

Abstract
Background
WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays) is a rare contiguous gene deletion syndrome with a 45% to 60% risk of developing Wilms tumor (WT). Currently, surveillance and treatment recommendations are based on limited evidence.
Methods
Clinical characteristics, treatments, and outcomes were analyzed for patients with WAGR and WT/nephroblastomatosis who were identified through International Society of Pediatric Oncology Renal Tumor Study Group (SIOP‐RTSG) registries and the SIOP‐RTSG network (1989‐2019). Events were defined as relapse, metachronous tumors, or death.
Results
Forty‐three patients were identified. The median age at WT/nephroblastomatosis diagnosis was 22 months (range, 6‐44 months). The overall stage was available for 40 patients, including 15 (37.5%) with bilateral disease and none with metastatic disease. Histology was available for 42 patients; 6 nephroblastomatosis without further WT and 36 WT, including 19 stromal WT (52.8%), 12 mixed WT (33.3%), 1 regressive WT (2.8%) and 2 other/indeterminable WT (5.6%). Blastemal type WT occurred in 2 patients (5.6%) after prolonged treatment for nephroblastomatosis; anaplasia was not reported. Nephrogenic rests were present in 78.9%. Among patients with WT, the 5‐year event‐free survival rate was 84.3% (95% confidence interval, 72.4%‐98.1%), and the overall survival rate was 91.2% (95% confidence interval, 82.1%‐100%). Events (n = 6) did not include relapse, but contralateral tumor development (n = 3) occurred up to 7 years after the initial diagnosis, and 3 deaths were related to hepatotoxicity (n = 2) and obstructive ileus (n = 1).
Conclusions
Patients with WAGR have a high rate of bilateral disease and no metastatic or anaplastic tumors. Although they can be treated according to existing WT protocols, intensive monitoring of toxicity and surveillance of the remaining kidney(s) are advised.
Lay Summary
WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays) is a rare genetic condition with an increased risk of developing Wilms tumor.
In this study, 43 patients with WAGR and Wilms tumor (or Wilms tumor precursor lesions/nephroblastomatosis) were identified through the international registry of the International Society of Pediatric Oncology Renal Tumor Study Group (SIOP‐RTSG) and the SIOP‐RTSG network. In many patients (37.5%), both kidneys were affected. Disease spread to other organs (metastases) did not occur.
Overall, this study demonstrates that patients with WAGR syndrome and Wilms tumor can be treated according to existing protocols. However, intensive monitoring of treatment complications and surveillance of the remaining kidney(s) are advised.
Keywords: aniridia; pediatric; predisposition; surveillance; treatment; WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays); Wilms tumor
Short abstract
Patients with WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays) and Wilms tumor/nephroblastomatosis, identified through the International Society of Pediatric Oncology Renal Tumor Study Group registries and network, have a high rate of bilateral disease and no metastatic or anaplastic tumors. Although they can be treated according to existing Wilms tumor protocols, intensive monitoring of toxicity and surveillance of the remaining kidney(s) are advised.
Introduction
WAGR syndrome (Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays) is caused by a rare contiguous germline gene deletion involving chromosome band 11p13. Children with WAGR syndrome have a 45% to 60% risk of developing Wilms tumor (WT). 1 , 2 , 3 Currently, surveillance and treatment recommendations for children with WAGR syndrome are based on limited evidence.
Historically, WAGR syndrome has played an important role in our understanding of WT genetics; it contributed to the identification of WT1, the first WT predisposition gene to be identified, in 1990. 4 , 5 , 6 , 7 , 8 , 9 The genetic diagnosis of WAGR syndrome requires the involvement of both WT1 and the aniridia gene PAX6 in the deletion, whereas patients with isolated PAX6 deletions are not at risk of developing WT. 3 The size of the deletion as well as the phenotype of patients with WAGR syndrome can vary widely, and they only partially depend on whether or not additional genes such as BDNF are involved. 10
One previous report, based on North American National Wilms Tumor Studies (NWTSs) 1 to 5, described the tumor characteristics, treatments, and outcomes of a cohort of patients with WAGR who developed WT and were treated according to consecutive NWTS protocols without preoperative chemotherapy. 2 However, the characteristics and outcomes of patients with WAGR and WT registered for the International Society of Pediatric Oncology Renal Tumor Study Group (SIOP‐RTSG) protocols, according to which preoperative chemotherapy is administered, have never been described. Currently, also in North American treatment protocols (Children's Oncology Group), preoperative chemotherapy is recommended for children with a genetic predisposition. 11 Here, we report the clinical and tumor characteristics and outcomes of patients with WAGR syndrome who developed WT and/or nephroblastomatosis and were identified through the 2 last SIOP‐RTSG protocol registries and the SIOP‐RTSG network in order to support surveillance and treatment recommendations.
Materials and Methods
Patients
In the SIOP‐RTSG studies (International Society of Pediatric Oncology [SIOP] 93‐01 12 and SIOP 2001 13 ), patients were prospectively registered from 1993 onward, in some countries up to and including 2019. These studies were not designed to register tumor predisposition syndromes, but the presence or absence of aniridia was recorded. The SIOP‐RTSG steering committee approved the research proposal for the current analysis, and we retrospectively identified all patients with aniridia in the SIOP 93‐01 and SIOP 2001 databases. Patients diagnosed before 2007 may have been previously reported by Van Heyningen et al, 3 but because this study did not describe WT characteristics or outcomes, we did not exclude patients with possible overlap.
By using SIOP study IDs, national and/or local principal investigators (PIs) were requested to confirm the diagnosis of WAGR and to complete missing data. Patients for whom the diagnosis of WAGR could not be confirmed were excluded from the analysis, whereas additional patients with WAGR and WT, identified by national and/or local PIs, were added to the series. They included 10 patients who were not in the central SIOP databases but were registered locally and 3 patients who were registered on prior or subsequent SIOP protocols (SIOP 9 14 and SIOP‐RTSG UMBRELLA 15 ).
For SIOP protocols, ethical approval was obtained from ethics committees of all participating countries, and written informed consent for participation was obtained from the parents or legal representatives of the patients. For those patients not registered in the central databases, national and/or local PIs confirmed that informed consent was obtained.
Additional Data Collection
National and/or local PIs were requested to complete an additional data collection form for each patient (see the supporting information) to obtain clinical information on various items, including the type of genetic testing, age at diagnosis of WAGR syndrome, presentation and symptoms of WT (symptomatic vs asymptomatic), birth weight, congenital abnormalities, cognitive impairment, chronic kidney disease status, and other clinical findings. Chronic kidney disease was defined as a decreased estimated glomerular filtration rate (eGFR; not further specified) and/or proteinuria 2+, at least on dipstick testing. End‐stage renal disease was defined as “requiring dialysis and/or kidney transplantation.”
Stage and Histology
Stage and histology were classified according to the SIOP‐RTSG staging system and histological classifications. 16 , 17 Bilateral disease was defined as synchronous bilateral WT, bilateral nephroblastomatosis, or WT with contralateral nephroblastomatosis. For the analysis of the overall stage at diagnosis, patients with metachronous tumors were considered unilateral if they had unilateral disease at their initial diagnosis. A local (abdominal) stage could be assigned only to patients with a diagnosis of WT. Nephroblastomatosis was defined as the presence of multiple or diffuse nephrogenic rests visible on imaging studies and, where possible, confirmed histologically. In the current SIOP‐RTSG histological classification, histological subtypes are assigned after preoperative chemotherapy and include completely necrotic WT (low risk); stromal, epithelial, mixed, or regressive WT and WT with focal anaplasia (intermediate risk); and diffuse anaplastic or blastemal‐type WT (high risk). For the majority of tumors registered in SIOP‐RTSG studies, a central pathology review was performed by national and/or regional pathology panels as well as the international SIOP‐RTSG pathology panel. 18 For the current analysis, the reviewed histological diagnosis was used if available.
Statistical Methods
Frequency distributions for age at diagnosis and tumor volume were analyzed nonparametrically. Event‐free survival and overall survival analysis was performed with the Kaplan‐Meier method. Patients diagnosed with nephroblastomatosis without further WT were excluded from the survival analysis, whereas patients who were initially diagnosed with nephroblastomatosis but went on to develop WT were included. The survival time was defined as the time from the diagnosis of WT to an event or last follow‐up. Events were defined as relapse, the development of metachronous tumors, or death.
Results
In the SIOP 93‐01 and SIOP 2001 databases, aniridia was recorded in 47 of 7842 patients with WT (0.6%). After further exploration, the diagnosis of WAGR could not be confirmed in 17 of the 47 patients, and they were, therefore, excluded: 12 did not have aniridia (they had been misclassified), and for the remaining 5 patients, we received no response from national/local PIs. Thirteen additional cases with WAGR and WT that were identified by national/local PIs but apparently were not registered in the central SIOP 93‐01 and SIOP 2001 databases were subsequently added to this study (Fig. 1). Overall, a total of 43 patients (18 phenotypic males and 25 phenotypic females) were included (Table 1). Patients had been treated according to the SIOP 93‐01 protocol (n = 8 [18.6%]), the SIOP 2001 protocol (n = 31 [72.1%]), the SIOP 9 protocol (n = 2 [4.7%]), or the SIOP‐RTSG UMBRELLA protocol (n = 1 [2.3%]). The treatment protocol was not specified for 1 patient (2.3%).
Figure 1.
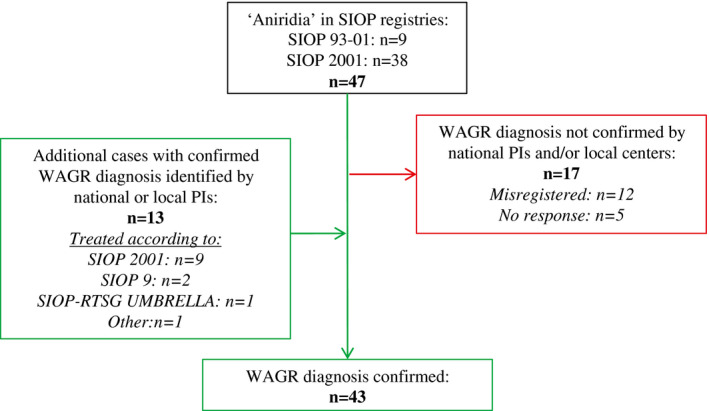
Inclusion of patients with WAGR syndrome and Wilms tumor/nephroblastomatosis based on aniridia registration in the SIOP 93‐01 and SIOP 2001 registries. PI indicates principal investigator; SIOP, International Society of Pediatric Oncology; SIOP‐RTSG, International Society of Pediatric Oncology Renal Tumor Study Group; WAGR, Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays.
TABLE 1.
Identified Patients With WAGR Syndrome and WT Based on the SIOP 93‐01 and SIOP 2001 Registries (n = 43)
| Patient No. | Protocol | Sex | Age at WT, mo | Unilateral or Bilateral | Abdominal Stage | Preoperative Treatment, wk | Surgery | Histology | Nephrogenic Rests | Notes | Follow‐Up, mo |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | SIOP 2001 | F | 15 | Unilateral | II | 4 | TN | Stromal‐type WT | ILNR | — | 96 |
| 2 | SIOP 2001 | F | 21 | Unilateral a | I | None | TN | Stromal‐type WT | ILNR | Contralateral tumor at age of 9 y | 119 |
| 3 | SIOP 2001 | F | 38 | NA | NA | 4 | TN and NSS | Nephroblastomatosis | ILNR | — | 5 b |
| 4 | SIOP 2001 | M | 14 | Bilateral | NA | 26 | TN and NSS | L: Nephroblastomatosis | ILNR | *Progression to blastemal‐type WT 12 mo after diagnosis | 38 |
| R: Nephroblastomatosis* | |||||||||||
| 5 | SIOP 2001 | M | 39 | Unilateral | I | 4 | TN | Regressive‐type WT | ILNR | — | 152 |
| 6 | SIOP 2001 | M | 18 | Bilateral | NA | 3 | None | NA | NA | Death due to hepatic failure 3 wk after diagnosis | 0 c |
| 7 | SIOP 2001 | F | 25 | Unilateral a | I | 4 | TN | Stromal‐type WT | No | Contralateral tumor at age of 3 y | 39 |
| 8 | SIOP 2001 | F | 25 | Bilateral | II | 5 | TN and NSS | L: Stromal‐type WT | ILNR | — | 114 |
| R: Nephroblastomatosis | |||||||||||
| 9 | SIOP 2001 | M | 36 | Unilateral | III | 6 | TN | Mixed‐type WT | ILNR | ESRD at age 16 y | 158 |
| 10 | SIOP 2001 | F | 13 | Unilateral | II | 4 | TN | Stromal‐type WT | ILNR | — | 104 |
| 11 | SIOP 2001 | M | 20 | Unilateral | I | 6 | NA | Stromal‐type WT | ILNR | — | 8 |
| 12 | SIOP 2001 | F | 23 | Unilateral | III | 4 | NA | Stromal‐type WT | No | — | 95 |
| 13 | SIOP 2001 | F | 12 | Bilateral | NA | 22 | NSS | L: Nephroblastomatosis (radiology only) | ILNR | — | 79 b |
| R: Nephroblastomatosis | |||||||||||
| 14 | SIOP 2001 | F | 14 | Unilateral | I | 4 | NSS | Mixed‐type WT | ILNR | — | 0 |
| 15 | SIOP 2001 | F | 37 | Unilateral | III | 4 | TN | Stromal‐type WT | No | — | 132 |
| 16 | SIOP 2001 | F | 35 | Unilateral | NA | 4 | NSS | Nephroblastomatosis | ILNR | Death, cause not specified | 9 b , c |
| 17 | SIOP 2001 | M | 26 | Bilateral | I | 8 | NSS | L: Mixed‐type WT | ILNR | — | 118 |
| R: Stromal‐type WT | |||||||||||
| 18 | SIOP 2001 | F | 9 | Bilateral | I | 5 | TN and NSS | L: Stromal‐type WT | No | — | 189 |
| R: Stromal‐type WT | |||||||||||
| 19 | SIOP 2001 | F | 23 | Unilateral | I | 4 | TN | Stromal‐type WT | No | — | 183 |
| 20 | SIOP 2001 | M | 22 | Unilateral | I | 4 | TN | Stromal‐type WT | ILNR | — | 142 |
| 21 | SIOP 2001 | F | 16 | Unilateral | III | 4 | TN | Stromal‐type WT | PLNR + ILNR | — | 0 |
| 22 | SIOP 2001 | M | 7 | Unilateral | I | 4 | NSS | Stromal‐type WT | ILNR | — | 95 |
| 23 | SIOP 9 | M | 20 | Bilateral | I | 4 | None | Mixed‐type WT (L/R not specified) | NA | Death due to hepatic failure 10 d after diagnosis | 0 c |
| 24 | SIOP 9 | M | 27 | Unilateral | I | 4 | NA | Mixed‐type WT | NA | — | 26 |
| 25 | SIOP 93‐01 | F | 11 | Bilateral | I | NA | NSS | L: Nephroblastomatosis | Yes | Obstructive ileus and death 19 mo after diagnosis | 19 c |
| R: Mixed‐type WT | |||||||||||
| 26 | SIOP 93‐01 | F | 27 | Unilateral | I | 4 | TN | Stromal‐type WT | Yes | — | 166 |
| 27 | SIOP 93‐01 | F | 36 | Bilateral | NA | NA | TN and NSS | L: Nephroblastomatosis | Yes | — | 155 b |
| R: Nephroblastomatosis | |||||||||||
| 28 | SIOP 93‐01 | F | 19 | Unilateral | I | NA | NSS | Mixed‐type WT | Yes | — | 91 |
| 29 | SIOP 93‐01 | M | 32 | Unilateral | NA | 4 | NA | Nephroblastomatosis | Yes | — | 34 b |
| 30 | SIOP 93‐01 | F | 16 | Unilateral | I | 4 | TN | Stromal‐type WT | No | — | 45 |
| 31 | SIOP 93‐01 | F | 31 | Unilateral | II | 4 | TN | Stromal‐type WT | No | — | 107 |
| 32 | SIOP 93‐01 | M | 26 | Bilateral | I | 4 | TN | L: No histology | NA | — | 96 |
| R: Indeterminable | |||||||||||
| 33 | SIOP 2001 | M | 12 | Unilateral | I | 4 | TN | Mixed‐type WT | ILNR | — | 45 |
| 34 | SIOP 2001 | M | 22 | NA | NA | NA | TN | Nephroblastomatosis | ILNR | SAE, no relapse | 141 b |
| 35 | SIOP 2001 | F | 23 | Unilateral | I | NA | TN | Stromal‐type WT | ILNR | — | 115 |
| 36 | SIOP 2001 | M | 44 | NA | NA | NA | NSS | Mixed‐type WT | PLNR | — | 50 |
| 37 | SIOP 2001 | M | 21 | Bilateral | NA | NA | TN and NSS | L: Nephroblastomatosis* | ILNR | *Progression to blastemal‐type WT 11 mo after diagnosis | 16 |
| R: Nephroblastomatosis | |||||||||||
| 38 | SIOP 2001 | M | 16 | Bilateral | NA | NA | NSS | L: Nephroblastomatosis (radiology only) | Yes | — | 11 |
| R: Stromal‐type WT | |||||||||||
| 39 | SIOP 2001 | F | 16 | Bilateral | I | 8 | NSS | L: Nephroblastomatosis | Yes | — | 148 |
| R: Nonanaplastic | |||||||||||
| 40 | Other | M | 28 | Unilateral | II | None | TN | Mixed‐type WT | No | — | 22 |
| 41 | SIOP 2001 | F | 6 | Bilateral | NA | 8 | TN and NSS | L: Nephroblastomatosis* | PLNR + ILNR | *Progression to stromal/mixed‐type WT 13 and 55 mo after initial diagnosis | 128 |
| R: Nephroblastomatosis* | |||||||||||
| 42 | SIOP‐RTSG UMBRELLA | F | 24 | Bilateral | II | 12 | TN | L: Stromal‐type WT | ILNR | — | 7 |
| R: Nephroblastomatosis (radiology only) | |||||||||||
| 43 | SIOP 2001 | F | 39 | Unilateral | I | None | NSS | Mixed‐type WT | NA | — | 110 |
Abbreviations: ESRD, end‐stage renal disease; F, female; ILNR, intralobar nephrogenic rests; L, left; M, male; NA, not available; NSS, nephron‐sparing surgery; PLNR, perilobar nephrogenic rests; R, right; SAE, serious adverse event; SIOP, International Society of Pediatric Oncology; SIOP‐RTSG, International Society of Pediatric Oncology Renal Tumor Study Group; TN, total nephrectomy; WAGR, Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays; WT, Wilms tumor.
Unilateral at diagnosis but developed metachronous disease.
Not included in the survival analysis (nephroblastomatosis only).
The patient died.
WAGR Diagnosis
For 38 of the 43 patients, the diagnosis of WAGR had been established by genetic testing, whereas the other 5 patients had been diagnosed with WAGR on the basis of their clinical characteristics alone. Details on the type of genetic testing were available for 16 cases, and the tests included fluorescence in situ hybridization (n = 10), array comparative genomic hybridization (n = 2), karyotyping (n = 2), a single‐nucleotide polymorphism array (n = 1), and quantitative polymerase chain reaction (n = 1). The exact span of the deletion was available for only 5 cases and ranged from 5 to 14 Mb in size. In 1 case, the deletion was mosaic (patient 33 in Table 1). The median age at the diagnosis of WAGR syndrome, available for 19 patients, was 2 months (range, 0‐47 months).
Presentation, Stage, and Preoperative Treatment
The median age at WT/nephroblastomatosis presentation was 22 months (range, 6‐44 months). The majority of the tumors were asymptomatic and were detected by surveillance (27 of 39 [69.2%]), whereas 12 patients (12 of 39 [30.8%]) presented with a palpable/visible abdominal mass and/or other symptoms such as hematuria. Among these 12 patients, 3 had been previously diagnosed with WAGR syndrome, and 2 had not yet been diagnosed with WAGR syndrome; for 7 patients, this information was not available. In 4 cases, the presence or absence of symptoms was not specified.
The overall stage was available for 40 patients. Fifteen patients (15 of 40 [37.5%]) had bilateral disease at diagnosis. This included bilateral nephroblastomatosis (n = 5; 3 progressed to WT on 1 or both sides), unilateral WT with contralateral nephroblastomatosis (n = 5), and bilateral WT (n = 2); in 3 patients with bilateral disease, it was not known whether they had bilateral WT or (a combination of WT and) nephroblastomatosis. None of the patients had metastatic disease. The local (abdominal) stage was available for 31 patients and included stage I for 21 (67.7%), stage II for 6 (19.4%), and stage III for 4 (12.9%).
Information regarding preoperative treatment (yes/no) was available for 42 patients; 39 of these patients (92.9%) received preoperative chemotherapy, including actinomycin D and vincristine in 30 cases and doxorubicin in 2 cases. The type of preoperative treatment was not available for the other 7 patients. The median duration of preoperative treatment was 8 weeks for bilateral cases (range, 4‐26 weeks; missing in 5 cases) and 4 weeks for unilateral cases (range, 4‐6 weeks; missing in 8 cases).
Tumor Volume and Response to Preoperative Treatment
The median tumor volume at diagnosis, available for 36 patients, was 46.5 mL (range, 1‐659 mL). Three patients presented with tumors larger than 500 mL, and all of these patients had been symptomatic at diagnosis. Tumors that were symptomatic at diagnosis had a median volume of 375 mL (range, 4.2‐659 mL), whereas tumors detected by surveillance had a median volume of 18 mL (range, 1‐396 mL; P = .001). For 28 patients, the response to preoperative chemotherapy was recorded; 14 patients showed a decrease in tumor volume (14 of 28 [50%]), 2 patients revealed a stable tumor volume (2 of 28 [7.1%]), and 12 patients revealed tumor growth (12 of 28 [42.9%]) during preoperative treatment. The histological subtype of tumors that increased in volume during preoperative treatment was the stromal type (n = 6), the mixed type (n = 2), or nephroblastomatosis (n = 4).
Surgery
Two patients with bilateral disease died of hepatic failure before surgery (patients 6 and 23 in Table 1). Among the other 13 patients with bilateral disease, 11 (85%) underwent nephron‐sparing surgery (NSS), including bilateral NSS (n = 4), unilateral NSS (no surgery on the other side; n = 2), and NSS preceded or followed by total nephrectomy on the contralateral side (n = 5). In 2 patients with bilateral disease, only a unilateral total nephrectomy was performed (patients 32 and 42 in Table 1). The type of surgery was specified for 21 patients with unilateral disease: NSS for 5 (23.8%) and total nephrectomy for 16 (76.2%).
Histological Subtype and Nephrogenic Rests
The histological subtype was available for 42 patients (with central review available for 32 of 42 [76.2%]). Six patients (6 of 42 [14.3%]) were diagnosed with nephroblastomatosis only. This was histologically confirmed in 5 patients; 1 patient with bilateral disease underwent unilateral resection (showing nephroblastomatosis), whereas the other kidney was diagnosed with nephroblastomatosis on the basis of imaging (patient 13 in Table 1). Three patients who were initially diagnosed with bilateral nephroblastomatosis on imaging experienced disease progression and were diagnosed with WT after histological assessment (11‐13 months after their first presentation; patients 4, 37, and 41 in Table 1).
Among patients with WT, the histological subtypes (n=36) included stromal WT in 19 (52.8%), mixed WT in 12 (33.3%), regressive WT in 1 (2.8%) and other/indeterminable WT in 2 (5.6%). Blastemal‐type WT occurred in 2 patients (5.6%) after prolonged treatment for nephroblastomatosis, whereas (focal or diffuse) anaplasia was not reported.
Upon histological assessment, nephroblastomatosis or nephrogenic rests were present in 30 of 38 patients (78.9%), including patients with intralobar (n = 20), perilobar (n = 1), or both intralobar and perilobar rests (n = 2). For the remaining 7 patients, the type of nephrogenic rests (intra‐ or perilobar) was not specified.
Survival and Events
Six patients with nephroblastomatosis only, without further (progression to) WT, were excluded from the survival analysis. Among these patients, 1 death (cause not specified) occurred after 9 months, and 1 serious adverse event (not further specified) occurred after 141 months of follow‐up.
Survival data were subsequently available for 37 patients with WT (including 1 patient without a histological diagnosis because the patient died before surgery) with a median follow‐up of 95 months (range, 0‐189 months). The estimated 5‐year event‐free survival rate was 84.3% (95% confidence interval, 72.4%‐98.1%; Fig. 2), and the overall survival rate was 91.2% (95% confidence interval, 82.1%‐100%; Fig. 3). Events occurred in 6 patients, including 3 patients (patients 2, 7, and 41 in Table 1) who developed metachronous contralateral tumors. All 3 patients had been treated for stromal‐type WT, and the contralateral tumors occurred 1, 7, and 3 years after the first tumor at the ages of 3, 9, and 5 years, respectively. These 3 patients were alive at last follow‐up.
Figure 2.

Kaplan‐Meier curve showing estimated event‐free survival and 95% confidence intervals for patients with WAGR syndrome and Wilms tumor (n = 37). WAGR indicates Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays.
Figure 3.
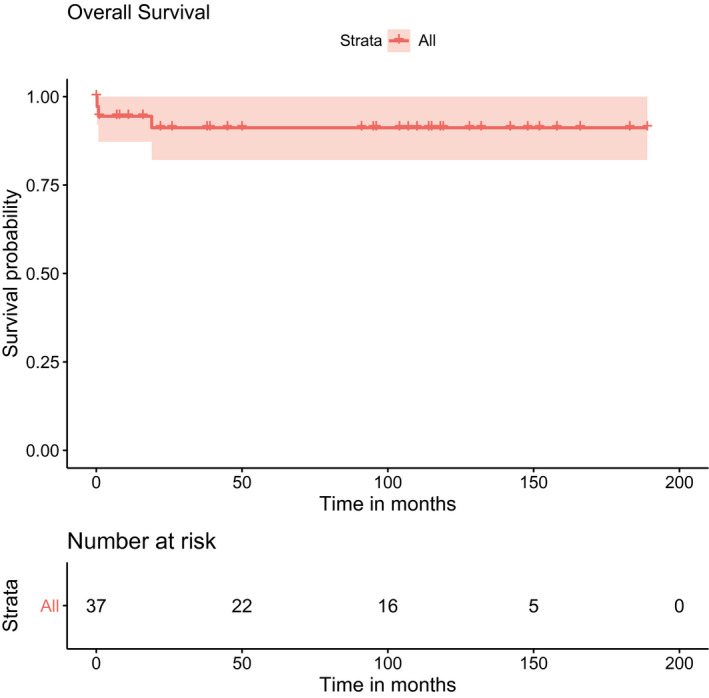
Kaplan‐Meier curve showing estimated overall survival and 95% confidence intervals for patients with WAGR syndrome and Wilms tumor (n = 37). WAGR indicates Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays.
Among patients with WT, 3 deaths occurred, including the deaths of 2 patients who died of hepatoxicity as a result of sinusoidal obstruction syndrome during preoperative chemotherapy (patients 6 and 23 in Table 1). In 1 of these patients, an incorrect dose (overdose) of actinomycin D had been administered (patient 23). The third patient died 19 months after diagnosis, and at this same date, obstructive ileus was registered as an event (patient 25 in Table 1). Because this patient was treated more than 20 years ago, we were unable to confirm the exact cause of death.
Chronic Kidney Disease
Data on chronic kidney disease were collected on the additional data collection form (see the supporting information) and were subsequently available for 20 patients. In 5 of these 20 patients (25%), a decreased eGFR, proteinuria (2+), or both were reported, with the age of onset varying from 3 to 16 years (time to onset, 2‐13 years after WT diagnosis). One of these patients had been treated for bilateral disease, whereas the other 4 patients had been treated for unilateral WT (unilateral nephrectomy in 3 cases and the surgery type not specified in the fourth case). One patient, treated for unilateral WT, was reported to have end‐stage renal disease at the age of 16 years (patient 9 in Table 1).
Additional Clinical Conditions
The additional data collection form (see the supporting information) was completed for 30 of the 43 patients, and for many items, the requested data could not be retrieved. Birth weight was available for 15 patients, and 3 of these patients (20%) were reported to have a birth weight below the 10th percentile for gestational age; the remaining 12 patients had a birth weight within the normal range. Congenital abnormalities other than aniridia, including ocular and genitourinary abnormalities as well as polydactyly (n = 2), macrocephaly (n = 1), Pierre‐Robin sequence (n = 1), and atrial septal defects (n = 2), were reported in 13 patients (Table 2).
TABLE 2.
Additional Findings in Patients With WAGR Syndrome and Wilms Tumor and/or Nephroblastomatosis as Reported on the Additional Data Collection Form (n = 30)
| Category | Finding | No. of Occurrences |
|---|---|---|
| Ocular findings other than aniridia | Cataracts | 10 |
| Peters anomaly | 2 | |
| Nystagmus | 5 | |
| Ocular hypotonia | 1 | |
| Retinal detachment | 2 | |
| Genitourinary findings | Cryptorchidism | 5 |
| Hypospadias | 2 | |
| Renal cysts | 1 | |
| Ovarian cysts | 1 | |
| Renal tissue disorganization | 1 | |
| Ureteric reflux | 1 | |
| Horseshoe kidney | 1 | |
| Neurological findings | Hypotonia | 1 |
| Hypertonia | 1 | |
| Epilepsy | 1 | |
| Metabolic findings | Obesity | 4 |
| Hypothyroidism | 1 | |
| Insulin resistance | 1 | |
| Pulmonary findings | Lung hypoplasia | 1 |
| Behavioral findings | Attention deficit (hyperactivity) disorder | 4 |
| Aggression | 1 | |
| Autism (spectrum) | 3 | |
| Sleep disturbances | 1 | |
| Musculoskeletal findings | Osteochondroma | 1 |
| Other | Hypertrophic pyloric stenosis | 1 |
| Mild pulmonary artery stenosis | 1 | |
| Polydactyly | 2 | |
| Macrocephaly | 1 | |
| Pierre‐Robin sequence | 1 | |
| Atrial septal defects | 2 |
Abbreviation: WAGR, Wilms tumor, aniridia, genitourinary anomalies, and range of developmental delays.
Note that each item was completed for only a subset of these patients, and for other patients, it was unclear whether the condition was absent or the requested data could not be retrieved.
Cognitive impairment was reported in 18 of 22 patients (81.8%) with available data. The 4 patients who were reported to have normal cognitive function were 2 to 16 years old at the last follow‐up. Additional clinical findings are summarized in Table 2. Other tumors such as gonadoblastoma were not reported.
Discussion
We identified 43 patients with confirmed WAGR syndrome and WT/nephroblastomatosis in the SIOP‐RTSG database and through their identification by national and/or local PIs within the SIOP‐RTSG network.
Overall, we demonstrated a high rate of bilateral disease (37.5%, including patients with bilateral or contralateral nephroblastomatosis, vs 6%‐7% in general cohorts 19 , 20 ) and no anaplastic tumors. Blastemal‐type WT, which is considered high‐risk after preoperative chemotherapy, was observed in only 2 patients after prolonged treatment for nephroblastomatosis. Metastatic disease was not observed in the current study; this was similar to the findings described by Breslow et al in 2003. 2
Event‐free and overall survival rates at 5 years after diagnosis appear to be similar to those described for nonsyndromic WT 19 , 20 except that relapses did not occur and mortality was exclusively due to non–tumor‐related causes. Because of the long‐term morbidity and mortality associated with the underlying syndrome, Breslow et al 2 reported that 20‐year overall survival was only 47.8% for patients with WAGR versus 85.5% for non‐WAGR patients, but our follow‐up data were insufficient to confirm this. Longer follow‐up data are also needed to reliably establish the risk of chronic kidney disease, which was reported in only 5 of 20 patients with available data in the current study but has previously been estimated to occur in 50% to 60% of patients with WAGR. 1 , 2 Our study was further limited by the fact that our data collection form did not specify the level of creatinine/eGFR changes required for the definition of chronic kidney disease.
Although 2 deaths were related to hepatic failure in our cohort of 42 patients, we are not aware of other studies reporting hepatotoxicity in patients with WAGR. Considering the fact that in 1 of the patients hepatic failure was related to an overdose of actinomycin D, we are uncertain about any potential association with the underlying WT1 defect. In addition to kidneys and other organs, WT1 protein is expressed in the developing liver. 21 If future studies report additional patients with WT1 aberrations and hepatotoxicity, this may warrant further investigation.
The difference in volume between tumors detected by surveillance and those that were symptomatic illustrates the benefit of surveillance, which enables a high rate of NSS even for unilateral cases (23.8%). Currently, different groups offer different WT surveillance recommendations in which surveillance is continued until the age of 5, 22 6, 1 7, 23 or 8 years. 24 On the basis of the current study, we would recommend that surveillance for WT be continued until the age of 5 years, at which 100% of the initial tumors were diagnosed. Notably, only approximately 90% were diagnosed before this age in the NWTS cohort. 2 , 25 Breslow et al 2 did not specify whether patients diagnosed at older ages had previously been under surveillance, and we hypothesize that they may have had nephroblastomatosis before their WT diagnosis.
For patients in whom WT or nephroblastomatosis has been previously diagnosed, an extended surveillance of the (remaining) kidney(s) may be warranted. We observed the occurrence of contralateral tumors up to 7 years after the initial diagnosis, with the latest occurrence at the age of 9 years; this suggests that nephrogenic rests in patients with WAGR carry a long‐lasting risk of progression to WT. It would be very useful to develop treatment modalities that can prevent this malignant transformation. A drug that has been suggested to induce differentiation of nephrogenic rests is retinoic acid (a metabolite of vitamin A), but clinical studies are limited to case reports. 26 , 27 In addition, metformin has been speculated to induce cell differentiation by inhibiting the mTOR pathway. 28 Although its potential role in cancer prevention is being studied in several adult populations, it has not been assessed in the context of nephrogenic rests and/or WT predisposition.
In patients with a genetic predisposition such as WAGR syndrome, preoperative chemotherapy is relevant to facilitate NSS. 11 However, we observed a high rate of progressive or nonresponsive tumors, which were frequently of the stromal subtype, as has been previously reported. 29 For patients suspected of bilateral nephroblastomatosis, it is challenging to decide whether or not, and at which time point, surgery should be performed, with the risks of disease progression being balanced against a loss of renal function. It has been suggested that a long period of pretreatment for bilateral nephroblastomatosis increases the risk of anaplastic WT and mortality, 30 but this was not observed in patients with WAGR syndrome. Although the risk of progression of (bilateral) nephroblastomatosis to WT appears to be high (3 of 5 patients), all 3 patients who experienced progression were alive and disease‐free at last follow‐up (1‐10 years after progression had occurred).
Gonadoblastoma, which has been occasionally reported in patients with WAGR syndrome, 1 was not reported in the current study, and the risk of developing gonadoblastoma appears to be lower with WAGR syndrome versus germline WT1 mutations, particularly in comparison with Frasier syndrome (intron 9 mutations), in which complete sex reversal (XY females) and gonadoblastoma are common. 31
Our study was limited by its retrospective design. The inclusion of 13 additional patients who were not registered in the central SIOP database may have introduced a bias; the exclusion of 5 patients for whom we were unable to get in contact with national/local PIs may have as well (notably, none of these 5 patients had metastatic or anaplastic disease, and all were alive and disease‐free at last follow‐up). For the collection of additional data, physicians had to rely on medical chart notes of patients who had been treated many years ago. A more complete picture of the phenotypic spectrum of WAGR syndrome can be achieved by involving parents, as has been previously shown, 1 and by recording both genetic and clinical features in prospective WT registries such as the SIOP‐RTSG UMBRELLA study, which is currently ongoing.
In conclusion, we confirm a lack of metastatic and anaplastic tumors and observe that patients with WAGR syndrome who develop WT and/or nephroblastomatosis can be successfully treated with current WT protocols. Our results illustrate the value of surveillance for enabling NSS and support the recommendation to continue surveillance until the age of 5 years, which can be further extended for patients with a prior diagnosis of WT/nephroblastomatosis. Because of the high rate of bilateral disease and the risk of contralateral tumor development and comorbidity, patients with WAGR syndrome need to be treated by multidisciplinary, expert teams.
Funding Support
Janna A. Hol is funded by Stichting Kinderen Kankervrij (award 278).
Conflict of Interest Disclosures
The authors made no disclosures.
Author Contributions
Janna A. Hol: Conceptualization and methodology, investigation, formal analysis, writing–original draft, and writing–review and editing. Marjolijn C. J. Jongmans: Conceptualization and methodology, investigation, supervision, and writing–review and editing. Hélène Sudour‐Bonnange: Investigation and writing–review and editing. Gema L. Ramírez‐Villar: Investigation and writing–review and editing. Tanzina Chowdhury: Investigation and writing–review and editing. Catherine Rechnitzer: Investigation and writing–review and editing. Niklas Pal: Investigation and writing–review and editing. Gudrun Schleiermacher: Investigation and writing–review and editing. Axel Karow: Investigation and writing–review and editing. Roland P. Kuiper: Supervision and writing–review and editing. Beatriz de Camargo: Investigation and writing–review and editing. Simona Avcin: Investigation and writing–review and editing. Danka Redzic: Investigation and writing–review and editing. Antonio Wachtel: Investigation and writing–review and editing. Heidi Segers: Investigation and writing–review and editing. Gordan M. Vujanic: Investigation and writing–review and editing. Harm van Tinteren: Conceptualization and methodology, investigation, and writing–review and editing. Christophe Bergeron: Investigation and writing–review and editing. Kathy Pritchard‐Jones: Investigation and writing–review and editing. Norbert Graf: Investigation and writing–review and editing. Marry M. van den Heuvel‐Eibrink: Conceptualization and methodology, supervision, and writing–review and editing.
Supporting information
Supplementary Material
Hol JA, Jongmans MCJ, Sudour‐Bonnange H, Ramírez‐Villar GL, Chowdhury T, Rechnitzer C, Pal N, Schleiermacher G, Karow A, Kuiper RP, de Camargo B, Avcin S, Redzic D, Wachtel A, Segers H, Vujanic GM, van Tinteren H, Bergeron C, Pritchard‐Jones K, Graf N, van den Heuvel‐Eibrink MM; for the International Society of Pediatric Oncology Renal Tumor Study Group . Clinical characteristics and outcomes of children with WAGR syndrome and Wilms tumor and/or nephroblastomatosis: The 30‐year SIOP‐RTSG experience. Cancer. 2021. 10.1002/cncr.33304
We thank the following physicians and researchers for providing data: Nicolas von der Weid (Basel, Switzerland), Cécile Dumesnil de Maricourt (Rouen, France), Stéphanie Proust (Angers, France), Dominique Plantaz and Cécile Perret (Grenoble, France), Caroline Thomas and Estelle Thebaud (Nantes, France), Cécile Boulanger and Nathalie Couteau (Toulouse, France), Arnauld C. Verschuur (Marseille, France), Sucheta J. Vaidya (Surrey, United Kingdom), Radna Minou Oostveen (London, United Kingdom), Reem Al‐Saadi (London, United Kingdom), Philip Maesa (Antwerp, Belgium), Jutte van der Werff‐ten Bosch (Brussels, Belgium), Michael Behrentz (Linköping, Sweden), Dragana Vujić (Belgrade, Serbia), and Henrik Hasle (Aarhus, Denmark).
Contributor Information
Janna A. Hol, Email: j.hol@prinsesmaximacentrum.nl.
for the International Society of Pediatric Oncology Renal Tumor Study Group (SIOP‐RTSG):
Nicolas von der Weid, Cécile Dumesnil de Maricourt, Stéphanie Proust, Dominique Plantaz, Cécile Perret, Caroline Thomas, Estelle Thebaud, Cécile Boulanger, Nathalie Couteau, Arnauld C. Verschuur, Sucheta J. Vaidya, Radna Minou Oostveen, Reem Al‐Saadi, Philip Maes, Jutte van der Werff‐ten Bosch, Michael Behrentz, Dragana Vujić, and Henrik Hasle
References
- 1. Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005;116:984‐988. [DOI] [PubMed] [Google Scholar]
- 2. Breslow NE, Norris R, Norkool PA, et al. Characteristics and outcomes of children with the Wilms tumour–aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2003;21:4579‐4585. [DOI] [PubMed] [Google Scholar]
- 3. Van Heyningen V, Hoovers JM, de Kraker J, Crolla JA. Raised risk of Wilms tumour in patients with aniridia and submicroscopic WT1 deletion. J Med Genet. 2007;44:787‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller RW, Fraumeni JF Jr, Manning MD. Association of Wilms's tumor with aniridia, hemihypertrophy and other congenital malformations. N Engl J Med. 1964;270:922‐927. [DOI] [PubMed] [Google Scholar]
- 5. Rose EA, Glaser T, Jones C, et al. Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms' tumor gene. Cell. 1990;60:495‐508. [DOI] [PubMed] [Google Scholar]
- 6. Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA. Homozygous deletion in Wilms tumours of a zinc‐finger gene identified by chromosome jumping. Nature. 1990;343:774‐778. [DOI] [PubMed] [Google Scholar]
- 7. Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 1990;60:509‐520. [DOI] [PubMed] [Google Scholar]
- 8. Pritchard‐Jones K, Fleming S, Davidson D, et al. The candidate Wilms' tumour gene is involved in genitourinary development. Nature. 1990;346:194‐197. [DOI] [PubMed] [Google Scholar]
- 9. Haber DA, Buckler AJ, Glaser T, et al. An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms' tumor. Cell. 1990;61:1257‐1269. [DOI] [PubMed] [Google Scholar]
- 10. Han JC, Thurm A, Golden Williams C, et al. Association of brain‐derived neurotrophic factor (BDNF) haploinsufficiency with lower adaptive behaviour and reduced cognitive functioning in WAGR/11p13 deletion syndrome. Cortex. 2013;49:2700‐2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ehrlich PF, Chi YY, Chintagumpala MM, et al. Results of treatment for patients with multicentric or bilaterally predisposed unilateral Wilms tumor (AREN0534): a report from the Children's Oncology Group. Cancer. 2020;126:3516‐3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Kraker J, Graf N, van Tinteren H, et al. Reduction of postoperative chemotherapy in children with stage I intermediate‐risk and anaplastic Wilms' tumour (SIOP 93‐01 trial): a randomised controlled trial. Lancet. 2004;364:1229‐1235. [DOI] [PubMed] [Google Scholar]
- 13. Pritchard‐Jones K, Bergeron C, de Camargo B, et al. Omission of doxorubicin from the treatment of stage II‐III, intermediate‐risk Wilms' tumour (SIOP WT 2001): an open‐label, non‐inferiority, randomised controlled trial. Lancet. 2015;386:1156‐1164. [DOI] [PubMed] [Google Scholar]
- 14. Tournade MF, Com‐Nougue C, de Kraker J, et al. Optimal duration of preoperative therapy in unilateral and nonmetastatic Wilms' tumor in children older than 6 months: results of the Ninth International Society of Pediatric Oncology Wilms' Tumor Trial and Study. J Clin Oncol. 2001;19:488‐500. [DOI] [PubMed] [Google Scholar]
- 15. Van den Heuvel‐Eibrink MM, Hol JA, Pritchard‐Jones K, et al. Position paper: rationale for the treatment of Wilms tumour in the UMBRELLA SIOP‐RTSG 2016 protocol. Nat Rev Urol. 2017;14:743‐752. [DOI] [PubMed] [Google Scholar]
- 16. Vujanic GM, Sandstedt B, Harms D, Kelsey A, Leuschner I, de Kraker J. Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood. Med Pediatr Oncol. 2002;38:79‐82. [DOI] [PubMed] [Google Scholar]
- 17. Boccon‐Gibod LA. Pathological evaluation of renal tumors in children: International Society of Pediatric Oncology approach. Pediatr Dev Pathol. 1998;1:243‐248. [DOI] [PubMed] [Google Scholar]
- 18. Vujanic GM, Gessler M, Ooms A, et al. The UMBRELLA SIOP‐RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol. 2018;15:693‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weirich A, Ludwig R, Graf N, et al. Survival in nephroblastoma treated according to the trial and study SIOP‐9/GPOH with respect to relapse and morbidity. Ann Oncol. 2004;15:808‐820. [DOI] [PubMed] [Google Scholar]
- 20. Sudour H, Audry G, Schleimacher G, Patte C, Dussart S, Bergeron C. Bilateral Wilms tumors (WT) treated with the SIOP 93 protocol in France: epidemiological survey and patient outcome. Pediatr Blood Cancer. 2012;59:57‐61. [DOI] [PubMed] [Google Scholar]
- 21. Armstrong JF, Pritchard‐Jones K, Bickmore WA, Hastie ND. The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech Dev. 1993;40:85‐97. [DOI] [PubMed] [Google Scholar]
- 22. Scott RH, Walker L, Olsen OE, et al. Surveillance for Wilms tumour in at‐risk children: pragmatic recommendations for best practice. Arch Dis Child. 2006;91:995‐999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kalish JM, Doros L, Helman LJ, et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. 2017;23:e115‐e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. International WAGR Syndrome Association . What is WAGR syndrome? Accessd July 22, 2020. https://wagr.org/what‐is‐wagr‐syndrome
- 25. Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet. 1998;79:268‐273. [DOI] [PubMed] [Google Scholar]
- 26. Friesenbichler W, Krizmanich W, Lakatos K, et al. Outcome of two patients with bilateral nephroblastomatosis/Wilms tumour treated with an add‐on 13‐cis retinoic acid therapy—case report. Pediatr Hematol Oncol. 2018;35:218‐224. [DOI] [PubMed] [Google Scholar]
- 27. Witt O, Hämmerling S, Stockklausner C, et al. 13‐cis retinoic acid treatment of a patient with chemotherapy refractory nephroblastomatosis. J Pediatr Hematol Oncol. 2009;31:296‐299. [DOI] [PubMed] [Google Scholar]
- 28. Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shamberger RC, Haase GM, Argani P, et al. Bilateral Wilms' tumors with progressive or nonresponsive disease. J Pediatr Surg. 2006;41:652‐657. [DOI] [PubMed] [Google Scholar]
- 30. Fürtwangler R, Schmolze M, Graber S, et al. Pretreatment for bilateral nephroblastomatosis is an independent risk factor for progressive disease in patients with stage V nephroblastoma. Klin Padiatr. 2014;226:175‐181. [DOI] [PubMed] [Google Scholar]
- 31. Gwin K, Cajaiba MM, Caminoa‐Lizarralde A, Picazo ML, Nistal M, Reyes‐Mugica M. Expanding the clinical spectrum of Frasier syndrome. Pediatr Dev Pathol. 2008;11:122‐127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material