaction was started by addition of 300 μL phosphate buffer (PB, 50 mM, pH 7.3). The solution was incubated with 800 rpm and at 37 °C in a thermomixer. An initial aliquot (25 μL) was taken directly and analyzed by analytical HPLC with UV detector. For compound containing d4T, λ=265 nm. Further aliquots were taken for monitoring the kinetic hydrolysis.
Enzyme‐catalyzed hydrolysis in CEM cell extracts
10 μL 50 mM DMSO stock solution of TriPPPro‐d4TTPs was diluted to 6.0 mM hydrolysis solution by addition of 73.3 μL DMSO. 7 different samples including 10 μL water and 10 μL hydrolysis solution were prepared. The reaction was started by addition of 50 μL human CEM cell extract and the mixture incubated with 800 rpm at 37 °C for different time periods of hydrolysis. The reactions were stopped by addition of 150 μL MeOH. The solution was kept on ice for 5 min followed by centrifugation for 5 min (13000 rpm). The supernatants were filtered (Chromafil® RC‐20/15 MS, 0.2 μm) and stored in liquid nitrogen. When testing, the samples were defrosted and injection volume with 80 μL was used for HPLC analysis. The calculation of t1/2 was performed analogously to that for the chemical hydrolysis studies.
Enzyme‐catalyzed hydrolysis in pig liver esterase (PLE)
10 μL 50 mM DMSO stock solution of TriPPPro‐d4TTPs was diluted to 6.0 mM hydrolysis solution by addition of 31.7 mL DMSO and 41.7 mL ultrapure water. Then 83.3 mL of the 6.0 mM solution was diluted with 125 μL DMSO and 833 μl 50 mM PB (pH 7.3). The reaction was started by addition of 62.5 mL of PLE in PB (3 mg/mL) and the mixture was incubated with 800 rpm at 37 °C in a thermomixer. At different times, aliquots (100 mL) were taken and the reaction was stopped by addition to 106 mL MeOH. The mixture was kept for 5 min on ice followed by centrifugation for 5 min (13000 rpm). The mixture was filtered (Chromafil RC‐20/15 MS, 0.2 mm) and stored in liquid nitrogen. When testing, the samples were defrosted and injection volume with 80 μL was used for HPLC analysis.
Preparation of the cell extracts
Human CD4 + T‐lymphocyte CEM cells were grown in RPMI‐1640‐based cell culture medium to a final density of ∼3×106 cells/mL. Then, cells were centrifuged for 10 min at 1,250 rpm at 4 C, washed twice with cold PB, and the pellet was resuspended at 108 cells/mL and sonicated (Hielscher Ultrasound Techn., 100 % amplitude, 3 ⋅ times for 10 sec) to destroy cell integrity. The resulting cell suspension was then centrifuged at 10000 rpm to remove cell debris, and the supernatant divided in aliquots before being frozen at −80 °C and used.
Antiviral assay against HIV
Inhibition of HIV‐1(IIIB)‐ and HIV‐2(ROD)‐induced cytopathicity in wild‐type CEM/0 and thymidine kinase‐deficient CEM/TK− cell cultures was measured in microtiter 96‐well plates containing ∼3×105 CEM cells/mL infected with 100 CCID50 of HIV per milliliter and containing appropriate dilutions of the test compounds. After 4–5 days of incubation at 37 °C in a CO2‐controlled humidified atmosphere, CEM giant (syncytium) cell formation was examined microscopically. The EC50 (50 % effective concentration) was defined as the compound concentration required to inhibit HIV‐induced giant cell formation by 50 %.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
C.M. is grateful to the Deutsche Forschungsgemeinschaft for financial support (ME 1161/15‐1). C.Z. and X.J. are grateful for CSC fellowships from the Chinese Ministry of Education. Open access funding enabled and organized by Projekt DEAL.
C. Zhao, X. Jia, D. Schols, J. Balzarini, C. Meier, ChemMedChem 2021, 16, 499.
References
- 1. Jordheim L. P., Durantel D., Zoulim F., Dumontet C., Nat. Rev. Drug Discovery 2013, 12, 447–464. [DOI] [PubMed] [Google Scholar]
- 2. Deval J., Drugs 2009, 69, 151–166. [DOI] [PubMed] [Google Scholar]
- 3. Cihlar T., Ray A. S., Antiviral Res. 2010, 85, 39–58. [DOI] [PubMed] [Google Scholar]
- 4. El Safadi Y., Vivet-Boudou V., Marquet R., Appl. Microbiol. Biotechnol. 2007, 75, 723–737. [DOI] [PubMed] [Google Scholar]
- 5. Burton J. R., Everson G. T., Clin. Liver Dis. 2009, 13, 453–465. [DOI] [PubMed] [Google Scholar]
- 6. De Clercq E., J. Clin. Virol. 2004, 30, 115–133. [DOI] [PubMed] [Google Scholar]
- 7. Balzarini J., Herdewijn P., De Clercq E., J. Biol. Chem. 1989, 264, 6127–6133. [PubMed] [Google Scholar]
- 8. Van Rompay A. R., Johansson M., Karlsson A., Pharmacol. Ther. 2000, 87, 189–198. [DOI] [PubMed] [Google Scholar]
- 9. Asahchop E. L., Wainberg M. A., Sloan R. D., Tremblay C. L., Antimicrob. Agents Chemother. 2012, 56, 5000–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meier C., Synlett 1998, 233–242. [Google Scholar]
- 11. Wagner C. R., Iyer V. V., McIntee E. J., Med. Res. Rev. 2000, 20, 417–451. [DOI] [PubMed] [Google Scholar]
- 12. Hecker S. J., Erion M. D., J. Med. Chem. 2008, 51, 2328–2345. [DOI] [PubMed] [Google Scholar]
- 13. Pradere U., Garnier-Amblard E. C., Coats S. J., Amblard F., Schinazi R. F., Chem. Rev. 2014, 114, 9154–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cahard D., McGuigan C., Balzarini J., Mini-Rev. Med. Chem. 2004, 4, 371–381. [DOI] [PubMed] [Google Scholar]
- 15. Li F. J., Maag H., Alfredson T., J. Pharm. Sci. 2008, 97, 1109–1134. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y., Gao Y., Wen X., Ma H., Asian J. Pharm. Sci. 2014, 9, 65–74. [Google Scholar]
- 17. Meier C., Eur. J. Org. Chem. 2006, 5, 1081–1102. [Google Scholar]
- 18. Krylov I. S., Kashemirov B. A., Hilfinger J. M., McKenna C. E., Mol. Pharm. 2013, 10, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meier C., Balzarini J., Antiviral Res. 2006, 71, 282–292. [DOI] [PubMed] [Google Scholar]
- 20. Jessen H. J., Schulz T., Balzarini J., Meier C., Angew. Chem. Int. Ed. 2008, 47, 8719–8722; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8847–8850. [Google Scholar]
- 21. Schulz T., Balzarini J., Meier C., ChemMedChem 2014, 9, 762–775. [DOI] [PubMed] [Google Scholar]
- 22. Weinschenk L., Gollnest T., Schols D., Balzarini J., Meier C., ChemMedChem 2015, 10, 891–900. [DOI] [PubMed] [Google Scholar]
- 23. Weinschenk L., Schols D., Balzarini J., Meier C., J. Med. Chem. 2015, 58, 6114–6130. [DOI] [PubMed] [Google Scholar]
- 24. Tan X. L., Chu C. K., Boudinot F. D., Adv. Drug Delivery Rev. 1999, 39, 117–151. [DOI] [PubMed] [Google Scholar]
- 25. Camarasa M. J., ChemMedChem 2018, 13, 1885–1889. [DOI] [PubMed] [Google Scholar]
- 26. Gollnest T., de Oliveira T. D., Schols D., Balzarini J., Meier C., Nat. Commun. 2015, 6, 8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gollnest T., de Oliveira T. D., Rath A., Hauber I., Schols D., Balzarini J., Meier C., Angew. Chem. Int. Ed. 2016, 55, 5255–5258; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5341–5344. [Google Scholar]
- 28. Meier C., Antiviral Chem. Chemother. 2017, 25, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jia X., Schols D., Meier C., J. Med. Chem. 2020, 63, 6003–6027. [DOI] [PubMed] [Google Scholar]
- 30. Sowa T., Quchi S., Bull. Chem. Soc. Jpn. 1975, 48, 2084–2090. [Google Scholar]
- 31.T. Gollnest, Ph.D. thesis, University of Hamburg, 2015.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary