

Abstract
This post hoc analysis assessed the benefit–risk profile of esketamine nasal spray + oral antidepressant (AD) induction and maintenance treatment in patients with treatment‐resistant depression (TRD). The Benefit–Risk Action Team framework was utilized to assess the benefit–risk profile using data from three induction studies and one maintenance study. Benefits were proportion of remitters or responders in induction studies and proportion of stable remitters or stable responders who remained relapse‐free in the maintenance study. Risks were death, suicidal ideation, most common adverse events (AEs), and potential long‐term risks. Per 100 patients on esketamine + AD vs. AD + placebo in induction therapy, 5–21 additional patients would remit and 14–17 additional patients would respond. In maintenance therapy, 19–32 fewer relapses would occur with esketamine. In both cases, there was little difference in serious or severe common AEs (primarily dissociation, vertigo, and dizziness). These findings support a positive benefit–risk balance for esketamine + AD as induction and maintenance treatment in patients with TRD.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Conventional antidepressants (ADs) are of limited use in treatment‐resistant depression (TRD). Esketamine nasal spray with oral ADs, in phase III studies, showed evidence of efficacy and safety in patients with TRD.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Using the data from five phase III studies, we assessed the treatment benefits vs. risks of esketamine + AD vs. AD + placebo as induction and maintenance treatment in TRD.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The present work further adds to the body of evidence, which suggests treatment benefits of esketamine nasal spray + AD in patients with TRD exceed risk when compared with treatment with AD alone.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The findings have implications for improved patient‐centric decision making and increased acceptance of decisions and improved adherence to treatment.
Benefit–risk assessment is an integral part of the regulatory approval process and is necessary throughout the lifecycle of a drug. 1 , 2 Assessing and weighing the potential benefits and risks of an investigational drug is critical for decision making by regulators, clinicians, and patients. 1 Over the past several decades, the methods of evaluating, interpreting, and communicating the benefit–risk profile of a drug have evolved and become more objective, consistent, and structured. 1 , 3 , 4 , 5 Various structured approaches, or benefit–risk frameworks, have been developed or adopted by regulatory agencies, public–private partnerships, and pharmaceutical companies, 3 including the Benefit–Risk Action Team (BRAT) framework. 6 , 7 , 8 , 9 The BRAT framework was developed by pharmaceutical companies collaborating under the Pharmaceutical Research and Manufacturer's Association. It offers a structured approach in evaluating and communicating the evidence for benefit–risk assessment, with a strong emphasis on end‐point selection and graphical communication of results. This method was utilized for assessing the benefit–risk profile of esketamine nasal spray + oral antidepressant (AD). It is important to note that this application of the BRAT framework neither replaces nor is intended to replace or comment on any decisions made by regulatory agencies.
Effective treatment of major depressive disorder (MDD) remains a challenge despite the availability of numerous ADs. 10 Up to one‐third of patients with MDD show an inadequate response to currently available medications and are considered as “treatment‐resistant.” 11 The definition of treatment‐resistant depression (TRD) varies; 12 however, a standardized definition of TRD is currently emerging and is often defined as failure of at least two treatment trials with ADs from the same or different pharmacologic classes, with adequate dose, duration, and compliance. 13 Compared with patients with MDD who respond to AD treatment, patients with TRD account for considerable disease burden, with a higher relapse rate and increased rate of suicide attempts. 10 , 14 , 15 TRD is associated with impairment in daily functioning, ability to work, and overall reduced health‐related quality of life. 10
Various treatment options and strategies for the management of TRD include electroconvulsive therapy (ECT), augmentation with lithium or atypical antipsychotics, switching ADs, and combining two ADs. However, current conventional ADs are of limited use due to delayed (4–6 weeks) onset of action, 16 and available treatments have safety concerns, especially for ECT, which may have cognitive adverse events (AEs). 17 Hence, there is a persistent need for additional safe and effective therapeutic options with novel mechanisms of action and with a rapid onset of effect.
Esketamine, the S‐enantiomer of ketamine, in conjunction with a newly initiated oral AD, was approved by the US Food and Drug Administration (FDA), and the European Medicines Agency (EMA), and several other Health Authorities in 2019 for the treatment of patients with TRD. 18 , 19 Esketamine’s efficacy and safety in patients with TRD was investigated in five phase III studies (three double‐blind induction studies, one maintenance study, and one open‐label safety study). The design, execution, and results of these studies are reported in detail elsewhere. 20 , 21 , 22 , 23 In addition to efficacy and safety outcomes of the individual clinical research studies, a thorough benefit–risk assessment across studies provides a systematic quantitative approach in balancing key efficacy and safety end points. In the present post hoc analysis, the principles of the BRAT framework were applied to conduct a structured benefit–risk assessment of esketamine + AD compared with AD + placebo as induction and maintenance treatment in TRD.
METHODS
The BRAT framework is a broadly accepted approach for benefit–risk assessment. 7 , 8 , 24 , 25 , 26 It is a process to facilitate the selection, organization, summarization, and communication of evidence for benefit–risk decision making. The BRAT framework comprises the following steps: (i) define the decision context, (ii) identify efficacy and safety outcomes that have an important effect on the benefit–risk balance, (iii) identify source data, (iv) customize the framework, (v) assess outcome importance, and (vi) display and interpret the key benefit–risk metrics (Table S1 ). 6 , 7 , 27 , 28 We utilized the BRAT framework because of its broad acceptance, its clear set of steps, and its strong emphasis on end‐point selection and graphical communication and interpretation of benefit–risk data.
Decision context
Up to one‐third of MDD patients have TRD. TRD is a serious and life‐threatening condition with high rates of individual and society‐level morbidity and a chronic disease course. Patients with TRD can be unable to work, maintain relationships, and in the most severe cases may become hospitalized or even commit suicide. All these concerns are more common and more severe in TRD than in MDD. 10 , 14 , 15 There also is considerable unmet medical need due to few available therapies and those available (e.g., ECT) being associated with considerable AEs. 17 To address use of esketamine for this medical need, the benefit–risk balance of esketamine + oral AD was evaluated in both induction (4 weeks) and maintenance therapy (16 weeks optimization and maintenance treatment of variable duration).
Identify efficacy and safety end points
For benefit–risk assessment of the induction studies, the beneficial end points were the proportion of remitters (Montgomery‐Åsberg Depression Rating Scale (MADRS) total score ≤ 12) or responders (≥ 50% reduction in MADRS total score) at the end of the double‐blind phase (definitions for all end points in Table S2 ). Remission and response were secondary efficacy end points not associated with formal statistical testing. Rather than the primary induction efficacy end point of change in MADRS total score, the secondary end points of remission and response were used for benefit–risk because these are clinically meaningful end points for psychiatrists that also allow for comparing proportions of beneficial events with those of harmful events. It is important to note that all remitters were also responders, given the study inclusion criteria of baseline MADRS ≥ 28, hence remission and response are not separate benefits but reflect different degrees of efficacy.
Although MADRS total score ≤ 10 is a commonly used definition for remission, 29 a definition of ≤ 12 has also been used in multiple published clinical studies. 30 , 31 In addition, the sponsor selected a definition of MADRS total score ≤ 12 based on data from a phase 0 study (data on file) suggesting that remote MADRS raters score slightly higher (an average of 2 points) than face‐to‐face raters when patients demonstrate lower overall symptom severity (i.e., MADRS total score < 12).
The beneficial end points for the maintenance phase were the proportion of stable remitters or stable responders in A Study of Intranasal Esketamine Plus an Oral Antidepressant for Relapse Prevention in Adult Participants With Treatment‐resistant Depression (SUSTAIN‐1) who remained relapse‐free. Relapse‐free proportion is used, rather than the primary efficacy end point of time to relapse, to allow for comparing proportions of beneficial events with harmful events.
For both induction and maintenance studies, safety outcomes (risks) included were death, incident suicidal ideation, and the most commonly observed adverse drug reactions (ADRs)— generally defined as AEs with an incidence of ≥ 10% in patients treated with esketamine + AD and greater than that for AD + placebo. The ADR frequency categorizations for all ADRs is based on Council for International Organizations of Medical Sciences (CIOMS) Working Group (Groups III and V) recommendations 32 using data from esketamine phase II and phase III TRD studies. Those ADRs at an incidence of ≥ 10% that are categorized as "very common" are reported as possibly associated with esketamine treatment most frequently and consistently across esketamine clinical studies. In addition, they represent the most typical safety profile of the product based on its pharmacological effects. Such events include dissociation, dizziness, nausea, sedation, headache, vertigo, dysgeusia, hypoesthesia, increased blood pressure, anxiety, and vomiting (Table S2 ). The severity for AEs was assessed by the investigator and graded as mild (symptoms were easily tolerated, caused minimal discomfort, and did not interfere with everyday activities); moderate (discomfort caused interference with normal activity); or, severe (extreme distress caused significant impairment of functioning or incapacitation, and prevented normal everyday activities).
Information on when an ADR is likely to begin and resolve relative to treatment administration is critical in understanding whether the ADRs are transient and manageable without clinical sequelae, when patients are either under medical supervision or under driving restrictions. All serious or severe common ADRs were thus characterized based on time to onset and resolution relative to the most recent dosing day. Specifically, serious or severe common ADRs were characterized by (i) onset and resolution on a dosing day, (ii) onset on a dosing day and resolution on a nondosing day, and (iii) onset on a nondosing day. ADRs that led to treatment discontinuation were also assessed. Additionally, given the observation of cognitive impairment and interstitial cystitis based on the ketamine abuse literature, these potential long‐term risks were also assessed. The value tree is presented in Figure S1 .
Source data selection
The benefit–risk assessment of esketamine as induction treatment used efficacy and safety data from the three short‐term phase III studies: A Study to Evaluate the Efficacy, Safety, and Tolerability of Fixed Doses of Intranasal Esketamine Plus an Oral Antidepressant in Adult Participants With Treatment‐resistant Depression (TRANSFORM‐1) (NCT02417064); 20 A Study to Evaluate the Efficacy, Safety, and Tolerability of Flexible Doses of Intranasal Esketamine Plus an Oral Antidepressant in Adult Participants With Treatment‐resistant Depression (TRANSFORM‐2) (NCT02418585); 21 and A Study to Evaluate the Efficacy, Safety, and Tolerability of Intranasal Esketamine Plus an Oral Antidepressant in Elderly Participants With Treatment‐resistant Depression (TRANSFORM‐3) (NCT02422186) 22 during the double‐blind phase. Assessment for maintenance treatment used data from the longer‐term phase III study, SUSTAIN‐1 (NCT02493868) 23 , during the maintenance phase.
In this quantitative assessment, the open‐label safety studies A Long‐term, Safety and Efficacy Study of Intranasal Esketamine in Treatment‐resistant Depression (SUSTAIN‐2) (NCT02497287) 33 and A Long‐term Safety Study of Esketamine Nasal Spray in Treatment‐resistant Depression (SUSTAIN‐3) (NCT02782104) were not used, as these were primarily safety and tolerability studies and there were no comparator groups. All studies included in this assessment were conducted in accordance with the principles of the Declaration of Helsinki. Approval from an Institutional Review Board was obtained in all participating centers, and all patients provided written informed consent.
Short‐term induction studies
The TRANSFORM‐1, 20 TRANSFORM‐2, 21 and TRANSFORM‐3 22 are randomized, double‐blind, active‐controlled studies aimed to evaluate efficacy in patients with TRD who did not respond to prior AD and switched to esketamine + a newly initiated oral AD vs. a newly initiated oral AD + placebo. These studies were nearly identical in design and inclusion/exclusion criteria (Figure 1a ). However, these studies had differences in age criteria (TRANFORM‐1 and 2: adults aged 18–64 years; TRANSFORM‐3: elderly aged ≥ 65 years), and in dosing regimen (fixed‐dose in TRANSFORM‐1; flexible‐dose in TRANSFORM‐2 and 3) (Figure 1a ). Patients in these studies were randomized into a double‐blind induction phase if they were nonresponders to 1–5 oral ADs in the current depressive episode at the start of screening/prospective observational phase. Patients were also eligible if they had nonresponse to oral ADs confirmed prospectively during the screening / observational phase. Nonresponse at the end of the screening / prospective observational phase was defined as ≤ 25% improvement in the MADRS total score from week 1 to week 4 and a MADRS total score of ≥ 28 on week 2 and week 4. The MADRS consists of 10 items that cover core depressive symptoms. Each item is scored from 0 (symptom is not present or normal) to 6 (severe or continuous presence of the symptom). A total score (0–60) is calculated by summing the scores of all 10 items. A higher score represents a more severe condition. These patients were eligible to enter a randomized double‐blind phase with fixed or flexible esketamine dosing along with AD (Figure 1a ). 20 , 21 , 22
Figure 1.
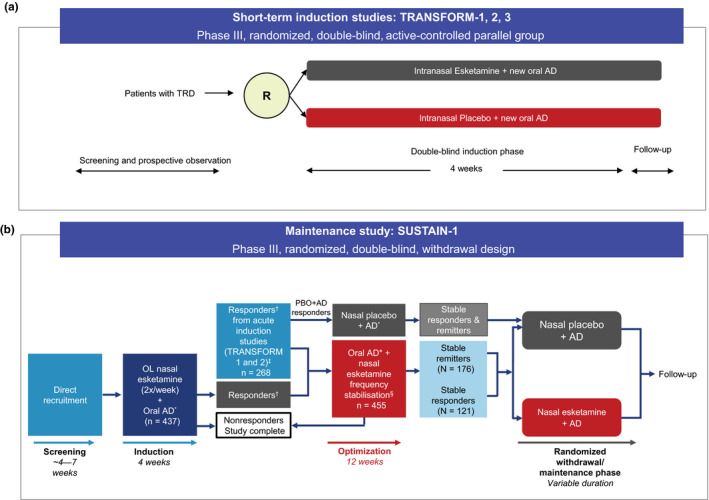
Esketamine phase III studies overview. (a) Short‐term induction studies (b) Maintenance study. Short‐term induction studies had same design but differed in dose and patients’ age: (i) TRANSFORM‐1: esketamine 56 or 84 mg fixed dose; age 18–64 years; (ii) TRANSFORM‐2: flexible dose; age: 18–64 years; (iii) TRANSFORM‐3: flexible dose; age: ≥ 65 years; *Duloxetine, escitalopram, sertraline, or venlafaxine extended‐release; †Responders defined as ≥ 50% reduction in the MADRS total score from baseline (Day 1 prerandomization) at the end of the 4‐week double‐blind induction phase of the acute 3001 and 3002 studies; ‡Responders who entered the optimization phase remained on the same intranasal study drug as taken in the induction phase; §Frequency of intranasal medication sessions was reduced to once weekly for 4 weeks, then individualized to weekly or every other week based on severity of depressive symptoms (lowest dosing frequency adequate to maintain remission (MADRS ≤ 12)). AD, antidepressant; MADRS, Montgomery‐Åsberg Depression Rating Scale; OL, open label; PBO, placebo; R, randomization; TRD, treatment‐resistant depression. [Colour figure can be viewed at wileyonlinelibrary.com]
Maintenance study
SUSTAIN‐1 was a double‐blind, randomized withdrawal design, relapse prevention study aimed to assess the safety and efficacy of continuation vs. discontinuation of esketamine, in presence of an ongoing oral AD, in patients who were in stable remission. 23
After achieving response to induction of esketamine in TRANFORM‐1 or TRANSFORM‐2 study, patients were transferred (transfer‐entry patients, n = 268) into the SUSTAIN‐1 study. In addition, patients were directly enrolled (direct‐entry patients, n = 437) into the SUSTAIN‐1 study after a screening phase and a 4‐week open‐label induction phase. Both patient populations met the same criteria for TRD, including prospective confirmation of nonresponse in the screening / prospective observational phase (Figure 1b ).
Direct‐entry and transfer‐entry patients who exhibited confirmed response at the end of the 4‐week induction phase entered a 12‐week optimization phase. At the end of the optimization phase, stable remitters and stable responders after 16 weeks of esketamine treatment entered the maintenance phase. Stable remitters were defined as patients with MADRS score ≤ 12 for ≥ 3 of the last 4 weeks, with one excursion (MADRS score > 12) or one missing MADRS assessment permitted at week 13 or 14 only, while stable responders were defined as patients with 50% reduction in MADRS score from baseline in each of the last 2 weeks of the optimization phase, but without achieving remission. Further details of study design and treatment have been described elsewhere. 23
Customizing the framework
The data sources from the clinical trial were sufficient to support all end points included. No customization was needed.
Assess outcome importance
Outcome importance was based on clinical judgement and was reinforced by a patient preference study conducted in TRD patients. 34
Display and interpret key benefit–risk metrics
The incidence proportions for each arm, risk differences and 95% confidence intervals (CIs) are summarized in effects tables. Risk differences and 95% CIs are also depicted in forest plots.
Statistical methods
For TRANSFORM‐1, TRANSFORM‐2, and TRANSFORM‐3, treatment comparisons were performed in all randomized patients who received one or more doses of study medication and one dose of oral AD during the double‐blind induction phase. For SUSTAIN‐1, treatment comparisons for efficacy end points were performed separately for randomized patients who were in stable remission (stable remitters) and who were stable responders (but not achieved stable remission) at the end of the optimization phase and who received one or more doses of study medication and one dose of oral AD during the maintenance phase. Safety analysis end points were analyzed in all randomized patients who received one or more doses of study medication or one dose of oral AD during the maintenance phase.
Risk differences of proportions were calculated for all efficacy and safety end points. In tables and figures below, proportions for each arm and risk differences are expressed per 100 patients. Thus, the risk difference for an event can be interpreted as the additional number of patients in a population of 100 who would experience that event when treated with esketamine + AD compared with being treated with AD + placebo. A negative value indicates that there were more occurrences of an outcome in the population treated with AD + placebo, and a positive value indicates more occurrences in a population treated with esketamine + AD. The 95% CIs are presented with risk differences to show statistical uncertainty. No statistical tests for these end points were applied nor any adjustments made for multiplicity. Observed case data were used for these analyses. An additional assessment in the subgroup of patients aged 65–74 years from TRANFORM‐3 was performed; however, a similar assessment was not performed for those > 75 years of age as there were too few patients (n = 21) in that age range.
RESULTS
Benefit–risk balance of induction treatment in adult patients
In TRANSFORM‐1, compared with AD + placebo, induction treatment with esketamine 56 mg + AD resulted in 15.2 (95% CI, 2.11 to 28.22) more responders per 100 patients and esketamine 84 mg + AD resulted in 14.2 (95% CI, 0.68 to 27.67) more responders per 100 patients at day 28. In TRANSFORM‐2, induction treatment with esketamine + AD at flexible dose resulted in 17.3 (95% CI, 4.01 to 30.60) more responders (Figure 2 and Table 1 ).
Figure 2.
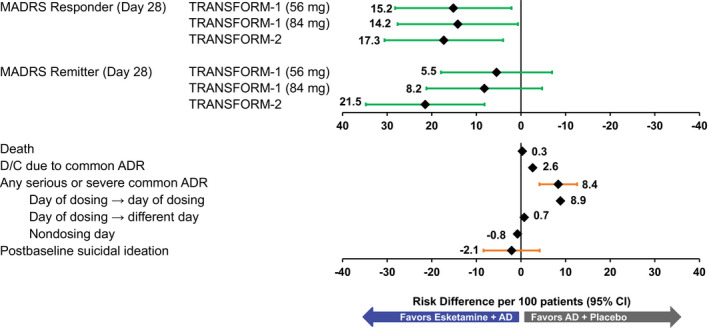
Benefit–risk assessment for short‐term treatment, risk differences (per 100 patients) in adults 18–64 years: TRANSFORM‐1 and 2 efficacy and pooled safety. No CI (confidence interval) provided if the number of events is 0 or 1 in either group. AD, antidepressants; ADR, adverse drug reaction; D/C, discontinuation; MADRS, Montgomery‐Åsberg Depression Rating Scale. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
Treatment comparison of efficacy and safety in induction phase (pooled studies TRANSFORM‐1 and 2)
|
Esketamine + AD Risk/100 Patients N = 343 |
AD + placebo Risk/100 Patients N = 222 |
Treatment Difference (Esketamine‐Placebo) |
|
|---|---|---|---|
|
(Esketamine + AD) – (AD + placebo) Risk Difference/100 Patients (95% CI) | |||
| Efficacy MADRS (Day 28) | |||
| Responders | |||
| TRANSFORM‐1 (56 mg) | 54.1 | 38.9 | 15.2 (2.11–28.22) |
| TRANSFORM‐1 (84 mg) | 53.1 | 14.2 (0.68–27.67) | |
| TRANSFORM‐2 | 69.3 | 52 | 17.3 (4.01–30.60) |
| Remitters | |||
| TRANSFORM‐1 (56 mg) | 36 | 30.6 | 5.5 (−6.98 to 17.94) |
| TRANSFORM‐1 (84 mg) | 38.8 | 8.2 (−4.76 to 21.20) | |
| TRANSFORM‐2 | 52.5 | 31 | 21.5 (8.17–34.78) |
| Safety | |||
| Death | 0.3 | 0 | 0.3 |
| Discontinuation due to common ADR a | 2.6 | 0 | 2.6 |
| Any serious or severe common ADR a | 12 | 3.6 | 8.4 (4.13–12.57) |
| Dissociation | 4.4 | 0 | 4.4 |
| Dizziness | 2.6 | 0.5 | 2.2 |
| Nausea | 1.5 | 0 | 1.5 |
| Sedation | 0.9 | 0.5 | 0.4 |
| Headache | 1.5 | 0.9 | 0.6 (−1.22 to 2.33) |
| Vertigo | 2.9 | 0.5 | 2.5 |
| Dysgeusia | 1.7 | 0 | 1.7 |
| Hypoesthesia | 0.6 | 0 | 0.6 |
| Blood pressure increased | 0 | 0 | 0 |
| Anxiety | 1.7 | 1.8 | −0.1 (−2.29 to 2.18) |
| Vomiting | 1.5 | 0 | 1.5 |
| Any serious or severe common ADR | |||
| Day of dosing → day of dosing | 10.2 | 1.4 | 8.9 (5.31 to 12.40) |
| Day of dosing → different day | 1.2 | 0.5 | 0.7 |
| Nondosing day | 1.5 | 2.3 | −0.8 (−3.12 to 1.53) |
| Postbaseline suicidal ideation |
N = 254 10.2 |
N = 162 12.3 |
−2.1 (−8.40 to 4.18) |
TRANSFORM‐1: esketamine 56 mg + AD, N = 115; esketamine 84 mg + AD, n = 114; AD + placebo, n = 113.
TRANSFORM‐2: esketamine + AD, n = 114; AD + placebo, n = 109.
MADRS total score ranges from 0 to 60; a higher score indicates a more severe condition. Negative change in score indicates improvement.
AD, antidepressant; ADR, adverse drug reaction; CI, confidence interval; MADRS, Montgomery‐Åsberg Depression Rating Scale; TRD, treatment‐resistant depression.
The following grouped terms with an incidence of ≥ 10% in TRD subjects treated with esketamine nasal spray + oral AD and greater than oral AD + placebo are regarded as common ADRs: dissociation, dizziness, nausea, sedation, headache, vertigo, dysgeusia, hypoesthesia, blood pressure increased, anxiety, and vomiting. No CI provided if the number of events is 0 or 1 in either group.
As noted in the Methods section, remission is a greater degree of benefit than response and provides an alternative view on efficacy. Compared with AD + placebo, esketamine + AD also resulted in a greater number of patients in remission at day 28, with 5.5 (95% CI, −6.98 to 17.94) and 8.2 (95% CI, −4.76 to 21.20) more remitters per 100 patients for the 56 mg dose and 84 mg dose, respectively in TRANSFORM‐1, and 21.5 (95% CI, 8.17 to 34.78) more remitters per 100 patients in TRANFORM‐2 (Figure 2 and Table 1 ).
The overall rates of serious or severe ADRs and the ADRs leading to discontinuation were higher in the esketamine + AD groups than for the AD + placebo groups for TRANSFORM‐1 and TRANSFORM‐2. Per 100 patients, esketamine + AD treatment vs. AD + placebo resulted in 2.6 more discontinuations due to a common ADR, and 8.4 (95% CI, 4.13 to 12.57) more serious or severe common ADRs (Figure 2 ). These events were predominantly dissociation, vertigo, and dizziness. The risk differences for serious or severe common ADRs that occurred and resolved on the day of dosing was 8.9 (95% CI, 5.31 to 12.40), the serious or severe common ADRs that occurred on day of dosing and resolved on a different day was 0.7, and those serious or severe common ADRs that occurred on a nondosing day was −0.8. Taken together, these results indicate that esketamine + AD was associated with ADRs that occur and resolve on the day of dosing, when patients are under medical supervision or driving restrictions, but there was no difference between esketamine + AD and AD + placebo beyond the day of dosing.
No severe AEs of increased blood pressure were reported in either study. The risk difference for death was 0.3; one death occurred in the esketamine arm of TRANSFORM‐2 from multiple injuries sustained in a road traffic accident and was not considered related to treatment. 20 Incident postbaseline suicidal ideation was numerically balanced between study arms −2.1 (95% CI, −8.40 to 4.18) per 100 patients. There was also no difference between treatment groups in any of the cognitive tests performed during TRANSFORM‐1 or TRANSFORM‐2.
Benefit–risk balance of induction treatment in elderly patients
The benefit–risk analysis for TRANSFORM‐3 in patients ≥ 65 years of age showed similar results to those of patients aged 18–64 years in TRANSFORM‐1 and TRANSFORM‐2. Compared with AD + placebo, esketamine + AD resulted in 13.7 (95% CI, −0.28 to 27.58) more responders and 10.8 (95% CI, −0.51 to 22.09) more remitters per 100 patients at day 28 (Table 2 ). For patients aged 65–74 years, both end points shifted slightly in favor of esketamine + AD, with 15.1 (95% CI, −0.08 to 30.27) more responders and 15.1 (95% CI, 2.53 to 27.66) more remitters per 100 patients treated with esketamine + AD (Figure 3 and Table 2 ).
Table 2.
Treatment comparison of efficacy and safety in induction phase (TRANSFORM‐3)
|
Esketamine + AD N = 72 Risk/100 Patients |
AD + placebo N = 65 Risk/100 Patients |
Treatment Difference (Esketamine + Oral AD) ‐ (Oral AD + Placebo) Risk Difference/100 Patients (95% CI) |
|
|---|---|---|---|
| Efficacy MADRS (Day 28) | |||
| Responder a (all) | 27 | 13.3 | 13.7 (−0.28 to 27.58) |
| Responder (65–74 years of age) | 28.3 | 13.2 | 15.1 (−0.08 to 30.27) |
| Remitters b (all) | 17.5 | 6.7 | 10.8 (−0.51 to 22.09) |
| Remitters (65–74 years of age) | 20.8 | 5.7 | 15.1 (2.53 to 27.66) |
| Safety | |||
| Death | 0 | 0 | 0 |
| Discontinuation due to common ADR c | 4.2 | 1.5 | 2.6 |
| Any serious or severe common ADR d | 4.2 | 3.1 | 1.1 (−5.15 to 7.33) |
| Dissociation | 0 | 0 | 0 |
| Dizziness | 0 | 1.5 | −1.5 |
| Nausea | 0 | 0 | 0 |
| Sedation | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 |
| Vertigo | 0 | 0 | 0 |
| Dysgeusia | 1.4 | 0 | 1.4 |
| Hypoesthesia | 0 | 0 | 0 |
| Blood pressure increased | 1.4 | 0 | 1.4 |
| Anxiety | 1.4 | 1.5 | −0.1 |
| Vomiting | 0 | 0 | 0 |
| Any serious or severe common ADR | |||
| Day of dosing → day of dosing | 1.4 | 0 | 1.4 |
| Day of dosing → different day | 0 | 0 | 0 |
| Nondosing day | 2.8 | 3.1 | −0.3 (−5.96 to 5.36) |
| Postbaseline suicidal ideation |
N = 58 13.8 |
N = 54 16.7 |
−2.9 (−16.20 to 10.45) |
MADRS total score ranges from 0 to 60; a higher score indicates a more severe condition. Negative change in score indicates improvement.
AD, antidepressant; ADR, adverse drug reaction; CI, confidence interval; MADRS, Montgomery‐Åsberg Depression Rating Scale; TRD, treatment‐resistant depression.
Responder is defined as the proportion of patients achieving at least 50% improvement in MADRS at Day 28.
Remitter is defined as the proportion of patients achieving MADRS total score of ≤ 12 at Day 28. (All patients had baseline MADRS > 28).
Responder (without remission) is defined as the proportion of patients achieving at least 50% improvement in MADRS and MADRS total score > 12 at Day 28.
The following grouped terms with an incidence of ≥ 10% in TRD subjects treated with intranasal esketamine + oral AD and greater than oral AD + placebo are regarded as common ADRs: dissociation, dizziness, nausea, sedation, headache, vertigo, dysgeusia, hypoesthesia, blood pressure increased, anxiety, and vomiting. No CI provided if the number of events is 0 or 1 in either group.
Figure 3.
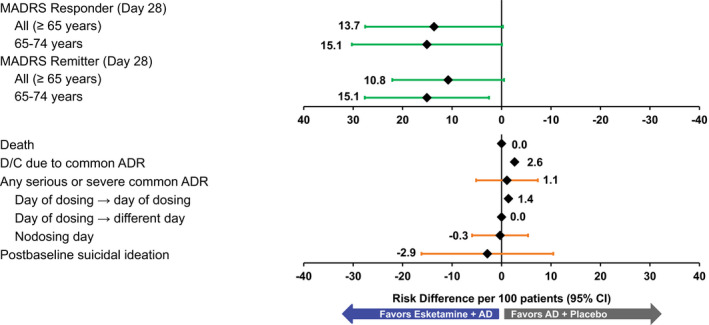
Benefit–risk assessment for short‐term treatment, risk differences (per 100 patients) in adults ≥ 65 years: TRANSFORM‐3. No CI (confidence interval) provided if the number of events is 0 or 1 in either group. AD, antidepressants; ADR, adverse drug reaction; D/C, discontinuation; MADRS, Montgomery‐Åsberg Depression Rating Scale. [Colour figure can be viewed at wileyonlinelibrary.com]
For the esketamine + AD group, there were 2.6 more discontinuations due to common ADRs per 100 patients and 1.1 more serious or severe common ADRs per 100 patients. Results for serious or severe common ADRs show the same pattern as seen with all patients: The risk difference for those that occurred and resolved on the day of dosing was 1.4, 0 for those that occurred on the day of dosing and resolved on a different day, and −0.3 for those that occurred on a nondosing day (Figure 3 and Table 2 ). One event of severe blood pressure increase occurred and resolved on the day of dosing in the esketamine + AD group. There were no deaths, and incident postbaseline suicidal ideation was numerically balanced between study arms: −2.9 (95% CI, −16.20 to 10.45) per 100 patients.
Benefit–risk balance of maintenance treatment
Among patients in SUSTAIN‐1 who had achieved stable remission after 16 weeks of treatment with esketamine + AD, randomized continuation with esketamine resulted in 18.7 (95% CI, 4.75 to 32.62) fewer relapses per 100 patients compared with discontinuing esketamine. Among patients who had achieved a stable response (but not remission) after 16 weeks of treatment with esketamine + AD, randomized continuation with esketamine resulted in 31.8 (95% CI, 15.16 to 48.48) fewer relapses per 100 patients compared with discontinuing esketamine (Figure 4 and Table 3 ). The primary reason for relapse was worsening depression manifesting as a deteriorating MADRS total score, with few patients meeting criteria for relapse based on a clinically relevant event. There were no meaningful differences in risks in SUSTAIN‐1 between stable remitters and stable responders entering the study, hence risk results were pooled.
Figure 4.
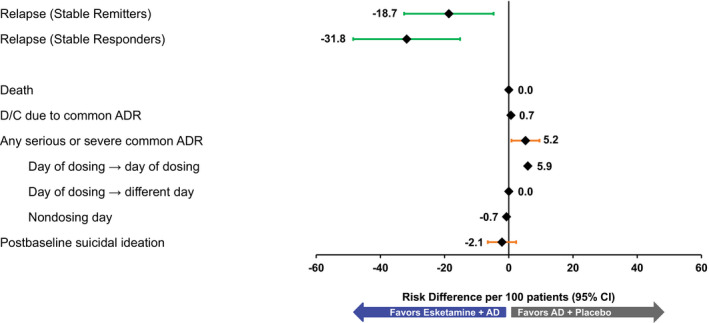
Benefit–risk assessment for maintenance treatment risk differences (per 100 patients): SUSTAIN‐1. No CI (confidence interval) provided if the number of events is 0 or 1 in either group. AD, antidepressants; ADR, adverse drug reaction; D/C, discontinuation. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 3.
Treatment comparison of efficacy and safety in maintenance phase: SUSTAIN‐1
| Safety |
Esketamine + AD N = 152 Risk/100 patients |
AD + Placebo N = 145 Risk/100 patients |
Treatment Difference (Esketamine + Oral AD) ‐ (Oral AD + Placebo) Risk Difference/100 patients (95% CI) |
|---|---|---|---|
| Efficacy MADRS (day 28) | |||
| Stable responder: relapse (all) a | 25.8 (N = 62) | 57.6 (N = 59) | −31.8 (−48.48 to −15.16) |
| Stable remitter: relapse (all) a | 26.7 (N = 90) | 45.3 (N = 86) | −18.7 (−32.62 to −4.75) |
| Safety | |||
| Death | 0 | 0 | 0 |
| Discontinuation due to common ADR a | 0.7 | 0 | 0.7 |
| Any serious or severe common ADR a | 6.6 | 1.4 | 5.2 (0.83 to 9.57) |
| Dissociation | 0.7 | 0 | 0.7 |
| Dizziness | 0.7 | 0 | 0.7 |
| Nausea | 0.7 | 0 | 0.7 |
| Sedation | 2.0 | 0 | 2.0 |
| Headache | 0.7 | 1.4 | −0.7 |
| Vertigo | 1.3 | 0 | 1.3 |
| Dysgeusia | 1.3 | 0 | 1.3 |
| Hypoesthesia | 0 | 0 | 0 |
| Blood pressure increased | 0 | 0 | 0 |
| Anxiety | 1.3 | 0 | 1.3 |
| Vomiting | 0 | 0 | 0 |
| Any serious or severe common ADR | |||
| Day of dosing → day of dosing | 5.9 | 0 | 5.9 |
| Day of doing → different day | 0 | 0 | 0 |
| Nondosing day | 0.7 | 1.4 | −0.7 |
| Postbaseline suicidal ideation |
N = 126 2.4 |
N = 133 4.5 |
−2.1 (−6.55 to 2.29) |
MADRS total score ranges from 0 to 60; a higher score indicates a more severe condition. Negative change in score indicates improvement.
AD, antidepressant; ADR, adverse drug reaction; CI, confidence interval; MADRS, Montgomery‐Åsberg Depression Rating Scale; TRD, treatment‐resistant depression.
The following grouped terms with an incidence of ≥ 10% in TRD subjects treated with intranasal esketamine + oral AD and greater than oral AD + placebo are regarded as common ADRs: dissociation, dizziness, nausea, sedation, headache, vertigo, dysgeusia, hypoesthesia, blood pressure increased, anxiety, and vomiting. No CI provided if the number of events is 0 or 1 in either group.
There were 0.7 more common ADRs leading to discontinuation and 5.2 (95% CI, 0.83 to 9.57) more serious or severe common ADRs per 100 patients receiving esketamine + AD. The risk differences for serious or severe common ADRs that occurred and resolved on the day of dosing was 5.9, serious or severe common ADRs that occurred on the day of dosing and resolved on a different day was 0, and those ADRs that occurred on a nondosing day was −0.7 (Figure 4 and Table 3 ). There were no deaths; incident postbaseline suicide ideation showed no meaningful difference (−2.1 (95% CI, −6.55 to 2.29) per 100 patients), and there was no difference between treatment groups in any of the cognitive tests performed.
Interstitial cystitis and changes in cognition were also considered as risks; however, there were no cases of interstitial cystitis. A battery of cognitive tests was performed, and the only difference observed in comparing treatment groups was a slowing of reaction time in the absence of any other change in cognitive performance in older patients (> 65 years of age), which was observed in the long‐term safety study SUSTAIN‐2. This observation could not be attributed to study medication and the clinical relevance and consequences have not been established. Overdose, abuse, or drug‐seeking behavior was not observed in any clinical study, and abuse potential is addressed in the comprehensive risk mitigation program.
DISCUSSION
Assessment of the benefit–risk profile of esketamine + AD is of particular interest owing to the high placebo effect in AD studies, use of an active control, and safety profile addressing both short‐term and long‐term AEs. The benefit–risk of esketamine + AD was assessed using the BRAT framework. Compared with AD + placebo, esketamine + AD in induction and maintenance treatment provided benefit of clinically meaningful response and remission among adults with TRD. The safety experience with esketamine + AD showed that the most common AEs (risks) that were severe or serious were primarily observed on the day of dosing. The differences between treatment groups of dosing day events that end on a nondosing day or that occur on nondosing days were both small. The events that were reported as serious or severe were transient in nature and mostly occurred and resolved within 2 hours of dosing without clinical sequelae, when patients are either under medical supervision (2 hours) or under driving restrictions (day of dosing).
The majority of the ADRs observed with esketamine + AD included common events such as dissociative symptoms, dizziness/vertigo, increased blood pressure, and sedation, occurred shortly after dosing and resolved on the same day while the patient was under the supervision of a healthcare professional. One death was observed across the four double‐blind controlled phase III studies and was due to multiple injuries sustained in a road traffic accident, which occurred 1 day after receiving a dose of esketamine and the investigator considered the event not related to esketamine use. Since patients receiving esketamine may experience immediate dissociative and sedative effect due to the underlying mechanism of action, there are safety concerns over potential for abuse, diversion, and misuse. To mitigate the risk of abuse and misuse, the FDA approved esketamine with a Risk Evaluation and Mitigation Strategy (REMS) program. 35 Under this provision, esketamine is made available only at medically supervised healthcare settings and hospitals that are certified in the REMS and patients self‐administer esketamine under the observation of a healthcare provider.
Based on the decision context, 5–21 additional patients remitting or 14–17 additional patients responding per 100 treated in this analysis, with symptom improvement starting to manifest in some patients within hours, is a considerable benefit that outweighs these transient adverse reactions (risks), particularly dissociation, vertigo, and dizziness, that are manageable and mitigated for adverse outcomes. Similar advantage in esketamine treatment was observed with elderly patients, especially in the subgroup of patients ≥ 65–74 years. Once response or remission has been achieved, the benefit observed with continued maintenance treatment of 19 to 32 fewer relapses per 100 patients over longer‐term therapy also outweighed the few serious or severe common ADRs. Overall, the analyses of evidence from phase III clinical studies suggest a positive benefit–risk balance for esketamine in combination with oral AD vs. AD + placebo in adults with TRD.
The occurrence of the short‐term AEs that occur on the day of dosing and generally last no more than 2 hours are potentially less impactful than reduction or relief of long‐lasting clinical depression. This perspective is reinforced by results from a patient preference study of both esketamine‐experienced and ketamine‐naive TRD patients, 34 , 36 which showed that patients regarded even partial relief of depression symptoms as much more important than the short‐term AE (i.e., dissociation and dizziness).
Implementation of the REMS program, which is designed to ensure safe use and mitigate the risks of serious adverse outcomes resulting from sedation, dissociation, and misuse of esketamine reinforces the positive benefit–risk profile of esketamine in patients with TRD.
The current assessment had several potential limitations: (i) Response and remission rates were used to allow comparing proportions of beneficial events with harmful events. For the longer maintenance studies, discontinuations can make proportions misleading. The small difference in discontinuation rates due to common ADRs mitigates this concern, but ideally exposure‐time rates could be assessed. (ii) Since all remitters were also responders in these studies, response and remission end points reflect different degrees of benefit, rather than distinct benefits. While distinct benefits are generally valuable, we found that using the mutually exclusive end‐point “responders who did not achieve remission” caused considerable confusion, and instead used response and remission end points to represent different degrees of efficacy. Alternative characterizations of the degree of efficacy based on one underlying assessment are not uncommon in benefit–risk assessment, such as the use of Psoriasis Area and Severity Index (PASI) measures PASI 50, PASI 75, and PASI 90 in psoriasis benefit–risk assessment. (iii) While very common in clinical studies, the dichotomization of continuous end points into those with and without a response (e.g., responder, nonresponder) masks the distribution of response or degree of benefit within each category. Additional analyses could represent this heterogeneity of response. (iv) The analyses include serious or severe common ADRs. ADRs of moderate severity could have also been considered in the risk assessment. They were prospectively excluded given our clinical judgment that the clinical impact of response and remission is much larger than that of the moderate, transitory ADRs observed. The patient preference study reinforced this perspective. 34 (v) The assessment of benefit–risk in patients > 75 years of age could not be carried out because of insufficient sample size. (vi) Further, dependency between benefit and risk end points was not considered, e.g., joint end points assessing whether those who benefited may have been more or less likely to have the risks. (vii) The frequencies of the benefits and risks in clinical trial population may differ from that in the general population of TRD patients. (viii) The benefit–risk assessment is limited to the time period of the studies conducted. Based on the long‐term SUSTAIN‐1 study 23 included in this analysis and another long‐term SUSTAIN‐2 (single‐arm, 1‐year duration) study, 33 the AD efficacy appears to be maintained and risks do not appear to increase. At present, there is no information available outside of the studies conducted.
In summary, in patients with TRD, esketamine nasal spray as induction and maintenance treatment along with a newly initiated oral AD provides an advantage in rapid onset of effect and sustained efficacy over the transient adverse effects that are medically manageable and resolved mainly on the day of treatment when there is medical supervision or there are driving restrictions. Taken together, the evidence supports a positive benefit–risk balance for esketamine + AD as a novel therapeutic option for this difficult‐to‐treat, potentially life‐threatening condition of TRD.
Funding
Janssen Research & Development, LLC, USA.
Conflict of Interest
All authors are employees of Janssen Research & Development, LLC.
Author Contribution
All authors wrote the manuscript and designed the research. B.L., E.G.K., and R.L. performed the research and analyzed the data. B.L. and E.G.K. contributed new reagents / analytical tools.
Clinical Trial Registration
TRANSFORM‐1: NCT02417064; TRANFORM‐2: NCT02418585; TRANFORM‐3: NCT02422186; and SUSTAIN‐1: NCT02493868.
Supporting information
Figure S1
Table S1
Table S2
Acknowledgments
The authors thank all patients for their participation in this study and acknowledge the collaboration and commitment of the investigators and their staff. Writing assistance was provided by Ramji Narayanan, M Pharm, ISMPP CMPP (SIRO Clinpharm Pvt. Ltd.) funded by Janssen Global Services and additional editorial support for this manuscript was provided by Ellen Baum, PhD (Janssen Global Services, LLC).
Data Availability Statement
The data sharing policy of the study sponsor, Janssen Pharmaceutical Companies of Johnson & Johnson, is available at https://www.janssen.com/clinical‐trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
References
- 1. US Food and Drug Administration . Structured approach to benefit‐risk assessment in drug regulatory decision‐making <http://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM329758.pdf> (2019). Accessed November 15, 2019.
- 2. Luteijn, J.M. et al State of the art in benefit‐risk analysis: medicines. Food Chem. Toxicol. 50, 26–32 (2012). [DOI] [PubMed] [Google Scholar]
- 3. Pignatti, F. et al Structured frameworks to increase the transparency of the assessment of benefits and risks of medicines: current status and possible future directions. Clin. Pharmacol. Ther. 98, 522–533 (2015). [DOI] [PubMed] [Google Scholar]
- 4. ICH harmonised guideline: revision of M4E guideline on enhancing the format and structure of benefit‐risk information in ICH, efficacy ‐ M4E(R2) <https://database.ich.org/sites/default/files/M4E_R2__Guideline.pdf> (2016). Accessed March 12, 2020.
- 5. Levitan, B. A concise display of multiple end points for benefit‐risk assessment. Clin. Pharmacol. Ther. 89, 56–59 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Coplan, P.M. , Noel, R.A. , Levitan, B.S. , Ferguson, J. & Mussen, F. Development of a framework for enhancing the transparency, reproducibility and communication of the benefit‐risk balance of medicines. Clin. Pharmacol. Ther. 89, 312–315 (2011). [DOI] [PubMed] [Google Scholar]
- 7. Hallgreen, C.E. et al Benefit‐risk assessment in a post‐market setting: a case study integrating real‐life experience into benefit‐risk methodology. Pharmacoepidemiol. Drug Saf. 23, 974–983 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Hughes, D. et al Recommendations report: recommendations for the methodology and visualisation techniques to be used in the assessment of benefit and risk of medicines <http://www.imi‐protect.eu/documents/HughesetalRecommendationsforthemethodologyandvisualisationtechniquestobeusedintheassessmento.pdf> (2015). Accessed November 15, 2019.
- 9. Levitan, B.S. et al Application of the BRAT framework to case studies: observations and insights. Clin. Pharmacol. Ther. 89, 217–224 (2011). [DOI] [PubMed] [Google Scholar]
- 10. Walker, E.R. , McGee, R.E. & Druss, B.G. Mortality in mental disorders and global disease burden implications: a systematic review and meta‐analysis. JAMA Psychiatry 72, 334–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rush, A.J. et al Acute and longer‐term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163, 1905–1917 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Trevino, K. , McClintock, S.M. , McDonald Fischer, N. , Vora, A. & Husain, M.M. Defining treatment‐resistant depression: a comprehensive review of the literature. Ann. Clin. Psychiatry 26, 222–232 (2014). [PubMed] [Google Scholar]
- 13. Brown, S. , Rittenbach, K. , Cheung, S. , McKean, G. , MacMaster, F.P. & Clement, F. Current and common definitions of treatment‐resistant depression: findings from a systematic review and qualitative interviews. Can. J. Psychiatry 64, 380–387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bergfeld, I.O. , Mantione, M. , Figee, M. , Schuurman, P.R. , Lok, A. & Denys, D. Treatment‐resistant depression and suicidality. J. Affect. Disord. 235, 362–367 (2018). [DOI] [PubMed] [Google Scholar]
- 15. DiBernardo, A. et al Humanistic outcomes in treatment resistant depression: a secondary analysis of the STAR*D study. BMC Psychiatry 18, 352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Machado‐Vieira, R. , Salvadore, G. , Luckenbaugh, D.A. , Manji, H.K. & Zarate Jr, C.A. Rapid onset of antidepressant action: a new paradigm in the research and treatment of major depressive disorder. J. Clin. Psychiatry 69, 946–958 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holtzheimer, P.E. Advances in the management of treatment‐resistant depression. Focus (Am. Psychiatr. Publ.) 8, 488–500 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. European Medical Agency . Spravato <https://www.ema.europa.eu/en/medicines/human/EPAR/spravato>. Accessed December 2, 2019.
- 19. Spravato [prescribing information]. (Janssen Pharmaceuticals, Titusville, NJ).
- 20. Fedgchin, M. et al Efficacy and safety of fixed‐dose esketamine nasal spray combined with a new oral antidepressant in treatment‐resistant depression: results of a randomized, double‐blind, active‐controlled study (TRANSFORM‐1). Int. J. Neuropsychopharmacol. 22, 616–630 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Popova, V. et al Efficacy and safety of flexibly dosed esketamine nasal spray combined with a newly initiated oral antidepressant in treatment‐resistant depression: a randomized double‐blind active‐controlled study. Am. J. Psychiatry 176, 428–438 (2019). [DOI] [PubMed] [Google Scholar]
- 22. Ochs‐Ross, R. et al Efficacy and safety of esketamine nasal spray plus an oral antidepressant in elderly patients with treatment‐resistant depression‐TRANSFORM‐3. Am. J. Geriatr. Psychiatry 28, 121–141 (2020). [DOI] [PubMed] [Google Scholar]
- 23. Daly, E.J. et al Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment‐resistant depression: a randomized clinical trial. JAMA Psychiatry 76, 893–903 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ziemssen, F. , Cruess, A. , Dunger‐Baldauf, C. , Margaron, P. , Snow, H. & Strain, W.D. Ranibizumab in diabetic macular oedema ‐ a benefit‐risk analysis of ranibizumab 0.5 mg PRN versus laser treatment. Eur. Endocrinol. 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levitan, B. , Markowitz, M. , Turkoz, I. , Fu, D.‐J. , Gopal, S. & Alphs, L. Benefit‐risk assessment of paliperidone oral extended‐release tablet versus monthly injectable for maintenance treatment of schizophrenia. Int. Clin. Psychopharmacol. 31, 315–322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Detke, H.C. , Lauriello, J. , Landry, J. & McDonnell, D.P. Within‐drug benefit‐risk evaluation of olanzapine long‐acting injection at one and two years of treatment. Int. J. Methods Psychiatr. Res. 23, 439–450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nixon, R. et al A case study using the PrOACT‐URL and BRAT frameworks for structured benefit risk assessment. Biom. J. 58, 8–27 (2016). [DOI] [PubMed] [Google Scholar]
- 28. Walker, S. , McAuslane, N. , Liberti, L. , Leong, J. & Salek, S. A Universal framework for the benefit‐risk assessment of medicines: is this the way forward? Ther. Innov. Regul. Sci. 49, 17–25 (2015). [DOI] [PubMed] [Google Scholar]
- 29. Zimmerman, M. , Posternak, M.A. & Chelminski, I. Derivation of a definition of remission on the Montgomery‐Asberg depression rating scale corresponding to the definition of remission on the Hamilton rating scale for depression. J. Psychiatr. Res. 38, 577–582 (2004). [DOI] [PubMed] [Google Scholar]
- 30. Kennedy, S.H. et al Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder: section 3. Pharmacological treatments. Can. J. Psychiatry 61, 540–560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Phillips, J.L. , Batten, L.A. , Tremblay, P. , Aldosary, F. & Blier, P. A Prospective, longitudinal study of the effect of remission on cortical thickness and hippocampal volume in patients with treatment‐resistant depression. Int. J. Neuropsychopharmacol. 18, pyv037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Council for International Organizations of Medical Sciences (CIOMS) . Guidelines for Preparing Core Clinical‐Safety Information on Drugs 2nd edn. (Report of CIOMS Working Groups III and V, Geneva, 1999). [Google Scholar]
- 33. Wajs, E. et al Esketamine nasal spray plus oral antidepressant in patients with treatment‐resistant depression: assessment of long‐term safety in a phase 3, open‐label study (SUSTAIN‐2). J. Clin. Psychiatry 81, 19m12891 (2020). [DOI] [PubMed] [Google Scholar]
- 34. Fairchild, A.O. et al Patient preferences for ketamine‐based antidepressant treatments in treatment‐resistant depression: results from a clinical trial and panel. Neurol. Psychiatry Brain Res. 47, 67–68 (2020). [Google Scholar]
- 35. US Food and Drug Administration . Approved risk evaluation and mitigation strategies (REMS): spravato (esketamine) <https://www.accessdata.fda.gov/scripts/cder/rems/index.cfm?event=IndvRemsDetails.page&REMS=386>. Accessed June 2, 2019.
- 36. Katz, E. et al The patient’s perspective in benefit‐risk: how a preference study supported the esketamine application for regulatory approval in treatment‐resistant depression. 59th Annual Meeting of the American Society of Clinical Psychopharmacology, Scottsdale, Arizona, May 28–31, 2019. Abstract #T73.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Data Availability Statement
The data sharing policy of the study sponsor, Janssen Pharmaceutical Companies of Johnson & Johnson, is available at https://www.janssen.com/clinical‐trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.