

Abstract
Coproporphyrin I (CPI) is an endogenous biomarker of OATP1B activity and associated drug‐drug interactions. In this study, a minimal physiologically‐based pharmacokinetic model was developed to investigate the impact of OATP1B1 genotype (c.521T>C), ethnicity, and sex on CPI pharmacokinetics and interindividual variability in its baseline. The model implemented mechanistic descriptions of CPI hepatic transport between liver blood and liver tissue and renal excretion. Key model parameters (e.g., endogenous CPI synthesis rate, and CPI hepatic uptake clearance) were estimated by fitting the model simultaneously to three independent CPI clinical datasets (plasma and urine data) obtained from white (n = 16, men and women) and Asian‐Indian (n = 26, all men) subjects, with c.521 variants (TT, TC, and CC). The optimized CPI model successfully described the observed data using c.521T>C genotype, ethnicity, and sex as covariates. CPI hepatic active was 79% lower in 521CC relative to the wild type and 42% lower in Asian‐Indians relative to white subjects, whereas CPI synthesis was 23% higher in male relative to female subjects. Parameter sensitivity analysis showed marginal impact of the assumption of CPI synthesis site (blood or liver), resulting in comparable recovery of plasma and urine CPI data. Lower magnitude of CPI‐drug interaction was simulated in 521CC subjects, suggesting the risk of underestimation of CPI‐drug interaction without prior OATP1B1 genotyping. The CPI model incorporates key covariates contributing to interindividual variability in its baseline and highlights the utility of the CPI modeling to facilitate the design of prospective clinical studies to maximize the sensitivity of this biomarker.
STUDY HIGHLIGHTS.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Reduced activity of OATP1B1 in homozygous carriers of c.521T>C resulted in higher plasma concentration of coproporphyrin I (CPI), an endogenous biomarker of OATP1B. Women showed lower CPI plasma concentrations than men.
WHAT QUESTION DID THIS STUDY ADDRESS?
To what extent do OATP1B1 c.521T>C, ethnicity, and sex affect CPI steady‐state baseline and the extent of CPI‐drug interaction? How does the assumption of CPI synthesis site affect model performance?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Modeling of CPI identified three covariates; decreased hepatic CPI uptake in 521CC relative to 521TT, Asian‐Indians relative to white subjects, and lower CPI synthesis in women relative to men. These results were mostly unaffected by the assumption on CPI synthesis site in the model. Theoretical simulation evaluated the impact of each covariate on CPI‐drug interaction risk.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Modeling of CPI maximizes the sensitivity of this biomarker to evaluate OATP1B1 interaction potential as early as in first‐in‐human studies and to facilitate the design of prospective interaction studies with corresponding clinical probes.
Hepatic uptake via OATP1B1 and 1B3 has been widely recognized as a rate‐limiting step in the clearance of anionic drugs, such as statins. 1 , 2 Considering the high degree of drug‐drug interactions (DDIs) associated with OATP1B1 and therapeutic implications, regulatory agencies suggest elucidating factors that can change pharmacokinetics (PKs) of OATP1B1 substrates (https://www.fda.gov/media/134581/download, https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf). Genetic polymorphism of SLCO1B1 coding OATP1B1, is one of the factors affecting exposure of OATP1B1 substrates. 3 , 4 Subjects with c.521T>C mutation (521CC), associated with reduced transporter activity, have higher exposure of OATP1B substrates relative to wild‐type allele (521TT). 5 In addition, 521CC subjects showed lower fold‐change in the area under the plasma concentration‐time curve ratio (AUCR) of statins in the presence of OATP1B inhibitors, 3 , 6 likely due to reduced contribution of OATP1B1 to the elimination in these individuals. In addition, several studies reported increased exposure of statins in Asians (mainly Japanese) relative to whites, 4 , 7 , 8 , 9 resulting in the reduced starting dose of OATP1B substrates (e.g., rosuvastatin) in this population. 10 Increased exposure of statins in Japanese was attributed to the intrinsic ethnic variability in OATP1B activity, in addition to genetic polymoprhism, 4 , 11 although some studies have challenged this hypothesis. 12 , 13 Recently, application of this assumption in letermovir physiologically‐based PK (PBPK) modeling explained ethnic difference in the PKs of this drug. 14
Recent years have seen an increased application of endogenous biomarkers as novel tools to investigate transporter function in vivo, with the primary aim to de‐risk transporter‐mediated DDIs in early drug development. 1 , 15 , 16 , 17 This trend has been particularly evident for OATP1B. Coproporphyrin I (CPI), a byproduct of heme synthesis, is one of the most promising endogenous biomarkers of OATP1B. It is a metabolically stable substrate of OATP1B1 and 1B3, as well as MRP2 and MRP3 transporters. 18 , 19 , 20 , 21 , 22 , 23 CPI‐drug interactions reported so far in human 22 , 24 , 25 , 26 , 27 , 28 and preclinical species 19 , 29 , 30 suggest that this biomarker is expected to closely follow the perpetrator concentration–time profile. In contrast to other OATP1B biomarkers, circadian rhythm or food intake are not likely to cause interindividual variability in its plasma baseline. 19 , 24
Several endogenous factors can affect the disposition of CPI. Hepatic uptake via OATP1B is the main elimination route of CPI (> 85%) 18 and increased CPI baseline was recently reported in OATP1B1 521CC, 25 , 31 whereas other genetic mutations of OATP1B (e.g., c.388A>G) showed no effect. 28 , 31 Higher proportion of CPI in urine in patients with MRP2 mutations (Dubin–Johnson syndrome) 32 were reported, with no strong evidence of altered plasma CPI concentrations. Ethnic differences in baseline CPI were not highlighted in previous studies including white and Asian subjects. 15 , 28 However, these studies did not consider OATP1B1 genotype in conjunction with ethnicity as a covariate. Changes in CPI synthesis may also alter CPI baseline. Increased urinary and fecal excretion of CPI was observed when hemogenesis is triggered by anemia 33 or hemolysis. 34 In addition, lower CPI baseline was reported in Japanese female subjects relative to male subjects, with no sex‐related differences in exposure of other OATP1B markers or probes, suggesting reduced CPI synthesis in women. 31
Several groups, including us, have reported CPI models with different complexities, ranging from a turnover model to more complex description of the processes in the liver, developed with the aim to facilitate quantitative understanding of CPI disposition. 18 , 35 , 36 In all modeling examples, hepatic and nonhepatic routes were considered, but there were inconsistencies in terms of CPI synthesis site; it was assumed to occur either in the blood (central) compartment 18 , 35 or liver, 36 with no clear consensus.
This study aimed to evaluate quantitatively the impact of genetic polymorphism of SLCO1B1 c.521T>C, ethnicity, and sex on interindividual variability in CPI baseline using the largest clinical dataset so far. To estimate the effect of these covariates and to account for the mechanistic description of hepatic transporter processes, the CPI model was extended from our previous work. 18 Parameter estimation was implemented by fitting the model simultaneously to three independent CPI clinical datasets, including white (n = 16, men and women) and Asian‐Indian (n = 26, neb) subjects and included all SLCO1B1 c.521T>C variants (521TT, TC, and CC). Comprehensive literature search on the available biological information related to CPI synthesis was performed and the impact of different assumptions of the CPI synthesis site was assessed by parameter sensitivity analysis using the extended CPI model. Finally, the impact of the covariates on CPI‐drug OATP1B‐mediated interaction potential was simulated to support optimal design of prospective clinical studies.
METHODS
Individual CPI clinical data
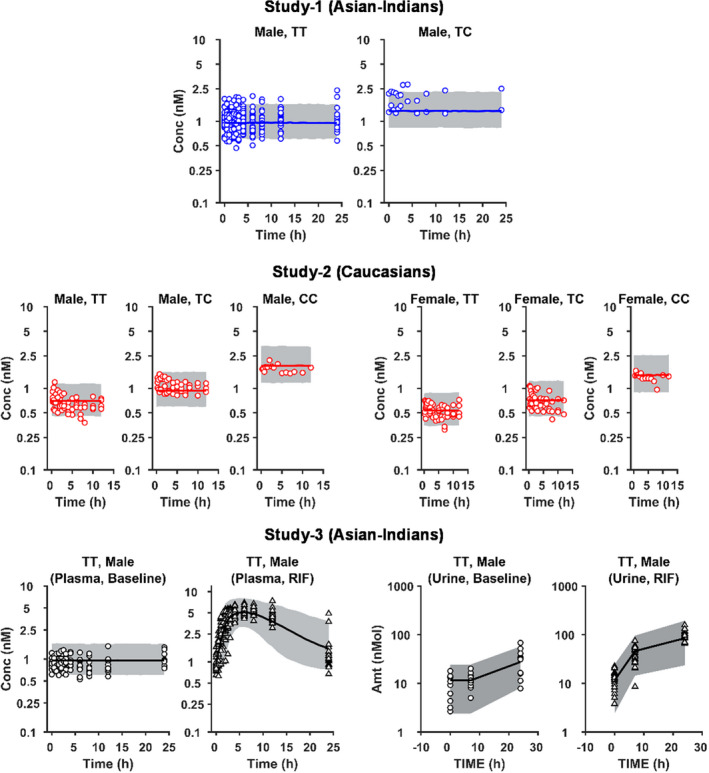
Individual CPI data were obtained from three clinical studies (Table 1 ). Shen et al. 37 (study 1) was conducted in 14 healthy male Asian‐Indians (521TT; n = 13, 521TC; n = 1) and CPI baseline plasma concentrations were measured over 24 hours (Supplementary Material Section S1 ). Yee et al. 25 (study 2) was conducted in 16 healthy Caucasians (521TT; n = 8, 521TC; n = 6, and 521CC; n = 2); only CPI concentration in plasma was measured on 1 occasion over 12 hours. Lai et al. 24 (study 3) was conducted in 12 healthy male Asian‐Indians (all 521TT) subjects and CPI concentrations in plasma and urine were measured over 24 hours on 3 occasions; 2 occasions involved interaction with rifampicin. Data on three main covariates (SLCO1B1 c.521T>C status, ethnicity, and sex) investigated here were available for all 42 subjects.
Table 1.
Clinical data used in CPI population PBPK model development
| No. subjects | Study 1 (Shen et al. 2019) 37 | Study 2 (Yee et al. 2019) 25 | Study 3 (Lai et al. 2019) 24 | |
|---|---|---|---|---|
| 14 | 8 | 8 | 12 | |
| Ethnicity | Asian‐Indians | Caucasian | Caucasian | Asian‐Indians |
| Sex | Male | Male | Female | Male |
| Study duration | 24h | 12h | 12h | 24h |
| SLCO1B1 c.521 Genotype | TT (n = 13), TC (n = 1) | TT (n = 4), TC (n = 3), CC (n = 1) | TT (n = 4), TC (n = 3), CC (n = 1) | TT (n = 12) |
| No. occasions; treatment |
Occ 1; Predose Occ 2; Furosemide |
Occ 1; Pravastatin | Occ 1; Pravastatin |
Occ 1; Rifampicin, Occ 2; Rosuvastatin Occ 3; Rifampicin + Rosuvastatin |
| No. plasma samples |
Occ 1; 155 (TT) and 12 (TC) Occ 2; 151 (TT) and 11 (TC) |
(TT); 58 (TC); 44 (CC); 15 |
(TT); 57 (TC); 41 (CC); 14 |
Occ 1; 144 Occ 2; 132 Occ 3; 132 |
| No. urine samples | 0 | 0 | 0 |
34 (pre‐treatment) 68 (post‐treatment) |
| Cave,base, nM |
TT; 1.07 ± 0.26 TC; 1.86 |
TT; 0.66 ± 0.11 TC; 1.08 ± 0.14 CC; 1.74 |
TT; 0.54 ± 0.05 TC; 0.69 ± 0.13 CC; 1.37 |
TT; 0.87 ± 0.16 |
Cave,base, coproporphyrin I concentration in plasma over whole study period in occasions without rifampicin treatment; CPI, coproporphyrin I; Occ, occasion.
CPI population PBPK model
The CPI model structure was based on our CPI model published previously, 18 but included a mechanistic description of the liver compartment (Figure 1 ). The model consists of five compartments, including blood (central), urine, liver vascular, liver tissue, and bile compartments. 38 The ordinary differential equations describing the concentration/amount in these compartments are:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where Cblood, CLV, CLT, Abile, and Aurine are the concentration (C) or amount (A) of CPI in the blood (central), liver vascular, liver tissue, bile, and urine compartments, respectively. The parameters ksyn, QH, Vc, VLV, VLT, CLR, CLpassive,u, CLactive,u, CLB, fub, and fuLT are rate of CPI synthesis, hepatic blood flow, volume of blood (central) compartment, volume of liver vascular, volume of liver tissue, renal clearance, hepatic passive clearance (unbound), hepatic active uptake clearance (unbound), biliary clearance, fraction unbound in blood, and faction unbound in liver tissue, respectively. Due to lack of corroborating evidence of CPI enterohepatic recirculation 29 and low bioavailability of labeled CPI in monkeys, 30 this process was assumed to be insignificant for the PK of CPI and was not considered in the current model development.
Figure 1.
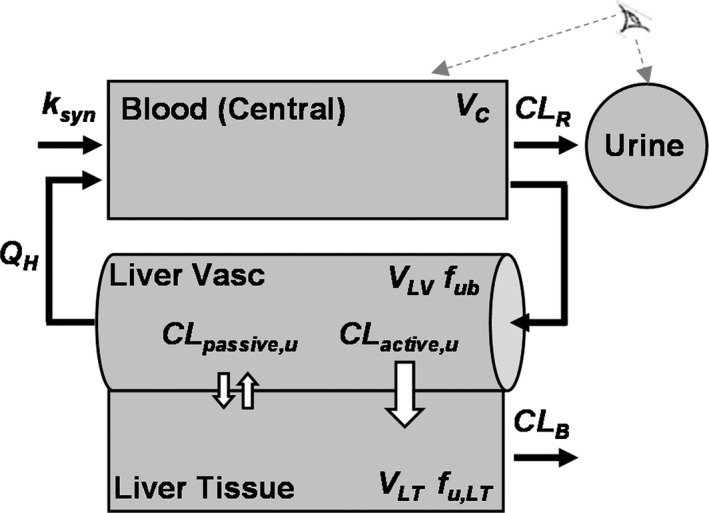
Structure of the coproporphyrin I minimal PBPK model. CLactive,u, hepatic uptake clearance (unbound); CLB, biliary clearance; CLpassive,u, hepatic passive clearance (unbound); CLR, renal clearance; fub, fraction unbound in the blood; fu,LT, fraction unbound in liver tissue; ksyn, endogenous synthesis rate; QH, hepatic blood flow; VC, volume of central compartment; VLV, volume of liver vascular; VLT, volume of liver tissue. An eye symbol represents observed compartments.
Competitive inhibition of OATP1B‐mediated CLactive,u by rifampicin was described by Eqs. 6 and 7.
| (6) |
| (7) |
where CRif is total rifampicin plasma concentration and Ki is the inhibition constant for OATP1B‐mediated CLactive,u. CRif was predicted using individual parameter estimates (posthoc) obtained during development of a population PK model for rifampicin, as reported previously. 18
Parameter estimation and covariate analysis
To improve the parameter identifiability, some CPI model parameters were fixed to in vitro determined or physiological values, namely in vitro CLpassive,u was scaled to in vivo CLpassive,u using hepatocellularity of 120 × 106 cells/g of liver, 38 and fu,LT was estimated from in vitro CPI uptake studies in human hepatocytes (Supplementary Material Section S2 ). fu,b was calculated from experimentally determined fraction unbound in plasma (fu,p), with labeled CPI measured in this study (Supplementary Material Section S2 ), and reported blood‐to‐plasma ratio (Table S4 ). QH and volumes of compartments (with the exception of the central compartment) were fixed to the physiological values reported previously. 38
Remaining model parameters were estimated using mixed‐effects modeling in NONMEM version 7.42 using ADVAN13 and first‐order conditional estimation with interaction method. The CPI model was fitted to all individual plasma and urine data simultaneously using combined proportional and additive residual error models for the residual unexplained variabilities of both plasma and urine data; simulated CPI blood concentrations were converted to plasma concentrations using blood‐to‐plasma ratio. Between subject variability (BSV) was estimated for ksyn, CLB, CLR, and Vc assuming log‐normal distribution of the parameters; between occasion variability (BOV) was estimated only for CLB because inclusion of BOV for other parameters did not improve model performance.
Three covariates were investigated in the CPI model using log‐likelihood ratio tests at a significance level of p < 0.05: the effects of SLCO1B1 c.521T>C and/or ethnicity on CLactive,u, and the effect of sex on ksyn. CLactive,u in 521TT subjects was represented as the reference value (CLactive,0), and the fraction change for polymorphic TC and CC allelic variants (COVGEN) were represented as follows:
| (8) |
where GEN is a dummy variable that takes the following values: GEN = 0 (521TT), 0.5 (521TC), and 1 (521CC). Here, the fractional change in 521TC was assumed to be half of the value for 521CC (an additive genetic effect). To keep COVGEN between 0 and 1, COVGEN was estimated indirectly using a surrogate variable of genetic effect (FRAX) shown in Eq. 9:
| (9) |
where FRAX can have any positive real value.
The effect of ethnicity on CLactive,u was also introduced as follows:
| (10) |
where CLactive,0 is for Caucian subjects with 521TT genotype (reference group), COVRACE is the fractional change in CLactive,u in Asian‐Indians relative to Caucasians, and RACE is a dummy variable that takes the value of 0 for Caucians and 1 for Asian‐Indians.
The effect of sex on CPI synthesis rate (ksyn,sex) was described as follows:
| (11) |
where COVSEX is the fractional change in ksyn in women relative to men, and SEX is a dummy variable that takes the value of 0 for men and 1 for women.
Data exploration and goodness‐of‐fit (GOF) plots were produced using MATLAB (R2017a version 9.2.0). GOF plots, such as observed data vs. population predicted (PRED) or individual PRED, conditionally weighted residual vs. TIME or population prediction were used to assess possible model misspecification. Visual predictive check (VPC) based on 5,000 simulated individuals using the covariate demographics (SLCO1B1 c.521T>C, ethnicity, and sex) was used to assess final model performance.
Location of CPI synthesis
The plausibility of the conflicting hypotheses on the site of the CPI synthesis (blood or liver) was assessed by comprehensive literature search. In addition to collation of biological evidence, the impact of the CPI synthesis site was evaluated by comparing estimated parameter values in modified CPI models when assuming different fractions of CPI synthesis in the liver (FLsyn, ranged from 0 to 1; i.e., 0–100% of ksyn in the liver).
| (12) |
| (13) |
Theoretical simulation of CPI‐drug interaction with different CLactive and ksyn
CPI PK parameters (baseline, AUCR, maximum concentration (Cmax) observed, and time of maximum concentration observed (Tmax)) were simulated with hypothetical OATP1B inhibitors using the developed CPI population PBPK model. Inhibitors were assumed to have the same PK as rifampicin, but varying OATP1B Ki (up to 10‐fold lower potency in inhibition). CLactive and ksyn in the model ranged from 10–125% or 50–125% relative to those in male Caucasians with 521TT, respectively, to capture the covariates estimated in this study. AUCR was calculated as a ratio of AUC0–24 with and without inhibitor. To evaluate changes derived from altered CPI disposition, PK of the inhibitor was assumed to be independent of the covariates.
Statistical analysis
CPI concentration in plasma over whole study period on occasions without rifampicin treatment (Cave,base) was calculated for each subject, and mean and SD of Cave,base were calculated for each subpopulation. All occasions without rifampicin were treated as baseline conditions because co‐administration of clinical probes (furosemide) had no effect on plasma CPI of 521TT subjects in study 1 (Figure S1 ).
RESULTS
Clinical studies evaluating CPI in different OATP1B1 genotype and ethnicity
Men with 521TC (1.86 nM) showed higher Cave,base than 521TT (1.07 ± 0.26 nM) in study 1; the same tendency was also observed in men (TT 0.66 ± 0.11 nM (n = 4), TC 1.08 ± 0.14 nM (n = 3), and CC 1.74 nM (n = 1)) and women (TT 0.54 ± 0.05 nM (n = 4), TC 0.69 ± 0.13 nM (n = 3), and CC 1.37 nM (n = 1)) in study 2. Female subjects showed lower Cave,base than men regardless of OATP1B1 genotype. Cave,base of male Caucasian subjects with 521TT (0.66 ± 0.11 nM in study 2) was lower than those in Asian‐Indians with 521TT genotype (1.07 ± 0.26 nM in study 1 and 0.87 ± 0.16 nM in study 3). Cave,base in male Asian‐Indians with 521TT was comparable between study 1 and study 3.
Development of CPI population PBPK model with multiple covariates
The value of CLpassive,u and fu,LT were fixed to those measured in vitro in human hepatocytes (scaled CLpassive,u = 8 L/h or equivalent to 0.76 µL/min/106 cells, and fu,LT = 0.19, details of in vitro studies in Supplementary Material Section S2 ). Experimentally determined CPI fu,p was 0.069. With these parameters fixed, population PBPK CPI model, accounting for the mechanistic liver description and the three covariates, successfully estimated all parameters with acceptable uncertainty (< 35% relative standard error except for additive residual error for CPI in urine; Table 2 ). The VPC showed adequate coverage of the data, with most of the data within acceptable range in GOF plots, despite moderately smaller conditionally weighted residual for higher PRED (plasma and urine; Figures 2 and 3 ).
Table 2.
CPI population PBPK model parameter estimates
| Parameter | kSYN in blood (FLsyn = 0) | kSYN in liver (FLsyn = 1) | ||||
|---|---|---|---|---|---|---|
| Fixeda | BSVb | BOVc | Fixeda | BSVb | BOVc | |
| System parameter | ||||||
| ksyn, nMol/h | 18.4 (11) | 10 (30) | ‐ | 33.1 (12) | 7.3 (87) | ‐ |
| CLB, L/h | 6.24 (24) | 38.2 (23) | 34.4 (21) | 5.28 (8) | 16 (28) | 14.2 (16) |
| CLR, L/h | 2.7 (6) | 12.7 (30) | ‐ | 2.7 (6) | 13.5 (28) | ‐ |
| VC, L | 11.9 (21) | 25.8 (29) | ‐ | 13 (16) | 24.9 (29) | ‐ |
| CLactive,0, L/h | 1397 (32) | ‐ | ‐ | 930 (15) | ‐ | ‐ |
| Ki, μM | 0.93 (7) | ‐ | ‐ | 1.44 (10) | ‐ | ‐ |
| Covariates | ||||||
| FRAX | 0.269 (13) | ‐ | ‐ | 0.486 (13) | ‐ | ‐ |
| COVGEN d | 0.788 | ‐ | ‐ | 0.673 | ‐ | ‐ |
| COVRACE | 0.417 (18) | ‐ | ‐ | 0.323 (16) | ‐ | ‐ |
| COVSEX | 0.232 (24) | ‐ | ‐ | 0.26 (21) | ‐ | ‐ |
| Residual unexplained variabilities | ||||||
| σprop (%) – plasma | 13.2 (5) | ‐ | ‐ | 13.3 (5) | ‐ | ‐ |
| σadd, nM – plasma | 0.001 FIXED | ‐ | ‐ | 0.001 FIXED | ‐ | ‐ |
| σprop (%) – urine | 34.8 (8) | ‐ | ‐ | 34.9 (8) | ‐ | ‐ |
| σadd, nMol – urine | 2.3 (50) | ‐ | ‐ | 2.18 (59) | ‐ | ‐ |
σprop, proportional residual error; σadd, additive residual error; BOV, between occasion variability; BSV, between subject variability; CLB, biliary clearance; CLR, renal clearance; CLuptake,0, hepatic active uptake clearance (CLactive,u) in white men with SLCO1B1 521TT genotype; COVGEN, fractional change in CLactive,u in SLCO1B1 521CC genotype; COVRACE, fractional change in CLactive,u in Asian‐Indian subjectss; COVsex, fractional change in ksyn in women relative to men; CPI, coproporphyrin I; FLsyn, fraction CPI synthesis in the liver; FRAX, surrogate variable of genetic effect; Ki, total rifampicin OATP1B1 inhibition constant (equivalent to 0.10 µM as unbound K i calculated with rifampicin fu,p of 0.11); ksyn, rate of coproporphyrin I synthesis; PBPK, physiologically‐based pharmacokinetic; Vc, volume of blood (central) compartment.
aThe population (fixed effect) parameters. Values within parentheses represent relative standard errors (RSE, %). bEstimated BSV (%) and its RSE (%). cEstimated BOV (%) and its RSE (%). dCalculated based on the population (fixed effect) parameter estimate of FRAX.
Figure 2.
Visual predictive check (VPC) for mechanistic population PBPK modeling of CPI plasma and urine data. Symbols, solid lines, and grey areas represent observed data, median population prediction, and 95% prediction intervals (n = 5,000), respectively. Simulations were performed for each subgroup, including subjects with different sex or SLCO1B1 c.521 (OATP1B1 transporter) genotype (521TT (TT), 521TC (TC), and 521CC (CC)) in three clinical studies. The CPI model with ksyn in the blood (central) compartment was used for the simulation. CPI, coproporphyrin I; PK, pharmacokinetic.
Figure 3.
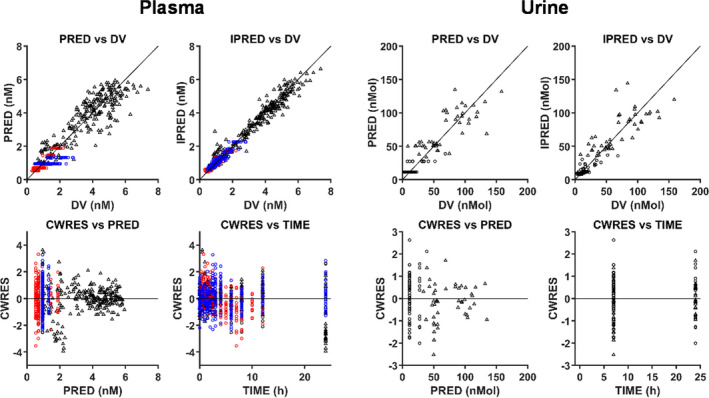
Goodness‐of‐fit (GOF) plots for mechanistic population PBPK modeling of CPI plasma and urine data. Colors represent clinical studies: blue = study 1; red = study 2; and black = study 3. Circles and triangles represent occasions with or without rifampicin administration, respectively. Solid lines are reference lines. The CPI model with ksyn in the blood (central) compartment was used for the simulation. CPI, coproporphyrin I; CWRES, conditional weighted residuals; DV, observed data; IPRED, individual prediction; PRED, population prediction.
Active process was a major contributor (> 99%) to CPI hepatic uptake; estimated CLactive,0 (for the reference 521 TT group) was 1,397 L/h (equivalent to 137 µL/min/106 cells), which was 175‐fold or 517‐fold higher than CLpassive,u or CLR (2.7 L/h), respectively. Estimated CLactive,0 was 2‐fold higher than the CPI CLactive obtained in vitro (64.8 µL/min/106 cells, Table S2 ). CLB (6.24 L/h) was lower than previously estimated (12.3 L/h) 18 ; however, this finding is not surprising considering the more mechanistic description of CPI hepatic disposition in this analysis, with the inclusion of CLactive,u. The estimated total rifampicin OATP1B Ki (0.93 µM, equivalent to 0.10 µM as unbound Ki calculated with rifampicin fu,p of 0.11 24 ) was consistent with our previous analysis (1.15 µM). 18 BSV of ksyn, CLB, CLR, and CLactive,u were < 40%.
The parameter estimation revealed 79% lower CLactive,u in 521CC relative to 521TT, which was slightly lower than in vitro‐estimated genotype effect based on comparison of CPI uptake in donors with 521TT and 521CC genotype (85.6 ± 6%; Table S3 ). Estimated CLactive,u in Asian‐Indians was 42% lower than that in Caucasians. Estimated CPI ksyn in female subjects was 23% lower than that in male subjects.
Location of CPI synthesis
Our comprehensive literature search on the location of CPI synthesis was not conclusive because of supportive information for both assumptions (details in Supplementary Material Section S4 ). In brief, CPI synthesis in blood was qualitatively suggested based on the parallel change in urinary CPI excretion and erythropoiesis activity in the blood. 33 , 34 Partial contribution of hepatic CPI synthesis seemed possible because an administered radiolabeled precursor of CPI, which is selectively taken into the hepatic heme synthesis pathway, was partially converted to CPI in humans. 39
Considering uncertainties above, parameter estimations with different values assumed for FLsyn (from 0 to 1) were investigated. VPC with each set of estimated parameters were visually indistinguishable (estimated values and VPC with FLsyn = 0 (synthesis in blood) and FLsyn = 1 (synthesis in liver) are shown in Table 2 and Figure S7 and S8, respectively). The model with CPI synthesis in the liver showed comparable parameter estimates to the model assuming CPI synthesis in the blood (central); moderate relative changes in ksyn (+80%), CLactive,0 (−33%), Ki (+55%), BSV in ksyn (−50%) and CLB (−82%), and BOV in CLB (−83%) were evident, with marginal changes in other parameters (±30%). Estimated parameters in the models with different contribution of CPI synthesis in liver (FLsyn = 0.25, 0.5, or 0.75) were between those estimated with FLsyn = 0 and 1 assumptions (Figure S9 ).
Theoretical simulation of CPI‐drug interaction in different covariate groups
CPI baseline and Cmax increased with decreasing CLactive,u (the effect of 521T>C or ethnicity), whereas opposite trend was seen with decrease in ksyn (the effect of sex; Figure 4 and Figure S10 ). Simulated AUCR with OATP1B inhibitor equivalent to rifampicin decreased with lower initial transporter activity (CLactive,u), whereas AUCR was insensitive to differences in ksyn. The effect of 521T>C genotype on CPI AUCR (25–40% decrease in 521CC relative to 521TT in white and Asian‐Indian subjects, respectively) was more evident than that of ethnicity (2–22% decrease in Asian‐Indian relative to Caucasian subjects in 521TT and CC, respectively). Moderate‐to‐weak OATP1B inhibitors showed the same trends, although the degree of interaction was smaller (details in Figure S11 ).
Figure 4.
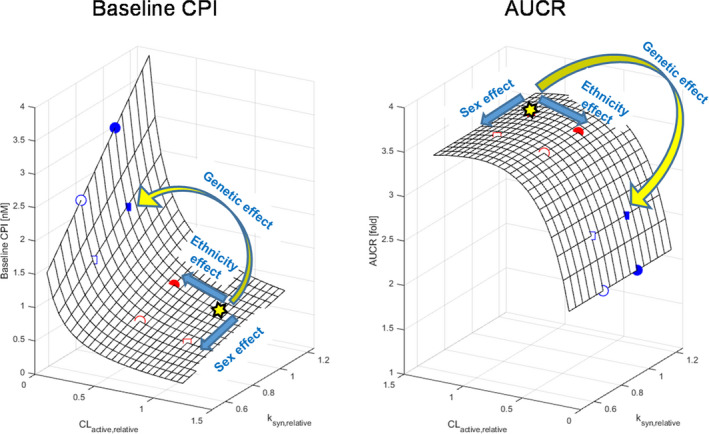
Theoretical simulation of CPI‐drug interaction across different CPI hepatic uptake clearance and synthesis rate. Simulation of CPI baseline and AUCR caused by the rifampicin equivalent OATP1B1 inhibitor with varying CPI hepatic uptake clearance and endogenous synthesis rate, represented as relative values (CLactive,u,relative and ksyn,relative) to those in male Caucasians with 521TT (yellow star), respectively. Symbols on surface plots represent subpopulations simulated with the population (fixed effect) parameters of each subpopulation; men (filled) and women (open), 521TT (red) and 521CC (blue), Caucasians (square), and Asian‐Indians (circle). Yellow and blue arrows indicate effects of three covariates on male white subjects with 521TT. AUCR, fold change in CPI AUC; CLactive, hepatic active clearance; CPI, coproporphyrin I; ksyn, zero‐order synthesis.
DISCUSSION
Utility of model‐informed drug development with CPI model
Mechanistic modeling has been increasingly used to improve quantitative understanding of the formation and elimination mechanisms of endogenous biomarkers for drug transporters; these efforts are more advanced for CPI/OATP1B in comparison to other endogenous biomarkers/transporters. One of the key applications of modeling approaches is to guide optimal design of OATP1B‐mediated interaction studies. 18 , 35 , 36 To that end, modeling and simulation studies have provided information on the adequate sample size to identify weak‐to‐moderate OATP1B inhibitors 18 and used estimated in vivo Ki based on CPI data to simulate the DDI between rifampicin/new inhibitors and statins. 35 , 36 , 40 One of the key advantages of CPI as an endogenous biomarker of OATP1B function in vivo is its relatively stable baseline in contrast to other biomarkers (e.g., fatty or bile acids and their conjugates). 15 Recent clinical studies have provided evidence of the effect of OATP1B1 genetic polymorphism 25 , 28 , 31 on CPI plasma concentration. Therefore, this study aimed to expand our previous CPI model 18 to incorporate multiple covariates on CPI disposition and investigate their impact on the evaluation of CPI‐drug interaction risk.
CPI population PBPK model was based on several assumptions. Certain parameters (e.g., CLpassive,u and fu,LT) were fixed to the values measured in vitro to improve parameter identifiability (Supplementary Material Section S2 ). Enterohepatic circulation of CPI was not considered in our model, supported by the recent studies. 29 , 30 For pragmatic reasons, concentration of rifampicin in the central compartment was used as inhibitory concentration affecting CPI CLactive,u; any potential rifampicin concentration gradient between blood and hepatic inlet was not considered. This assumption may potentially underestimate rifampicin OATP1B Ki, but the covariates estimated as relative changes are not likely to be sensitive to this assumption. Inhibition of MRP2 by rifampicin was not considered due to the lack of bile data for verification, as discussed previously. 18 Biological evidence for the location of CPI synthesis was inconclusive (Supplementary Material Section S4 ) and CPI plasma exposure is likely to be derived from both liver and blood. Parameter estimation with assumed CPI synthesis either in the blood or liver resulted in comparable parameters (Table 2 ) and recovery of plasma data. These findings are not surprising and highlight that plasma data in isolation (or combined with urine) are not sufficiently sensitive/informative to differentiate the CPI synthesis site, in particular, any contribution of the liver.
Covariates affecting CPI hepatic active uptake clearance
In our study, OATP1B1 521T>C was considered a sole allelic variant that affects CPI disposition, due to limited clinical evidence of the effect of other polymorphisms (e.g., c.388A>G). 28 , 31 The implementation of the genetic effect in the model was based on relatively small number of subjects with c.521T>C mutation (9 subjects with 521TC or CC), but the overall trends are in line with a recently published study. 31 The 521CC has resulted in 79% lower CPI CLactive,u relative to 521TT, in agreement with clinical OATP1B1 probes. 11 , 41 The 521CC effect was slightly lower compared with in vitro evaluation (80–92%; Table S3 ). This slight discrepancy can be rationalized by large interindividual differences in protein expression of OATP1B 42 , 43 and small number of hepatocyte donors used in the in vitro evaluation. Considering available dataset, this genetic effect on CPI exposure was mainly based on the data reported in Caucasians. Ethnic differences in the degree of genetic effect are not likely considering comparable effects of 521T>C mutation reported for clinical OATP1B probes between Caucasians and Asians. 41
The literature evidence of ethnic differences in the activity of OATP1B relative to Caucasians was reported for Japanese, 11 but not for Asian‐Indians due to limited number of clinical studies in this population. Two clinical studies reported higher (up to 63%) AUC of rosuvastatin in Asian‐Indians relative to Caucasians 7 , 9 ; however, BCRP 421G>A status of those subjects was not reported. Therefore, those results require careful interpretation, as rosuvastatin is a substrate of OATP1B1 and BCRP and genetic polymorphism of both transporters affects its plasma exposure. 7 , 8 , 9 , 44 Birmingham et al. 9 reported moderately (> 26%) higher rosuvastatin AUC in Asian‐Indians relative to Caucasians regardless of OATP1B1 and BCRP genotype, implying the existence of intrinsic ethnic variability in OATP1B activity between the two populations. Therefore, the ethnicity effect in the CPI model was assumed on CLactive,u and 42% lower CLactive,u was estimated for Asian‐Indians relative to Caucasians. We excluded the possibility of the ethnicity effect on ksyn because of comparable blood hemoglobin levels reported for Asian‐Indians and Caucasins, 45 suggesting minimal differences in CPI synthesis between these two populations. Reported mean CPI Cave,base in Japanese (men, 521TT) of 0.63–0.77 nM 26 , 31 is comparable to Caucasians (Table 1 ) and in contrast to the proposed ethnic differences in OATP1B activity between Japanese and Caucasians. 11 Lower hemoglobin has been reported in Japanese relative to Caucasians, which may imply lower CPI synthesis in Japanese. 46
Covariates affecting CPI synthesis rate
In Caucasian subjects, lower Cave,base was observed in women relative to men regardless of OATP1B1 genotype (Table 1 ). Comparable exposure of other endogenous biomarkers of OATP1B (e.g., fatty acids) between men and women in the same subjects (data not shown) suggested no sex effect on CLactive,u, but reduced CPI synthesis in women. These findings are supported by the comparable protein expression of hepatic OATP1B between men and women 42 and lower blood hemoglobin in women. 47 Although this study used all the available individual CPI data, it is worth noting that small sample size in some subpopulations in this study (e.g., 521CC) may bias the estimation of covariates, as suggested by power calculations in previous studies. 18 Further studies with larger sample size, including different demographics (e.g., OATP1B1 genotype, ethnicity, or sex) would address these questions and validate/confirm assumptions made here with regard to CPI synthesis or intrinsic ethnic differences in OATP1B activity.
Prospective application of the developed CPI model
As CPI is a surrogate marker of OATP1B functional activity in vivo, the population PBPK model developed here provides a powerful and informative tool for the prospective evaluation of OATP1B‐mediated interaction risk. This approach is particularly valuable for exploration of interaction risk in subpopulations that are often not considered in clinical studies or in populations with multiple co‐existing covariates. Simulations of such what‐if scenarios are where a model‐informed drug development approach exerts its true potential. Theoretical simulations of CPI‐drug interactions illustrated complex interplay between biomarker synthesis and OATP1B‐mediated uptake, with high sensitivity of AUCR to changes in CLactive,u, but not to ksyn (Figure 4 ). Decrease in CLactive,u reduces the proportion of CPI eliminated via OATP1B, which is compensated by increased elimination of CPI into urine (data not shown), resulting in decreased AUCR. Relatively greater effect of 521T>C genotype on CLactive,u (−79% in 521CC relative to 521TT) than that of ethnicity (−42% in Asian‐Indian relative to white subjects) results in more pronounced impact of the genetic effect on the CPI interaction (AUCR). Simulated decreased AUCR in 521CC is consistent with data reported with clinical OATP1B probes 3 , 6 and suggests the risk of underestimation of the magnitude of transporter‐mediated interaction if these subjects are included in a CPI‐drug interaction study, which is more likely to happen in clinical studies performed without prior genotyping (e.g., first‐in‐human study) or in populations with higher prevalence of 521CC. 48 In contrast, any changes in CPI synthesis did not affect fraction transported via OATP1B and simulated AUCR, highlighting no risk of underestimation of the OATP1B interaction magnitude if female subjects are included in the study. As exemplified here, the modeling of CPI gives us prospective insights of clinical output and enables optimal clinical design to increase likelihood of success. Furthermore, the physiological description of the liver processes in the CPI model enables the extension of the model to other special populations (e.g., evaluation of the changes in OATP1B activity in the chronic kidney disease population). 49 , 50
In conclusion, this study evaluated the impact of the genotype of OATP1B1 c521T>C, ethnicity, and sex on CPI baseline and its OATP1B‐mediated interaction potential. For the first time, this revised CPI population PBPK model implemented reduced active uptake due to OATP1B1 polymorphism, ethnic differences in active uptake between Asian‐Indians and Caucasians, and reduced CPI synthesis in women. Modeling highlighted that assumption of the CPI synthesis site (blood, liver, or both) has marginal impact on the simulated plasma exposure of this endogenous biomarker ; availability of liver concentrations would be necessary to allow differentiation of these sites. The CPI PBPK model proposed here aims to facilitate the design of prospective clinical studies with OATP1B perpetrator drugs to maximize the sensitivity of this biomarker.
Funding
H.T. was financially supported by a fellowship grant from Asahi Kasei Pharma Corporation. S.B. was supported by a PhD studentship from the Biotechnology and Biological Sciences Research Council, UK (BB/L502376/1) and UCB, UK.
Conflict of interest
The authors declared no competing interests for this work.
Author contributions
H.T., S.B., K.M., H.S., K.O., and A.G. wrote the manuscript. H.T., S.B., H.S., K.O., and A.G. designed the research. H.T., S.B. and Y.Z. performed the research. H.T., S.B., Y.Z., and K.O. analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
Acknowledgments
The authors thank Prof. Kathleen M. Giacomini and Dr. Sook Wah Yee (University of California, San Francisco, California, USA) for sharing individual clinical data of coproporphyrin I.
References
- 1. Chu, X. et al Clinical probes and endogenous biomarkers as substrates for transporter drug‐drug interaction evaluation: perspectives from the International Transporter Consortium. Clin. Pharmacol. Ther. 104, 836–864 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Ménochet, K. , Kenworthy, K.E. , Houston, J.B. & Galetin, A. Simultaneous assessment of uptake and metabolism in rat hepatocytes: a comprehensive mechanistic model. J. Pharmacol. Exp. Ther. 341, 2–15 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yee, S.W. et al Influence of transporter polymorphisms on drug disposition and response: a perspective from the International Transporter Consortium. Clin. Pharmacol. Ther. 104, 803–817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsamandouras, N. et al Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population‐modeling approach. Clin. Pharmacol. Ther. 96, 90–100 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Link, E. et al SLCO1B1 variants and statin‐induced myopathy ‐ a genomewide study. N. Engl. J. Med. 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 6. He, Y.J. et al Rifampicin alters atorvastatin plasma concentration on the basis of SLCO1B1 521T>C polymorphism. Clin. Chim. Acta 405, 49–52 (2009). [DOI] [PubMed] [Google Scholar]
- 7. Lee, E. et al Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin. Pharmacol. Ther. 78, 330–341 (2005). [DOI] [PubMed] [Google Scholar]
- 8. Birmingham, B.K. et al Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: a class effect? Eur. J. Clin. Pharmacol. 71, 341–355 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Birmingham, B.K. et al Rosuvastatin pharmacokinetics and pharmacogenetics in Caucasian and Asian subjects residing in the United States. Eur. J. Clin. Pharmacol. 71, 329–340 (2015). [DOI] [PubMed] [Google Scholar]
- 10. US Food and Drug Administration . Prescribing information for CRESTOR <https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/021366s033lbl.pdf>.
- 11. Tomita, Y. , Maeda, K. & Sugiyama, Y. Ethnic variability in the plasma exposures of oatp1b1 substrates such as HMG‐CoA reductase inhibitors: a kinetic consideration of its mechanism. Clin. Pharmacol. Ther. 94, 37–51 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Wu, H.F. et al Rosuvastatin pharmacokinetics in Asian and white subjects wild type for both OATP1B1 and BCRP under control and inhibited conditions. J. Pharm. Sci. 106, 2751–2757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li, R. & Barton, H.A. Explaining ethnic variability of transporter substrate pharmacokinetics in healthy Asian and Caucasian subjects with allele frequencies of OATP1B1 and BCRP: a mechanistic modeling analysis. Clin. Pharmacokinet. 57, 491–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo, Y. et al Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin. Pharmacol. Ther. 104, 865–889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barnett, S. et al Comprehensive evaluation of the utility of 20 endogenous molecules as biomarkers of OATP1B inhibition compared with rosuvastatin and coproporphyrin I. J. Pharmacol. Exp. Ther. 368, 125–135 (2019). [DOI] [PubMed] [Google Scholar]
- 16. Rodrigues, A.D. , Taskar, K.S. , Kusuhara, H. & Sugiyama, Y. Endogenous probes for drug transporters: balancing vision with reality. Clin. Pharmacol. Ther. 103, 434–448 (2018). [DOI] [PubMed] [Google Scholar]
- 17. Jones, N.S. et al Complex DDI by fenebrutinib and the use of transporter endogenous biomarkers to elucidate the mechanism of DDI. Clin. Pharmacol. Ther. 107, 269–277 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barnett, S. et al Gaining mechanistic insight into coproporphyrin I as endogenous biomarker for OATP1B‐mediated drug‐drug interactions using population pharmacokinetic modeling and simulation. Clin. Pharmacol. Ther. 104, 564–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen, H. et al Coproporphyrins I and III as functional markers of OATP1B activity: in vitro and in vivo evaluation in preclinical species. J. Pharmacol. Exp. Ther. 357, 382–393 (2016). [DOI] [PubMed] [Google Scholar]
- 20. Shen, H. et al Comparative evaluation of plasma bile acids, dehydroepiandrosterone sulfate, hexadecanedioate, and tetradecanedioate with coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab. Dispos. 45, 908–919 (2017). [DOI] [PubMed] [Google Scholar]
- 21. Bednarczyk, D. & Boiselle, C. Organic anion transporting polypeptide (OATP)‐mediated transport of coproporphyrins I and III. Xenobiotica 46, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Kunze, A. , Ediage, E.N. , Dillen, L. , Monshouwer, M. & Snoeys, J. Clinical investigation of coproporphyrins as sensitive biomarkers to predict mild to strong OATP1B‐mediated drug‐drug interactions. Clin. Pharmacokinet. 57, 1559–1570 (2018). [DOI] [PubMed] [Google Scholar]
- 23. Gilibili, R.R. et al Coproporphyrin‐I: a fluorescent, endogenous optimal probe substrate for ABCC2 (MRP2) suitable for vesicle‐based MRP2 inhibition assay. Drug Metab. Dispos. 45, 604–611 (2017). [DOI] [PubMed] [Google Scholar]
- 24. Lai, Y. et al Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug‐drug interactions mediated by organic anion transporting polypeptide inhibition. J. Pharmacol. Exp. Ther. 358, 397–404 (2016). [DOI] [PubMed] [Google Scholar]
- 25. Yee, S.W. et al Organic anion transporter polypeptide 1B1 polymorphism modulates the extent of drug‐drug interaction and associated biomarker levels in healthy volunteers. Clin. Transl. Sci. 12, 388–399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mori, D. et al Dose‐dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin. Pharmacol. Ther. 107, 1004–1013 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takehara, I. et al Comparative study of the dose‐dependence of OATP1B inhibition by rifampicin using probe drugs and endogenous substrates in healthy volunteers. Pharm. Res. 35, 138 (2018). [DOI] [PubMed] [Google Scholar]
- 28. Shen, H. et al Further studies to support the use of coproporphyrin I and III as novel clinical biomarkers for evaluating the potential for organic anion transporting polypeptide 1B1 and OATP1B3 inhibition. Drug Metab. Dispos. 46, 1075–1082 (2018). [DOI] [PubMed] [Google Scholar]
- 29. Takehara, I. , Watanabe, N. , Mori, D. , Ando, O. & Kusuhara, H. Effect of rifampicin on the plasma concentrations of bile acid‐O‐sulfates in monkeys and human liver‐transplanted chimeric mice with or without bile flow diversion. J. Pharm. Sci. 108, 2756–2764 (2019). [DOI] [PubMed] [Google Scholar]
- 30. Gu, X. et al Absorption and disposition of coproporphyrin I (CPI) in cynomolgus monkeys and mice: pharmacokinetic evidence to support the use of CPI to inform the potential for OATP inhibition. Drug Metab. Dispos. 48, 724–734 (2020). [DOI] [PubMed] [Google Scholar]
- 31. Mori, D. et al Effect of OATP1B1 genotypes on plasma concentrations of endogenous OATP1B1 substrates and drugs, and their association in healthy volunteers. Drug Metab. Pharmacokinet. 34, 78–86 (2019). [DOI] [PubMed] [Google Scholar]
- 32. Koskelo, P. & Mustajoki, P. Altered coproporphyrin‐isomer excretion in patients with the Dubin‐Johnson syndrome. Int. J. Biochem. 12, 975–978 (1980). [DOI] [PubMed] [Google Scholar]
- 33. Watson, C.J. The erythrocyte coproporphyrin: variation in respect to erythrocyte protoporphyrin and reticulocytes in certain of the anemias. Arch. Intern. Med. 86, 797–809 (1950). [DOI] [PubMed] [Google Scholar]
- 34. Dobriner, K. & Rhoads, C.P. The porphyrins in health and disease. Physiol. Rev. 20, 416–468 (1940). [Google Scholar]
- 35. Yoshida, K. , Guo, C. & Sane, R. Quantitative prediction of OATP‐mediated drug‐drug interactions with model‐based analysis of endogenous biomarker kinetics. CPT Pharmacometrics Syst. Pharmacol. 7, 517–524 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshikado, T. et al PBPK modeling of coproporphyrin I as an endogenous biomarker for drug interactions involving inhibition of hepatic OATP1B1 and OATP1B3. CPT Pharmacometrics Syst. Pharmacol. 7, 739–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen, H. et al Evidence for the validity of pyridoxic acid (PDA) as a plasma‐based endogenous probe for OAT1 and OAT3 function in healthy subjects. J. Pharmacol. Exp. Ther. 368, 136–145 (2019). [DOI] [PubMed] [Google Scholar]
- 38. Gertz, M. , Tsamandouras, N. , Säll, C. , Houston, J.B. & Galetin, A. Reduced physiologically‐based pharmacokinetic model of repaglinide: impact of OATP1B1 and CYP2C8 genotype and source of in vitro data on the prediction of drug‐drug interaction risk. Pharm. Res. 31, 2367–2382 (2014). [DOI] [PubMed] [Google Scholar]
- 39. Shimizu, Y. , Ida, S. , Naruto, H. & Urata, G. Excretion of porphyrins in urine and bile after the administration of delta‐aminolevulinic acid. J. Lab. Clin. Med. 92, 795–802 (1978). [PubMed] [Google Scholar]
- 40. Cheung, K.W.K. et al GDC‐0810 pharmacokinetics and transporter‐mediated drug interaction evaluation with an endogenous biomarker in the first‐in‐human, dose escalation study. Drug Metab. Dispos. 47, 966–973 (2019). [DOI] [PubMed] [Google Scholar]
- 41. Li, R. Estimating in vivo fractional contribution of OATP1B1 to human hepatic active uptake by mechanistically modeling pharmacogenetic data. AAPS J. 21, 69 (2019). [DOI] [PubMed] [Google Scholar]
- 42. Badée, J. , Achour, B. , Rostami‐Hodjegan, A. & Galetin, A. Meta‐analysis of expression of hepatic organic anion‐transporting polypeptide (OATP) transporters in cellular systems relative to human liver tissue. Drug Metab. Dispos. 43, 424–432 (2015). [DOI] [PubMed] [Google Scholar]
- 43. Peng, K.W. , Bacon, J. , Zheng, M. , Guo, Y. & Wang, M.Z. Ethnic variability in the expression of hepatic drug transporters: absolute quantification by an optimized targeted quantitative proteomic approach. Drug Metab. Dispos. 43, 1045–1055 (2015). [DOI] [PubMed] [Google Scholar]
- 44. Lee, C.A. et al Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug‐drug interaction study design. Drug Metab. Dispos. 43, 490–509 (2015). [DOI] [PubMed] [Google Scholar]
- 45. Pasupula, D.K. & Reddy, P.S. When is a south Indian really anemic? Indian J. Clin. Biochem. 29, 479–484 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miyazaki, Y. et al Differing clinical features between Japanese and Caucasian patients with myelodysplastic syndromes: analysis from the International Working Group for Prognosis of MDS. Leuk. Res. 73, 51–57 (2018). [DOI] [PubMed] [Google Scholar]
- 47. Yamada, M. , Wong, F.L. & Suzuki, G. Longitudinal trends of hemoglobin levels in a Japanese population ‐ RERF’s adult health study subjects. Eur. J. Haematol. 70, 129–135 (2003). [DOI] [PubMed] [Google Scholar]
- 48. Lam, Y.‐W.F. & Scott, S.A. Pharmacogenomics (Elsevier, New York, NY, 2019). 10.1002/cpt.1998. [e‐pub ahead of print]. [DOI] [Google Scholar]
- 49. Tan, M.L. et al Effect of chronic kidney disease on nonrenal elimination pathways: a systematic assessment of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP. Clin. Pharmacol. Ther. 103, 854–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tatosian, D.A. et al A microdose cocktail to evaluate drug interactions in patients with renal impairment. Clin. Pharmacol. Ther. (2020). 10.1002/cpt.1998. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material