

Significance Statement
C3 glomerulopathy (C3G) is a rare, progressive kidney disease, characterized by alternative pathway hyperactivation and glomerular complement deposition. Animal models are valuable to explore modulators of C3G progression. A severe C3G mouse model was developed by replacing the mouse C3 gene with the human equivalent. The humanized C3 mice mimic pathologic features of patients with C3G, potentially due to dysregulated interaction of human C3 protein with mouse complement regulators. A C5-blocking antibody showed that C5 dominates pathogenesis of humanized C3 mice. C3b- and complement factor B–blocking antibodies provide benefit, indicating that alternative-pathway hyperactivation drives pathology in these mice. The humanized model exhibits rapid, severe renal disease, offering the opportunity to genetically and pharmacologically dissect critical contributors to complement-driven renal pathology.
Keywords: C3 glomerulopathy, mouse model, humanized C3 mice, accelerated kidney disease, C2 KO, liver phenotype, C5, C3b, CFH mAbs
Visual Abstract

Abstract
Background
C3 glomerulopathy (C3G) is characterized by the alternative-pathway (AP) hyperactivation induced by nephritic factors or complement gene mutations. Mice deficient in complement factor H (CFH) are a classic C3G model, with kidney disease that requires several months to progress to renal failure. Novel C3G models can further contribute to understanding the mechanism behind this disease and developing therapeutic approaches.
Methods
A novel, rapidly progressing, severe, murine model of C3G was developed by replacing the mouse C3 gene with the human C3 homolog using VelociGene technology. Functional, histologic, molecular, and pharmacologic assays characterize the presentation of renal disease and enable useful pharmacologic interventions in the humanized C3 (C3hu/hu) mice.
Results
The C3hu/hu mice exhibit increased morbidity early in life and die by about 5–6 months of age. The C3hu/hu mice display elevated biomarkers of kidney dysfunction, glomerulosclerosis, C3/C5b-9 deposition, and reduced circulating C3 compared with wild-type mice. Administration of a C5-blocking mAb improved survival rate and offered functional and histopathologic benefits. Blockade of AP activation by anti-C3b or CFB mAbs also extended survival and preserved kidney function.
Conclusions
The C3hu/hu mice are a useful model for C3G because they share many pathologic features consistent with the human disease. The C3G phenotype in C3hu/hu mice may originate from a dysregulated interaction of human C3 protein with multiple mouse complement proteins, leading to unregulated C3 activation via AP. The accelerated disease course in C3hu/hu mice may further enable preclinical studies to assess and validate new therapeutics for C3G.
C3 glomerulopathy (C3G) is a spectrum of disorders encompassing C3 GN and dense deposit disease. About 50% of patients with C3G progress to ESKD within 10 years after diagnosis, with limited treatment options. The underlying cause of C3G pathology is dysregulated alternative pathway (AP) complement activation as a result of acquired or genetic defects. The acquired defects include the development of C3 nephritic factors, autoantibodies which stabilize the AP C3 convertase. The genetic defects include either mutations in the C3 convertase components—complement component C3 and complement factor B (CFB)—or regulatory proteins—such as complement factor H (CFH), CFH-related 1, 2, 3, and 5, complement factor I (CFI), and CD46.1–4 Histologically, kidneys of patients with C3G show abundant glomerular C3 deposition in the absence of Igs, and a range of pathologic features, such as mesangial matrix expansion, membrane and endocapillary proliferation with crescent formation.1,5 Histologic evidence of membrane attack complex (MAC) deposition suggests terminal activation of complement component C5.6–9
CFH is a critical negative regulator of AP; it controls complement activation both by decreasing the formation and increasing the dissociation of AP C3 convertase.2 CFH mutations, leading to its loss of function or deficiency, were discovered in patients with C3G.10–14 Unchecked complement activation in these patients leads to the depletion of the circulatory C3 levels. Interestingly, a spontaneous mutation identified in CFH in pigs results in CFH deficiency and a lethal C3G-like phenotype.15
Mice genetically deficient in CFH (CFH−/−) were developed by Pickering et al.16 and have been a valuable tool to understand the mechanisms underlying C3G. Similar to patients with C3G, the CFH−/− mice present with unregulated AP complement activation, glomerular pathologies consistent with C3 and MAC deposition with minimal Igs, mesangial matrix expansion, and electron-dense deposits. CFH−/− mice do not show early mortality, and the development of ESKD can take several months to manifest. Mice with deletion of both CFH and complement factor P present with a fatal C3G phenotype, suggesting multiple complement regulatory proteins may need to be altered to accelerate the phenotype in mice.17–19 Moreover, an acute hemolytic syndrome–associated point mutation in murine C3 in mice led to the robust development of chronic thrombotic microangiopathy (TMA) with thrombocytopenia, hemolysis, hematuria, and kidney failure.20 Here, we report the generation of a mouse model with a striking C3G-like phenotype that displays early ESKD/mortality, which was developed by replacing the mouse C3 gene with the equivalent human C3 gene. The robust phenotype observed in these mice may be due to a lack of a crosstalk of human C3 protein with multiple mouse complement regulators.
Methods
Animal Studies
All animal studies were carried out per the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Regeneron Pharmaceuticals Institutional Animal Care and Use Committee. Only littermate wild-type (WT) mice were used as controls.
Generation of C3hu/hu Mice
The C3hu/hu mice were generated using VelociGene technology.21 Briefly, the mouse C3 gene, including the 5′ regulatory elements and all of the coding exons 1 through 41, was replaced with the human C3 gene, including the 5′ regulatory elements, and all of the coding exons 1 through 41.
First, a targeted, 25-kb deletion of the mouse C3 gene was generated in mouse embryonic stem (ES) cells (50% 129; 50% B6) by replacement of coding exons 2 through 41 with a lacZ-loxP-neo-loxP cassette. The resulting mouse ES cells are heterozygous C3 knockout (KO). Second, a targeting construct was generated, containing mouse C3 upstream and downstream homology arms flanking a human C3 sequence extending from 9-kb upstream of coding exon 1 to 1.5-kb downstream of the polyA signal. A loxP-hyg-loxP cassette was inserted at the 3′ end of the human sequence. Electroporation of this targeting construct into heterozygous mouse C3 KO ES cells resulted in the replacement of 30.6 kb of mouse sequence, including 6.6 kb of 5′ regulatory region and exons 1–41 of mouse C3 (GRCm38 genome coordinates chromosome 17: 57,204,017–57,234,702), with 53.5 kb of human sequence, including 9 kb of 5′ regulatory region, exons 1–41 of human C3, and 1.5 kb of sequence downstream of the polyA signal (GRCh38 genome coordinates chromosome 19: 6,676,325–6,729,720). Targeted, heterozygous, humanized, C3 ES cell clones were introduced into an eight-cell-stage mouse embryo. F0 mice, wholly derived from the donor ES cell and bearing the humanized C3 gene, were identified by genotyping for loss of mouse allele and gain of human allele using a modification-of-allele assay.21 A TaqMan quantitative PCR assay was used to confirm the human C3 gene sequence replaced the deleted mouse C3 gene sequence in the humanized allele. The same assay was used to assay DNA purified from tail biopsy specimens for mice derived from the targeted ES cells to determine their C3 genotypes and confirm that the humanized C3 allele had transmitted through the germ line. Two mice heterozygous for the replacement were bred to generate a mouse that is homozygous for the replacement of the endogenous mouse C3 gene by the human C3 gene.
Generation of C3HET/WT/C2 KO Mice
The C3HET/WT/C2 KO mice were generated by electroporating 2 μg of the C2-targeting vector, which replaces exons 4–6 with a LacZ reporter self-deleting Neo cassette, 10 μg of each of two guide RNA (gRNA) plasmids (sequences below), and 5 μg Cas9 plasmid into C3HET/WT ES cells. TaqMan quantitative PCR assays were used to confirm that the C2 gene was deleted and the vector targeted correctly. The same assays were used to assay DNA purified from tail biopsy specimens for mice derived from the targeted ES cells to determine their C2 genotypes.
The gRNA sequences used for targeting were as follows: 5′ C2 gRNA, CCGCTGCTCCTCCTCAAATA (targets mouse C2 exon 4); and 3′ C2 gRNA, GGTTCAGGTGACCCGAGCGC (targets mouse C2 exon 6).
Human C3, Exon 3, Single-Nucleotide-Polymorphism Target Vector Sequencing
The human complement C3 bacterial artificial chromosome (BAC) targeting vector (200 ng) was Sanger sequenced in separate reactions using the forward primer (5′-GAACAGACCCCTGACAATG-3′) and the reverse primer (5′- ACCACCTTCTCCACCACTTG-3′). Primers flanked the sequence of interest in exon 3 at approximately 70-bp upstream and 60-bp downstream, respectively. Sequencing results were trimmed and aligned to the reference in Sequencher to confirm the single-nucleotide-polymorphism (SNP) variant in the BAC vector.
CFH or CFI Administration in C3hu/hu Mice
C3hu/hu mice were administered with a single dose of normal human serum purified CFH or CFI (Complement Technology, Inc.) intravenously. Serial blood samples were collected to measure human C3 levels.
Assays
Serum C3 concentrations were determined with the Complement C3 Human ELISA kit and Complement C3 Mouse ELISA kit (Abcam), as per the manufacturer’s directions. Serum C5 concentrations were determined with the Complement C5 Mouse ELISA kit (Abcam), as per the manufacturer’s directions. Serum chemistry was performed using the ADVIA Chemistry XPT system (Siemens Healthineers). A complete blood count was done using Genesis Hematology Analyzer (Oxford Science, Inc.). Serum- or urine-free hemoglobin was measured using the Hemoglobin Assay Kit (Sigma-Aldrich) according to the manufacturer’s protocol. Urinary albumin, urinary creatinine (Exocell, Inc.), BUN (BioAssay Systems), and serum cystatin C (sCysC; BioVendor) were measured according to the manufacturers’ instructions. Urinary C5a levels were measured in C3hu/hu mice using the Mouse Complement Component C5a DuoSet (R&D Systems), according to the manufacturer’s protocol. Urine was collected for 16–18 hours using diuresis cages (Tecniplast).
Histopathology Assessment
Histopathology analysis of the kidney sections was performed as described below. In brief, sections stained with Periodic acid–Schiff (PAS) and hematoxylin and eosin (H&E) were imaged in an Aperio AT2 Slide Scanner with the 40× objective. All of the kidney sections were observed, in a blinded fashion, for lesions in the glomerulus, tubules, interstitium, and blood vessels. The lesions were scored for severity as zero, normal; one, minimal; two, mild; three, moderate; and four, severe. The lesions scored for glomerular pathology (for all of the glomeruli in a section) were glomerular sclerosis, glomerular atrophy, endocapillary proliferation, membranoproliferation, mesangial cell hypertrophy, leukocyte infiltration, crescent formation, fibrin deposition, fibrinoid necrosis of the capillary tufts, and glomerular thrombosis. The lesions scored for the tubules were tubular necrosis, degeneration, regeneration, tubulitis, tubular proteinosis with or without dilation, and tubular atrophy. The lesions scored for the interstitium were mononuclear infiltration and fibrosis; for the blood vessels, the lesions scored were arteriosclerosis, vascular thrombosis, and vasculitis. The scores for each of these components were summed up as a total pathology score for each mouse. For measurements of expansion of mesangial cell matrix, at least 25 glomeruli per section were subjected to measurement of the PAS-positive area. An OD-based threshold was applied to each glomerular tuft, using a modification of the Indica Labs Area Quantification algorithm, separating nuclear staining (dark blue) and background (light pink) from Schiff-positive matrix (bright purple). Matrix and total tuft area were measured and reported, along with the mesangial cell matrix, as a percentage of the tuft area.
Electron microscopy for glomerular-pathology assessment was performed as described elsewhere.16 Briefly, cortical kidney-tissue samples from either WT (n=7) or C3hu/hu (n=7) mice were fixed in glutaraldehyde. The fixed samples were processed for sectioning and staining by a contract research organization (Probetex Inc., San Antonio, TX) to capture images and assess histopathology. The histologic assessment included measures of podocyte foot process effacement, glomerular basement membrane (GBM) thickness, qualitative assessment of glomerular endothelial cell abnormalities, and the presence and location of glomerular electron-dense deposits. Formalin-fixed livers were embedded in paraffin. Sections (3-μm thick) were made from formalin-fixed, paraffin-embedded, liver tissues and then stained with H&E. The stained sections were imaged in an Aperio AT2 Slide Scanner with the 40× objective. An examination of liver pathologies was performed in a blinded fashion.
Generation and Characterization of C5 mAb
The C5 mAb was generated by immunizing C5 KO mice with a recombinant mouse C5 protein. The binding was confirmed by coating Reacti-Bind 96-well polystyrene plates (Pierce) with mouse C5 protein. Plates were blocked with BSA, anti-C5 or isotype mAb was titrated, and binding was detected with a polyclonal goat anti-mouse antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch). The signal was developed with 3,3',5,5'-tetramethylbenzidine, stopped with 2N sulfuric acid, and OD at 450 nm was read on a spectrophotometer.
Classic pathway (CP) and AP hemolysis assays were conducted with normal human serum, as described below. The pharmacodynamic properties of the mAb were examined by administering C57BL6 (Taconic Biosciences) with a single dose of either anti-C5 mAb (5 or 30 mg/kg) or isotype control, mouse IgG1 (30 mg/kg), subcutaneously. The efficacy of the anti-C5 mAb was examined by conducting CP hemolysis ex vivo at 1, 3, 8, or 14 days postdosing.
Hemolysis Assays
Hemolysis assays were performed as previously reported with slight modifications.22
CP Hemolysis Assay
The desired number of sheep red blood cells (SRBCs; Colarado Serum Company) were washed in gelatin veronal buffer (GVB)+++ (Boston BioProducts) and resuspended at 1×109 cells/ml. To sensitize the SRBCs, a total of 1×109 per milliliter of SRBCs were mixed with an equal volume of the 1:50 diluted rabbit anti-sheep hemolysin (Rockland Immunochemicals) at a dilution of 1.5 mg/ml at 37°C for 20 minutes. Sensitized SRBCs were diluted to 2×108 cells/ml in GVB++ before using in hemolysis assay. For in vitro hemolysis assays to test antibody dose response, pooled normal mouse serum was diluted to 10% in GVB++ buffer. For the ex vivo hemolysis assay, individual serum from antibody-dosed mice was diluted to 10% in GVB++ buffer. Round-bottomed, 96-well plates were used to measure hemolysis activity. A total of 100 μl of sensitized SRBCs (2×108 cells/ml) were plated into 96-well plate, followed by the addition of 100 μl of diluted serum samples. Cells were gently mixed and incubated at 37°C for 60 minutes. A total of 100 μl of the supernatant was transferred to a fresh 96-well, flat-bottomed plate and read at an absorbance of 412 nm on a Spectramax microplate reader.
AP Hemolysis Assay
The desired number of rabbit RBCs (Colarado Serum Company) were washed in GVB–magnesium ion/EGTA buffer and resuspended at 2×108 cells/ml. For the in vitro hemolysis assay to test antibody dose response, normal mouse serum was diluted to 10% in GVB–magnesium ion/EGTA buffer (Boston BioProducts). Round-bottomed, 96-well plates were used to measure hemolysis activity. A total of 100 μl rabbit RBCs (2×108 cells/ml) were plated into 96-well plates at 37°C, followed by the addition of 100 μl of diluted serum. Cells were gently mixed and incubated at 37°C for 60 minutes. After incubation time, the cells were spun down by centrifugation at 1250 × g at 4°C. A total of 100 μL of the supernatant was transferred to a fresh, 96-well, flat-bottomed plate and read at 412 nm on a Spectramax microplate reader.
Calculation of Percentage Hemolysis
The percentage of hemolysis was calculated using the absorbance values, with the following equation:
Antibody Treatment in C3hu/hu Mice
C3hu/hu mice were treated with either a mouse anti-mouse C5 mAb or an isotype control antibody, mouse IgG1, starting at approximately 8 weeks of age. WT mice were treated with isotype control. In some experiments, C3hu/hu mice were treated with either a mouse anti-mouse C5 mAb or an isotype control mAb, mouse IgG1, starting at approximately 8 weeks of age until 24 weeks of age (16 weeks of treatment), and then mice were followed without any treatment. In another experiment, one group of mice were treated with C5 mAb until 24 weeks of age (16 weeks of treatment), and another group were treated until 32 weeks of age (24 weeks of treatment) before stopping the treatment. The C5 mAb or isotype control (mouse IgG1) was administered at 50 mg/kg, subcutaneously, three times per week.
For experiments involving anti-C3b mAb and anti-CFB mAb, humanized camelid C3b mAb (clone 9611) and murine CFB mAb (clone 1379) were made on the basis of the published sequences.23–25 Comprehensive characterization data were available for these antibodies in the aforementioned references. C3hu/hu mice were treated with either an anti-C3b mAb or an isotype control mAb, human IgG1, for anti-C3b study, with either an anti-CFB mAb or an isotype control mAb, mouse IgG1, for anti-CFB study, starting at approximately 7 weeks of age. The antibodies were administered at 50 mg/kg for anti-C3b study and 100 mg/kg for anti-CFB study, subcutaneously, three times per week. The treatment continued until 17 weeks of age for both studies.
Immunofluorescence Analysis
The frozen sections of kidney or liver tissue from C3hu/hu and WT mice were assessed for the presence of C3 deposition and the C5b-9 MAC per the following protocol. In brief, sagittal sections were made from optimal cutting temperature compound–embedded frozen kidney. A mouse mAb (clone 755; Abcam) was used to detect C3 deposition, and a rabbit polyclonal anti–C5b-9 primary antibody (Abcam) was used to detect C5b-9 deposition. Visualization was achieved with Alexa Fluor 647–conjugated goat anti-mouse IgG2b (Jackson ImmunoResearch) and Alexa Flour 647–conjugated donkey anti-rabbit (Jackson ImmunoResearch) secondary antibodies. Separate sections were stained with Alexa Fluor 647–conjugated, donkey anti-mouse IgG alone. The analysis of fibrin deposition in the frozen kidney sections was done using FITC-conjugated, anti–human fibrin/fibrinogen IgG (Dako, Carpinteria, CA), as described earlier.26 For additional slides, primary antibodies were substituted with isotype controls and subjected to the same staining protocols; these showed minimal or negative signal (data not shown). Stained slides were imaged on the Zeiss Axioscan and were quantified in Indica Halo.
RNA Preparation, Sequencing, Read Mapping, and Downstream Analysis
Total RNA was extracted from the mouse tissue samples using the MagMAX kit (Life Technologies, Carlsbad, CA). Strand-specific RNA-sequencing (RNA-seq) libraries were prepared from 500 ng RNA using the KAPA Stranded mRNA-Seq Kit (KAPA Biosystems). Twelve-cycle PCR was performed to amplify libraries. Sequencing was performed on an Illumina HiSeq2000 system (Illumina) by multiplexed, single-read runs with 33 cycles. Raw sequence data (BCL files) were converted to the FASTQ format via Illumina Casava 1.8.2. Reads were decoded on the basis of their barcodes, and read quality was evaluated with FastQC. For quantification of C3 expression in the humanized C3 mice (Figure 1B), a customized mouse genome was built by concatenating the human C3 genomic sequence with the reference mouse genome (GRCm38), wherein the original mouse C3 locus was masked with Ns. The gene structure annotations of the mouse C3 gene were replaced by the human counterparts accordingly in the customized transcriptome. Reads from WT and humanized C3 mice were mapped to the standard (National Center for Biotechnology Information’s GRCm38 genome and RefSeq transcriptome) and the customized mouse genome and the transcriptome, respectively, using ArrayStudio software (OmicSoft, Cary, NC), allowing two mismatches. Reads mapped to the sense-strand exons of a gene were summed at the gene level. The statistical significance of differentially expressed genes was assessed using the DESeq package (version 1.26),27 with default parameters. Genes were considered significantly perturbed if they had an adjusted P value (false discovery rate) <0.05, and absolute fold change ≥1.5. Pathway enrichment analysis was performed using the fgsea package (Sergushichev AA: An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 10.1101/060012), using the Hallmark set of gene signatures from MsigDB.28 The sequencing data are accessible at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE150838.
Figure 1.
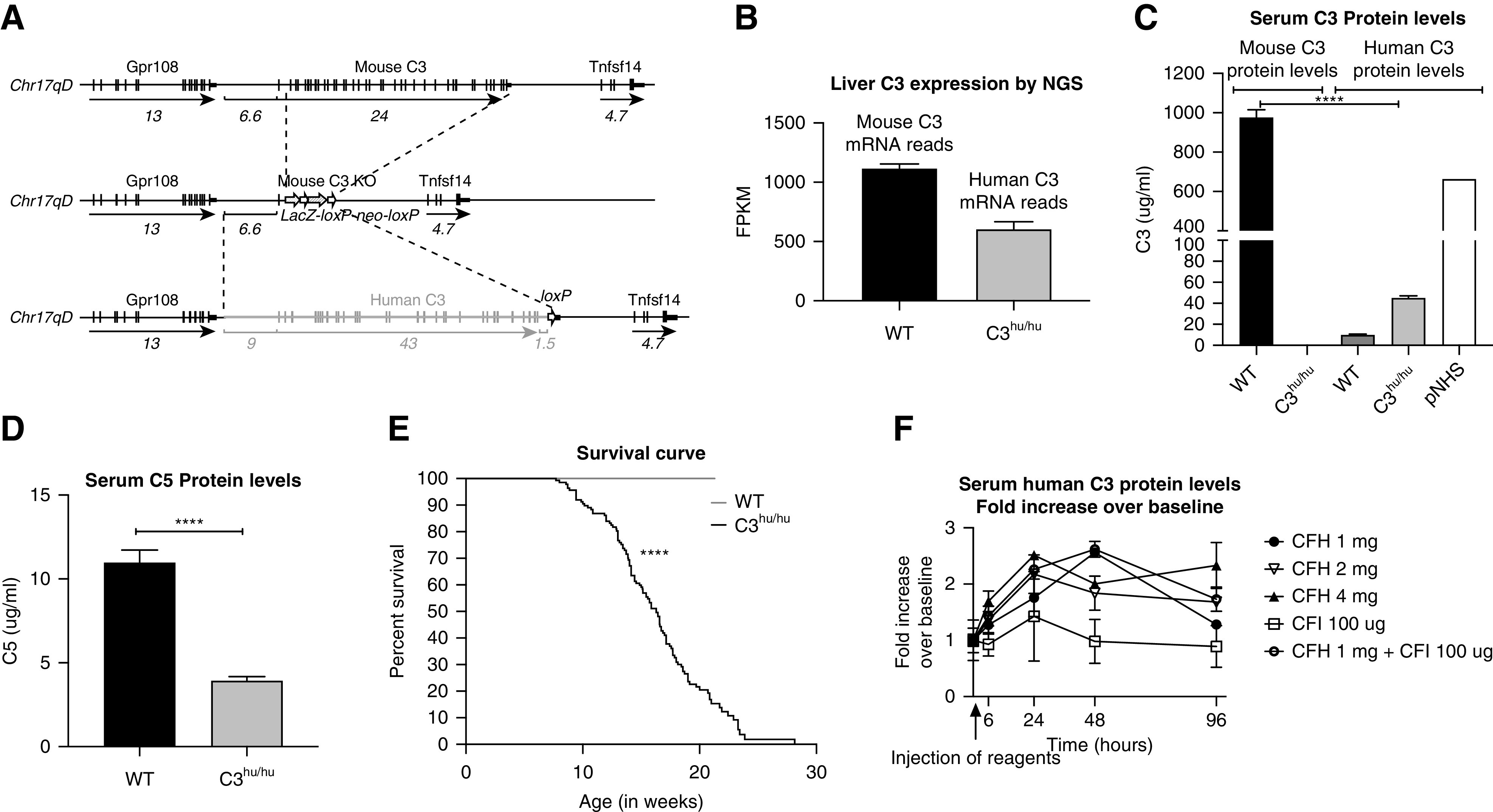
Generation of C3hu/hu mice. (A) An illustration, not to scale, of the mouse C3, mouse C3 KO, and humanized C3 genomic loci. In two sequential targeting steps, the mouse C3 gene, including 6.6 kb of 5′ regulatory region and the entire coding region from exon 1 through exon 41, is deleted and replaced by the human C3 gene, including 9 kb of 5′ regulatory region, the entire coding region from exon 1 through exon 41, 1.5 kb of sequence downstream of the polyA signal, and a loxP site. Horizontal black lines indicate mouse sequence; the horizontal double gray line indicates human sequence; vertical black lines indicate exons; black boxes indicate 3′ untranslated regions; white arrows indicate loxP sites; sequence lengths in kilobases are indicated below the sequences. (B) Expression of the C3 gene in WT (n=3) and C3hu/hu (n=3) by next-generation sequencing (NGS). (C) Serum C3 levels in WT (n=8) and C3hu/hu (n=16). (D) Serum C5 levels in WT (n=7) and C3hu/hu (n=12). (E) Graph showing survival curve of WT (n=17) and C3hu/hu (n=137) mice. (F) Graph showing results of serum C3 levels postadministration of 1 mg CFH (n=3), 2 mg CFH (n=4), 4 mg CFH (n=3), CFI (n=2), or CFH and CFI (n=3) in C3hu/hu mice. ****P<0.001, versus WT. Chr17, chromosome 17; FPKM, fragments per kilobase of transcript per million mapped reads; pNHS, pooled normal human serum (BioIVT).
Blood Smears
Peripheral blood smears were prepared and stained using the Diff Quik Staining Kit (Polysciences, Inc.) as described before.29 Images were captured using a 40× objective for the schistocyte analysis.
Statistical Analyses
Survival curves were analyzed with the GraphPad Prism software, using the Mantel–Cox log-rank test. The two-way ANOVA mixed analysis with Sidak multiple comparisons was used for time-course studies. One-way ANOVA with the Tukey multiple comparisons test was used to compare three groups for a single time-point analysis. The unpaired t test with Welch correction was used when comparing two groups. A P value of <0.05 was considered significant. Data were expressed as mean±SEM, unless otherwise specified.
Results
Development of C3hu/hu Mice
The C3hu/hu mice were generated using VelociGene technology.21 Briefly, the human C3 gene containing 5′ regulatory elements and all of the coding exons 1 through 41 of the human C3 gene replaced the murine C3 gene locus, spanning 5′ regulatory elements and all of the coding exons 1 through 41 (Figure 1A; detailed description in the Methods section). To confirm the type of C3 variant that went into C3hu/hu mice, we sequenced the targeting vector because of a known SNP variant in exon 3 of the human complement C3 gene. The difference, “CGC” or “GGC,” results in an amino acid change from arginine to glycine (R102G) changing the C3S variant to a C3F variant.30 Sequencing results confirmed the BAC vector contained the CGC (arginine) variant (C3S) used in the mouse humanization (Supplemental Figure 1, A–C). The human C3 mRNA expression in the liver of the C3hu/hu mice was approximately 600 fragments per kilobase of transcript per million mapped reads, which was about half that of the mouse C3 mRNA expression in WT littermates (Figure 1B). The circulatory human C3 protein levels in the C3hu/hu mice were only about 4.5% compared with the circulatory mouse C3 protein levels in WT controls (Figure 1C). As expected, the mouse C3 protein was not detected in C3hu/hu mice. The human C3 protein detected in WT mice appears to be the background for the human C3 ELISA kit. In addition to lower C3 levels, serum C5 levels were also lower in the C3hu/hu mice compared with WT controls, consistent with the hypothesis of complement overactivation and consumption of C3 and C5 in the C3hu/hu mice (Figure 1D). Spontaneous mortality was observed in C3hu/hu mice, with a median age of survival of about 16 weeks (Figure 1E). Unless otherwise specified, male mice homozygous for human C3 (C3hu/hu) were used for phenotyping experiments.
Recovery in Circulatory C3 Levels with Administration of Human CFH Protein
C3 KO mice are grossly normal,31 suggesting the low C3 levels observed in C3hu/hu mice may not be the cause of mortality. The low circulatory C3 levels could be due to lack of crosstalk between human C3 protein and mouse complement regulatory proteins, resulting in complement overactivation and subsequent C3 consumption. To test this hypothesis, we examined if administration of purified human fluid-phase regulatory proteins CFH or CFI improves circulatory C3 levels in C3hu/hu mice. A single injection of 1 mg of purified human CFH with or without human CFI (100 μg) led to a 2.5-fold elevation in circulatory C3 levels within 48 hours postadministration. Increasing CFH dosage to 2 or 4 mg per mouse did not further elevate the C3 levels (Figure 1F). CFI administration alone did not lead to an elevation of circulatory C3 levels. These data suggest CFH, but not CFI, may be one of the critical mouse regulatory proteins that is unable to bind/block human C3 protein activation in the fluid phase in C3hu/hu mice.
Local Complement Activation in the Kidneys of C3hu/hu Mice
Patients with C3G show increased activation of C3 via AP, leading to systemic C3 consumption, renal C3 deposition, and local complement activation. We examined local complement activation in the kidneys of C3hu/hu mice. C3hu/hu mice, but not WT littermates, showed intense staining for C3 and C5b-9 (MAC) deposition in the glomeruli, both in the capillary wall and in the mesangium (Figure 2, A–D). Measurement of staining showed a significant increase in C3 and C5b-9 in the glomeruli of C3hu/hu mice compared with WT mice (Figure 2, G and H). No IgG deposition was observed in the glomeruli of WT (Figure 2E), whereas little IgG deposition was detected in C3hu/hu mice (Figure 2F), which could be secondary to tissue damage and subsequent neoepitope generation or vascular leak. These data suggest local complement activation and deposition occur in the kidneys of C3hu/hu mice.
Figure 2.
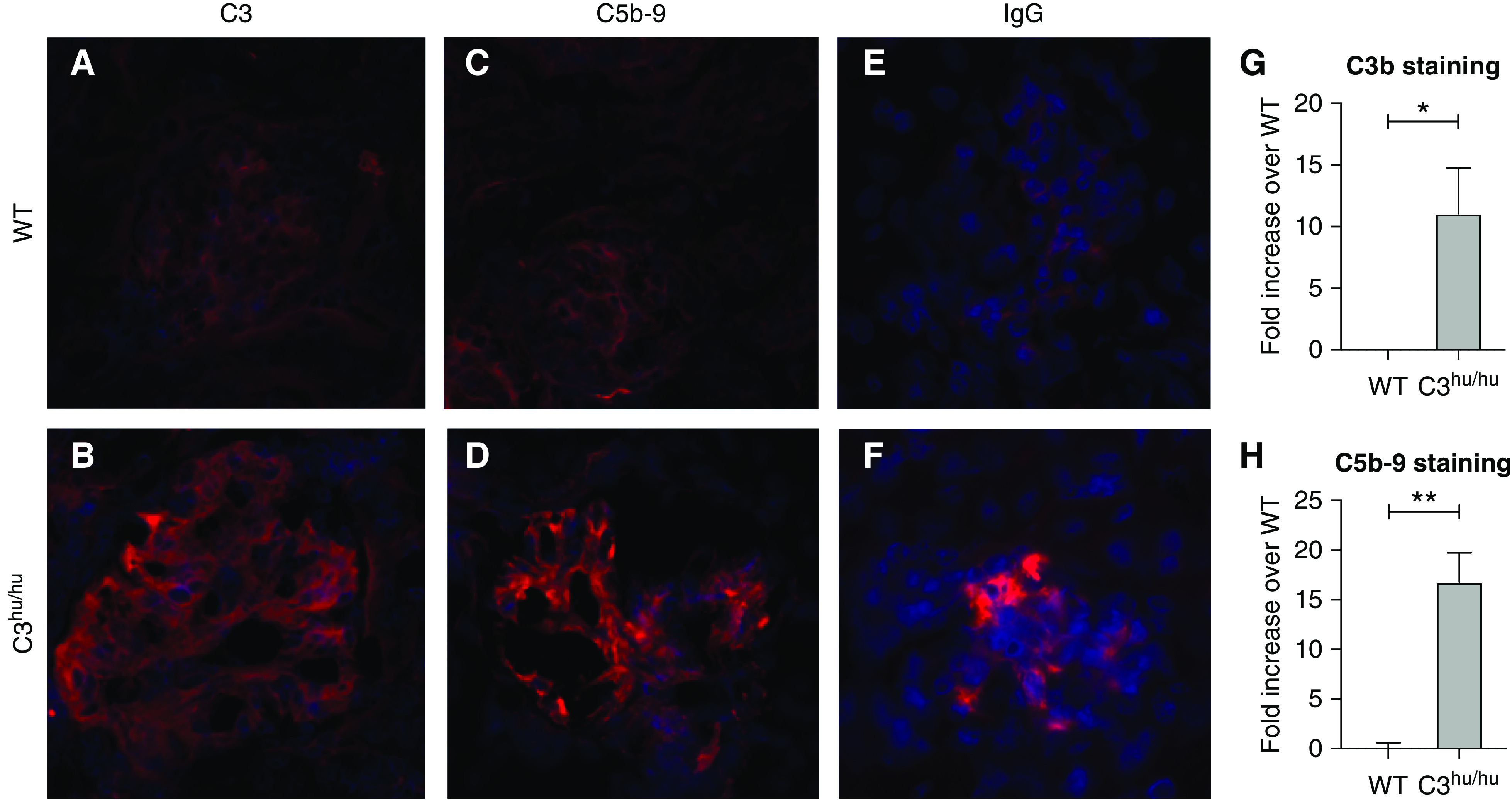
Increased staining of C3, MAC, and IgG in the kidneys of 13-week-old WT (n=6) and C3hu/hu (n=11) mice. Representative images showing cross-sections of kidney tissue stained for (A and B) human C3 protein (red), and (C and D) C5b-9 MAC (red). (E and F) IgG (red) of WT and C3hu/hu mice, respectively. (G and H) Graphs showing fold increase in C3b and C5b-9 staining, respectively, in C3hu/hu mice compared with WT mice. *P<0.05, **P<0.01.
Kidney Pathology in C3hu/hu Mice
Next, we examined if systemic dysregulation and local complement activation in the C3hu/hu mice lead to kidney injury/failure. Histopathology analysis of H&E images of the kidney sections from the C3hu/hu mice revealed multiple glomerular lesions, including membranoproliferative glomerulopathy (Figure 3C), hypertrophy of mesangial cells (Figure 3D), segmental to global glomerular sclerosis (Figure 3E), fibrin deposition with necrosis and leukocyte infiltration (Figure 3, F and G), and epithelial crescents (Figure 3G).
Figure 3.
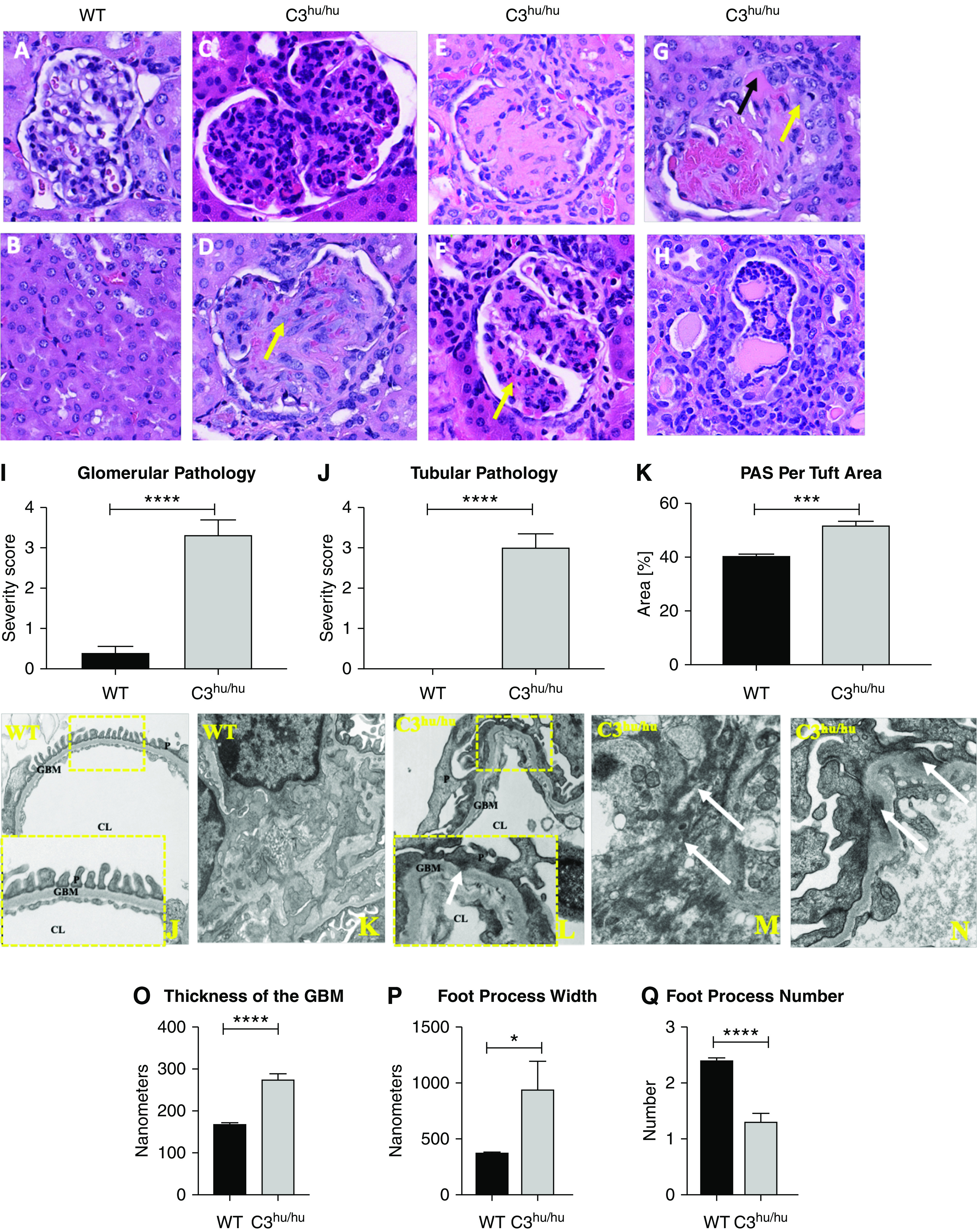
Histopathology of the kidneys from C3hu/hu mice. Representative images (×40) and pathology score of H&E-stained kidney sections from WT (n=8) and C3hu/hu (n=12) mice of 13- to 16-week-old mice. (A) Normal glomerulus from WT mouse. (B) Normal tubulointerstitium from WT mouse. (C) Membranoproliferative glomerulopathy in a C3hu/hu mouse. (D) Glomerulus from a C3hu/hu mouse showing hypertrophy (yellow arrow) of mesangial cells. (E) Glomerulus from a C3hu/hu mouse showing global sclerosis of the glomerular capillary tufts. (F) Glomerulus from a C3hu/hu mouse showing fibrinoid necrosis of the glomerular capillary tufts. (G) Glomerulus from a C3hu/hu mouse showing fibrin (black arrow) and epithelial crescent (yellow arrow). (H) Tubular segment from a C3hu/hu mouse with epithelial cell degeneration, necrosis, and inflammatory cells in the lumen. (I) Glomerular pathology score in WT and C3hu/hu mice. (J) Tubular pathology score in WT and C3hu/hu mice. (K) Graphs showing quantification of PAS staining in the glomeruli. (J and K) Electron micrographs of a glomerular capillary loop (higher magnification in inset) and mesangium, respectively, from WT mice. The capillary loop shows normal ultrastructure where podocytes have regular interdigitating foot processes placed on the outer aspect of an intact GBM. The endothelial lining shows normally placed fenestrae. The mesangium is normal in appearance, where cells occupy a thinly distributed matrix. (L) Electron micrographs of capillary loops (higher magnification in inset) of glomeruli from C3hu/hu mice. The capillary loops illustrate abnormal ultrastructure where podocyte foot processes are effaced, resting on a thickened GBM, often showing subepithelial protrusions (white arrow). The GBM contains electron-lucent, subendothelial spaces (rarefactions), with or without flocculent material within them. (M and N) Electron-dense deposits in the mesangium and subepithelial space, respectively (white arrows). (O–Q) Graphs representing quantification of GBM thickness, foot process width, and foot process number, respectively. WT, n=7; C3hu/hu, n=7. *P<0.05, ***P<0.001, ****P<0.0001, versus WT. CL, capillary lumen; P, podocyte.
The C3hu/hu mice also had tubular and interstitial changes consistent with tubular necrosis, degeneration, and tubulitis (Figure 3H). The WT mice showed no appreciable glomerular or tubular pathologies (Figure 3, A and B). Both glomerular and tubular injury scores were significantly higher in the C3hu/hu mice compared with WT mice (Figure 3, I and J, respectively). There was also a significant increase in mesangial matrix expansion, as measured by PAS staining (Figure 3K).
Electron-microscopy image analysis revealed that the C3hu/hu mice had marked glomerular pathology, including podocyte effacement, thickening and contortion of the GBM, and endothelium with a reduction of fenestrae (Figure 3L). The GBM contains electron-lucent subendothelial spaces (rarefactions), with or without flocculent material. The mesangium was expanded, and both mesangial and subepithelial electron-dense deposits were observed (Figure 3, M and N). Quantification of pathologies revealed a significant increase in GBM thickness and podocyte foot process width, and a decrease in foot process number in the C3hu/hu mice compared with the WT mice (Figure 3, O–Q).
Functional analysis revealed the C3hu/hu mice presented with significant elevation of BUN and sCysC, markers of glomerular filtration, indicating failing kidneys (Figure 4, A and B). The C3hu/hu mice also showed significantly elevated urinary albumin (normalized to urinary creatinine), indicating glomerular injury (Figure 4C). Gene-expression profiling of C3hu/hu mouse kidneys by RNA-seq showed large-scale dysregulation, with genes involved in energy metabolism and oxidative phosphorylation consistently downregulated, consistent with ongoing kidney injury (Figure 4D). By contrast, upregulated genes tended to be associated with extracellular matrix, wound healing, immune, inflammatory, coagulation, and complement pathways. Thus, these findings suggest uncontrolled complement activation in C3hu/hu mice likely leads to glomerular injury/pathology and failing of the kidneys.
Figure 4.
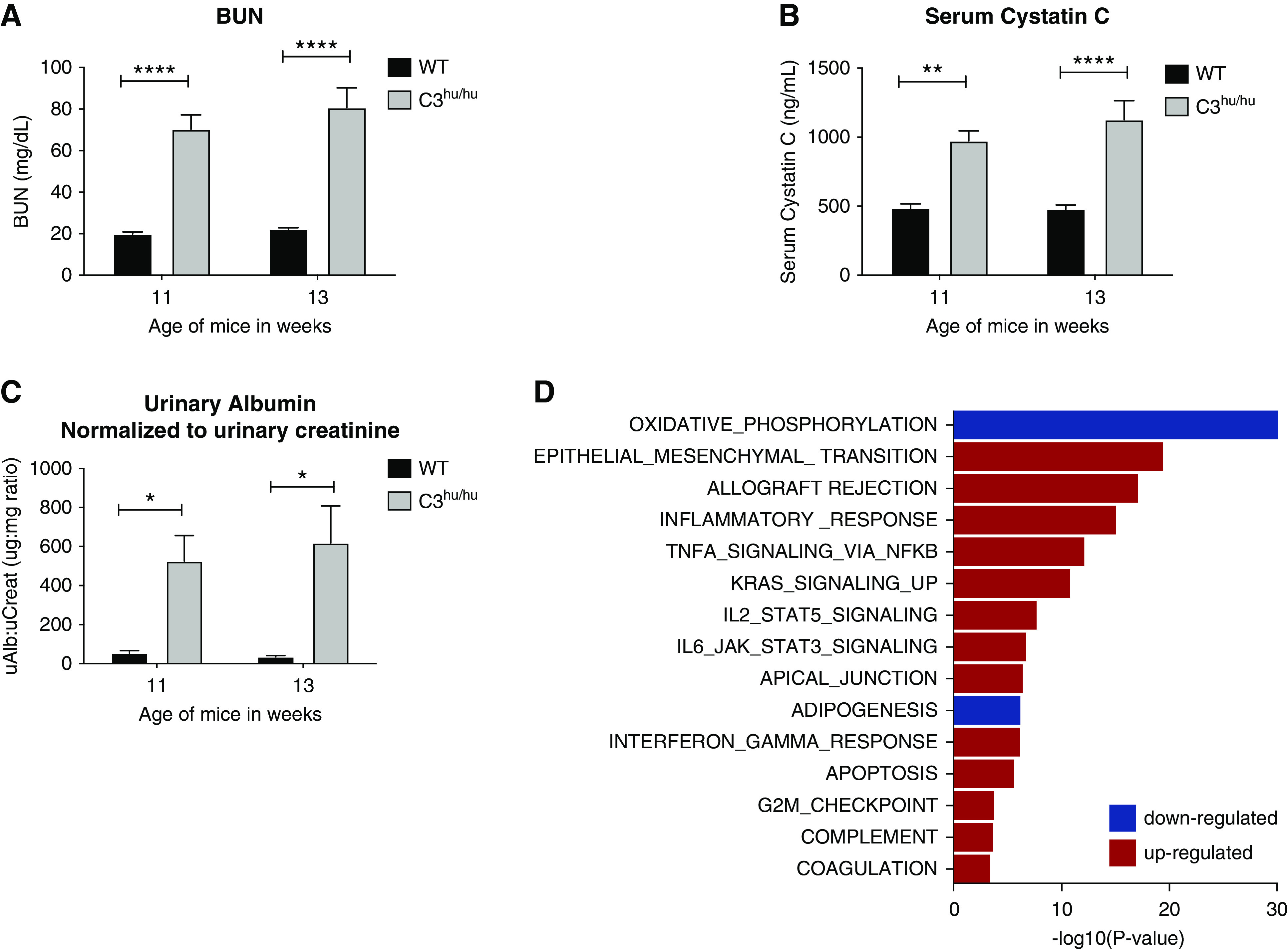
Functional and molecular characteristics of C3hu/hu mice. (A and B) Graphs depicting the results of BUN and sCysC, respectively, in WT (n=6) and C3hu/hu mice (n=9). (C) Graph depicting the results of urinary albumin per day and urinary albumin normalized to creatinine, respectively, in WT (n=6) and C3hu/hu mice (n=9). (D) Gene-set enrichment analysis31 of genes differentially expressed between C3hu/hu mice and their WT controls. Graph shows subset of the MsigDB hallmark gene sets with adjusted P value for enrichment <10−3. uAlb, urinary albumin; uCreat, urinary creatinine. *P<0.05, **P<0.01, ****P<0.0001.
Liver Pathology in C3hu/hu Mice
The C3hu/hu mouse livers showed inflammation in the portal region, bile duct hyperplasia, and C3 deposition (Supplemental Figure 2, B and D). The C3hu/hu mice also showed significant elevation of liver injury markers, alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase (Supplemental Figure 2, E–G). The complement deposition and liver damage observed in the C3hu/hu mice are consistent with the other mouse models of C3G.19,32
Examining for TMA Phenotype in C3hu/hu Mice
We next examined if C3hu/hu mice present with the TMA phenotype due to overactivation of the complement. As shown in Supplemental Figure 3, there is a significant increase in the platelet number in the C3hu/hu mice compared with WT mice. Although there was no change in hematocrit, a slight, but significant, decrease was observed in RBCs and total hemoglobin in the C3hu/hu mice. Moreover, the serum levels of free hemoglobin were not altered in the C3hu/hu mice. The significant increase in urinary hemoglobin levels in the C3hu/hu mice suggests hematuria. The fibrin deposition in the kidney was consistent with the other fatal C3G models.33 Lastly, schistocytes were not observed in the blood smears of the C3hu/hu mice. These data indicate the C3hu/hu mice do not present with classic signs of TMA.
Phenotype Rescue of C3hu/hu Mice with Blockade of C5
Activation of terminal complement component C5 could be a driver of pathogenesis in C3hu/hu mice. To address this question, we have treated C3hu/hu mice with a mouse C5-blocking mAb of isotype IgG1. The anti-C5 mAb binds to mouse C5 and blocks its function in in vitro CP and AP hemolysis assays (Supplemental Figure 4, A–C). A single subcutaneous dose of 30 mg/kg anti-C5 mAb blocked CP hemolysis for up to 3 days (Supplemental Figure 4D). The C3hu/hu mice administered with anti-C5 mAb significantly improved survival rate (Figure 5A). At the end of the study, only three out of 16 C3hu/hu mice on isotype-control treatment survived, compared with 13 out of 16 mice on anti-C5 mAb treatment. The C3hu/hu mice dosed with anti-C5 mAb showed significant improvement in body weight and kidney function, as measured by BUN and sCysC, compared with an isotype control (Figure 5, B–D). Once the anti-C5 mAb treatment was withdrawn after 16 weeks of treatment, the mice succumbed to the disease, indicating a requirement for continuous C5 blockade (Figure 5E). In an additional experiment (Figure 5F), two anti-C5 mAb treatment groups, with one group treated for 16 weeks and another for 24 weeks, were compared. Although a significant mortality benefit was observed during the treatment period in both cases, once the treatment was stopped, the mice died rapidly. Interestingly, there was about 30% mortality with anti-C5 mAb in the 24-week treatment regimen, suggesting anti-C5 mAb treatment, although offering a significant survival benefit, does not completely rescue the mortality. The pathologic assessment showed a significant reduction in glomerular hypertrophy, glomerular and tubular injury score, and neutrophil and macrophage infiltration with anti-C5 mAb (Figure 6, A–O). There is also a nonsignificant decrease in mesangial matrix expansion (data not shown). As expected, anti-C5 mAb significantly reduced urinary C5a levels (Figure 6P).
Figure 5.
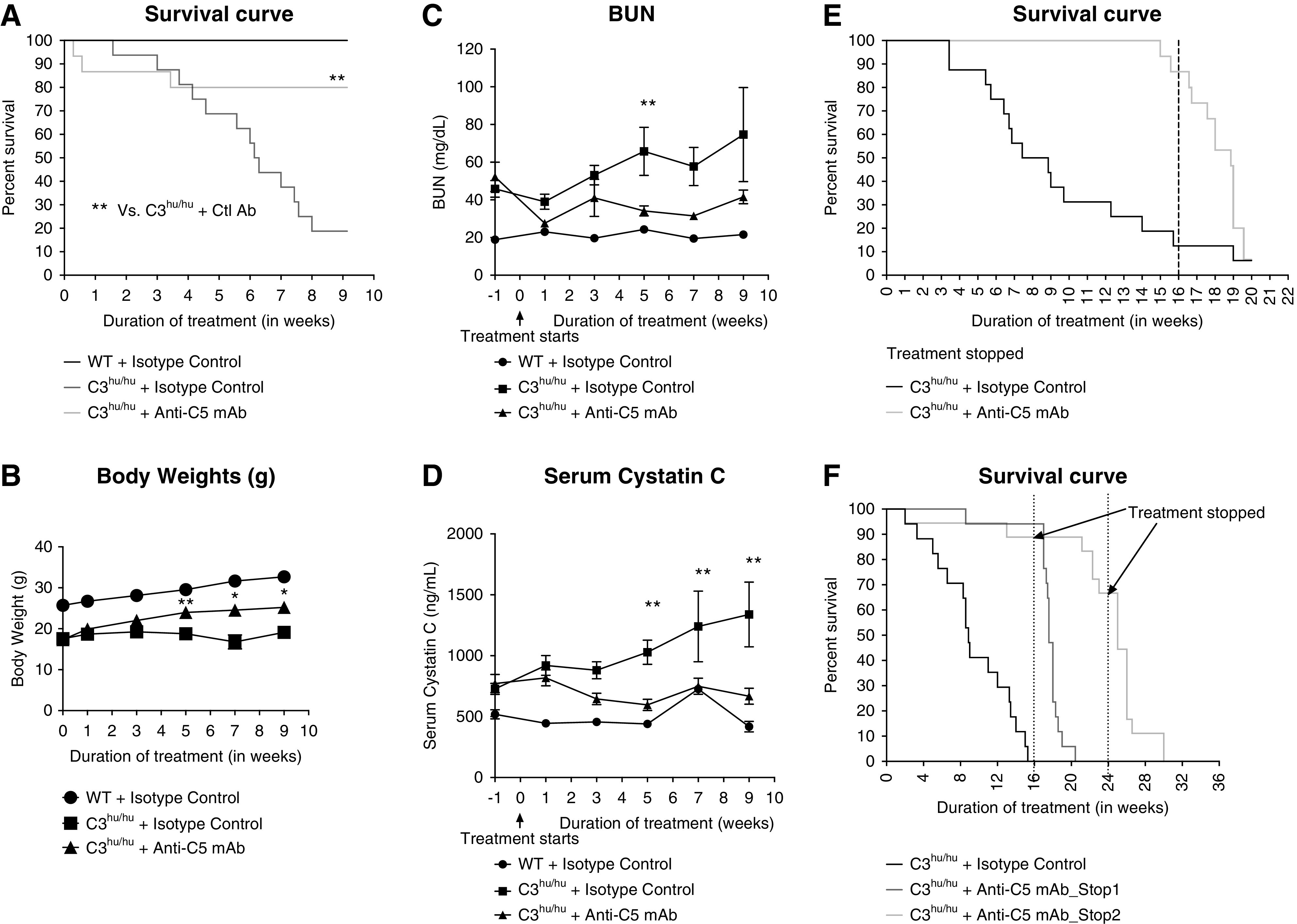
Anti-C5 mAb improves survival, body condition, and kidney function in C3hu/hu mice. Shown are the survival curves of (A) WT and isotype control (n=5), C3hu/hu and isotype control (n=16), and C3hu/hu and anti-C5 mAb (n=16); (E) C3hu/hu and isotype control (n=16), C3hu/hu and anti-C5 mAb (n=15), and (F) C3hu/hu and isotype control (n=17), C3hu/hu and anti-C5 mAb_Stop1 (n=17), and C3hu/hu and Anti-C5 mAb_Stop2 (n=17). (B) Body weight and (C and D) BUN and sCysC of WT mice treated with isotype control and C3hu/hu mice treated with isotype control or anti-C5 mAb. For the experiment in (E), treatment stopped after 16 weeks. For the experiment in (F), one group treatment stopped at 16 weeks (stop 1) and another group treatment stopped at 24 weeks (stop 2). *P<0.05, **P<0.005, C3hu/hu and anti-C5 mAb versus C3hu/hu and isotype control.
Figure 6.
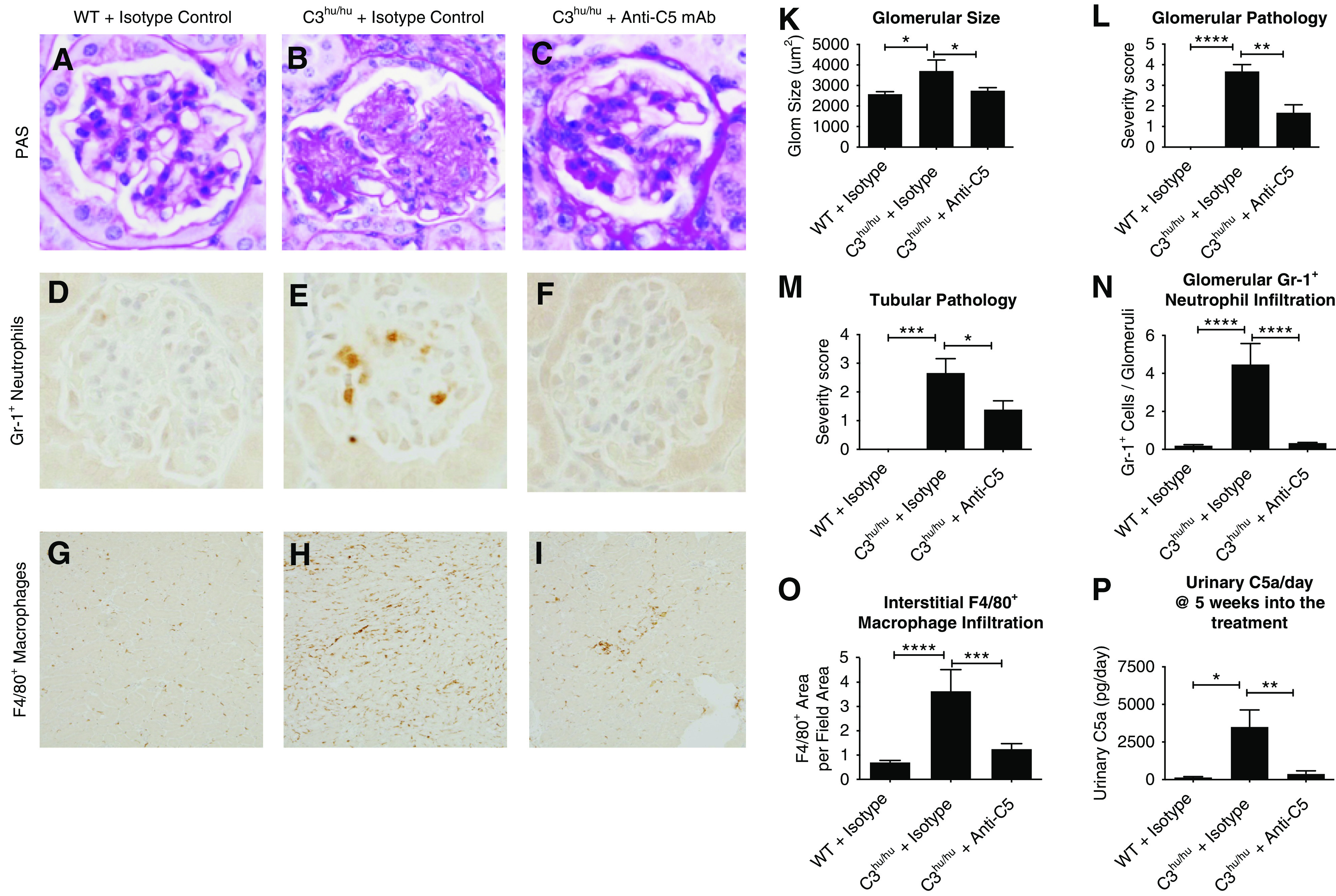
Anti-C5 mAb offers histopathology benefit in C3hu/hu mice. (A–I) Representative histopathology images of WT mice treated with isotype control (n=10), and C3hu/hu mice treated with isotype control (n=6) or anti-C5 mAb M1M17628N (n=18). (A–C) Images from PAS-stained sections. (D–F) Images from Gr-1–stained sections for neutrophils. (G–I) Images from F4/80+-stained sections for macrophages. (K) Graph showing quantification of glomerular (glom) size. (L and M) Graphs showing glomerular and tubular pathology scores. (N) Graph showing quantification of Gr-1+ neutrophils. (O) Graph showing quantification of F4/80+ macrophages. (P) Graph showing urinary C5a levels. WT mice treated with isotype control (n=5), and C3hu/hu mice treated with isotype control (n=10) or anti-C5 mAb M1M17628N (n=12). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Anti-C5 mAb treatment almost completely abrogated the gene-expression changes observed in the C3hu/hu mice. Of the 3443 genes differentially expressed between C3hu/hu and WT mice, only 129 were still perturbed after anti-C5 treatment, and with markedly diminished effect sizes (Figure 7A). mRNA levels of genes involved in extracellular matrix remodeling, immune signaling, cytokines, complement, and diverse white blood cell markers all resembled WT expression, in contrast to the dramatic upregulation observed in the isotype-treated C3hu/hu mice (Figure 7B). Energy and lipid metabolism gene expression was also rescued and attained WT levels, with few exceptions (e.g., Apobec2). These data indicate that activation of C5 likely plays a predominant role in kidney injury and inflammation, leading to kidney failure and subsequent death of C3hu/hu mice.
Figure 7.
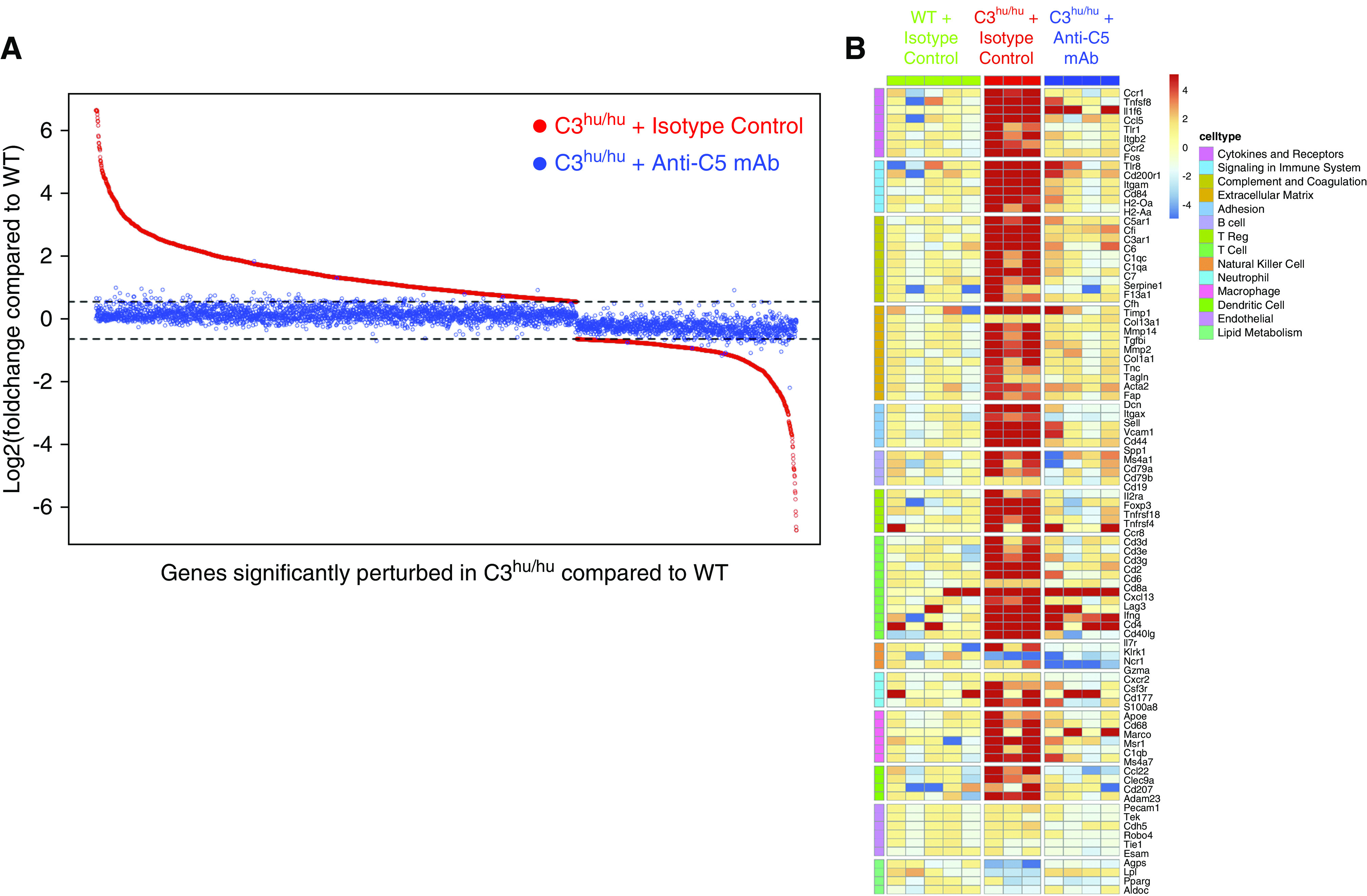
Anti-C5 mAb rescues molecular signature in C3hu/hu mice. (A) Scatterplot demonstrating that anti-C5 antibody treatment reversed disease signature genes in C3hu/hu mice. Shown are log2 fold changes of all 3443 genes differentially expressed between isotype-treated C3hu/hu and WT mice (red dots), sorted by diminishing fold change. Each gene’s corresponding fold change when comparing C5 mAb-treated C3hu/hu mice with WT is plotted in blue at the same position along the x axis. Horizontal dotted lines indicate 50% up- or downregulation, corresponding to our fold change cutoff for identifying differential expression. (B) Heatmap showing representative genes (rows) for perturbed pathways and immune cell types. Each column represents an individual animal, and each cell represents an individual gene-expression measurement, normalized separately for each gene to the median of WT controls. Red and blue colors represent up- and downregulated genes, respectively. All pathways (although not all genes) of interest were perturbed in the C3hu/hu and isotype control mice, and then subsequently attenuated by anti-C5 mAb treatment.
Phenotype Rescue of C3hu/hu Mice with Blockade of AP C3 Convertase
We next asked if blocking AP activation with anti-C3b or CFB mAbs in C3hu/hu mice offers protection. We first tested these mAbs in vitro to confirm their functional blockade. Anti-C3b mAb blocked hemolysis in normal human serum via AP, but not CP, confirming its specificity (Supplemental Figure 5, A and B, respectively). Anti-CFB mAb blocked AP hemolysis in both normal human and mouse sera (Supplemental Figure 5C). We then conducted in vivo experiments with these mAbs in C3hu/hu mice. Similar to anti-C5 mAb, we observed a significant improvement in survival rate with both anti-C3b and anti-CFB mAbs (Figure 8, A and B). In the anti-C3b mAb study, only three out of 14 C3hu/hu mice on isotype control mAb treatment survived, compared with 13 out of 14 mice on anti-C3b mAb treatment. Similarly, in the anti-CFB mAb study, only three out of 17 C3hu/hu mice treated with isotype control mAb survived, compared with 16 out of 17 mice treated with anti-CFB mAb. The mortality in mice heterozygous for human C3 (C3hu/WT) was similar to C3hu/hu mice, with a median age of survival of about 15 weeks. When C2 was deleted from C3hu/WT mice, the survival curve was not significantly different from C3hu/WT alone, suggesting classic and lectin pathways are not responsible for the phenotype observed. Both anti-C3b and anti-CFB mAbs also improved kidney function, as measured by BUN, in C3hu/hu mice compared with control antibody treatment (Supplemental Figure 6, A and B). Both anti-C3b and anti-CFB mAbs also significantly rescued circulatory C3 and C5 levels in C3hu/hu mice compared with an isotype control. As expected, anti-C5 mAb significantly rescued circulatory C5 levels, but does not affect circulatory C3 levels (Supplemental Figure 7). Altogether, these data indicate that AP activation is the predominant driver of disease in C3hu/hu mice.
Figure 8.
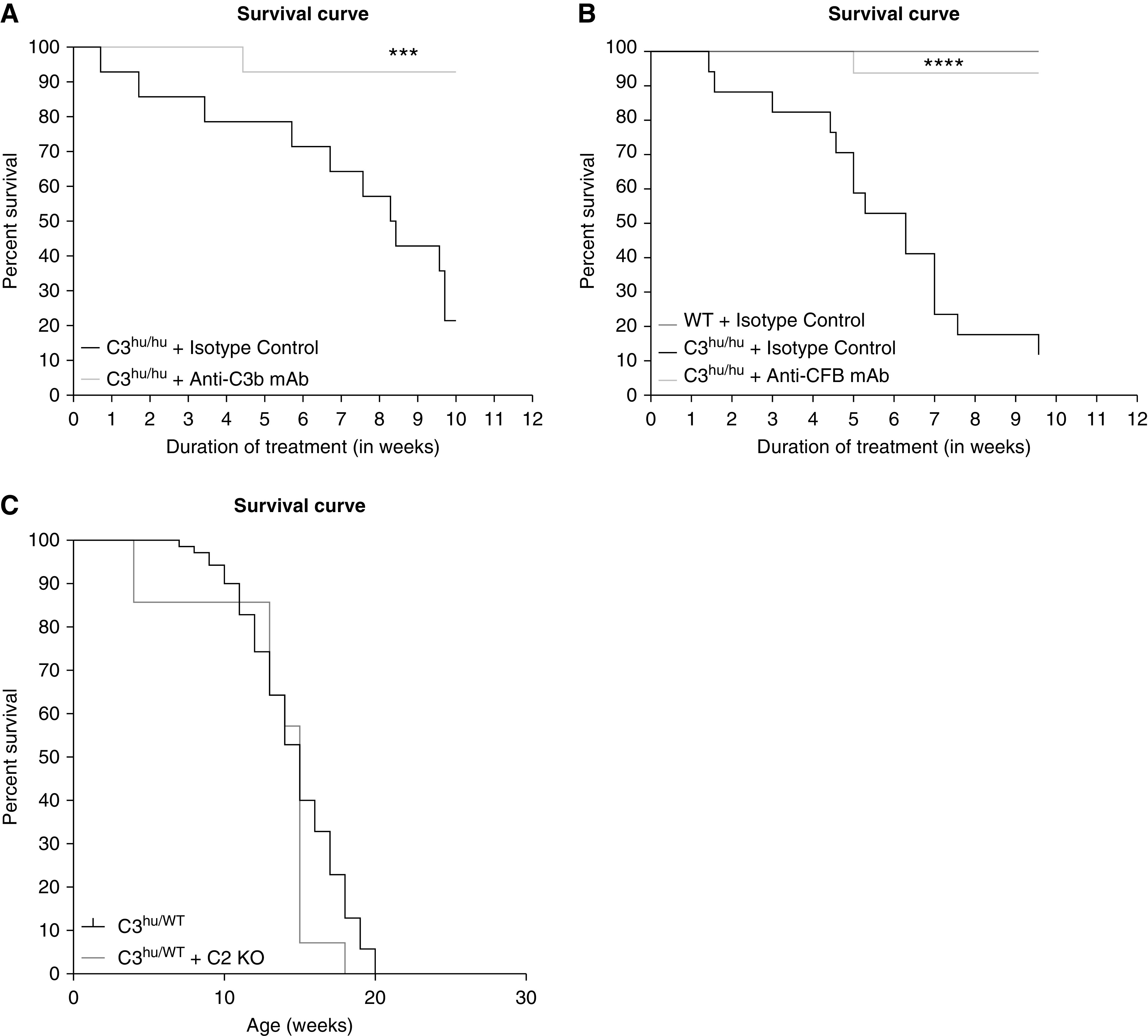
The AP C3 convertase blocking mAbs, anti-C3b, or anti-CFB improve survival rate in C3hu/hu mice. (A) Survival curve with anti-C3b mAb (C3hu/hu and isotype control, n=14; C3hu/hu and anti-C3b, n=14). (B) Survival curve with anti-CFB mAb (WT and isotype control, n=4; n=C3hu/hu and isotype control, n=17; C3hu/hu and anti-C3b, n=16). (C) Survival curve with C2 KO (C3hu/WT, n=70; C3hu/WT and C2 KO, n=14). ***P<0.001, ****P<0.0001, versus C3hu/hu and isotype control.
Discussion
C3G is a devastating kidney disease with poor prognosis and limited treatment options.1,34–36 CFH−/− mice are a classic C3G model that had improved our understanding of the disease over the last several years.16,37,38 Novel C3G models may further help advance the knowledge of the disease and gain insight into mechanisms and therapeutic strategies in complement-driven kidney diseases. Here, we showed that our C3hu/hu mice present a C3G-like disease with a faster progression to ESKD, potentially due to spontaneous hyperactivation of complement via AP.
The complement system, comprising >30 proteins, predominantly functions to remove pathogens and immune complexes in a highly regulated manner. Any mechanisms leading to an imbalance between the activation and regulation of the complement pathways can lead to potential damage to the host tissue.39,40 Using VelociGene technology, we are able to make large and precise genome modifications in mouse, including replacing the full coding sequence of mouse genes of interest with the gene encoding human homolog in the native mouse locus. This provides unique opportunities to test human therapeutics in mice.21 One caveat is that, in a tightly regulated system with multiple interacting proteins and highly coordinated crosstalk, these substitutions can result in dysregulation of complex pathways due to interspecies incompatibility. Previously, we discovered that mice “humanized” with the human C5 gene were functionally C5 deficient, because mouse C3 convertase is unable to cleave human C5.41 We have shown that adding human C3 protein to WT mouse serum enhances hemolysis, suggesting hyperactivation of the pathway.41 We also noted that human C3 protein, when administered in large doses (1 mg per mouse) in WT mice, was consumed within hours from circulation, suggesting spontaneous activation (data not shown).
We observed spontaneous mortality when the mouse C3 gene was replaced with that of the human C3 gene. The median age of survival was similar for mice either homozygous or heterozygous for human C3, suggesting humanizing one C3 allele may be sufficient for the lethal phenotype. The circulatory C3 protein levels were lower in C3hu/hu mice, similar to CFH−/− mice and as observed in patients with C3G.16,34 The circulatory C5 levels were also lower in the C3hu/hu mice, consistent with overactivation of the complement system. The lower C3 protein levels in C3hu/hu mice could be a result of the hyperactivation of AP, as suggested by C3b- and CFB-blocking antibody studies. The decrease in circulatory C3 levels (in the absence of hyperactivation) could also be, in part, due to lower C3 mRNA expression, on the basis of the observation that C3 gene expression in C3hu/hu mice is only a third of the levels in WT mice. However, our findings showed that the administration of a single dose of human CFH protein, but not CFI, in these mice led to increased circulatory C3 levels. These data suggest defective crosstalk between human C3 and mouse CFH protein, leading to fluid-phase dysregulation of AP. A mere fluid-phase dysregulation may not explain the accelerated disease in C3hu/hu mice, because CFH−/− mice manifest the disease very slowly. Hence, we presume that both fluid- and membrane-phase dysregulation may occur in C3hu/hu mice. Indeed, previous data show defective crosstalk between mouse regulatory protein, Crry, and human C3b.42 Interestingly, genetic deletion of complement factor P in CFH−/− and mutant mice also led to accelerated disease in CFH−/− mice.17–19 However, it is not completely clear how a lack of complement factor P, a positive regulator, and CFH, a negative regulator, led to severe phenotype in these mice, but it may be a result of a selective increase in the function of C3 convertase.
Two major C3 polymorphic allotypes exist: C3S and C3F.30 The names were derived on the basis of their electrophoretic mobility on agarose gels, with C3F moving faster compared with C3S. The C3F possesses a g.C>G polymorphism at position 304 in exon 3, leading to substitution of glycine instead of arginine (R102G). The C3F variant is associated with several diseases, including IgA nephropathy, C3G, and age-related macular degeneration.30,43 C3F showed higher complement activation owing to its low affinity to CFH.44 Our Sanger sequencing data from exon 3 of the BAC confirmed the C3hu/hu mice contain the C3S variant. These data indicate the phenotype exhibited by C3hu/hu mice is not due to the presence of the unstable C3F variant.
In our study, we observed complement activation in the kidneys of C3hu/hu mice. The glomerular pathologies, the elevation of kidney failure markers, proteinuria, and hematuria were consistent with patients with C3G. The C3hu/hu mice do not show platelet or hematocrit reduction, schistocytes, or increased serum-free hemoglobin, ruling out TMA as a driver of the phenotype. The decline in RBCs and total hemoglobin could be secondary to CKD. The fibrin deposition in the kidney was similar to published fatal models of C3G.33 Increased C3 and MAC deposition in the glomeruli and elevated urinary C5a levels suggest activation of both central and terminal complement components in the kidney. Systemic complement activation can also result in complement deposition and local complement activation in multiple organs. Recent data show C3 deposition and liver injury in mouse models of C3G.19,32 Consistent with these observations, the C3hu/hu mice also showed liver inflammation, C3 deposition, and elevation of liver injury markers.
Anti-C5 mAb treatment offered survival and functional, histopathologic, and molecular recovery. These data suggest C5 may play a predominant role in the pathology in these mice. Our preliminary data showed the average survival time was not altered by knocking out C2 in C3hu/WT mice, suggesting the classic or lectin pathway may not be the underlying cause of the phenotype. The survival and functional benefit observed by specifically blocking AP C3 convertase, with anti-C3b or anti-CFB, indicates AP activation is the primary driver of the disease in these mice. Further work is needed to gain a more detailed, mechanistic understanding of the phenotype presented in the C3hu/hu mice, and the relative contributions of the many complex components and regulators of the complement pathway. In conclusion, we have developed C3hu/hu mice that show a C3G-like phenotype with a rapid disease progression. This novel model of C3G may represent an additional tool for preclinical studies to evaluate and compare new therapies. Finally, the C3hu/hu mouse can serve as a valuable in vivo model to test human C3-directed treatments.
Disclosures
All authors are employees of Regeneron Pharmaceuticals, Inc. Y. Bai, K. Devalaraja-Narashimha, G. Halasz, C. Huang, T. Huang, T. Kaplan, A. Latuszek, Y. Luo, L. Macdonald, S. MacDonnell, J. McWhirter, K. Meager, L.G. Morton, A. Murphy, A. Muthuswamy, W. Olson, S. Prasad, Y. Wei, G. Yancopoulos, and B. Zambrowicz report having an ownership interest in Regeneron Pharmaceuticals. H. Gartner reports having an ownership interest in Bristol Meyers Squibb Pharmaceuticals, Gilead Pharmaceuticals, and Lexicon Pharmaceuticals. S. MacDonnell is a scientific advisor or member of the New York Academy of Sciences. K. Meagher and L.G. Morton report having patents and inventions with Regeneron Pharmaceuticals. L.G. Morton reports receiving research funding and speaker’s bureau fees from Regeneron Pharmaceuticals. S. Prasad reports having patents and inventions with Avigen and COR Therapeutics. G. Yancopoulos is a scientific advisor or member of Regeneron. All remaining authors have nothing to disclose.
Funding
This work was funded by Regeneron Pharmaceuticals.
Supplementary Material
Acknowledgments
We would like thank Michel Naum for cloning large DNA targeting vectors for eukaryotic cells, Dr. Vera Voronina for targeting, Mei Huang and Dr. Wojtek Auerbach for ES cell culture, Craig Grant for ES cell screening, Lakeisha Esau for mouse genotyping, Jan Roos and William Poueymirou for colony management and cohort delivery, Dr. Kalyani Penta for coordinating antibody development and characterization, and Dr. Min Ni and Dr. Yueming Ding for managing the RNA-seq workflow.
Dr. Kishor Devalaraja-Narashimha wrote much of the manuscript; Dr. Kishor Devalaraja-Narashimha and Dr. Lori G. Morton conceived the concept and designed the experiments; Mr. Yifan Luo, Ms. Cong Huang, Mr. John OBrien, Dr. Karoline Meagher, and Ms. Sarah Casanova produced much of the data; Mr. Theodore Kaplan, Mr. Chien Liu, Dr. Anantharaman Muthuswamy, and Dr. Scott MacDonnell performed histopathology analysis; Ms. Rebecca Peyser Boiarsky, Dr. Yu Bai, and Dr. Gabor Halasz performed molecular profiling analysis; Ms. Adrianna Latuszek and Dr. Ying Hu performed in vitro hemolysis assays for anti-C5 mAb; Mr. Hans Gartner performed Sanger sequencing; Dr. Yi Wei led RNA-seq by next-generation sequencing; Dr. Tammy Huang and Dr. William Olson led antibody development and characterization; Dr. Brian Zambrowicz led mice generation; Dr. John McWhirter designed the mice; and Dr. William Olson, Dr. Brian Zambrowicz, Dr. Lynn Macdonald, Dr. Srinivass Prasad, Dr. Andrew Murphy, Dr. George Yancopoulos, Dr. Lori G. Morton, and Dr. Kishor Devalaraja-Narashimha provided critical analysis of the data.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020050698/-/DCSupplemental.
Supplemental Figure 1. Sequencing human C3 target vector to confirm CGC SNP variant coding for Arg102.
Supplemental Figure 2. Liver pathology in C3hu/hu mice.
Supplemental Figure 3. TMA phenotype data.
Supplemental Figure 4. Binding and functional characterization of anti-C5 mAb.
Supplemental Figure 5. Functional characterization of anti-C3b and anti-CFB mAbs.
Supplemental Figure 6. C3b and CFB mAb effects on kidney function in C3hu/hu mice.
Supplemental Figure 7. C5, C3b and CFB mAb effects on C3 and C5 levels in C3hu/hu mice.
References
- 1.Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al.: C3 glomerulopathy: Consensus report. Kidney Int 84: 1079–1089, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S, et al.: The role of complement in C3 glomerulopathy. Mol Immunol 67: 21–30, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Servais A, Frémeaux-Bacchi V, Lequintrec M, Salomon R, Blouin J, Knebelmann B, et al.: Primary glomerulonephritis with isolated C3 deposits: A new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet 44: 193–199, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spartà G, Gaspert A, Neuhaus TJ, Weitz M, Mohebbi N, Odermatt U, et al.: Membranoproliferative glomerulonephritis and C3 glomerulopathy in children: Change in treatment modality? A report of a case series. Clin Kidney J 11: 479–490, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook HT, Pickering MC: Histopathology of MPGN and C3 glomerulopathies. Nat Rev Nephrol 11: 14–22, 2015 [DOI] [PubMed] [Google Scholar]
- 6.Herlitz LC, Bomback AS, Markowitz GS, Stokes MB, Smith RN, Colvin RB, et al.: Pathology after eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol 23: 1229–1237, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Quintrec M, Lionet A, Kandel C, Bourdon F, Gnemmi V, Colombat M, et al.: Eculizumab for treatment of rapidly progressive C3 glomerulopathy. Am J Kidney Dis 65: 484–489, 2015 [DOI] [PubMed] [Google Scholar]
- 8.Barbour TD, Ruseva MM, Pickering MC: Update on C3 glomerulopathy. Nephrol Dial Transplant 31: 717–725, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nester CM, Smith RJ: Complement inhibition in C3 glomerulopathy. Semin Immunol 28: 241–249, 2016 [DOI] [PubMed] [Google Scholar]
- 10.Ault BH, Schmidt BZ, Fowler NL, Kashtan CE, Ahmed AE, Vogt BA, et al.: Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem 272: 25168–25175, 1997 [DOI] [PubMed] [Google Scholar]
- 11.Dragon-Durey MA, Frémeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, Deschenes G, et al.: Heterozygous and homozygous factor h deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: Report and genetic analysis of 16 cases. J Am Soc Nephrol 15: 787–795, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Levy M, Halbwachs-Mecarelli L, Gubler MC, Kohout G, Bensenouci A, Niaudet P, et al.: H deficiency in two brothers with atypical dense intramembranous deposit disease. Kidney Int 30: 949–956, 1986 [DOI] [PubMed] [Google Scholar]
- 13.Rusai K, Zaller V, Szilagyi A, Kain R, Prohaszka Z, Cook HT, et al.: A rare case: Childhood-onset C3 glomerulonephritis due to homozygous factor H deficiency. CEN Case Rep 2: 234–238, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Servais A, Noël LH, Dragon-Durey MA, Gübler MC, Rémy P, Buob D, et al.: Heterogeneous pattern of renal disease associated with homozygous factor H deficiency. Hum Pathol 42: 1305–1311, 2011 [DOI] [PubMed] [Google Scholar]
- 15.Jansen JH, Høgåsen K, Harboe M, Hovig T: In situ complement activation in porcine membranoproliferative glomerulonephritis type II. Kidney Int 53: 331–349, 1998 [DOI] [PubMed] [Google Scholar]
- 16.Pickering MC, Cook HT, Warren J, Bygrave AE, Moss J, Walport MJ, et al.: Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet 31: 424–428, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Ruseva MM, Vernon KA, Lesher AM, Schwaeble WJ, Ali YM, Botto M, et al.: Loss of properdin exacerbates C3 glomerulopathy resulting from factor H deficiency. J Am Soc Nephrol 24: 43–52, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams AL, Gullipalli D, Ueda Y, Sato S, Zhou L, Miwa T, et al.: C5 inhibition prevents renal failure in a mouse model of lethal C3 glomerulopathy. Kidney Int 91: 1386–1397, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Herbert AP, et al.: Combination of factor H mutation and properdin deficiency causes severe C3 glomerulonephritis. J Am Soc Nephrol 24: 53–65, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith-Jackson K, Yang Y, Denton H, Pappworth IY, Cooke K, Barlow PN, et al.: Hyperfunctional complement C3 promotes C5-dependent atypical hemolytic uremic syndrome in mice. J Clin Invest 129: 1061–1075, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, et al.: High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol 21: 652–659, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Matsuzawa T, Ikarashi Y: Haemolysis of various mammalian erythrocytes in sodium chloride, glucose and phosphate-buffer solutions. Lab Anim 13: 329–331, 1979 [DOI] [PubMed] [Google Scholar]
- 23.Etemad-Gilbertson B, Guild BC, Kim Y-I, Klagge I, Kraus A, Roguska M, et al. : Compositions and methods for antibodies targeting complement protein C3b. World Intellectual Property Organization publication no. WO/2010/136311, 2010. Available at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010136311&tab=PCTBIBLIO&_cid=P11-KEEJJ7-16341-1. Accessed November 3, 2020
- 24.Holers VM, Thurman JM, Taube C, Gelfand EW, Gilkeson GS: Inhibition of factor B, the alternative complement pathway and methods related thereto. World Intellectual Property Organization publication no. WO/2005/077417, 2005. Available at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2005077417&tab=PCTBIBLIO&_cid=P11-KEEK2D-21031-1. Accessed November 3, 2020
- 25.Emlen W, Holers VM, Flynn P: Humaneered anti-factor B antibody. US Patent and Trademark Office no. 20080299114 A1, 2008. Available at: http://patft.uspto.gov/netacgi/nph-Parser?Sect2=PTO1&Sect2=HITOFF&p=1&u=/netahtml/PTO/search-bool.html&r=1&f=G&l=50&d=PALL&RefSrch=yes&Query=PN/7964705. Accessed November 3, 2020
- 26.Ueda Y, Mohammed I, Song D, Gullipalli D, Zhou L, Sato S, et al.: Murine systemic thrombophilia and hemolytic uremic syndrome from a factor H point mutation. Blood 129: 1184–1196, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S: Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P; The Molecular Signatures Database: The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vernon KA, Ruseva MM, Cook HT, Botto M, Malik TH, Pickering MC: Partial complement factor H deficiency associates with C3 glomerulopathy and thrombotic microangiopathy. J Am Soc Nephrol 27: 1334–1342, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delanghe JR, Speeckaert R, Speeckaert MM: Complement C3 and its polymorphism: Biological and clinical consequences. Pathology 46: 1–10, 2014 [DOI] [PubMed] [Google Scholar]
- 31.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC: Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A 92: 11490–11494, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski J, Renner B, Pickering MC, Serkova NJ, Smith-Jones PM, Clambey ET, et al.: Complement factor H-deficient mice develop spontaneous hepatic tumors. J Clin Invest 130: 4039–4054, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Van Lookeren Campagne M, Katschke KJ Jr., Gullipalli D, Miwa T, Ueda Y, et al.: Prevention of fatal C3 glomerulopathy by recombinant complement receptor of the Ig superfamily. J Am Soc Nephrol 29: 2053–2059, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbour TD, Pickering MC, Cook HT: Recent insights into C3 glomerulopathy. Nephrol Dial Transplant 28: 1685–1693, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nester CM, Smith RJ: Treatment options for C3 glomerulopathy. Curr Opin Nephrol Hypertens 22: 231–237, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riedl M, Thorner P, Licht C: C3 glomerulopathy. Pediatr Nephrol 32: 43–57, 2017 [DOI] [PubMed] [Google Scholar]
- 37.Pickering MC, Warren J, Rose KL, Carlucci F, Wang Y, Walport MJ, et al.: Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc Natl Acad Sci U S A 103: 9649–9654, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose KL, Paixao-Cavalcante D, Fish J, Manderson AP, Malik TH, Bygrave AE, et al.: Factor I is required for the development of membranoproliferative glomerulonephritis in factor H-deficient mice. J Clin Invest 118: 608–618, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holers VM: Complement and its receptors: New insights into human disease. Annu Rev Immunol 32: 433–459, 2014 [DOI] [PubMed] [Google Scholar]
- 40.Morgan BP, Harris CL: Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 14: 857–877, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Latuszek A, Cao J, Mujica A, Wiegand S, McWhirter J, Murphy A, MacDonald L: Humanized C5 and C3 animals, World Intellectual Property Organization publication no. WO/2015/171523, 2015. Available at: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015171523&tab=PCTBIBLIO. Accessed November 3, 2020
- 42.Kim YU, Kinoshita T, Molina H, Hourcade D, Seya T, Wagner LM, et al.: Mouse complement regulatory protein Crry/p65 uses the specific mechanisms of both human decay-accelerating factor and membrane cofactor protein. J Exp Med 181: 151–159, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez E, Nan R, Li K, Gor J, Perkins SJ: A revised mechanism for the activation of complement C3 to C3b: A molecular explanation of a disease-associated polymorphism. J Biol Chem 290: 2334–2350, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heurich M, Martínez-Barricarte R, Francis NJ, Roberts DL, Rodríguez de Córdoba S, Morgan BP, et al.: Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci U S A 108: 8761–8766, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.