

Summary
Heat‐stressed Arabidopsis plants release heterochromatin‐associated transposable element (TE) silencing, yet it is not accompanied by major reductions of epigenetic repressive modifications. In this study, we explored the functional role of histone H1 in repressing heterochromatic TEs in response to heat stress.
We generated and analyzed RNA and bisulfite‐sequencing data of wild‐type and h1 mutant seedlings before and after heat stress.
Loss of H1 caused activation of pericentromeric Gypsy elements upon heat treatment, despite these elements remaining highly methylated. By contrast, nonpericentromeric Copia elements became activated concomitantly with loss of DNA methylation. The same Copia elements became activated in heat‐treated chromomethylase 2 (cmt2) mutants, indicating that H1 represses Copia elements through maintaining DNA methylation under heat.
We discovered that H1 is required for TE repression in response to heat stress, but its functional role differs depending on TE location. Strikingly, H1‐deficient plants treated with the DNA methyltransferase inhibitor zebularine were highly tolerant to heat stress, suggesting that both H1 and DNA methylation redundantly suppress the plant response to heat stress.
Keywords: Arabidopsis thaliana, CMT2, DNA methylation, H1, heat stress, transposable element
Introduction
Transposable elements (TEs) constitute a large proportion of many eukaryotic genomes, including plants (McClintock, 1984; SanMiguel et al., 1996; Kumar & Bennetzen, 1999; Havecker et al., 2004). TEs lead to genetic variability and are therefore considered a major driving force for genome evolution (Mirouze & Paszkowski, 2011; Lisch, 2013; Belyayev, 2014; Grandbastien, 2015). Nevertheless, their ability to transpose can induce mutations and overall genetic instability, which has enforced the evolution of a complex epigenetic regulatory network to repress TE activation and transposition (Slotkin & Martienssen, 2007; Lisch, 2009; Zemach et al., 2010; Gutzat & Mittelsten Scheid, 2012; Marí‐Ordóñez et al., 2013). As a consequence, in plants, most TEs are quiescent and both transcriptionally and transpositionally inactive (Ito et al., 2011; Baubec et al., 2014). However, under stress conditions, such as seasonal and daily temperature changes, TEs can be activated and increase genetic instability (Pecinka et al., 2010; Tittel‐Elmer et al., 2010; Ito et al., 2011; Bucher et al., 2012; Cavrak et al., 2014), yet only a small proportion of TEs become active under stress (Dubin et al., 2018), revealing that there are so far largely unexplored mechanisms maintaining TE repression under stress conditions.
DNA methylation in the regulatory region of genes and TEs is crucial for silencing (Zemach et al., 2010; Jones, 2012; Schübeler, 2015; Zhang et al., 2018). Plants have DNA methylation in all three cytosine contexts (CG, CHG and CHH; H = A, T, C) (Feng et al., 2010; Law & Jacobsen, 2010; Zemach et al., 2013; Bewick & Schmitz, 2017), which are established and maintained by different mechanisms. The establishment and maintenance of CHH methylation are mediated by the DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) that is targeted by 24‐nt small interfering RNAs in the RNA‐directed DNA methylation (RdDM) pathway, which is mainly active on euchromatic TEs (Law & Jacobsen, 2010; Matzke & Mosher, 2014). In heterochromatic histone H1‐containing regions, CHH methylation is maintained by CHROMOMETHYLASE 2 (CMT2), which can be recruited by lysine 9 dimethylation on histone H3 (H3K9me2) (Stroud et al., 2014). Access of CMT2 to heterochromatic regions is mediated by the chromatin remodeller DECREASE IN DNA METHYLATION 1 (DDM1) (Zemach et al., 2013; Lyons & Zilberman, 2017). Thus, DDM1 together with CMT2 mediate CHH methylation in pericentromeric regions, whereas DRM2 targets mainly euchromatic regions (Zemach et al., 2013). CG and CHG methylation are established by the RdDM pathway, but the maintenance of CG methylation occurs by METHYLTRANSFERASE 1 (MET1) and CHG methylation by CMT3 (Law & Jacobsen, 2010; Du et al., 2015; Bewick & Schmitz, 2017; Zhang et al., 2018). CMT3 can be recruited by H3K9me2 (Law & Jacobsen, 2010; Du et al., 2012, 2015). H3K9me2 and CHG methylation facilitate each other through regulatory feedback loops (Du et al., 2012, 2014).
Heterochromatic regions are generally compact chromatin structures that are enriched for DNA methylation, H3K9me2 (Grewal & Jia, 2007; Vaillant & Paszkowski, 2007; Roudier et al., 2009) and histone H1 (Zemach et al., 2013; Rutowicz et al., 2019; Choi et al., 2020). H1 impedes the access of CMT2, causing increased DNA methylation on heterochromatic TEs upon H1 depletion (Zemach et al., 2013; Lyons & Zilberman, 2017). Loss of H1 causes only a few TEs to be upregulated, but the dual loss of H1 and DNA methylation causes strong TE activation, revealing that DNA methylation and H1 cooperatively maintain heterochromatic TEs in an inaccessible and silent state (Choi et al., 2020). In the vegetative cell of pollen, natural depletion of H1 allows access of the DNA glycosylase DEMETER to heterochromatic TEs, causing their activation by DNA demethylation (He et al., 2019).
Temperature shifts from cold to high temperature cause transcriptional activation of genes and heterochromatic TEs in Arabidopsis (Grandbastien, 1998; Lämke & Bäurle, 2017). H2A.Z eviction leads to gene expression by affecting DNA accessibility upon high temperature, whereas the loss of DNA methylation on TEs enhances TE activation in response to heat (Kumar & Wigge, 2010; Tittel‐Elmer et al., 2010; Cavrak et al., 2014). Resetting of this stress‐induced epigenetic state on TEs requires the redundant action of DDM1 and MORPHEUS' MOLECULE 1 (MOM1) and CHROMATIN ASSEMBLY FACTOR 1 (CAF1) (Pecinka et al., 2010; Iwasaki & Paszkowski, 2014). Given the connection between DDM1 and H1 and the enrichment of H1 on heterochromatic TEs, we hypothesized that H1 has a functional role in repressing TEs during temperature stress.
We explored this question using the h1.1 h1.2 double mutants and found that H1 is required in particular for TE repression in response to heat stress. We discovered that the response to H1 loss differs depending on TE location; although pericentromeric GYPSY TEs became activated in heat‐treated h1 mutants despite maintaining high levels of DNA methylation, nonpericentromeric COPIA elements became activated concomitantly with loss of DNA methylation. Many of those activated COPIA elements also became activated in heat‐treated cmt2 mutants, suggesting that H1 repressed COPIA elements through maintaining DNA methylation. Heat‐induced COPIA elements previously were shown to be strongly activated in DNA methylation impaired mutants upon heat treatment (Ito et al., 2011; Thieme et al., 2017). Most strikingly, H1‐deficient plants treated with the DNA methyltransferase inhibitor zebularine were highly tolerant to heat stress, suggesting that both H1 and DNA methylation redundantly suppress the plant response to heat stress.
Materials and Methods
Plant material, growth conditions, heat stress and chemical treatments
All wild‐type and mutant plants were in the Arabidopsis thaliana accession Columbia (Col‐0) background. The h1.1‐1 (SALK_128430C) and h1.2‐2 (GK‐116E08) plant lines were described previously (Rutowicz et al., 2015); h1.1‐1 h1.2‐2 double mutants were generated by crossing (H1, histone 1). The cmt2‐5 (SAIL_906_G03) and cmt3‐11 (SALK_148381) mutants were described previously (Chan et al., 2006; Shen et al., 2014) (CMT, CHROMOMETHYLASE 2).
Plants were grown on plates with 1/2 Murashige & Skoog medium including vitamins (Duchefa, Haarlem, The Netherlands) and 1% sucrose. Zebularine‐treated plants were grown on ½MS medium supplemented with 40 µM zebularine (Abcam, Cambridge, UK). Plates were sealed with micropore tape and stratified for 2 d at 4°C in the dark. The plates were then transferred to a growth chamber with a 16 h : 8 h, light (110 µmol m−2 s−1, 22°C) : dark (20°C) photoperiod. Ten‐day‐old seedlings were used for RNA extraction. For heat stress treatments, we incubated 10‐d‐old seedlings at 4°C for 1 h followed by 37°C for 24 h in the dark, as published previously (Shen et al., 2014). The survival rate of heat‐treated plants was determined by incubating 10‐d‐old seedlings in the dark at 4°C for 1 h followed by 37°C for 36 h and then removing to normal conditions for 2 d. Seedlings were defined as lethal if they showed bleaching of shoot apices and leaves.
RNA Isolation and quantitative reverse transcription (qRT)‐PCR
RNA extraction was performed using the MagMAX™ Plant RNA Isolation Kit (Thermo Fisher Scientific, Göteborg, Sweden) followed by cDNA synthesis using a RevertAid first‐strand cDNA synthesis kit (Thermo Fisher Scientific). Experiments were performed in biological triplicates. Quantitative PCR with gene‐specific primers (Supporting Information Table S1) was performed using a HOT FIREPol EvaGreen qPCR Supermix (Solis biodyne, Tartu, Estonia) according to the manufacturer’s instructions. The reactions were performed using the 'CFX connect' Real‐time PCR cycler detection system (Bio‐Rad). For qPCR analysis of ONSEN copy numbers, ACTIN2 was used to normalize DNA levels.
RNA sequencing
Except for the h1 samples from where only duplicates were generated, RNA of biological triplicates was extracted (50 mg seedlings per replicate) using a RNeasy Plant Mini Kit (Qiagen). Libraries were generated using DNA‐free RNA with the TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Sequencing was performed on an Illumina HiSeq2000 in 50‐bp single‐end mode. The correlation between replicates is shown Fig. S1 and details on the data in Table S2. A comparison between our RNA‐seq data and previously published data (Choi et al., 2020) of h1 mutants under nonheat conditions is shown in Fig. S2.
DNA Isolation and bisulfite sequencing
For a single replicate, four seedlings of wild‐type or the h1 mutants were collected before and after heat stress. Around 500 ng genomic DNA was extracted using a MagJET Plant Genomic DNA Kit (Thermo Fisher Scientific). Libraries for two biological replicates were prepared by Novogene (Hongkong, China) and sequenced on an Illumina HiSeq2000 in 150‐bp paired‐end mode.
Bioinformatic analysis
For RNA analysis, untrimmed reads were mapped to the TAIR10 Arabidopsis reference genome using Star (v2.5.3.a, (Dobin et al., 2013)). Expression counts were generated using the R function summarizeOverlaps from the package htseq in union mode on exons from the reference transcriptome AtRTD (Zhang et al., 2017). Differential expression analysis was performed using the R/deseq2 (v1.20.0, (Love et al., 2014)). Genes with a log2 fold change ≥ 1 or ≤ −1 and Bonferroni‐adjusted P‐value (padj) ≤ 0.05 were considered to be differentially expressed. Likewise, expression counts along transposable elements (TEs) were generated using summarizeOverlaps from R/htseq in union mode along the coordinates of the 31 189 TAIR10 annotated TEs. Differential expression analysis was performed using DESeq2, using the same threshold values as for gene transcripts.
For DNA methylation analysis, the 150‐bp‐long pair‐end reads were first quality trimmed by removing the first five bases from the 5′ end and the last 20 bases from the 3′ end. Reads were mapped to the reference genome TAIR10 in PE mode (‐‐score_min L, 0, −0.6) using Bismark v0.16.3. Mapped reads were deduplicated and cytosine methylation values calculated using the Bismark Methylation Extractor. Methylation reports were pooled for both replicates for further analyses.
Differentially methylated regions (DMRs) in each context were calculated only for demethylation in 50‐bp bins considering as the fractional methylation threshold the bins with differences below the 1st decile. Those were parsed if they passed a fisher test (P < 0.01).
Published datasets were processed as follows: H1 chromatin affinity purification (ChAP) reads from GSE122394 (Choi et al., 2020) control libraries were mapped to the TAIR10 genome using bowtie2. Coverage was calculated using the coverage function from R/nucleR (Flores & Orozco, 2011) and normalized by the total number of mapped reads. The normalized input signal was subtracted from the H1.1 and H1.2 ChAP signal. Small RNA reads from GSE116067 (Tan et al., 2018) were mapped to the TAIR10 genome using bowtie (‐v 0‐best). Pooled mapped reads were separated into two categories (21/22‐ and 24‐nucleotides‐long) and remapped using shortstack (Johnson et al., 2016). The alignments were normalized by converting coverage values to reads per million values (Wang et al., 2020).
Pericentrometic heterochromatin was considered to span the regions between the following coordinates: Chr1: 11 500 020–17 696 331; Chr2: 1100 003–7192 918; Chr3: 10 298 763–17 289 015; Chr4: 1500 001–2300 002; Chr4: 2800 003–6300 004 and Chr5: 8999 997–5982 772 (Copenhaver et al., 1999).
Results
Absence of H1 affects gene and TE activation after heat stress
Histone H1 is abundant throughout the whole genome and mediates global nucleosome positioning in plants and animals, but only few genes and TEs become deregulated when H1 is deficient (Geeven et al., 2015; Rutowicz et al., 2019; Choi et al., 2020). In plants, heat stress causes dispersal of heterochromatin, similar to H1‐deficient mutants (Pecinka et al., 2010; Rutowicz et al., 2019). We therefore addressed the question of whether H1 has a role in antagonizing heat stress. Because H1.1 and H1.2 are constitutively expressed and highly redundant (Rutowicz et al., 2019; Choi et al., 2020), we used h1.1 h1.2 double mutants, henceforth referred to as h1 mutants. Expression levels of H1.3 are very low under normal conditions (Ascenzi & Gantt, 1997; Rutowicz et al., 2015) and H1.3 was not induced by heat stress (Fig. S3), justifying the use of the h1 mutants to explore the role of H1 in the heat response. We generated RNA‐seq profiling data of wild‐type and h1 mutants before and after heat stress (4°C for 1 h followed by 37°C for 24 h in dark conditions). It was previously shown that loss of H1 activates gene expression but only weakly derepresses TEs (Rutowicz et al., 2019; Choi et al., 2020), we therefore focused initially on upregulated genes in h1 mutants. We found that 301 genes (log2 fold change ≥ 1 and Bonferroni‐adjusted P‐value (padj) ≤ 0.05) were upregulated in h1 mutants compared to wild‐type (Fig. 1a; Table S3). Surprisingly, only 116 genes (log2 fold change ≥ 1 and Bonferroni‐adjusted P‐value (padj) ≤ 0.05) were activated in h1 after heat stress compared to heat‐treated wild‐type (Fig. 1a; Table S3). Many genes (265), including 68 heat‐responsive genes in wild‐type that were activated in h1, were not changed or became downregulated in h1 upon heat (Figs 1a,c, S4a,b), suggesting that heat attenuates the effect of loss of H1 on genes. In order to test whether H1 functions as a repressor after heat stress, we analyzed previously published H1 enrichment data (Choi et al., 2020) on upregulated genes and downregulated genes in h1 mutants upon heat stress. We found that upregulated genes in heat‐treated h1 mutants were significantly more enriched for H1 than downregulated genes (Fig. S5), indicating that H1 has mainly a repressive role after heat stress. We identified three over‐represented gene ontology (GO) functional categories among the upregulated genes in h1 mutants upon heat stress, corresponding to carbohydrate binding, hydrolase activity and nucleoside‐triphosphatase (Fig. S6). Consistent with previous findings (Choi et al., 2020), we found only few (13) TEs upregulated in h1 mutants, but this number substantially increased (86 upregulated TEs) in h1 upon heat compared to heat‐treated wild‐type (Fig. 1b,d; Table S4). Most of these TEs also were heat responsive in wild‐type (Figs 1d, S4c). This reveals a repressive role of H1 on TEs in response to heat stress. Previous work revealed that loss of H1 together with loss of DNA methylation causes TE de‐repression (Choi et al., 2020). To understand the H1 repressive mechanism in response to heat, we generated bisulfite‐sequencing data of wild‐type and h1 mutants with and without heat treatment. We found that CG methylation levels in the 2‐kb upstream region of the 265 upregulated genes in h1 mutants without heat treatment (Fig. 1a) did not significantly change compared to wild‐type, irrespective of heat treatment (Fig. 1e). By contrast, CHG and CHH methylation decreased in h1 mutants without heat treatment, and also decreased to similar levels in wild‐type and h1 mutants upon heat treatment (Fig. 1f,g). Loss of CHH methylation in h1 was partly attenuated by heat stress, providing a possible explanation why many genes became activated in h1, but not in heat‐treated h1 mutants (Fig. 1a,c). We identified CHG and CHH differentially methylated regions (DMRs) in h1 mutants with and without heat treatment. We focussed on the 2‐kb upstream region of those 265 genes that were upregulated in untreated h1 mutants but did not change expression upon heat treatment (Fig. 1a,h). Upon heat treatment, we found that 49 genes gained hypermethylated CHG or CHH regions (Hyper DMRs), whereas 81 genes lost hypomethylated regions (Hypo DMRs; Fig. 1h) in h1 mutants, which restored CHG or CHH methylation to wild‐type levels upon heat (Fig. 1f,g), consistent with fewer genes being activated in h1 mutants after heat stress. Together, our data reveal that heat treatment and loss of H1 both caused a reduction of CHG and CHH methylation (Fig. 1f,g), likely a consequence of opened chromatin structure (Zemach et al., 2013; Rutowicz et al., 2019). However, heat treatment of h1 mutants partly restored CHH methylation (Fig. 1g,h), indicating that heat ameliorates the effect of loss of H1 on genes.
Fig. 1.
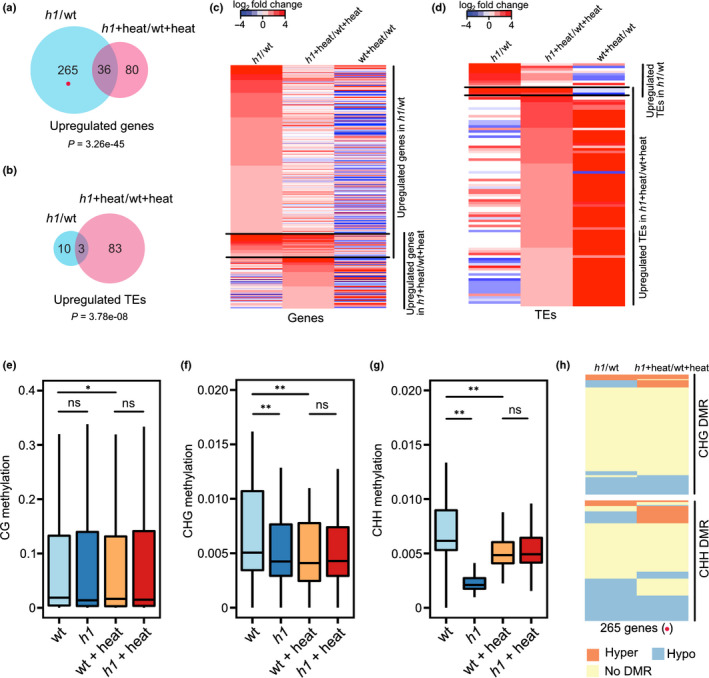
Histone H1 is required for transposable element (TE) repression after heat stress inArabidopsis. Venn diagrams show upregulated genes (log2‐fold change ≥ 1, adjusted P‐value (padj) ≤ 0.05, (a)) and TEs (log2‐fold change ≥ 1, padj ≤ 0.05, (b)) in h1 mutants vs wild‐type (h1/WT) and h1 plus heat vs WT plus heat (h1 + heat/WT + heat). The red dot in (a) represents the same group of genes marked in panel (h). Heat maps show upregulated genes (c) and TEs (d) in h1 mutants vs WT and h1 plus heats WT plus heat. Averaged CG (e), CHG (f) and CHH (g) methylation in the −2 kb to transcription start site (TSS) region of 265 upregulated genes in h1 mutants vs WT but not in h1 plus heats WT plus heat are shown in four different conditions of WT, h1, WT plus heat (WT + heat), and h1 plus heat (h1 + heat). *, P < 0.05; **, P < 0.01; ns, not significant (Wilcoxon test). The lower and upper hinges of the boxplots correspond to the first and third quartiles of the data, the black lines within the boxes mark the median. CHG and CHH DMRs (h) in the −2 kb to TSS region of 265 upregulated genes in h1 vs WT. Hyper and hypo refer to hyper‐ or hypomethylated regions, respectively, present in the upstream region of genes in h1 vs WT or h1 plus heat vs WT plus heat.
Gain of DNA methylation is not sufficient to repress GYPSY TEs in h1 mutants after heat stress
In contrast with genes that were less affected in heat‐treated h1 mutants compared to heat‐treated wild‐type plants, we found increased numbers of TEs upregulated in heat‐stressed h1 mutants compared to heat‐stressed wild‐type plants (Fig. 1b,d). In order to understand the role of H1 on TE repression in response to heat, we analyzed H1 enrichment data (Choi et al., 2020) to test whether upregulated TEs differ in their H1 enrichment compared to nonupregulated TEs. Indeed, we found that in wild‐type, upregulated TEs in heat‐treated h1 mutants were more strongly enriched for H1 compared to TEs not affected by heat (Fig. 2a). Those upregulated TEs also had higher wild‐type methylation levels in all three sequence contexts compared to nonupregulated TEs (Fig. 2b), consistent with H1 being associated with heavily methylated heterochromatic TEs (Rutowicz et al., 2015; Choi et al., 2020). Upregulated TEs belonged to different TE families, among which LTR/GYPSY (P = 2.81e‐24), LTR/COPIA (P = 9.88e‐05) and DNA/En‐Spm (P = 0.0045) families were significantly enriched (Fig. 2c) (LTR, long terminal repeat). Among the most enriched retrotransposons, LTR/COPIA TEs were mildly increased in h1 mutants after heat stress, whereas LTR/GYPSY TEs were more strongly upregulated (Fig. 2d). We analyzed DNA methylation levels of all GYPSY TEs and found that they gained DNA methylation in all sequence contexts in h1 compared to wild‐type, independent of being subjected to heat stress or not (Fig. 2e–g). This is consistent with previous findings revealing that absence of H1 causes gain of methylation in heterochromatic regions, but loss of methylation in euchromatic regions (Zemach et al., 2013; Lyons & Zilberman, 2017). Upregulated GYPSY TEs in h1 upon heat followed this trend and likewise gained DNA methylation of all three sequence contexts in h1 mutants vs WT both before and after heat stress (Fig. 2h–j). This suggests that in the absence of H1, high levels of DNA methylation are not sufficient to repress a subset of TEs in response to heat stress. Thus, H1 has an indispensable and DNA methylation‐independent repressive function in response to heat stress.
Fig. 2.
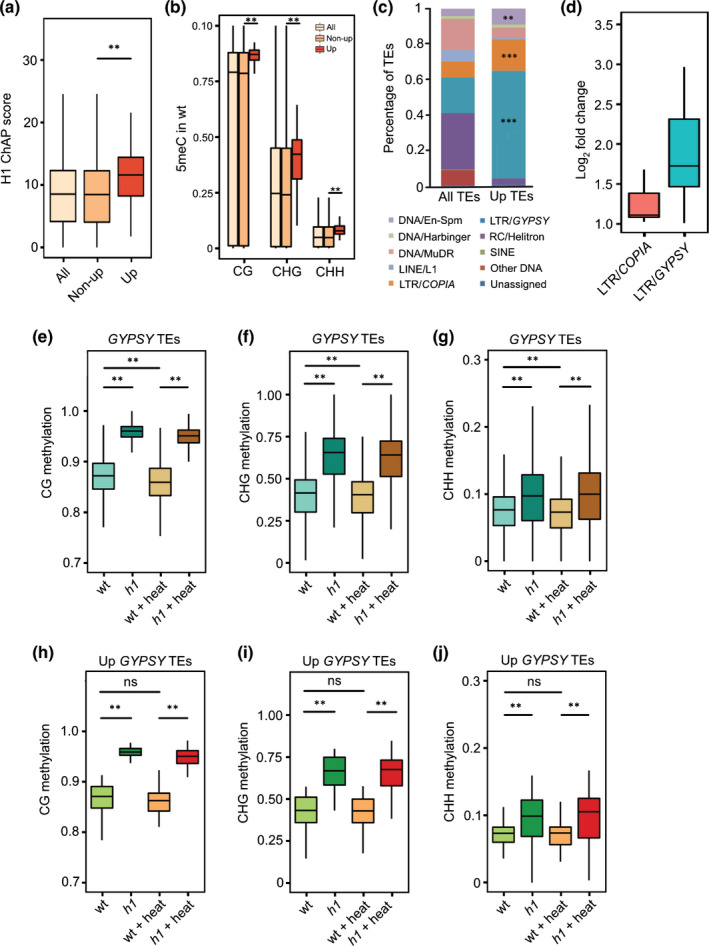
Histone H1‐mediated repression of GYPSY transposable elements (TEs) upon heat occurs independently of DNA methylation in Arabidopsis. Wild‐type (WT) H1 enrichment (a) on all, nonupregulated and upregulated TEs in h1 plus heat vs WT plus heat. **, P < 0.01 (Wilcoxon test). ChAP, chromatin affinity purification. H1 ChAP score was defined as the average ChAP coverage from start to end of the TE. Wild‐type DNA methylation level (CG, CHG and CHH, (b)) on all, nonupregulated and upregulated TEs in h1 plus heat vs WT plus heat. **, P < 0.01 (Wilcoxon test). Percentages of TEs are classified in different families (c). Up TEs refer to the upregulated TEs in h1 plus heat vs WT plus heat. **, P < 0.01; ***, P < 0.001 (hypergeometric test). Expression level (log2‐fold change, (d)) of upregulated long terminal repeat (LTR)/COPIA and LTR/GYPSY elements in h1 plus heat vs WT plus heat. Averaged CG (e), CHG (f) and CHH (g) methylation level on all GYPSY TEs in WT, h1, WT plus heat (WT + heat) and h1 plus heat (h1 + heat). Averaged CG (h), CHG (i) and CHH (j) methylation on upregulated GYPSY TEs in h1 plus heat vs WT plus heat in four different conditions of WT, h1, WT plus heat and h1 plus heat. **, P < 0.01; ns, not significant (Wilcoxon test). The lower and upper hinges of the boxplots correspond to the first and third quartiles of the data, the black lines within the boxes mark the median.
H1 and DNA methylation cooperatively control a subset of COPIA elements in response to heat
Upregulated COPIA elements in h1 mutants upon heat were mainly COPIA78/ONSEN elements, including the eight full‐length heat responsive ONSEN TEs (Ito et al., 2013; Cavrak et al., 2014; Pietzenuk et al., 2016). In contrast with GYPSY TEs that strongly gained DNA methylation in h1 mutants after heat stress, there were only mild changes in DNA methylation on COPIA TEs (Fig. 3a–c). However, those heat‐responsive COPIA78/ONSEN elements that became upregulated upon heat stress, had increased DNA methylation levels in h1 mutants without heat treatment, but experienced a strong loss of CG and CHG methylation upon heat treatment, independent of the presence or absence of H1 (Fig. 3d–f). Stronger activation of COPIA78/ONSEN elements in h1 mutants upon heat compared to heat‐treated wild‐type suggests that under heat stress, DNA methylation and H1 cooperatively repress a subset of COPIA TEs.
Fig. 3.
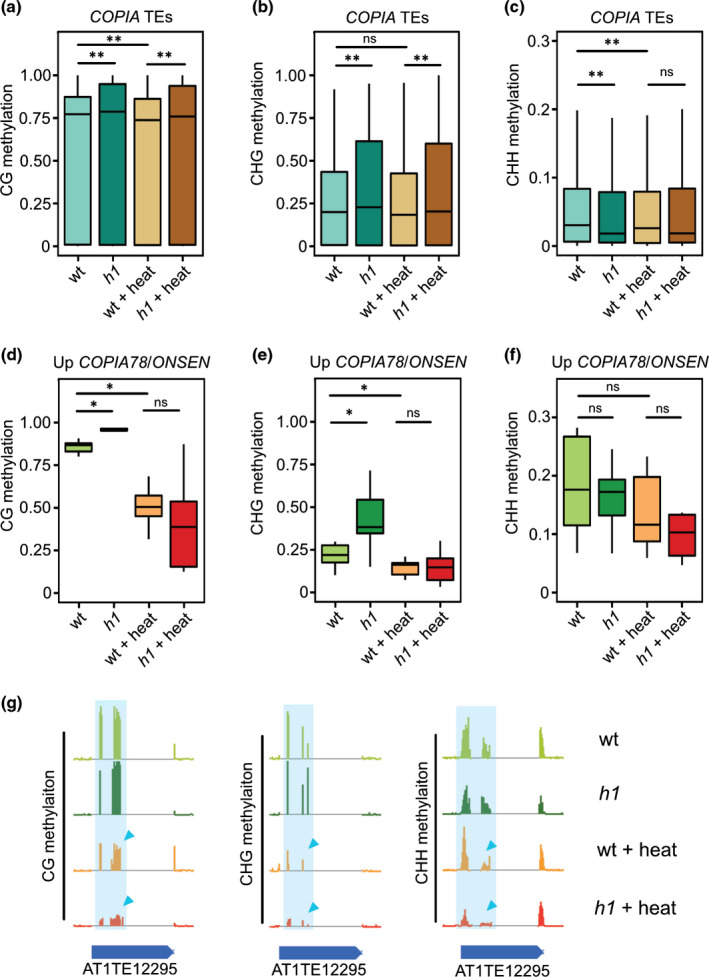
Histone H1 represses COPIA78/ONSEN transposable elements (TEs) upon heat through maintaining DNA methylation in Arabidopsis. Averaged CG (a), CHG (b) and CHH (c) methylation on all COPIA TEs in wild‐type (WT), h1, wt plus heat (WT + heat) and h1 plus heat (h1 + heat). Averaged CG (d), CHG (e) and CHH (f) methylation on upregulated COPIA78/ONSEN elements in h1 plus heat vs WT plus heat in four different conditions of WT, h1, WT plus heat, and h1 plus heat. *, P < 0.05; **, P < 0.01; ns, not significant (Wilcoxon test). The lower and upper hinges of the boxplots correspond to the first and third quartiles of the data, the black lines within the boxes mark the median. Example of CG, CHG and CHH methylation levels (g) on ONSEN 1 (AT1TE12295). Triangle points to the decreased DNA methylation region.
Among those upregulated COPIA TEs were three COPIA78/ONSEN elements (ONSEN 1 (AT1TE12295), ONSEN 2 (AT3TE92522) and ONSEN 3 (AT5TE15240)) that had more strongly reduced DNA methylation in all three sequence contexts in heat‐treated h1 mutants compared to heat‐treated wild‐type (Fig. 3g, S7). Heat treatment in combination with impaired epigenetic repression previously was shown to cause ONSEN transposition (Ito et al., 2011; Thieme et al., 2017), however, the copy numbers of ONSEN remained the same in the second generation of heat‐treated h1 mutants as in wild‐type, revealing that H1 does not control ONSEN transposition (Fig. S8).
CMT2 controls COPIA78/ONSEN element silencing during heat stress
Natural CMT2 variation was shown to correlate with CHH methylation variation and temperature seasonality in Arabidopsis, and cmt2 mutants are more heat tolerant (Shen et al., 2014; Dubin et al., 2015). Because CMT2 acts on H1 containing loci (Zemach et al., 2013), we explored the connection between H1 and CMT2 under heat stress. We generated transcriptome data of cmt2 mutants before and after heat stress. Only 26 TEs became activated in cmt2 mutants upon heating (Fig. 4a; Table S4). Among those, six TEs belonged to the GYPSY family, whereas 13 TEs belonged to the COPIA78/ONSEN subfamily. Among the upregulated TEs, 15 overlapped with TEs upregulated in heat‐treated h1 mutants (Fig. 4a), including 12 COPIA78/ONSEN TEs. The COPIA78/ONSEN elements were mildly upregulated in heat‐treated h1 mutants but became strongly upregulated in heat‐treated cmt2 mutants (Fig. 4b), revealing that CMT2, like H1, plays a major repressive role on TEs of the COPIA78/ONSEN subfamily in response to heat stress. Heat stress‐induced expression of COPIA78/ONSEN also was observed in RNA‐directed DNA methylation (RdDM) mutants (Ito et al., 2011), suggesting that COPIA78/ONSEN elements are common targets of the CMT2 and RdDM pathways. Consistently, upregulated COPIA elements strongly accumulated 24‐nt siRNAs, differing from nonupregulated COPIA elements and Gypsy elements (Fig. 4c). The RdDM pathway targets the edges of long TEs, whereas CMT2 is required for TE body methylation on long heterochromatic TEs (Zemach et al., 2013). In agreement with the dual regulation of COPIA78 elements by both pathways, upregulated COPIA elements were significantly larger than upregulated GYPSY elements (Fig. 4d). Upregulated COPIA and GYPSY elements also differed in their chromosomal location: whereas upregulated GYPSY TEs were concentrated in the pericentromeric region, upregulated COPIA TEs were mainly dispersed along the chromosome arms (Fig. 4e), which may explain their different mode of regulation.
Fig. 4.
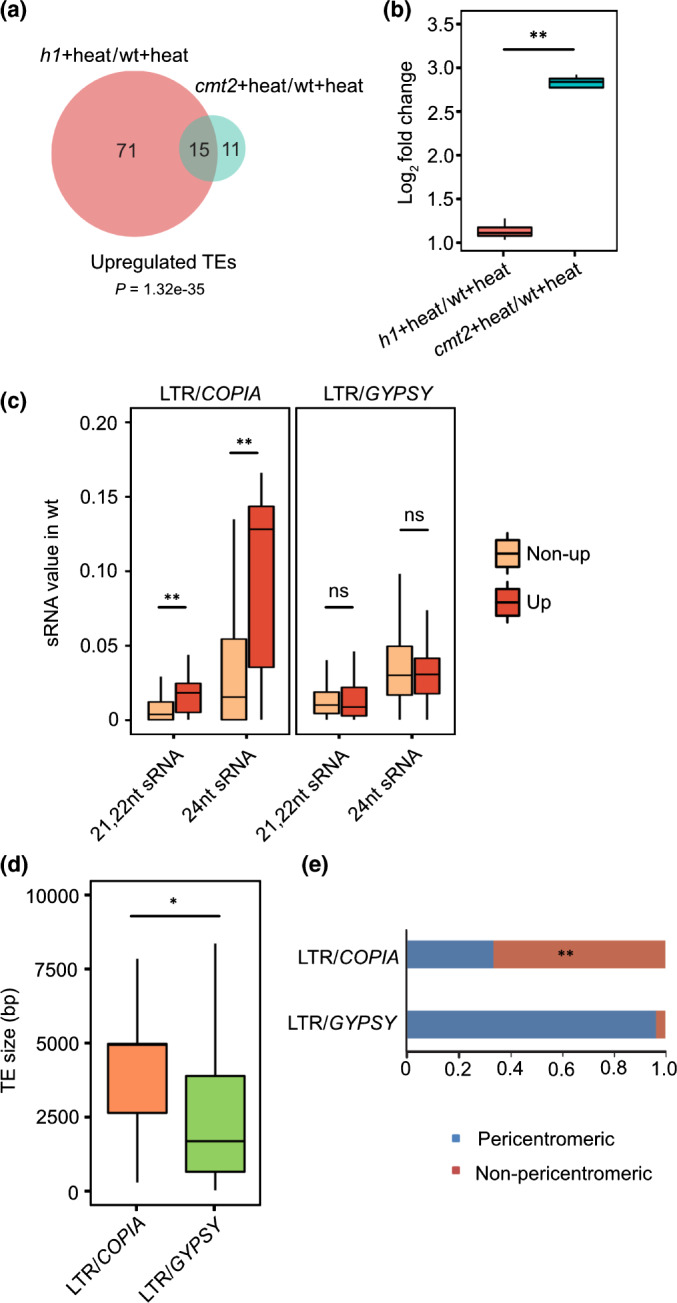
CHROMOMETHYLASE 2 (CMT2) is required for COPIA78/ONSEN transposable element (TE) repression under heat conditions in Arabidopsis. Venn diagram (a) shows upregulated TEs in h1 plus heat vs wild‐type (WT) plus heat (h1 + heat/WT + heat) and cmt2 plus heats wt plus heat (cmt2 + heat/WT + heat). Boxplot (b) shows the expression level of commonly upregulated COPIA78/ONSEN elements in h1 plus heat vs WT plus heat and cmt2 plus heat vs WT plus heat. **, P < 0.01 (Wilcoxon test). Boxplot (c) shows WT sRNA value on upregulated and nonupregulated long terminal repeat (LTR)/COPIA and LTR/GYPSY elements in h1 plus heat vs WT plus heat. **, P < 0.01; ns, not significant (Wilcoxon test). Boxplot (d) shows the size of upregulated LTR/COPIA TEs and LTR/GYPSY TEs in h1 plus heat vs WT plus heat. *, P < 0.05 (Wilcoxon test). The lower and upper hinges of the boxplots correspond to the first and third quartiles of the data, the black lines within the boxes mark the median. The percentages of upregulated LTR/COPIA and LTR/GYPSY‐type TEs in h1 plus heat vs WT plus heat are classified by location (e). Upregulated COPIA elements are significantly enriched in nonpericentromeric regions compared to all LTR/COPIA TEs. **, P < 0.01 (hypergeometric test).
H1‐deficient plants treated with zebularine have improved heat‐stress tolerance
In order to further explore the interaction between H1‐mediated TE repression and DNA methylation, we grew wild‐type and h1 plants on media supplemented with zebularine, a widely used inhibitor of DNA methyltransferases (Griffin et al., 2016). Consistent with the transcriptome data, COPIA78/ONSEN expression increased upon heat treatment and was further increased in h1 mutants upon heat as tested by qRT‐PCR (Fig. 5a,b; note different axis scale). Zebularine treatment increased COPIA78/ONSEN expression upon heat treatment, but the effect was similar in wild‐type and h1 mutants (Fig. 5b), consistent with the strong effect of cmt2 mutants on COPIA78/ONSEN expression upon heat treatment (Fig. 4b). By contrast, one GYPSY TE expression was increased in zebularine‐treated wild‐type and h1 mutants, and strongly increased in heat‐treated h1 mutants compared to wild‐type (Fig. 5c,d), indicating a synergistic repressive effect of H1 and DNA methylation on GYPSY repression upon heat treatment.
Fig. 5.
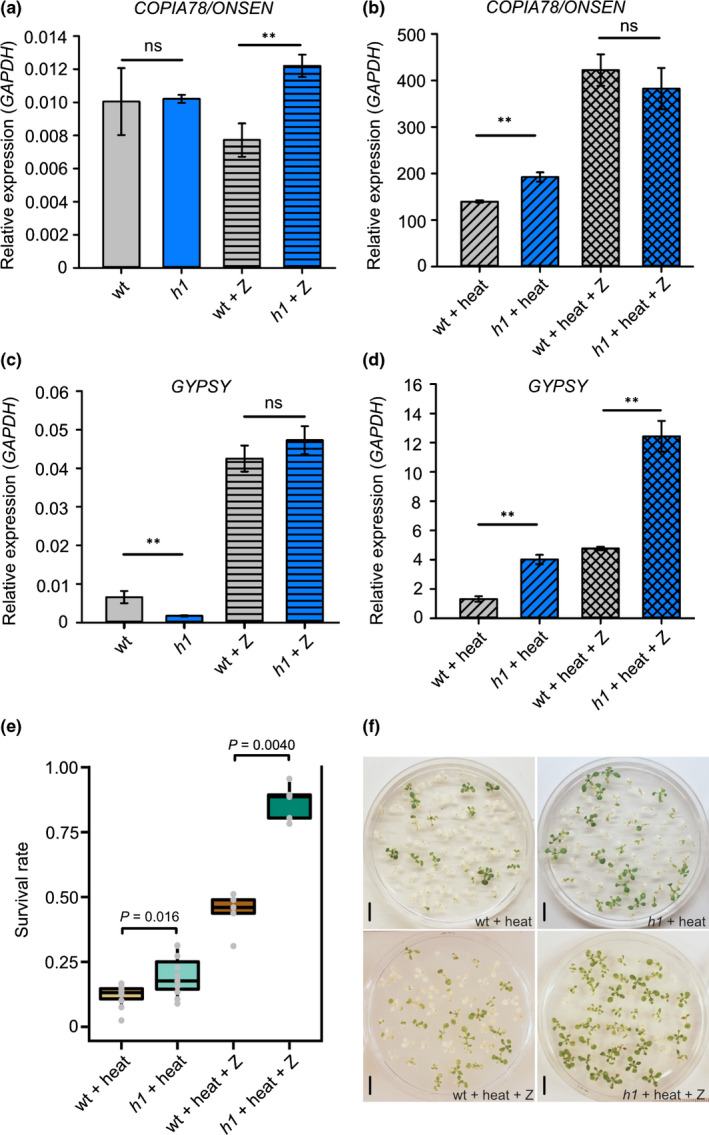
Loss of histone H1 and DNA hypomethylation increase Arabidopsis heat tolerance. COPIA78/ONSEN (a) and GYPSY (c) transcripts in wild‐type (WT), h1, WT plus 40 µm zebularine (WT + Z) and h1 plus 40 µm zebularine (h1 + Z). COPIA78/ONSEN (b) and GYPSY (d) transcripts in wt plus heat (WT + heat), h1 plus heat (h1 + heat), WT plus heat and 40 µm zebularine (WT + heat + Z) and h1 plus heat and 40 µm zebularine (h1 + heat + Z). Error bars represent 1 SD. **, P < 0.01; ns, not significant (Student’s t‐test). Survival rate (e) of seedlings of WT, h1 mutants, WT plus 10 µm zebularine and h1 plus 10 µm zebularine after 36 h heat stress and 48 h recovery under long day conditions. Each of three biological replicates corresponds to ≥ 90 seedlings. The lower and upper hinges of the boxplots correspond to the first and third quartiles of the data, the black lines within the boxes mark the median. Representative pictures (f) of plates with WT, h1 mutants, WT plus 10 µm zebularine and h1 plus 10 µm zebularine after 36 h heat stress and 48 h recovery under long day conditions. Bars, 1 cm.
In order to functionally explore the impact of H1 on the temperature‐stress response, we tested the sensitivity of wild‐type and h1 mutants to severe heat stress (1 h at 4°C followed by 36 h at 37°C in dark conditions) by measuring the survival rate of heat‐treated plants after returning to normal conditions for 2 d. The leaves of most plants gradually became yellow and bleached and the plants finally died during these two days, resulting in ≤ 20% survival rate of wild‐type plants. The h1 mutants had a significantly higher survival rate (1.6‐fold greater; P = 0.016, Wilcoxon signed‐rank test) than wild‐type (Fig. 5e,f). Strikingly, however, when the zebularine was present, the survival rate of wild‐type and h1 mutants strongly increased after heat stress, reaching c. 50% in wild‐type and c. 86% in h1 mutants (Fig. 5e,f), revealing a so far unknown potential of hypomethylation in combination with H1 deficiency to antagonize heat stress. We assessed whether the increased heat tolerance was a consequence of increased transposition rates of COPIA78/ONSEN TEs in h1 mutants treated with zebularine and heat; however, we did not observe increased ONSEN copy numbers in the second generation of heat‐ and zebularine‐treated h1 mutants compared to wild‐type (Fig. S9).
Discussion
In this work, we explored the function of the linker histone H1 in response to heat stress in plants and discovered a novel role of H1 in modulating the plant response to heat. Although loss of H1 causes only a mild effect under normal conditions (Rutowicz et al., 2015, 2019), we found that under heat stress, loss of H1 strongly activated transposable elements (TEs), mainly GYPSY and COPIA elements. Stress treatment of the h1.3 mutant previously was reported to cause increase of CHH methylation at defined loci and reduced gene expression (Rutowicz et al., 2015). Likewise, we observed that decreased CHH methylation in the 2‐kb upstream region of upregulated genes in h1 mutants was partly restored in heat‐treated h1, suggesting that increased CHH methylation accounts for the dampened gene expressed in stressed h1 mutants. Histone H1 together with DNA methylation were shown to jointly suppress aberrant intragenic transcription (Choi et al., 2020). Thus, it isalso possible that the formation of aberrant transcripts in h1 at least in part account for reduced transcript levels.
In heterochromatic regions of Arabidopsis, H1 impedes DNA methyltransferase accessibility to chromatin, leading to increased DNA methylation upon H1 depletion (Zemach et al., 2013; Lyons & Zilberman, 2017). Although we found this effect very pronounced on GYPSY TEs, this effect was much weaker on COPIA TEs, indicating distinct regulation of both types of TEs by H1 (Fig. 6). In heat‐treated h1 mutants, gain of DNA methylation was not sufficient to repress GYPSY TEs, demonstrating a key repressive role for H1 on GYPSY TEs. Depletion of DNA methylation by zebularine treatment caused strongly increased expression of GYPSY TEs in heat‐treated h1 mutants, exceeding the upregulation in heat‐treated h1 without zebularine, revealing that H1 and DNA methylation synergistically repress GYPSY TEs under heat, consistent with previous work (Choi et al., 2020).
Fig. 6.
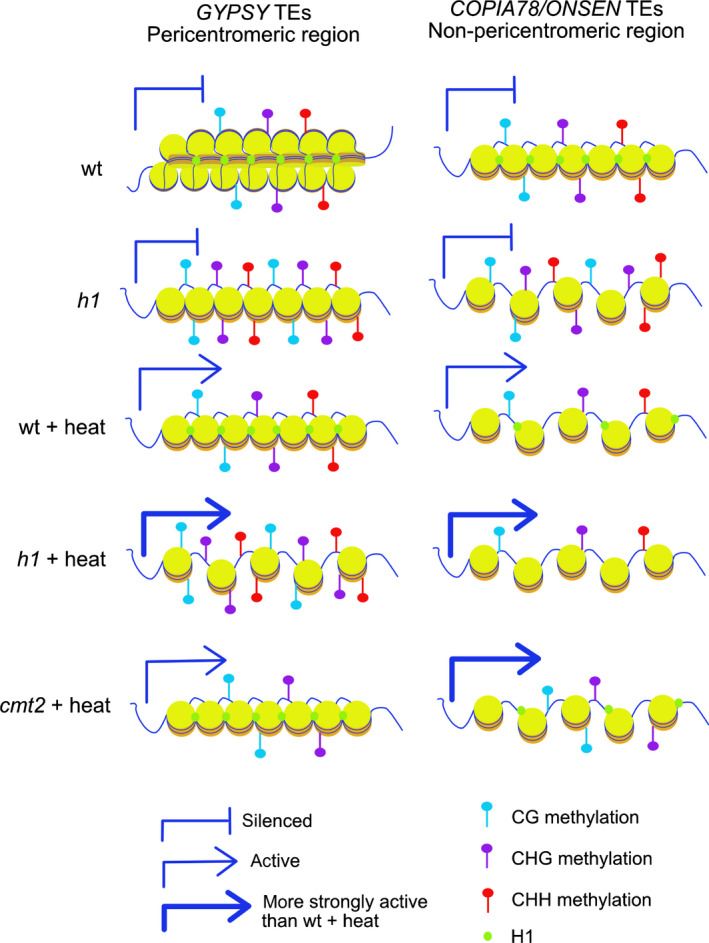
Model depicting the role of histone H1 and DNA methylation in selective repression of GYPSY and COPIA78/ONSEN retrotransposons in Arabidopsis. Upregulated GYPSY transposable elements (TEs) in heat‐treated h1 mutants are located in pericentromeric regions, whereas upregulated COPIA78/ONSEN elements are dispersed in the chromosome arms. In h1 mutants, chromatin structure is opened and the two groups of TEs gain DNA methylation and remain silenced. After heat stress of wild‐type plants (WT + heat), DNA methylation level remains unchanged on upregulated GYPSY TEs, but decrease on upregulated COPIA78/ONSEN elements, thus likely contributing to their activation. Opened chromatin structure is likely sufficient to induce expression of GYPSY TEs. In heat‐treated h1 mutants (h1 + heat), GYPSY TEs gain DNA methylation, which, however, is not sufficient for repression and GYPSY TEs become more strongly expressed compared to heat‐treated WT. By contrast, COPIA78/ONSEN TEs lose DNA methylation in heat‐treated h1 mutants, which contributes to their upregulation, because COPIA78/ONSEN TEs, but not GYPSY TEs, are strongly activated in heat‐treated chromomethylase 2 (cmt2) mutans (cmt2 + heat).
Like GYPSY TEs that gained DNA methylation upon loss of H1, heat‐induced COPIA TEs also gained DNA methylation in h1 mutants. However, In contrast with GYPSY TEs, which maintained high levels of DNA methylation in h1 mutants upon heat treatment, COPIA elements lost DNA methylation upon heat treatment in wild‐type and h1 mutants. Because COPIA elements became more strongly activated in heat‐treated h1 mutants than heat‐treated wild‐type, we conclude that the combination of reduced DNA methylation and loss of H1 caused transcriptional activation of COPIA elements upon heat. Nevertheless, most of the heat induced COPIA elements also were activated in heat‐treated chromomethylase 2 (cmt2) mutants, indicating that a complete loss of CHH methylation has the activating effect on the same COPIA elements as loss of H1 under heat.
The difference in chromosomal location may explain the different regulatory impact of DNA methylation on GYPSY and COPIA element expression in response to heat. GYPSYelements are located in compacted heterochromatic regions and possibly rely on Microrchidia (MORC) proteins for repression. MORC proteins act in heterochromatic regions and enforce silencing of TEs independently of DNA methylation, likely by affecting chromatin structure (Moissiard et al., 2012). Heat causes loss of chromocenter organization, similar to the effect caused by loss of H1 (Pecinka et al., 2010; Rutowicz et al., 2019; Choi et al., 2020). Thus, the combination of H1 loss together with heat may cause a synergistic effect, opening heterochromatic pericentromeric regions and activating GYPSY TE expression. By contrast, nonpericentromeric COPIA elements affected by heat, likely rely on the combination of CMT2 and H1 for stable repression (Choi et al., 2020).
Most strikingly, we found that depletion of DNA methylation by zebularine enhanced heat tolerance and that this effect was strongly enhanced in h1 mutants. Previous work revealed that loci controlling adaptive responses to the environment are frequent targets of TE insertions (Ito et al., 2016; Quadrana et al., 2016). COPIA TEs contain heat responsive factor binding elements (HREs) that confer heat response to the nearby genes (Ito et al., 2011; Cavrak et al., 2014; Pietzenuk et al., 2016; Thieme et al., 2017), and transcriptional activation of COPIA78/ONSEN elements correlates with environmental heat stress in most species of the Brassicaceae (Ito et al., 2013). CHH methylation‐deficient cmt2 mutants are more heat‐tolerant than wild‐type (Shen et al., 2014), consistent with the upregulated COPIA type TEs in both cmt2 and h1 mutants upon heat stress. We speculate that reduced DNA methylation and loss of H1 exposes heat‐responsive elements that are epigenetically silenced under normal conditions. The combination of loss of H1 and reduced DNA methylation acts cooperatively on COPIA and GYPSY TE activation, possibly explaining the strongly enhanced heat tolerance of zebularine‐treated h1 seedlings. Whether or not this effect is heritable remains to be explored.
Author contributions
LH conceived the study; SL and JdJ designed and performed the experiments; MST‐A and JS‐G analyzed high‐throughput sequencing data; and SL and CK interpreted the data and wrote the manuscript. All authors approved the final version of the manuscript.
Data availability
The RNA and bisulfite‐sequencing data in this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE152402.
Supporting information
Fig. S1 Correlation analysis of RNA‐seq samples.
Fig. S2 Comparison of data in this study with previously published data (Choi et al., 2020).
Fig. S3 H1.3 expression does not change in heat‐treated h1 mutants compared to heat‐treated WT.
Fig. S4 A substantial proportion of upregulated genes in h1 mutants and TEs in h1 upon heat are heat‐responsive in WT.
Fig. S5 H1 represses gene expression after heat stress.
Fig. S6 Gene ontology terms of upregulated genes in h1 plus heat vs WT plus heat.
Fig. S7 Decreased CG, CHG and CHH methylation on upregulated Copia78/ONSEN elements.
Fig. S8 H1 does not affect ONSEN remobilization upon heat.
Fig. S9 ONSEN does not amplify in the progeny of heat and zebularine‐treated WT and h1 mutants.
Table S1 Primers used in the manuscript.
Table S2 Mapping statistics of RNA sequencing data.
Table S3 Lists of deregulated genes in h1 mutants before and after heat stress.
Table S4 Lists of upregulated TEs in h1 mutants and cmt2 mutants before and after heat stress.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
This work was funded by the Swedish Research Council VR (grant no. 2014‐05822 to LH), the Swedish Research Council Formas (grant no. 2016‐00961 to LH) and by the Knut‐and‐Alice‐Wallenberg Foundation (grant no. 2012.0087 to LH). RNA‐seq sequencing was performed by the SNP&SEQ Technology Platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory. The SNP&SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
References
- Ascenzi R, Gantt JS. 1997. A drought‐stress‐inducible histone gene in Arabidopsis thaliana is a member of a distinct class of plant linker histone variants. Plant Molecular Biology 34: 629–641. [DOI] [PubMed] [Google Scholar]
- Baubec T, Finke A, Mittelsten Scheid O, Pecinka A. 2014. Meristem‐specific expression of epigenetic regulators safeguards transposon silencing in Arabidopsis . EMBO Reports 15: 446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyayev A. 2014. Bursts of transposable elements as an evolutionary driving force. Journal of Evolutionary Biology 27: 2573–2584. [DOI] [PubMed] [Google Scholar]
- Bewick AJ, Schmitz RJ. 2017. Gene body DNA methylation in plants. Current Opinion in Plant Biology 36: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucher E, Reinders J, Mirouze M. 2012. Epigenetic control of transposon transcription and mobility in Arabidopsis . Current Opinion in Plant Biology 15: 503–510. [DOI] [PubMed] [Google Scholar]
- Cavrak VV, Lettner N, Jamge S, Kosarewicz A, Bayer LM, Scheid OM. 2014. How a retrotransposon exploits the plant’s heat stress response for its activation. PLoS Genetics 10: e1004115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW‐L, Henderson IR, Zhang X, Shah G, Chien JS‐C, Jacobsen SE. 2006. RNAi, DRD1, and histone methylation actively target developmentally important non‐CG DNA methylation in Arabidopsis . PLoS Genetics 2: e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Lyons DB, Kim MY, Moore JD, Zilberman D. 2020. DNA methylation and histone H1 jointly repress transposable elements and aberrant intragenic transcripts. Molecular Cell 77: 310–323.e7. [DOI] [PubMed] [Google Scholar]
- Copenhaver GP, Nickel K, Kuromori T, Benito M‐I, Kaul S, Lin X, Bevan M, Murphy G, Harris B, Parnell LD et al 1999. Genetic definition and sequence analysis of Arabidopsis centromeres. Science 286: 2468–2474. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Groth M, Feng S, Hale CJ, Li S, Vashisht AA, Gallego‐Bartolome J, Wohlschlegel JA, Patel DJ et al 2014. Mechanism of DNA methylation‐directed histone methylation by KRYPTONITE. Molecular Cell 55: 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Johnson LM, Jacobsen SE, Patel DJ. 2015. DNA methylation pathways and their crosstalk with histone methylation. Nature Reviews Molecular Cell Biology 16: 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhong X, Bernatavichute YV, Stroud H, Feng S, Caro E, Vashisht AA, Terragni J, Chin HG, Tu A et al 2012. Dual binding of chromomethylase domains to H3K9me2‐containing nucleosomes directs DNA methylation in plants. Cell 151: 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin MJ, Mittelsten Scheid O, Becker C. 2018. Transposons: a blessing curse. Current Opinion in Plant Biology 42: 23–29. [DOI] [PubMed] [Google Scholar]
- Dubin MJ, Zhang P, Meng D, Remigereau M‐S, Osborne EJ, Paolo Casale F, Drewe P, Kahles A, Jean G, Vilhjálmsson B et al 2015. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. eLife 4: e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, Chen P‐Y, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME et al 2010. Conservation and divergence of methylation patterning in plants and animals. Proceedings of the National Academy of Sciences, USA 107: 8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Orozco M. 2011. nucleR: a package for non‐parametric nucleosome positioning. Bioinformatics 27: 2149–2150. [DOI] [PubMed] [Google Scholar]
- Geeven G, Zhu Y, Kim BJ, Bartholdy BA, Yang S‐M, Macfarlan TS, Gifford WD, Pfaff SL, Verstegen MJAM, Pinto H et al 2015. Local compartment changes and regulatory landscape alterations in histone H1‐depleted cells. Genome Biology 16: 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandbastien M‐A. 1998. Activation of plant retrotransposons under stress conditions. Trends in Plant Science 3: 181–187. [Google Scholar]
- Grandbastien M‐A. 2015. LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochimica et Biophysica Acta (BBA) ‐ Gene Regulatory Mechanisms 1849: 403–416. [DOI] [PubMed] [Google Scholar]
- Grewal SIS, Jia S. 2007. Heterochromatin revisited. Nature Reviews Genetics 8: 35–46. [DOI] [PubMed] [Google Scholar]
- Griffin PT, Niederhuth CE, Schmitz RJ. 2016. A comparative analysis of 5‐azacytidine‐ and zebularine‐induced DNA demethylation. G3: Genes, Genomes, Genetics 6: 2773–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzat R, Mittelsten Scheid O. 2012. Epigenetic responses to stress: triple defense? Current Opinion in Plant Biology 15: 568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havecker ER, Gao X, Voytas DF. 2004. The diversity of LTR retrotransposons. Genome Biology 5: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Vickers M, Zhang J, Feng X. 2019. Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. eLife 8: e42530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Gaubert H, Bucher E, Mirouze M, Vaillant I, Paszkowski J. 2011. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 472: 115–119. [DOI] [PubMed] [Google Scholar]
- Ito H, Kim J‐M, Matsunaga W, Saze H, Matsui A, Endo TA, Harukawa Y, Takagi H, Yaegashi H, Masuta Y et al 2016. A stress‐activated transposon in Arabidopsis induces transgenerational abscisic acid insensitivity. Scientific Reports 6: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Yoshida T, Tsukahara S, Kawabe A. 2013. Evolution of the ONSEN retrotransposon family activated upon heat stress in Brassicaceae. Gene 518: 256–261. [DOI] [PubMed] [Google Scholar]
- Iwasaki M, Paszkowski J. 2014. Identification of genes preventing transgenerational transmission of stress‐induced epigenetic states. Proceedings of the National Academy of Sciences, USA 111: 8547–8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NR, Yeoh JM, Coruh C, Axtell MJ. 2016. Improved placement of multi‐mapping small RNAs. G3: Genes, Genomes, Genetics 6: 2103–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Reviews Genetics 13: 484–492. [DOI] [PubMed] [Google Scholar]
- Kumar A, Bennetzen JL. 1999. Plant retrotransposons. Annual Review of Genetics 33: 479–532. [DOI] [PubMed] [Google Scholar]
- Kumar SV, Wigge PA. 2010. H2A.Z‐containing nucleosomes mediate the thermosensory response in Arabidopsis . Cell 140: 136–147. [DOI] [PubMed] [Google Scholar]
- Lämke J, Bäurle I. 2017. Epigenetic and chromatin‐based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biology 18: 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JA, Jacobsen SE. 2010. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics 11: 204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D. 2009. Epigenetic regulation of transposable elements in plants. Annual Review of Plant Biology 60: 43–66. [DOI] [PubMed] [Google Scholar]
- Lisch D. 2013. How important are transposons for plant evolution? Nature Reviews Genetics 14: 49–61. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DB, Zilberman D. 2017. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. eLife 6: e30674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marí‐Ordóñez A, Marchais A, Etcheverry M, Martin A, Colot V, Voinnet O. 2013. Reconstructing de novo silencing of an active plant retrotransposon. Nature Genetics 45: 1029–1039. [DOI] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA. 2014. RNA‐directed DNA methylation: an epigenetic pathway of increasing complexity. Nature Reviews Genetics 15: 394–408. [DOI] [PubMed] [Google Scholar]
- McClintock B. 1984. The significance of responses of the genome to challenge. Science 226: 792–801. [DOI] [PubMed] [Google Scholar]
- Mirouze M, Paszkowski J. 2011. Epigenetic contribution to stress adaptation in plants. Current Opinion in Plant Biology 14: 267–274. [DOI] [PubMed] [Google Scholar]
- Moissiard G, Cokus SJ, Cary J, Feng S, Billi AC, Stroud H, Husmann D, Zhan Y, Lajoie BR, McCord RP et al 2012. MORC family ATPases required for heterochromatin condensation and gene silencing. Science 336: 1448–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecinka A, Dinh HQ, Baubec T, Rosa M, Lettner N, Scheid OM. 2010. Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis . The Plant Cell 22: 3118–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietzenuk B, Markus C, Gaubert H, Bagwan N, Merotto A, Bucher E, Pecinka A. 2016. Recurrent evolution of heat‐responsiveness in Brassicaceae COPIA elements. Genome Biology 17: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrana L, Bortolini Silveira A, Mayhew GF, LeBlanc C, Martienssen RA, Jeddeloh JA, Colot V. 2016. The Arabidopsis thaliana mobilome and its impact at the species level. eLife 5: e15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier F, Teixeira FK, Colot V. 2009. Chromatin indexing in Arabidopsis: an epigenomic tale of tails and more. Trends in Genetics 25: 511–517. [DOI] [PubMed] [Google Scholar]
- Rutowicz K, Lirski M, Mermaz B, Teano G, Schubert J, Mestiri I, Kroteń MA, Fabrice TN, Fritz S, Grob S et al 2019. Linker histones are fine‐scale chromatin architects modulating developmental decisions in Arabidopsis . Genome Biology 20: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutowicz K, Puzio M, Halibart‐Puzio J, Lirski M, Kotliński M, Kroteń MA, Knizewski L, Lange B, Muszewska A, Śniegowska‐Świerk K et al 2015. A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis . Plant Physiology 169: 2080–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SanMiguel P, Tikhonov A, Jin Y‐K, Motchoulskaia N, Zakharov D, Melake‐Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z et al 1996. Nested retrotransposons in the intergenic regions of the Maize genome. Science 274: 765–768. [DOI] [PubMed] [Google Scholar]
- Schübeler D. 2015. Function and information content of DNA methylation. Nature 517: 321–326. [DOI] [PubMed] [Google Scholar]
- Shen X, Jonge JD, Forsberg SKG, Pettersson ME, Sheng Z, Hennig L, Carlborg Ö. 2014. Natural CMT2 variation is associated with genome‐wide methylation changes and temperature seasonality. PLoS Genetics 10: e1004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. 2007. Transposable elements and the epigenetic regulation of the genome. Nature Reviews Genetics 8: 272–285. [DOI] [PubMed] [Google Scholar]
- Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. 2014. Non‐CG methylation patterns shape the epigenetic landscape in Arabidopsis . Nature Structural & Molecular Biology 21: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L‐M, Zhang C‐J, Hou X‐M, Shao C‐R, Lu Y‐J, Zhou J‐X, Li Y‐Q, Li L, Chen S, He X‐J. 2018. The PEAT protein complexes are required for histone deacetylation and heterochromatin silencing. The EMBO Journal 37: e98770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieme M, Lanciano S, Balzergue S, Daccord N, Mirouze M, Bucher E. 2017. Inhibition of RNA polymerase II allows controlled mobilisation of retrotransposons for plant breeding. Genome Biology 18: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tittel‐Elmer M, Bucher E, Broger L, Mathieu O, Paszkowski J, Vaillant I. 2010. Stress‐induced activation of heterochromatic transcription. PLoS Genetics 6: e1001175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant I, Paszkowski J. 2007. Role of histone and DNA methylation in gene regulation. Current Opinion in Plant Biology 10: 528–533. [DOI] [PubMed] [Google Scholar]
- Wang Z, Butel N, Santos‐González J, Borges F, Yi J, Martienssen R, Martinez G, Köhler C. 2020. Polymerase IV plays a crucial role in pollen development in Capsella . The Plant Cell 32: 950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh P‐H, Coleman‐Derr D, Eshed‐Williams L, Thao K, Harmer SL, Zilberman D. 2013. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1‐containing heterochromatin. Cell 153: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. 2010. Genome‐wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–919. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lang Z, Zhu J‐K. 2018. Dynamics and function of DNA methylation in plants. Nature Reviews Molecular Cell Biology 19: 489–506. [DOI] [PubMed] [Google Scholar]
- Zhang R, Calixto CPG, Marquez Y, Venhuizen P, Tzioutziou NA, Guo W, Spensley M, Entizne JC, Lewandowska D, ten Have S et al 2017. A high quality Arabidopsis transcriptome for accurate transcript‐level analysis of alternative splicing. Nucleic Acids Research 45: 5061–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Correlation analysis of RNA‐seq samples.
Fig. S2 Comparison of data in this study with previously published data (Choi et al., 2020).
Fig. S3 H1.3 expression does not change in heat‐treated h1 mutants compared to heat‐treated WT.
Fig. S4 A substantial proportion of upregulated genes in h1 mutants and TEs in h1 upon heat are heat‐responsive in WT.
Fig. S5 H1 represses gene expression after heat stress.
Fig. S6 Gene ontology terms of upregulated genes in h1 plus heat vs WT plus heat.
Fig. S7 Decreased CG, CHG and CHH methylation on upregulated Copia78/ONSEN elements.
Fig. S8 H1 does not affect ONSEN remobilization upon heat.
Fig. S9 ONSEN does not amplify in the progeny of heat and zebularine‐treated WT and h1 mutants.
Table S1 Primers used in the manuscript.
Table S2 Mapping statistics of RNA sequencing data.
Table S3 Lists of deregulated genes in h1 mutants before and after heat stress.
Table S4 Lists of upregulated TEs in h1 mutants and cmt2 mutants before and after heat stress.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The RNA and bisulfite‐sequencing data in this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE152402.