

Abstract
Background and Aims
G protein–coupled receptor (GPR) 55 is a putative cannabinoid receptor, and l‐α‐lysophosphatidylinositol (LPI) is its only known endogenous ligand. Although GPR55 has been linked to energy homeostasis in different organs, its specific role in lipid metabolism in the liver and its contribution to the pathophysiology of nonalcoholic fatty liver disease (NAFLD) remains unknown.
Approach and Results
We measured (1) GPR55 expression in the liver of patients with NAFLD compared with individuals without obesity and without liver disease, as well as animal models with steatosis and nonalcoholic steatohepatitis (NASH), and (2) the effects of LPI and genetic disruption of GPR55 in mice, human hepatocytes, and human hepatic stellate cells. Notably, we found that circulating LPI and liver expression of GPR55 were up‐regulated in patients with NASH. LPI induced adenosine monophosphate–activated protein kinase activation of acetyl–coenzyme A carboxylase (ACC) and increased lipid content in human hepatocytes and in the liver of treated mice by inducing de novo lipogenesis and decreasing β‐oxidation. The inhibition of GPR55 and ACCα blocked the effects of LPI, and the in vivo knockdown of GPR55 was sufficient to improve liver damage in mice fed a high‐fat diet and in mice fed a methionine‐choline–deficient diet. Finally, LPI promoted the initiation of hepatic stellate cell activation by stimulating GPR55 and activation of ACC.
Conclusions
The LPI/GPR55 system plays a role in the development of NAFLD and NASH by activating ACC.
Abbreviations
- ACC
acetyl‐CoA carboxylase
- ACTA2
actin α2
- AMPK
adenosine monophosphate–activated protein kinase
- AST
aspartate aminotransferase
- CB1
cannabinoid type 1 receptor
- CoA
coenzyme A
- COL1α1
collagen α1
- COL1α2
collagen α2
- CPT1A
carnitine palmitoyl transferase 1a
- ECS
endocannabinoid system
- GPR
G protein–coupled receptor
- HFD
high‐fat diet
- HSC
hepatic stellate cell
- LPI
l‐α‐lysophosphatidylinositol
- MBOAT7
membrane‐bound O‐acyltransferase domain–containing 7
- MCD
methionine‐choline–deficient
- NAFL
nonalcoholic fatty liver
- NAFLD
NAFL disease
- NAS
NAFLD activity score
- NASH
nonalcoholic steatohepatitis
- NEFA
nonesterified fatty acid
- pACC
phosphorylated ACC
- pAMPKα
phosphorylated AMPKα
- shGPR55
shRNA against GPR55
- shRNA
short hairpin RNA
- TGFβ1
transforming growth factor β1
Nonalcoholic fatty liver (NAFL) disease (NAFLD) is a major global health threat because of its growing incidence and prevalence. It is becoming the leading cause of liver disease and despite the many efforts to find therapeutic targets for this disease, there is no established therapy yet.( 1 ) In this regard, G protein–coupled receptors (GPRs) are the most intensively studied drug targets, given their heavy involvement in human pathophysiology and their pharmacological flexibility. In fact, approximately 34% of all approved drugs act in some way through GPRs,( 2 , 3 ) reflecting the relevance of activating or inhibiting signal transduction cascades by modulating the activity of GPRs in drug discovery.
GPR55 is, in addition to the classical cannabinoid type 1 receptor (CB1) and classical cannabinoid type 2 receptor,( 4 , 5 , 6 ) considered a putative cannabinoid receptor.( 7 ) The endocannabinoid system (ECS) is involved in a wide range of biological processes; however, one of the aspects that has attracted more attention over the last years is undoubtedly its ability to modulate energy metabolism. GPR55 is widely distributed in rodent and human tissues( 8 ) and mediates the actions of its endogenous ligand l‐α‐lysophosphatidylinositol (LPI)( 9 , 10 , 11 , 12 ) and to a much lesser extent mediates the actions of anandamide and 2‐arachydonoyl‐glycerol.( 13 ) LPI belongs to the class of lysophospholipids and is generated by phosphatidylinositol hydrolysis through the action of the calcium‐dependent phospholipase A2( 14 ) and calcium‐independent phospholipase A1.( 15 ) GPR55 is implicated in numerous physiological actions, including reproduction, angiogenesis, apoptosis, inflammation, and energy homeostasis, among others.( 8 , 16 ) In the gastrointestinal tract, the activation of GPR55 resulted in the inhibition of neurogenic contractions in the mouse intestine( 17 ) and is also involved in colon motility.( 18 ) Regarding the role of the LPI/GPR55 system in metabolism, the LPI/GPR55 system has been positively associated with human obesity because LPI induces the expression of lipogenic genes in explants of visceral adipose tissue from patients with obesity.( 19 ) In the pancreas, the exposure of rodent islets to GPR55 agonists enhanced insulin secretion and improved glucose tolerance.( 20 , 21 ) Similarly, the stimulation of human islets with GPR55 ligands also potentiates insulin levels.( 22 )
GPR55 mRNA has also been described in the liver of humans( 19 ) and mice( 20 ); however, the hepatic function of the LPI/GPR55 system is still unknown. Herein, we sought to investigate the potential role of this system in the development of NAFLD. We found that circulating LPI and liver GPR55 expression levels were increased in patients and animal models with NAFLD. LPI augmented lipid content by inducing de novo lipogenesis and decreasing β‐oxidation, whereas down‐regulation of GPR55 and acetyl–coenzyme A (CoA) carboxylase (ACC) α blocked the effects of LPI. The knockdown of GPR55 was sufficient to improve liver damage in mice fed a high‐fat diet (HFD) and mice fed a methionine‐choline–deficient (MCD) diet. Finally, LPI promoted the initiation of hepatic stellate cell (HSC) activation by stimulating GPR55 and ACC. These results highlight the importance of LPI and GPR55 in the development of NAFL and nonalcoholic steatohepatitis (NASH).
Participants and Methods
Human liver samples
These samples were obtained from patients at the University Hospital of Salamanca, Santa Cristina University Hospital in Madrid (Spain) and Hospital Clinico Universidad Catlica (Chile). Baseline characteristics of each cohort and exclusion criteria are described in Supporting Participants and Methods. Informed consent in writing was obtained from each patient and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the appropriate institutional review committee.
Animals and diets
Male C57BL/6J were used and fed with different diets as described in Supporting Participants and Methods. Animal protocols were approved by the Committee at the University of Santiago de Compostela and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals.”
For further details please see Supporting Participants and Methods.
Results
Hepatic GPR55 Is Overexpressed in Human and Murine NAFL and NASH
Analysis of liver biopsies from patients with NAFLD and obesity (with a body mass index [BMI] > 35 kg/m2) revealed elevated mRNA expression of GPR55 compared with individuals without obesity and NAFLD (Fig. 1A). (Characterization of cohort 1 is shown in Supporting Table S1.) GPR55 was similarly increased in patients with obesity and NAFL or NASH compared with individuals without obesity and without liver damage (Fig. 1A). Further, hepatic GPR55 mRNA was significantly elevated in another cohort of individuals with a BMI < 30 kg/m2 and NAFLD compared with individuals without liver disease (Fig. 1B) and showed a positive correlation with NAFLD activity score (NAS) (Supporting Fig. S1). (Characterization of cohort 2 is shown in Supporting Table S2.) Finally, serum analysis of LPI was conducted in a third cohort of patients with obesity and NAFLD or NASH. (Characterization of cohort 3 shown in Supporting Table S3.) Our results revealed higher levels of LPI 16:0, 18:1, and 18:1 isomers in patients with NASH compared with those with only fatty liver (Fig. 1C). Furthermore, we found that these species of LPI were increased as inflammation and ballooning scores augmented (Supporting Fig. S2A). When patients from cohort 3 were separated as with diabetes versus without diabetes, we found that serum levels of these species of LPI did not show any difference (Supporting Fig. S2B), suggesting that changes in LPI species are independent of dysfunctions in glucose metabolism.
FIG. 1.
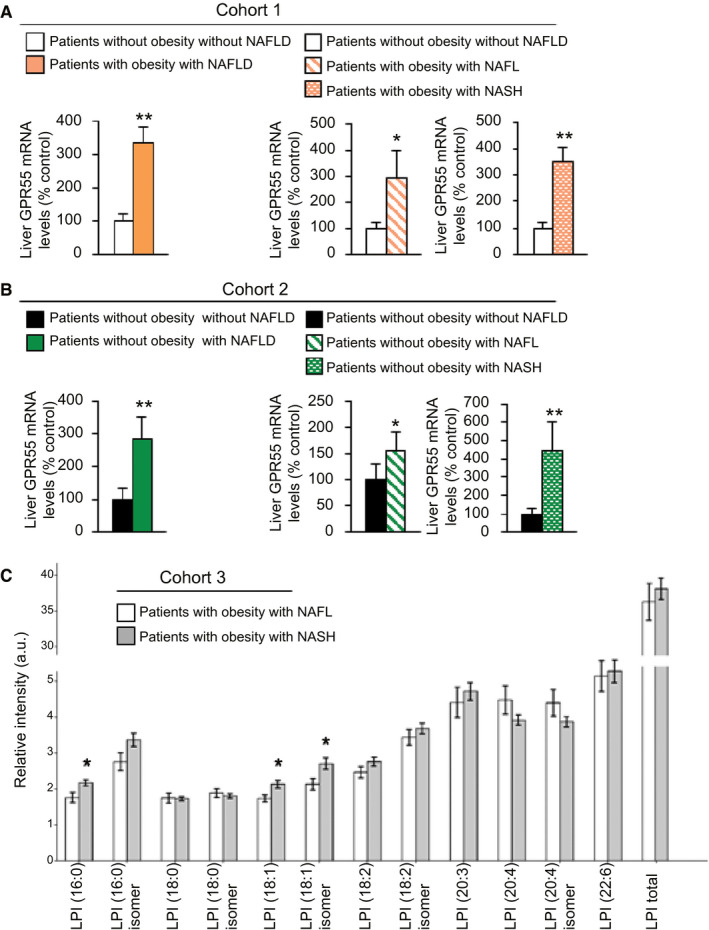
Hepatic GPR55 mRNA is up‐regulated in patients with NAFLD. GPR55 mRNA levels in the liver of (A) participants without obesity and without NAFLD (n = 11) or participants with obesity and NAFLD (n = 35) (cohort 1), either grouped or considering the NAFL (n = 8) and NASH (n = 27) stages in two different groups, and (B) patients without obesity and without (n = 18) or with (n = 21) NAFLD (cohort 2), either grouped or considering the NAFL (n = 13) and NASH (n = 8) stages in two different groups. (C) Serum levels of the different species of LPI in patients with NASH (n = 78) compared with patients with fatty liver without NASH (n = 31) (cohort 3). HPRT was used to normalize mRNA levels. Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05 and **P < 0.01. Abbreviations: GPR 55, G protein–coupled receptor 55; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; LPI, l‐α‐lysophosphatidylinositol; NAFL, nonalcoholic fatty liver; NAFLD, NAFL disease; NASH, nonalcoholic steatohepatitis.
We next studied the expression of GPR55 in livers from mice fed an HFD with 45%‐kcal fat content, a choline‐deficient HFD (CD‐HFD), a very HFD (vHFD) with 60%‐kcal fat content, and an MCD diet at different times. In all of these animal models, we detected the expected histological alterations and increases of both mRNA and protein levels of GPR55 (Fig. 2A‐D). GPR55 mRNA and protein levels were also augmented in isolated hepatocytes from mice fed an MCD compared with mice fed a chow diet (Fig. 2E). Finally, we used the human hepatic cell line THLE2 and treated these hepatocytes with oleic acid. This treatment significantly increased GPR55 mRNA expression compared with hepatocytes treated with bovine serum albumin (BSA) (Fig. 2F). These results indicate that GPR55 expression is consistently elevated in conditions of steatosis and steatohepatitis in both humans and rodents.
FIG. 2.
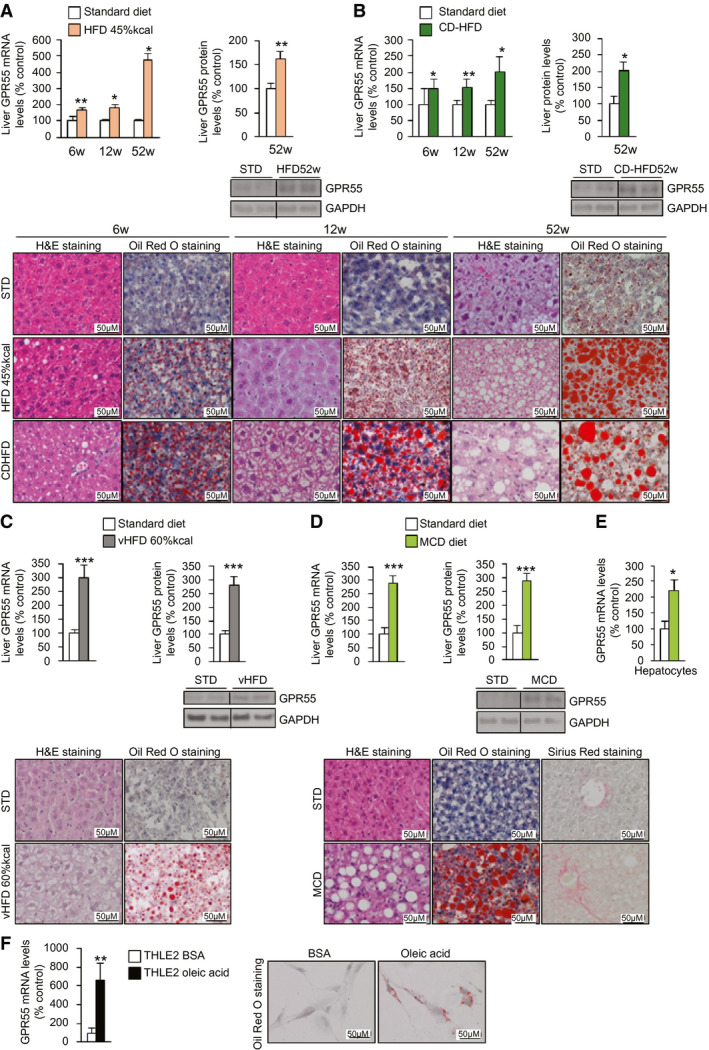
Hepatic GPR55 is increased in different models of NAFLD in vivo and in vitro. GPR55 mRNA levels, GPR55 protein levels, H&E staining, and Sirius Red staining in the liver of (A) mice fed an HFD with 45%‐kcal fat content or an STD for 6, 12, and 52 weeks (n = 5‐14); (B) mice fed a CD‐HFD or STD for 6, 12, and 52 weeks (n = 5‐10); (C) mice fed a vHFD with 60%‐kcal fat content or an STD for 10 weeks (n = 9‐10); and (D) mice fed an MCD diet or an STD for 4 weeks (n = 7‐10 per group). (E) GPR55 mRNA levels in hepatocytes isolated from the liver of mice fed an MCD diet or STD for 4 weeks (n = 5 per group). (F) GPR55 mRNA levels in THLE2 cells cultured in oleic acid or BSA (n = 6). GAPDH and HPRT were used to normalize protein and mRNA levels. Dividing lines indicate splicing in the same gel. Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: BSA, bovine serum albumin; CD‐HFD, choline‐deficient HFD; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GPR55, G protein–coupled receptor 55; H&E, hematoxylin and eosin; HFD, high‐fat diet; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; MCD, methionine‐choline–deficient; NAFLD, nonalcoholic fatty liver disease; STD, standard diet; vHFD, very HFD.
LPI Increases Fatty Acid Synthesis and Decreases β‐Oxidation in Hepatocytes
LPI (10 µM), the endogenous ligand of GPR55,( 9 , 10 , 11 ) increased GPR55 mRNA expression after 1 hour in both THLE2 (Fig. 3A) and HepG2 (Supporting Fig. S3A) cells. After 24 hours of its administration, LPI incrementally altered the lipid content in both cell lines (Fig. 3C and Supporting Fig. S3B). At the molecular level in the two cell lines, we found diminished levels of phosphorylated ACC (pACC) and phosphorylated adenosine monophosphate–activated protein kinase (AMPK) α (pAMPKα), ACC's main regulator, and diminished protein levels of carnitine palmitoyl transferase 1a (CPT1A) (Fig. 3B and Supporting Fig. S3B), suggesting an activation of ACC and thus a stimulation of de novo fatty acid synthesis and inhibition of fatty acid oxidation, respectively. The used dose of LPI did not alter protein levels of apoptotic markers (Supporting Fig. S3C). To further characterize whether the effects of LPI were mediated by GPR55, we silenced the receptor and found that LPI was not able to increase the lipid content (Fig. 3C and Supporting Fig. S3D). Consistent with this, although LPI decreased protein levels of pAMPKα and pACC, it failed to do so after GPR55 silencing (Fig. 3C and Supporting Fig. S3D). Given that GPR55 was mediating LPI‐induced lipid deposition, we next studied whether this receptor could be relevant for the deposition of lipids induced by a different stimulus. Oleic acid–induced lipid content was strongly attenuated after the inhibition of GPR55 (Fig. 3D). Finally, because LPI activates ACC, we next investigated whether the inhibition of ACC would affect LPI’s effects. LPI failed to increase lipid content in THLE2 and HepG2 cells after silencing of ACCα (Fig. 3E and Supporting Fig. S3E).
FIG. 3.
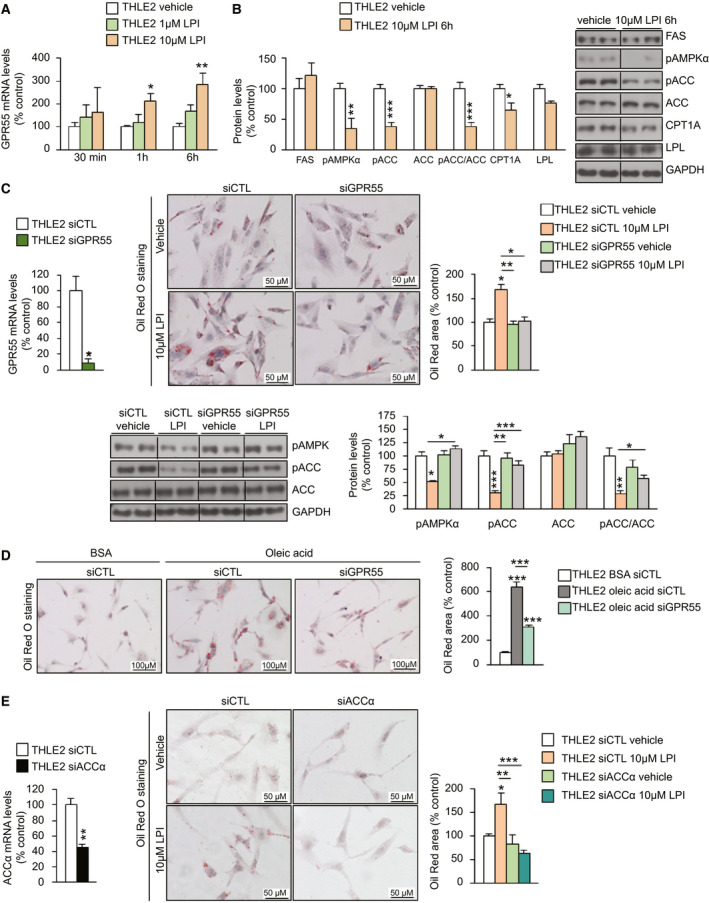
GPR55 regulates lipid accumulation in human hepatocytes through pACC. (A) GPR55 mRNA in THLE2 hepatic cells treated with different doses of LPI (n = 6) (B) Protein levels of markers of lipid metabolism in cells following administration of 10 µM LPI for 6 hours (n = 6). (C) mRNA levels of GPR55 in THLE2 cells transfected with siGPR55 or siCTL for 48 hours (n = 3 per group). Oil Red O staining of THLE2 cells after silencing GPR55, treated with vehicle or LPI. Lipids were quantified and normalized to the total number of nuclei per field (n = 6). Protein levels of pAMPKα, pACC, and ACC were measured (n = 5‐6). (D) Oil Red O staining in THLE2 cells cultured in oleic acid (1 mM) after the down‐regulation of GPR55 (n = 6). (E) mRNA levels of ACCα in THLE2 cells transfected with siACCα or siCTL for 48 hours (n = 5‐6). Oil Red O staining of THLE2 cells after silencing ACCα, treated with vehicle or LPI (n = 6). GAPDH and HPRT were used to normalize protein and mRNA levels. Dividing lines indicate splicing in the same gel. Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: ACC, acetyl–coenzyme A carboxylase; BSA, bovine serum albumin; CPT1a, carnitine palmitoyl transferase 1a; FAS, Fas cell‐surface death receptor; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GPR55, G protein–coupled receptor 55; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; LPI, l‐α‐lysophosphatidylinositol; LPL, lipoprotein lipase; pACC, phosphorylated ACC; pAMPKα, phosphorylated adenosine monophosphate–activated protein kinase α; siACCα, small interfering RNA ACCα; siCTL, small interfering RNA control; siGPR55, small interfering RNA GPR55.
Because these results indicated that LPI and GPR55 were modulating fatty acid synthesis and oxidation, we next directly measured these parameters using [3H]acetate and [14C]palmitate, respectively, and detected that LPI increased de novo triglyceride, diglyceride, free fatty acid, and phospholipids lipogenesis (Fig. 4A and Supporting Fig. S4A), whereas LPI decreased β‐oxidation (Fig. 4B and Supporting Fig. S4B). These LPI‐induced effects were blocked when GPR55 was silenced (Fig. 4). An important event that occurs during development of NAFLD/NASH is the reduction in mitochondrial respiratory‐chain activity.( 23 ) To investigate whether this decrease in β‐oxidation alters mitochondrial respiration, we monitored the oxygen consumption rate (OCR) in cells treated with LPI. We found a significant reduction in the OCR 1 hour after the administration of LPI. This effect was blunted when etomoxir, a CPT1A inhibitor, was provided 30 minutes before the LPI (Supporting Fig. S4C).
FIG. 4.
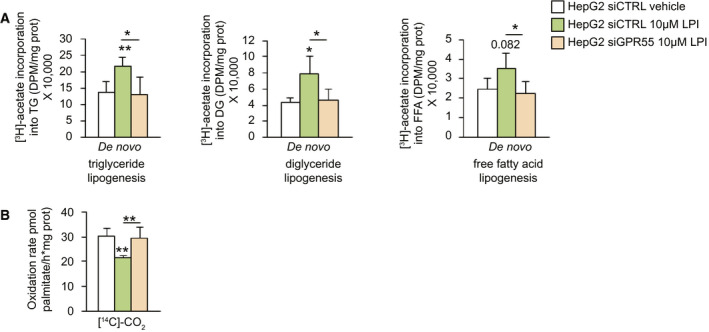
LPI inhibits lipogenesis and promotes β‐oxidation in hepatocytes through GPR55. De novo (A) triglyceride, diacylglyceride, and fatty acid lipogenesis and (B) palmitate oxidation in HepG2 human cells treated with vehicle or LPI and down‐regulating GPR55 (n = 4‐5). Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05 and **P < 0.01. Abbreviations: GPR55, G protein–coupled receptor 55; LPI, l‐α‐lysophosphatidylinositol; siCTRL, small interfering RNA control; siGPR55, small interfering RNA GPR55.
LPI Induces Liver Steatosis in Mice Through GPR55 and ACC
C57BL/6J mice were treated with LPI (0.5 mg/kg) intraperitoneally for 7 days. As reported before,( 19 ) this dose of LPI did not affect body weight (Supporting Fig. S5A) or food intake (Supporting Fig. S5B). Herein, we detected changes in neither liver mass nor aspartate aminotransferase (AST) or cholesterol, but serum triglycerides and nonesterified fatty acids (NEFAs) were increased after LPI treatment (Supporting Fig. S5C). The hepatic lipid content, GPR55 mRNA expression, and fatty acid synthase (FAS) protein levels in LPI‐treated mice were significantly higher than in vehicle‐treated mice (Supporting Fig. S5D,E), whereas the levels of pAMPKα, the ratio of pACC to ACC, and the levels CPT1A were reduced (Supporting Fig. S5F).
To test whether GPR55 was mediating the in vivo effects of LPI, we performed the same pharmacological treatment but added a group of mice, which previously had a lentivirus encoding short hairpin RNA (shRNA) against GPR55 (shGPR55) injected into the tail vein. Although LPI increased GPR55 expression, shGPR55‐treated mice showed a marked decrease in GPR55 expression in the liver (Fig. 5A). The LPI‐induced lipid and triglyceride content in the liver was blunted in shGPR55‐treated mice (Fig. 5B). Moreover, the reduction of pAMPKα and pACC caused by LPI did not occur in shGPR55‐treated mice (Fig. 5C). Consistent with the activation of ACC by LPI, the levels of malonyl‐CoA in the liver were increased by LPI and reduced after the knockdown of GPR55 (Fig. 5D).
FIG. 5.
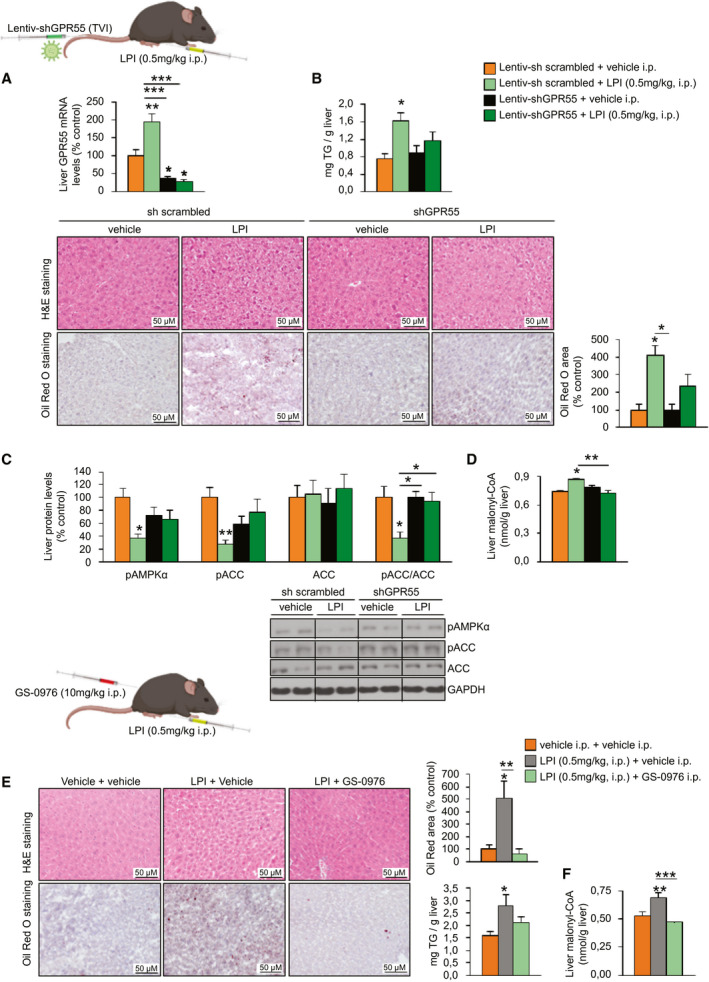
LPI induces hepatic steatosis in a GPR55‐dependent and ACC‐dependent manner in vivo. C57BL/6J mice fed a standard diet randomly received a TVI with Lentiv‐sh scrambled or Lentiv‐shGPR55 and 3 weeks later were treated with either vehicle or LPI (0.5 mg/kg) for 7 days (n = 6). (A) GPR55 mRNA levels in the liver. (B) H&E staining (upper panel) and Oil Red O staining (lower panel) of liver sections. Lipids were quantified and normalized to the total number of nuclei per field. Hepatic triglyceride content was also directly measured. (C) Hepatic protein levels of pAMPKα, pACC, and ACC. (D) Hepatic malonyl‐CoA levels. C57BL/6J mice fed a standard diet were treated daily with the ACC inhibitor GS‐0976 at a dose of 10 mg/kg (or vehicle) for 7 days and were co‐administered either vehicle or LPI (0.5 mg/kg) for the same period (n = 8). (E) H&E staining (upper panel) and Oil Red O staining (lower panel) of liver sections. Lipids were quantified in the Oil Red O staining sections. Hepatic triglyceride content was also directly measured. (F) Hepatic malonyl‐CoA levels. HPRT and GAPDH were used to normalize, respectively, mRNA and protein levels. Dividing lines indicate splicing in the same gel. Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: ACC, acetyl‐CoA carboxylase; CoA, coenzyme A; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GPR55, G protein–coupled receptor 55; H&E, hematoxylin and eosin; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; i.p., intraperitoneally; Lentiv‐shGPR55, lentiviral vector encoding short hairpin RNA against GPR55; Lentiv‐sh scrambled, lentiviral vector encoding scrambled short hairpin RNA; LPI, l‐α‐lysophosphatidylinositol; pACC, phosphorylated ACC; pAMPKα, phosphorylated adenosine monophosphate–activated protein kinase α; TVI, tail‐vein injection.
Next, we assessed whether ACC was also playing a relevant role as a mediator of LPI‐induced actions in the liver. For this, we treated one group of mice with LPI and treated another group with LPI and the ACC inhibitor GS‐0976, which is in clinical trials for the treatment of NAFLD.( 24 ) We injected GS‐0976 intraperitoneally at a dose of 10 mg/kg for 1 week as reported( 25 ) and found that it blocked the hepatic effects of LPI on lipid accumulation, triglyceride content, and malonyl‐CoA (Fig. 5E).
Knockdown of Hepatic GPR55 Ameliorates HFD‐Induced Steatosis
After demonstrating that the pharmacological activation of GPR55 induced liver steatosis, we next performed the opposite experimental paradigm. For this, mice were fed a vHFD with 60%‐kcal fat intake for 10 weeks. In the sixth week, mice underwent tail‐vein injection of a lentivirus encoding shGPR55 while continuing with the HFD for 4 more weeks until being sacrificed. The efficiency of the infection was assessed by a significant reduction in GPR55 mRNA and protein levels when compared with scrambled shRNA–treated mice (Fig. 6A). Body weight and food intake remained unchanged after the knockdown of hepatic GPR55 (Fig. 6B). Serum levels of AST, triglycerides, and NEFAs were significantly lower compared with those of the control group (Fig. 6C). Hepatic lipid content, triglycerides, and F4/80 were reduced after the knockdown of GPR55 (Fig. 6D). Consistent with this, the inhibition of GPR55 in the liver inactivated ACC, as demonstrated by increased pAMPKα and pACC levels and down‐regulated malonyl‐CoA levels and CPT1A (Fig. 6E). In agreement with the amelioration in liver steatosis, levels of different markers of endoplasmic reticulum (ER) stress, inflammation, and apoptosis (Fig. 6F) were significantly reduced after the suppression of hepatic GPR55.
FIG. 6.
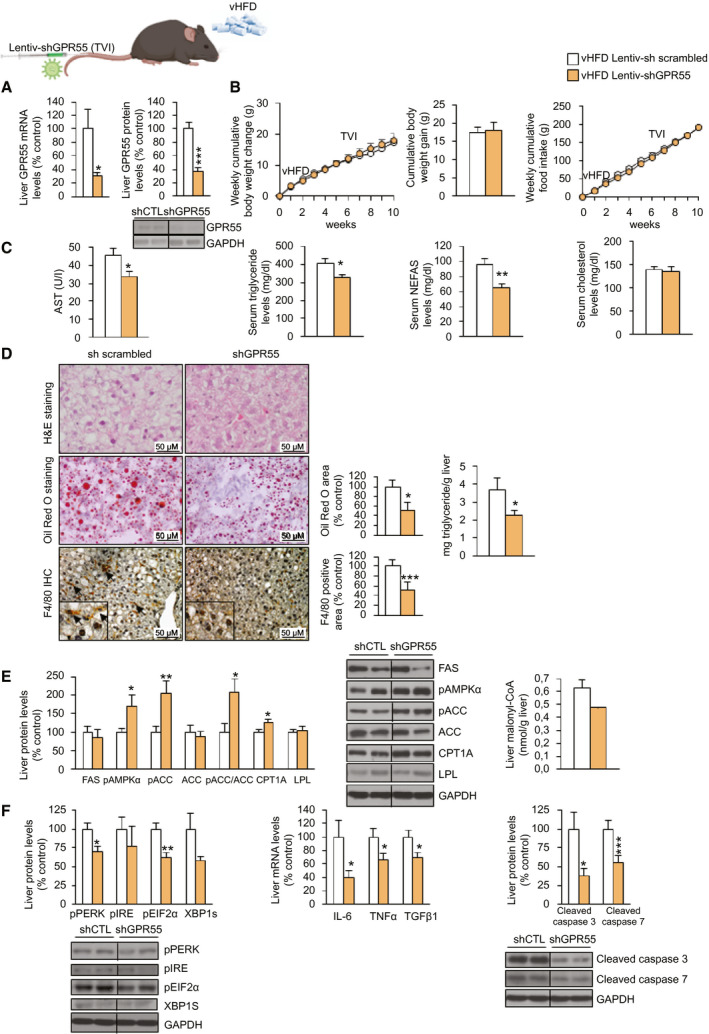
The down‐regulation of hepatic GPR55 ameliorates NAFL induced by a vHFD (60%‐kcal fat content) in mice. C57BL/6J mice fed a vHFD diet for 6 weeks randomly received a TVI with Lentiv‐sh scrambled or Lentiv‐shGPR55, maintaining the vHFD for 4 weeks more. (A) GPR55 mRNA levels in the liver. (B) Body‐weight change and food intake. (C) Serum levels of AST, triglycerides, NEFAs, and cholesterol. (D) H&E staining (upper panel), Oil Red O staining (middle panel), and F4/80‐immunohistochemistry staining (lower panel) of liver sections. Lipids were quantified in the Oil Red O staining sections, and the F4/80‐positive area was quantified in the F4/80‐immunohistochemistry staining sections. Hepatic triglyceride content was also directly measured. (E) Hepatic levels of proteins involved in lipid metabolism and hepatic levels of malonyl‐CoA. (F) Protein levels of ER stress markers, gene expression of inflammatory markers, and cleaved isoforms of apoptotic markers caspase 3 and caspase 7 in the liver. HPRT and GAPDH were used to normalize mRNA and protein levels, respectively. Dividing lines indicate splicing in the same gel. Data are presented as mean ± SEM (n = 7). Statistical differences are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: AST, aspartate aminotransferase; CoA, coenzyme A; CPT1a, carnitine palmitoyl transferase 1a; ER, endoplasmic reticulum; FAS, fatty acid synthase; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GPR55, G protein–coupled receptor 55; H&E, hematoxylin and eosin; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; i.p., intraperitoneally; Lentiv‐shGPR55, lentiviral vector encoding short hairpin RNA against GPR55; Lentiv‐sh scrambled, lentiviral vector encoding scrambled short hairpin RNA; NAFL, nonalcoholic fatty liver; NEFA, nonesterified fatty acid; pEIF2α, phosphorylated eukaryotic initiation factor 2α; pIRE, phosphorylated iron‐responsive element; pPERK, phosphorylated protein kinase R‐like ER kinase; shCTL, short hairpin RNA control; shGPR55, short hairpin RNA against GPR55; TVI, tail‐vein injection; vHFD, very high‐fat diet; XBP1S, X‐box binding protein 1S.
Knockdown of Hepatic GPR55 Ameliorates MCD Diet–Induced and CCl4‐Induced Steatohepatitis
Because GPR55 mRNA expression in the liver was up‐regulated in humans and rodents with NASH, we evaluated the potential of the inhibition of GPR55 to ameliorate NASH. To test this, we used mice fed an MCD diet, which induces macrovesicular steatosis and is widely used in NASH research.( 26 ) Mice underwent tail‐vein injection of a lentivirus encoding shGPR55 and were fed an MCD diet 4 weeks later for 4 weeks until being sacrificed. The efficiency of the infection was assessed by the significant reduction in GPR55 mRNA and protein levels when compared with scrambled shRNA–treated mice (Supporting Fig. S6A). Although liver mass and serum levels of triglycerides, NEFAs, and cholesterol were not affected by the inhibition of hepatic GPR55 (Supporting Fig. S6B), circulating AST was significantly lower compared with that of the control group (Fig. 7A). In line with this, the inhibition of GPR55 reduced hepatic lipid and triglyceride content, hydroxyproline, F4/80, and collagen (Fig. 7B,C) and inactivated ACC, as demonstrated by increased pAMPKα and pACC levels and decreased malonyl‐CoA and collagen mRNA (Fig. 7C). According to the improvement in liver condition, levels of different markers of ER stress, inflammation, fibrosis, and apoptosis were significantly reduced after the suppression of hepatic GPR55 (Supporting Fig. S6C‐F).
FIG. 7.
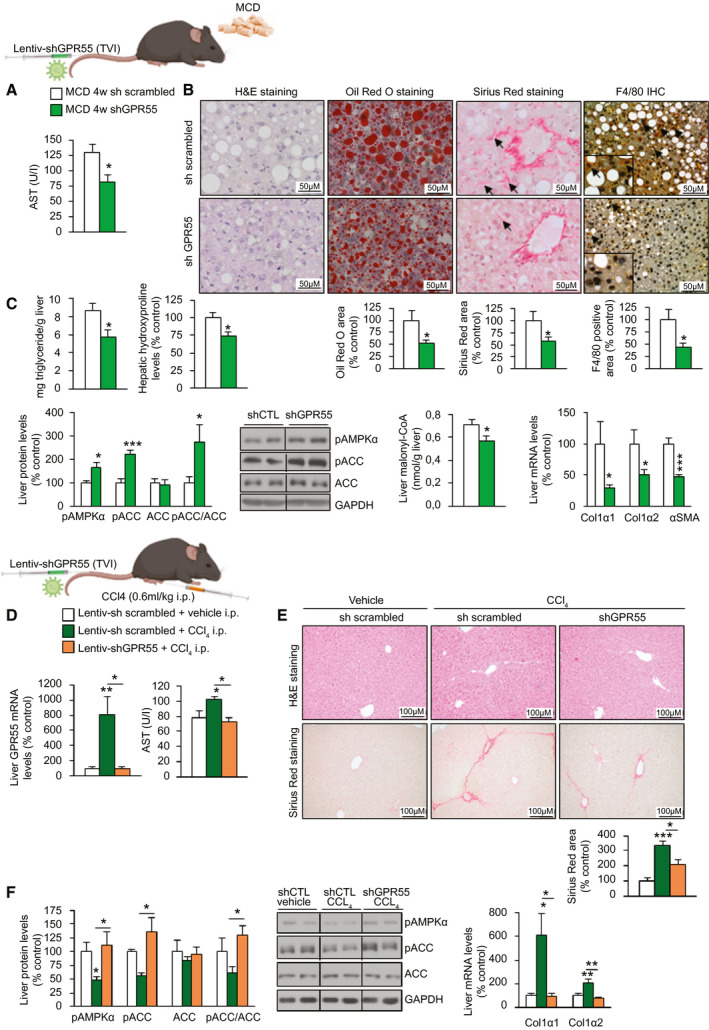
Hepatic silencing of GPR55 ameliorates NASH induced by an MCD diet and fibrosis induced by CCl4 in mice. C57BL/6J mice randomly received a TVI with Lentiv‐shGPR55 or Lentiv‐sh scrambled and 4 weeks later were fed an MCD diet for 4 weeks (n = 7). (A) Serum levels of AST. (B) H&E staining (first panel), Oil Red O staining (second panel), Sirius Red staining (third panel), and F4/80‐immunohistochemistry staining (fourth panel) of liver sections. Staining areas were quantified. (C) Triglyceride content; levels of hydroxyproline; protein levels of pAMPKα, pACC, and ACC; levels of malonyl‐CoA; and mRNA levels of fibrosis markers in the liver. C57BL/6J mice were treated with CCl4 (0.6 mL/kg administered i.p.) or vehicle once a week for 6 weeks, receiving a TVI with Lentiv‐shGPR55 or Lentiv‐sh scrambled during the first week. (D) GPR55 mRNA levels in the liver and serum levels of AST (n = 8). (E) H&E staining (upper panel) and Sirius Red staining (lower panel) of liver sections. Collagen deposition was quantified in the Sirius Red staining sections (n = 8). (F) Protein levels of pAMPKα, pACC, and ACC and mRNA levels of fibrosis markers in the liver (n = 4‐8). HPRT and GAPDH were used to normalize mRNA and protein levels, respectively. Dividing lines indicate splicing in the same gel. Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: ACC, acetyl‐CoA carboxylase; AST, aspartate aminotransferase; CoA, coenzyme A; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GPR55, G protein–coupled receptor 55; H&E, hematoxylin and eosin; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; i.p., intraperitoneally; Lentiv‐shGPR55, lentiviral vector encoding short hairpin RNA against GPR55; Lentiv‐sh scrambled, lentiviral vector encoding scrambled short hairpin RNA; MCD, methionine‐choline–deficient; NASH, nonalcoholic steatohepatitis; pACC, phosphorylated ACC; pAMPKα, phosphorylated adenosine monophosphate–activated protein kinase α; shCTL, short hairpin RNA control; shGPR55, short hairpin RNA against GPR55; sh scrambled, scrambled short hairpin RNA; TVI, tail‐vein injection.
Next, we used a second model of fibrosis, similar to CCl4‐induced liver fibrosis, in which we also injected a lentivirus encoding shGPR55. We found that after 6 weeks, CCl4 induced the expression of GPR55 (Fig. 7D). This effect was concomitant with higher levels of serum AST, a higher level of collagen in the liver, and decreased pAMPKα and pACC (Fig. 7D‐F). However, all of these CCl4‐induced actions were blocked in mice with GPR55 knockdown in the liver (Fig. 7D‐F). Overall, these results indicate that genetic inhibition of hepatic GPR55 ameliorates fibrosis.
The LPI/GPR55 System Modulates the Activation of HSCs
After demonstrating that (1) GPR55 expression is induced in the liver of mice fed an MCD diet (Fig. 2D) and that (2) GPR55 knockdown in the liver can ameliorate MCD diet–induced and CCl4‐induced NASH (Fig. 7), we investigated whether the LPI/GPR55 system could mediate the mechanisms governing HSC activation, which is a key step in liver fibrosis. We found that GPR55 is highly expressed in primary HSCs of wild‐type mice and in the LX‐2 human HSC line compared with THLE2 cells (Fig. 8A). Furthermore, GPR55 mRNA expression was also up‐regulated in isolated HSCs of mice fed an MCD diet (Fig. 8A). Because LX‐2 cells have been extensively characterized and retain key features of hepatic stellate signaling, we used this cell line for this aim. LPI (10 µM) increased GPR55 mRNA expression as early as 1 hour after treatment (Fig. 8B), and after 12 hours, LPI was able to increase the expression of actin α2 (ACTA2), collagen α1 (COL1α1), and collagen α2 (COL1α2), markers of HSC activation and extracellular matrix production, and also stimulated cell proliferation (Fig. 8C). Given that lipid metabolism is essential for the state of HSCs, we also evaluated the effects of LPI on ACC. In results similar to the findings shown in liver and hepatocytes, LPI decreased pAMPKα and pACC (Fig. 8C). Moreover, LX‐2 cells treated with LPI showed increased lipid content (Fig. 8D).
FIG. 8.
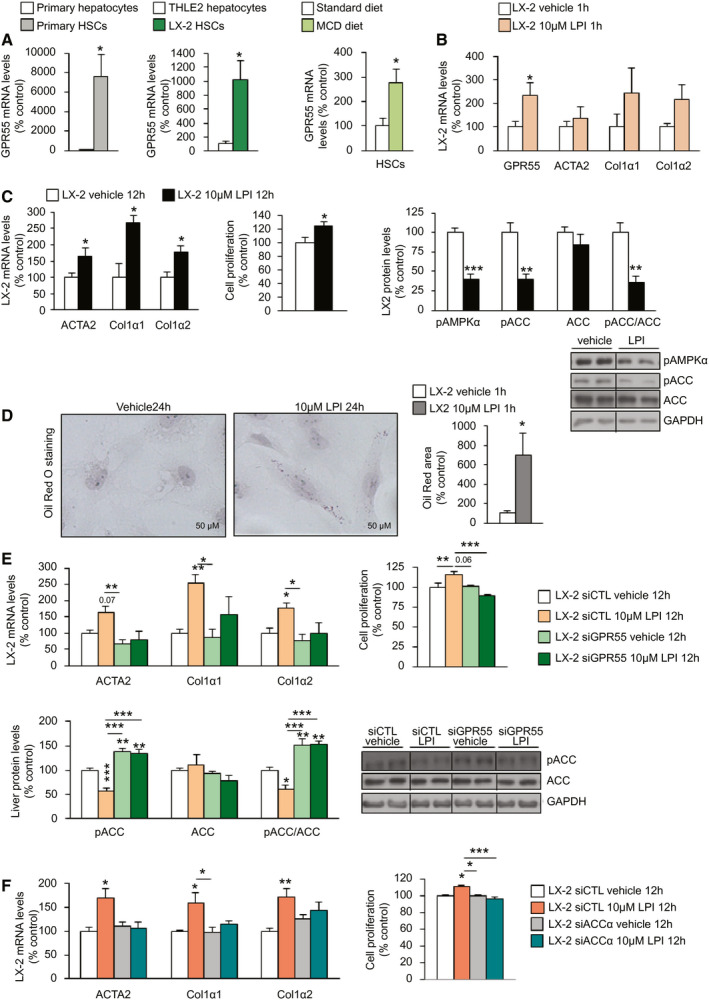
LPI activates LX‐2 HSCs through GPR55 and ACC. (A) GPR55 mRNA levels in primary HSCs compared with primary hepatocytes obtained from mice fed a standard diet and in LX‐2 human HSCs compared with THLE2 cells (n = 4‐8); HSCs isolated from the liver of mice fed an MCD diet or a standard diet for 4 weeks (n = 2‐4). (B) GPR55, ACTA2, COL1α1, and COL1α2 mRNA expression in LX‐2 cells after the administration of LPI (n = 8). (C) ACTA2, COL1α1, and COL1α2 mRNA expression; relative proliferation measured by MTT test; and protein levels of pAMPKα, pACC, and ACC in LX‐2 cells treated with LPI or vehicle for 12 hours (n = 6‐12). (D) Oil Red O staining of LX‐2 cells showing staining of lipids after the administration of LPI for 24 hours (n = 6). The right graph shows total lipid content normalized to the total number of nuclei per field. (E) ACTA2, COL1α1, and COL1α2 mRNA expression and relative proliferation and protein levels of pACC and ACC in LX‐2 cells transfected with siGPR55 or scrambled siRNA and treated with 10 µM LPI or vehicle for 12 hours. (F) ACTA2, COL1α1, and COL1α2 mRNA expression and relative proliferation in LX‐2 cells transfected with siACCα or scrambled siRNA (n = 6) and treated with vehicle or LPI for 12 hours. HPRT and GAPDH were used to normalize mRNA and protein levels, respectively. Dividing lines indicate spliced bands from the same gel. Data are presented as mean ± SEM. Statistical differences are denoted by *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: ACC, acetyl–coenzyme A carboxylase; ACTA2, actin α2; COL1α1, collagen α1; COL1α2, collagen α2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; GPR55, G protein–coupled receptor 55; HPRT, hypoxanthine‐guanine phosphoribosyltransferase; HSC, hepatic stellate cell; LPI, l‐α‐lysophosphatidylinositol; MCD, methionine‐choline–deficient; MTT, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; pACC, phosphorylated ACC; pAMPKα, phosphorylated adenosine monophosphate–activated protein kinase α; siACCα, siRNA ACCα; siCTL, siRNA control; siGPR55, siRNA GPR55; siRNA, small interfering RNA.
Subsequently, we silenced GPR55 in LX‐2 cells (Supporting Fig. S7A) and found that the LPI‐induced expression of ACTA2, COL1α1, COL1α2, and pACC, as well as cell proliferation, were abolished when GPR55 was silenced in LX‐2 cells (Fig. 8E). To evaluate the relevance of ACC mediating the actions of LPI in LX‐2 cells, we next silenced ACCα in LX‐2 cells (Supporting Fig. S7B) and found that this also caused the blockade of LPI‐induced expression of ACTA2, COL1α1, and COL1α2, as well as cell proliferation (Fig. 8F).
As expected, the treatment with transforming growth factor β1 (TGFβ1), a key factor contributing to the initiation of HSC activation,( 27 ) significantly increased mRNA expression of ACTA2, COL1α1, and COL1α2, as well as increasing GPR55 levels in LX‐2 cells (Supporting Fig. S7C). Thus, we next investigated whether GPR55 could modulate the effects of TGFβ1 and found that the silencing of GPR55 was sufficient to partially block TGFβ1 induction of ACTA2, COL1α1, and COL1α2 (Supporting Fig. S7D). Despite GPR55 partially blunting the effects of TGFβ1, the treatment of LX‐2 cells with LPI caused a slight but not significant increase of TGFβ1 (Supporting Fig. S7E), suggesting that even though both LPI and TGFβ1 require GPR55 to promote a profibrotic state, there is not mechanistic link between these two molecules.
Discussion
The findings presented herein show that GPR55 is overexpressed in the liver of patients and animal models with NAFLD and that LPI‐induced GPR55 activation promotes lipid storage, thereby participating in the progression of liver steatosis and steatohepatitis. We also demonstrated that the knockdown of hepatic GPR55 ameliorates HFD‐induced NAFL and both MCD diet–induced and CCl4‐induced NASH. Pathologic overactivation of the ECS is associated with the development of dyslipidemia, obesity, and type 2 diabetes.( 4 , 28 , 29 ) These actions of the ECS have been mainly attributed to the activation of central and peripheral CB1 receptors.( 30 ) HFD increased the hepatic levels of the endocannabinoid anandamide CB1 receptors and basal rates of fatty acid synthesis, and the latter is reduced by CB1 blockade.( 29 , 31 ) More specifically, the activation of hepatic CB1 stimulates de novo lipogenesis through induction of the lipogenic transcription factor sterol regulatory element binding protein 1c and its target enzymes ACC and FAS.( 29 ) Our present results indicate that hepatic activation of GPR55 by its endogenous ligand LPI is also able to promote lipid deposition in the liver by its actions on both hepatocytes and HSCs.
Some types of circulating lysophospholipids have emerged as signaling molecules with important effects on inflammation and fatty liver disease.( 32 , 33 , 34 ) In line with this, our present results indicate that LPI levels are increased in patients with NASH. The mechanisms by which LPI triggers lipid accumulation seem to involve alterations in hepatic fatty acid metabolism because LPI is also able to increase de novo lipogenesis by decreasing the phosphorylation of ACC, which leads to more active ACC in both hepatocytes and HSCs. Furthermore, we also found that LPI inhibited β‐oxidation, which is consistent with the decreased levels of CPT1A that catalyze the primary regulated step in overall mitochondrial fatty acid oxidation in the liver mitochondrial outer membrane. Our current data are in agreement with those of a previous study showing that postprandial lysophospholipids suppress hepatic fatty acid oxidation( 35 ) and promote hepatic inflammatory and fibrotic transcriptional changes.( 36 ) In this sense, it has been recently reported that membrane‐bound O‐acyltransferase domain–containing 7 (MBOAT7), which encodes an acyltransferase enzyme that specifically esterifies arachidonyl‐CoA to LPI lipids, is involved in NAFLD. In particular, Mboat7 knockdown caused liver‐disease progression and was associated with a significant accumulation of 16:0 and 18:1 LPI species in the liver( 36 ); importantly, these were the two species we found to be positively correlated with NAS. Therefore, these two studies involved LPIs as important mediators in the development of NAFLD.
The LPI receptor GPR55 is increased in the liver not only in mice fed an HFD but also in mice exposed to different diets, such as the CD‐HFD and the MCD diet, or in mice exposed to treatments like CCl4, representing models of steatosis and NASH respectively. Importantly, the results are in agreement with human data because GPR55 levels were equally up‐regulated in the liver of patients with obesity and with only steatosis or with NASH. This up‐regulation is independent of obesity because patients without obesity and with NAFLD also showed higher expression of GPR55. Overall, these results support the hypothesis that increased levels of endogenous LPI and GPR55 are early events in the progression of liver disease by causing dyslipidemia and metabolic dysregulation of the liver. It is important to highlight that the data on GPR55 in the liver are homogenous in both rodents and humans. This is not the case for GPR55 in white adipose tissue, where its mRNA expression was decreased in rodents with obesity but increased in the adipose tissue of patients with obesity.( 19 ) Moreover, although the LPI/GPR55 system induced lipogenic gene expression in human adipose tissue, it failed to promote adipocyte differentiation in mouse‐derived 3T3‐L1 cells.( 19 )
In agreement with the stimulation of lipid storage induced by LPI in two different human hepatic cell lines (THLE2 and HepG2) and rodents, the knockdown of hepatic GPR55 ameliorated oleic acid–induced, LPI‐induced, and HFD‐induced steatosis, as well as MCD diet–induced and CCl4‐induced NASH. This is consistent with the results of a previous report indicating that direct injection of exogenous LPIs can rapidly increase the expression of inflammatory and fibrotic genes in HFD‐fed mice with Mboat7 knockdown.( 36 ) Targeting specific tissues is a relevant issue because GPR55 is widely expressed in metabolic tissues.( 8 , 37 ) Several studies have reported on the metabolic phenotype of mice lacking GPR55, showing inconsistent results. Although two studies have found increased adiposity and insulin resistance in GPR55‐deficient mice,( 38 , 39 ) others have reported that their tendency toward increased obesity was not significant.( 40 ) Moreover, differences in body composition were also not reproducible.( 38 , 39 ) From among these studies assessing the lack of GPR55, one has shown that GPR55 deficiency was associated with impaired insulin signaling in the liver.( 38 ) However, it is likely that all GPR55‐expressing organs differentially contribute to the phenotype of these global knockout mice, and the lack of GPR55 thus does not prove that hepatic GPR55 was contributing to this insulin‐signaling impairment. Although we failed to detect any difference in glucose tolerance after the inhibition of hepatic GPR55 in mice fed an HFD (data not shown), we found a clear reduction in the lipid content, as well as a decreased expression of inflammatory and apoptotic markers. Our virogenetic tool showed that with just a partial (60%‐70%) reduction in GPR55 levels in the liver of mice, it is possible to achieve a clear‐cut amelioration of liver damage in different models of NAFLD and NASH. These in vivo results were consistent with results obtained in human hepatic cell lines, indicating that the LPI/GPR55 system can directly regulate lipid metabolism in the liver.
Because GPR55 expression was augmented in both isolated hepatocytes and HSCs from mice challenged with MCD diet–induced NASH, an important point was to investigate whether the LPI/GPR55 pathway was relevant in both cell types. Our findings indicate that LPI increases lipid content in hepatocytes, an effect that was blunted by silencing GPR55. In addition, LPI increased the expression of profibrotic genes, such as ACTA2, COL1α1, and COL1α2, an effect that was again suppressed when GPR55 was silenced in HSCs. These in vitro findings agree with the amelioration of fibrosis when GPR55 was knocked down in mice fed an MCD diet or treated with CCl4. Therefore, these results indicate that the activation of GPR55 is involved in fibrosis.
ACC is a biotin‐dependent enzyme that catalyzes the irreversible carboxylation of acetyl‐CoA to produce malonyl‐CoA, therefore providing the substrate for the biosynthesis of fatty acids. Preclinical studies have demonstrated that the lack or inhibition of ACCα and ACCβ reduced steatosis.( 25 , 41 , 42 , 43 , 44 ) Clinical trials showed that GS‐0976, an inhibitor of ACC, also reduced hepatic steatosis and fibrosis markers in patients with NASH.( 24 , 45 ) Taking into account that (1) pACC is regulated by LPI/GPR55, (2) genetic silencing of ACC blunted the effects of LPI in hepatocytes and HSCs, (3) mice treated GS‐0976 did not show LPI‐induced steatosis, and (4) GPR55 was consistently expressed in mice and patients with NAFLD, our results suggest that the LPI/GPR55 system modulates hepatic lipid metabolism by activating ACC. Our results obtained in human hepatocytes and animal models indicate that the mechanism mediating that activation involves AMPKα, the main enzymatic upstream regulator of ACC. Indeed, an inability to describe how LPI/GPR55 regulates the phosphorylation of AMPKα is a limitation of the present study, and the potential role of some upstream modulators of AMPK, such as liver kinase B1 and/or calmodulin‐dependent protein kinase kinase β, just to name a few, should be further assessed.
Although the promotion of steatosis by LPI is an event somewhat expected for a molecule activating ACC and thereby inducing de novo lipogenesis in hepatocytes, the consequences of the activation of ACC in HSCs are still not understood. Quiescent HSCs contain ample triglycerides( 46 ) and express ACC and other genes involved in de novo lipogenesis,( 47 ) suggesting that de novo lipogenesis was operative during quiescence and restrained during activation of HSCs.( 47 , 48 ) However, a recent report has shown that activated HSC have increased levels of ACCα.( 49 ) In addition, another study has demonstrated a direct role for ACC in driving hepatic fibrosis through the activation of HSCs into collagen‐secreting myofibroblasts, whereas ACC inhibition had an antifibrotic activity.( 50 ) Our results indicated that the activation of the LPI/GPR55 system triggered ACC activity and the expression of profibrotic markers in HSCs, and these effects were blocked after silencing ACC in these cells. Thus, our results support a role for ACC in the activation of HSCs.
In summary, these results indicate that (1) GPR55 expression is increased in the liver of patients with liver damage and in animal models of liver damage; (2) the endogenous ligand of GPR55 called LPI is, notably, positively correlated with the NAS in humans and causes steatosis in a GPR55 manner; (3) the knockdown of hepatic GPR55 ameliorates LPI‐induced and HFD‐induced NAFLD, as well as MCD‐induced and CCl4‐induced NASH; and (4) the LPI/GPR55 system activates ACC through AMPK to cause liver disease.
Author Contributions
M.F.F., U.F., M.J.G.‐R., N.D.S.L., X.B., A.G.‐R., C.A., M.I.‐L., T.C.D., M.V.‐R., A.S., V.G.‐O., E.N., C.I., B.P., D.B., C.F., M.T., J.L.T., L.H.‐C., O.B., J.P.A., F.B., D.G., and M.F. contributed to the conception and design of the study, acquisition of data, and analysis and interpretation of data. M.L., C.D., M.M., M.L.M.‐C., M.A., C.G.‐M., J.M.M., P.A., and R.N. contributed to drafting the article and revising it critically for important intellectual content.
Supporting information
Supporting Material
Supported by grants from the Fondo Europeo de Desarrollo Regional (FEDER)/Ministerio de Ciencia, Innovación y Universidades (MCIU)/Agencia Estatal de Investigación (AEI) (C.D.: BFU2017‐87721; M.L.: RTI2018–101840‐B‐I00; R.N.: BFU2015‐70664R; A.G.‐R.: PI16/00823; C.G.‐M.: PI17/00535), Xunta de Galicia (M.L.: 2015‐CP079 and 2016‐PG068; R.N.: 2015‐CP080 and 2016‐PG057), Fundación Banco Bilbao Vizcaya Argentaria (BBVA; to R.N.), Fundación Atresmedia (M.L. and R.N.), European Foundation for the Study of Diabetes (R.N.), and Fundación Francisco Cobos (A.G.‐R.). MCIU/AEI/FEDER, European Union, (RTI2018‐095134‐B‐100 to P.A.) provided aid to support the research groups of Sistema Universitario Vasco (IT971‐16 to P.A). MCIU provided SAF2017‐87301‐R and RTI2018‐096759‐1‐100, which were integrated into the Plan Estatal de Investigación Científica y Técnica e Innovación and were cofinanced with FEDER (to M.L.M.‐C. and T.C.D. respectively), and La Caixa Foundation Program and 2018 Fundación BBVA Grants for Scientific Research Teams (to M.L.M.‐C.). The research leading to these results has also received funding from the European Community’s H2020 Framework Programme under the following grant: European Research Council Synergy Grant 2019‐WATCH‐810331 to R.N. Centro de Investigación Biomédica en Red (CIBER) de Fisiopatología de la Obesidad y Nutrición and CIBER de Enfermedades Hepáticas y Digestivas are initiatives of the Instituto de Salud Carlos III (ISCIII) of Spain, which is supported by FEDER funds, Gilead Sciences International Research Scholars Program in Liver Disease (to MVR), PI16/01548 (to MM) and the Red de Trastornos Adictivos‐RTA (RD16/0017/0023). This article was partially supported by grants from the Fondo Nacional de Desarrollo Científico y Tecnológico grants 1191145 (to M.A.), 1200227 to JPA and 1191183 (to F.B.) and by the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT, AFB170005, CARE Chile UC, Basal Centre for Excellence in Science and Technology; to M.A.). We thank MINECO for the Severo Ochoa Excellence Accreditation provided to the Center for Cooperative Research in Biosciences (SEV‐2016‐0644).
Potential conflict of interest: Dr. Alonso is employed by OWL. Dr. Iruarrizaga‐Lejarreta is employed by OWL. Dr. Mato consults for, advises, and owns stock in OWL. He consults for and advises Abbott. He consults for Galmed.
References
Author names in bold designate shared co‐first authorship.
- 1. LaBrecque DR, Abbas Z, Anania F, Ferenci P, Khan AG, Goh KL, et al.; World Gastroenterology Organisation . World Gastroenterology Organisation global guidelines: nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Clin Gastroenterol 2014;48:467‐473. [DOI] [PubMed] [Google Scholar]
- 2. Hauser AS, Chavali S, Masuho I, Jahn LJ, Martemyanov KA, Gloriam DE, et al. Pharmacogenomics of GPCR drug targets. Cell 2018;172:41‐54.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hauser AS, Attwood MM, Rask‐Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 2017;16:829‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev 2006;27:73‐100. [DOI] [PubMed] [Google Scholar]
- 5. Cota D. Role of the endocannabinoid system in energy balance regulation and obesity. Front Horm Res 2008;36:135‐145. [DOI] [PubMed] [Google Scholar]
- 6. Pagotto U, Cervino C, Vicennati V, Marsicano G, Lutz B, Pasquali R. How many sites of action for endocannabinoids to control energy metabolism? Int J Obes (Lond) 2006;30(Suppl. 1):S39‐S43. [DOI] [PubMed] [Google Scholar]
- 7. Baker D, Pryce G, Davies WL, Hiley CR. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol Sci 2006;27:1‐4. [DOI] [PubMed] [Google Scholar]
- 8. Tuduri E, Imbernon M, Hernandez‐Bautista RJ, Tojo M, Ferno J, Dieguez C, et al. GPR55: a new promising target for metabolism? J Mol Endocrinol 2017;58:R191‐R202. [DOI] [PubMed] [Google Scholar]
- 9. Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 2007;362:928‐934. [DOI] [PubMed] [Google Scholar]
- 10. Henstridge CM, Balenga NA, Ford LA, Ross RA, Waldhoer M, Irving AJ. The GPR55 ligand L‐alpha‐lysophosphatidylinositol promotes RhoA‐dependent Ca2+ signaling and NFAT activation. FASEB J 2009;23:183‐193. [DOI] [PubMed] [Google Scholar]
- 11. Oka S, Kimura S, Toshida T, Ota R, Yamashita A, Sugiura T. Lysophosphatidylinositol induces rapid phosphorylation of p38 mitogen‐activated protein kinase and activating transcription factor 2 in HEK293 cells expressing GPR55 and IM‐9 lymphoblastoid cells. J Biochem 2010;147:671‐678. [DOI] [PubMed] [Google Scholar]
- 12. Alhouayek M, Masquelier J, Muccioli GG. Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends Pharmacol Sci 2018;39:586‐604. [DOI] [PubMed] [Google Scholar]
- 13. Sharir H, Abood ME. Pharmacological characterization of GPR55, a putative cannabinoid receptor. Pharmacol Ther 2010;126:301‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Billah MM, Lapetina EG. Formation of lysophosphatidylinositol in platelets stimulated with thrombin or ionophore A23187. J Biol Chem 1982;257:5196‐5200. [PubMed] [Google Scholar]
- 15. Kobayashi T, Kishimoto M, Okuyama H. Phospholipases involved in lysophosphatidylinositol metabolism in rat brain. J Lipid Mediat Cell Signal 1996;14:33‐37. [DOI] [PubMed] [Google Scholar]
- 16. Choi JW, Lee CW, Chun J. Biological roles of lysophospholipid receptors revealed by genetic null mice: an update. Biochim Biophys Acta 2008;1781:531‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ross GR, Lichtman A, Dewey WL, Akbarali HI. Evidence for the putative cannabinoid receptor (GPR55)‐mediated inhibitory effects on intestinal contractility in mice. Pharmacology 2012;90:55‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li K, Fichna J, Schicho R, Saur D, Bashashati M, Mackie K, et al. A role for O‐1602 and G protein‐coupled receptor GPR55 in the control of colonic motility in mice. Neuropharmacology 2013;71:255‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moreno‐Navarrete JM, Catalán V, Whyte L, Diaz‐Arteaga A, Vázquez‐Martinez R, Rotellar F, et al. The L‐alpha‐lysophosphatidylinositol/GPR55 system and its potential role in human obesity. Diabetes 2012;61:281‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romero‐Zerbo SY, Rafacho A, Diaz‐Arteaga A, Suárez J, Quesada I, Imbernon M, et al. A role for the putative cannabinoid receptor GPR55 in the islets of Langerhans. J Endocrinol 2011;211:177‐185. [DOI] [PubMed] [Google Scholar]
- 21. McKillop AM, Moran BM, Abdel‐Wahab YH, Flatt PR. Evaluation of the insulin releasing and antihyperglycaemic activities of GPR55 lipid agonists using clonal beta‐cells, isolated pancreatic islets and mice. Br J Pharmacol 2013;170:978‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song SLB, Baker D, Huang GC, Amiel SA, King AJ, Bowe JE, et al. Islet GPR55 is coupled to increased insulin secretion and decreased apoptosis [Abstract 378]. Diabetologia 2012;55(Suppl. 1):S1‐S538. [Google Scholar]
- 23. Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013;58:1497‐1507. [DOI] [PubMed] [Google Scholar]
- 24. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS‐0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology 2018;155:1463‐1473.e1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goedeke L, Bates J, Vatner DF, Perry RJ, Wang T, Ramirez R, et al. Acetyl‐CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology 2018;68:2197‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today 2017;22:1707‐1718. [DOI] [PubMed] [Google Scholar]
- 27. Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, Brenner DA. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 1999;30:77‐87. [DOI] [PubMed] [Google Scholar]
- 28. Engeli S, Böhnke J, Feldpausch M, Gorzelniak K, Janke J, Batkai S, et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes 2005;54:2838‐2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osei‐Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Bátkai S, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet‐induced obesity. J Clin Invest 2005;115:1298‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cota D, Marsicano G, Tschöp M, Grübler Y, Flachskamm C, Schubert M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest 2003;112:423‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bergholm R, Sevastianova K, Santos A, Kotronen A, Urjansson M, Hakkarainen A, et al. CB(1) blockade‐induced weight loss over 48 weeks decreases liver fat in proportion to weight loss in humans. Int J Obes (Lond) 2013;37:699‐703. [DOI] [PubMed] [Google Scholar]
- 32. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
- 33. Tanaka N, Matsubara T, Krausz KW, Patterson AD, Gonzalez FJ. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 2012;56:118‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lehmann R, Franken H, Dammeier S, Rosenbaum L, Kantartzis K, Peter A, et al. Circulating lysophosphatidylcholines are markers of a metabolically benign nonalcoholic fatty liver. Diabetes Care 2013;36:2331‐2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Labonté ED, Pfluger PT, Cash JG, Kuhel DG, Roja JC, Magness DP, et al. Postprandial lysophospholipid suppresses hepatic fatty acid oxidation: the molecular link between group 1B phospholipase A2 and diet‐induced obesity. FASEB J 2010;24:2516‐2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Helsley RN, Varadharajan V, Brown AL, Gromovsky AD, Schugar RC, Ramachandiran I, et al. Obesity‐linked suppression of membrane‐bound O‐acyltransferase 7 (MBOAT7) drives non‐alcoholic fatty liver disease. eLife 2019;8:e49882 10.7554/eLife.49882. https://elifesciences.org/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Henstridge CM, Brown AJ, Waldhoer M. GPR55: metabolic help or hindrance? Trends Endocrinol Metab 2016;27:606‐608. [DOI] [PubMed] [Google Scholar]
- 38. Lipina C, Walsh SK, Mitchell SE, Speakman JR, Wainwright CL, Hundal HS. GPR55 deficiency is associated with increased adiposity and impaired insulin signaling in peripheral metabolic tissues. FASEB J 2019;33:1299‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meadows A, Lee JH, Wu CS, Wei Q, Pradhan G, Yafi M, et al. Deletion of G‐protein‐coupled receptor 55 promotes obesity by reducing physical activity. Int J Obes (Lond) 2016;40:417‐424. [DOI] [PubMed] [Google Scholar]
- 40. Bjursell M, Ryberg E, Wu T, Greasley PJ, Bohlooly YM, Hjorth S. Deletion of Gpr55 results in subtle effects on energy metabolism, motor activity and thermal pain sensation. PLoS One 2016;11:e0167965 10.1371/journal.pone.0167965. http://www.plosone.org/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mao J, DeMayo FJ, Li H, Abu‐Elheiga L, Gu Z, Shaikenov TE, et al. Liver‐specific deletion of acetyl‐CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci U S A 2006;103:8552‐8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choi CS, Savage DB, Abu‐Elheiga L, Liu ZX, Kim S, Kulkami A, et al. Continuous fat oxidation in Aetyl–CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Natl Acad Sci U S A 2007;104:16480‐16485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, et al. Reversal of diet‐induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl‐CoA carboxylases 1 and 2. J Clin Invest 2006;116:817‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lally JSV, Ghoshal S, DePeralta DK, Moaven O, Wei L, Masia R, et al. Inhibition of Acetyl‐CoA carboxylase by phosphorylation or the inhibitor ND‐654 suppresses lipogenesis and hepatocellular carcinoma. Cell Metab 2019;29:174‐182.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lawitz EJ, Coste A, Poordad F, Alkhouri N, Loo N, McColgan BJ, et al. Acetyl‐CoA carboxylase inhibitor GS‐0976 for 12 weeks reduces hepatic de novo lipogenesis and steatosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2018;16:1983‐1991.e3. [DOI] [PubMed] [Google Scholar]
- 46. Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Goodman DS. Biochemical characteristics of isolated rat liver stellate cells. Hepatology 1987;7:1224‐1229. [DOI] [PubMed] [Google Scholar]
- 47. She H, Xiong S, Hazra S, Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem 2005;280:4959‐4967. [DOI] [PubMed] [Google Scholar]
- 48. Wallace MC, Friedman SL, Mann DA. Emerging and disease‐specific mechanisms of hepatic stellate cell activation. Semin Liver Dis 2015;35:107‐118. [DOI] [PubMed] [Google Scholar]
- 49. de Oliveira da Silva B, Alberici LC, Ramos LF, Silva CM, da Silveira MB, Dechant CRP, et al. Altered global microRNA expression in hepatic stellate cells LX‐2 by angiotensin‐(1‐7) and miRNA‐1914‐5p identification as regulator of pro‐fibrogenic elements and lipid metabolism. Int J Biochem Cell Biol 2018;98:137‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vijayakumar A, Ghoshal S, Kusam S, Sulfab M, Newstrom D, Wang T, et al. ACC supports metabolic reprogramming in hepatic stellate cells during pathological fibrosis in the liver Presented at: EMBO Workshop on Organ Crosstalk in Energy Balance and Metabolic Disease; April 3‐11, 2019; Cadiz, Spain. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Material