

Abstract
Objective.
Previous studies in an experimental synovitis model in rats determined that administration of glutamate and aspartate into the joint produces hyperalgesic responses, while their receptor antagonists provide protection against the development of a hyperalgesic state. We examined concentrations of amino acids in synovial fluid (SF) to determine if increases might be relevant to human joint pathology.
Methods.
One hundred forty-four repository SF samples from patients undergoing diagnostic or therapeutic arthrocentesis and 14 SF samples from 7 cadavers were analyzed by high pressure liquid chromatography and compared as arthritic and control cohorts.
Results.
Compared to the average concentrations from the autopsy cases, the excitatory amino acids (EAA) glutamate and aspartate in SF from patients with synovitis were 54 and 28 times higher, respectively. Increases for all other amino acids ranged from 3 to 18-fold. The values for glutamate and aspartate were significantly higher than the mean increase for other amino acids compared using unpaired t tests (p < 0.0001). The mean ratio of glutamate and aspartate elevations over the mean increase for other amino acids was 4-fold and 2-fold, respectively. The EAA were highest in Reiter’s, infectious arthropathies, and systemic lupus erythematosus, but did not appreciably segregate to diagnosis or SF white blood cell count.
Conclusion.
Our data provide evidence of increased glutamate and aspartate in the SF of humans with active arthritis, suggesting that glutamate mediated events may contribute to the pathogenesis of human arthritic conditions.
Key Indexing Terms: GLUTAMATE, ASPARTATE, SYNOVIAL FLUID, ARTHRITIS, HYPERALGESIA
It is known that neuronal influences might contribute to the disease progression and deformities seen in autoimmune diseases such as rheumatoid arthritis (RA). The contribution of neuronal mediators to the pathogenesis of arthropathy is evidenced by the unilateral sparing of inflammation and chronic synovitis in the paralytic limbs of patients after stroke1. In several cases, it was determined that patients who developed a stroke or paralytic injury to one side of the body did not develop inflammatory or associated degenerative arthritis on the involved side, compared to disease development or progression on the nonparalyzed side2. In 2 cases, the patients developed stroke after the onset of severe chronic changes of RA, and over the next several years experienced incomplete reversal of the arthritic deformities2,3. Subsequent case reports describe stroke victims with cessation of gouty attacks on the affected side4 and one case where the patient experienced a lessening of involvement of scleroderma of the affected side, suggesting other inflammatory or degenerative processes might be affected by neuronal activity5. Another report has described lessening of Heberden’s nodes associated with hemiplegia6
Although much is known immunologically about peripheral inflammatory initiators such as trauma, pathogens, and antigen-antibody complexes, fewer studies have addressed neurogenic mechanisms involved in inflammatory conditions. It was shown using a knee joint kaolin/carrageenan inflammation model in rats that development of both the sensitized nociceptive state (hyperalgesia) and the inflammatory response itself rely on intact neuronal communication between the knee joint and spinal cord7. It was shown that edema formation and hyperalgesic nociceptive responses were reduced significantly by either lesioning peripheral nerves or dorsal roots or by blocking neuronal activity in the spinal cord7–9. Continuous activation of peripheral afferent fibers by noxious stimulation results in hyperexcitability of dorsal horn neurons that subsequently produce inappropriate, antidromic (away from the central nervous system) firing of primary afferent fibers10,11. This aberrant outgoing electrical activity may trigger the release of neurotransmitter substances that are measurable in the periphery, such as glutamate, substance P, and calcitonin gene related peptide12–14. These transmitter substances contribute to inflammation directly or indirectly through activation of specific receptors found on the peripheral nerves15–17. Our studies have shown that hyperalgesia and allodynia can be initiated by exogenous application of glutamate and aspartate directly into the joint space, and administration of excitatory amino acid (EAA) receptor antagonists directly into inflamed joints attenuates this increase in nociceptive behavioral responsiveness18. Thus, the persistent nociceptive changes seen in this inflammatory pain model may be the result of both peripheral and central neuronal sensitization mechanisms contributing to a cycle of persistent nociception.
To determine if this animal model has relevance to human synovitis, we obtained selected amino acid concentrations of SF derived from 144 patients and analyzed these values in the context of their clinical profiles. The amino acid concentrations in the SF were compared to disease diagnosis and SF white blood cell (WBC) count values obtained from retrospective chart review. The measureable concentrations of EAA glutamate and aspartate in active synovitis were greater by far than the concentrations of other amino acids for all diagnoses. Large concentrations of these 2 EAA in human SF would activate articular afferents generating painful input, as in the rat synovitis model described above. Over prolonged periods of intense activation and exposure to the toxic effects of excess glutamate, this may initiate central neurogenic mechanisms that override protective, restorative mechanisms19. This may contribute to tissue damage and chronic nociceptive activation and perhaps indirectly to the inflammation itself. Determination of high glutamate/aspartate content in SF may identify a subset of patients who might benefit from additional therapy using N-methyl-D-aspartate (NMDA) and non-NMDA EAA receptor antagonists applied directly into the joint, as in our experimental studies18
MATERIALS AND METHODS
Patient sample selection.
SF were collected at UTMB-Galveston from patients who underwent diagnostic or therapeutic arthrocenteses from on-site clinics or in consultation from 1983 to 1998. The Gulf Coast Arthritis Registry, Serologies, maintains these discarded samples as part of a serology/fluids repository at the University of Texas Medical Branch. A total of 144 samples were separately analyzed. The SF were collected under sterile conditions and aliquoted in glass tubes with no preservatives or in tubes containing sodium citrate and stored at −20°C under sterile conditions. For this study, 1 ml aliquots were transferred to microcentrifuge tubes and analyzed under blinded conditions. Retrospective chart reviews of these patients were carried out to independently confirm the diagnoses using criteria endorsed by the American College of Rheumatology20. The disease categories include RA, gout, osteoarthritis (OA), pseudogout, Reiter’s syndrome, psoriatic arthritis, infectious arthropathy, trauma, systemic lupus erythematosus (SLE), and juvenile arthritis. In most cases, patients had sought medical attention for painful, swollen joints and the chart confirmed the joint to be painful, swollen, erythematous, and warm to touch. Joints were considered septic or infected by bacteria if an organism could be cultured from SF. Eleven SF samples were excluded from diagnostic analyses as the diagnosis could not be confirmed from the data available.
Cadaver samples.
There are no available published data for amino acids levels in normal human joints, nor would ethical standards allow obtaining these specimens. To correct for this limitation of SF control values from humans, SF was aspirated from the knee joints of 7 recently deceased patients awaiting autopsy. The range in age was 3–68 years, the causes of death included cerebral hemorrhage (n = 2), stroke (n = 1), cardiac arrest in renal failure (n = 1), and respiratory failure (n = 3). Time of harvest after death was pronounced was between 3 and 15 h. No autopsy case was known to have recent arthritic diagnosis or complaints attributable to arthritis while alive, nor was active joint pathology noted in the chart at the time of death.
High performance liquid chromatography (HPLC).
Waters 717 plus autosampler (Waters, Milford, MA, USA), Beckman 114M solvent delivery pumps (Beckman, Fullerton, CA, USA), Bioanalytical Systems FL-45 fluorescence detector (excitation 250 nm, emission 456; Bioanalytical Systems, West Lafayette, IA, USA), and Waters Millennium software were used for ammo acid determination by HPLC. Standard HPLC protocol was modified for dilutions. The SF samples were pelleted, and in some cases diluted in millipore distilled deionized water. Samples (100 μl) were injected into the HPLC analyzer to quantify amino acid concentrations. Reagent solutions (all millipore filtered) included sodium borate (0.01 M, pH 8.95) and were run at 25°C.
Experimental standards and quality control.
Internal amino acid controls included homocysteine and norleucine. The recommended internal control is norleucine, as homocysteine competes with another protein peak in RA samples. Internal sample controls were set up in every set of 25 samples as quality controls for each run and among the separate runs (total = 36). Duplicate samples showed < 10% variance in HPLC runs. A total of 180 SF samples were run in 5 separate sets, to test a total of 144 patients. In addition, controls in triplicate were set up that included sodium heparin, citrate, and ethylenediamine-tetraacetic acid (EDTA), to determine if these tube additives would affect the measurement of SF amino acid concentrations.
A separate set of controls was used to analyze the contribution of blood in the SF samples using serial dilutions of SF and whole blood and serum, and to measure the effect of freeze-thawing on the tubes. About 90% of the samples were frozen and thawed × 2 (10% × 3) before HPLC analysis. Corrected for dilutional effect, there was no effect of additional EDTA, heparin, citrate, blood, or freeze-thawing (× 5) on amino acid determinations (data not shown). When analyzed for age of repository sample, there were no qualitative or quantitative differences in sample concentrations.
Statistics.
Significant changes in amino acid concentrations were determined using the unpaired t test. A p value < 0.05 was considered significant.
RESULTS
Clinical and inflammatory profiles of patient SF.
Of 144 SF samples, 133 had medical records available and sufficient for confirmation of diagnoses and SF WBC counts. Table 1 depicts the diagnosis for each sample and the (SF) WBC count for these cases. The majority of the cases were RA (n = 48), OA (n = 24), and gout (n = 25). The percentages of inflammatory joint fluid samples were derived from the number of samples with available WBC counts (Table 1, last column). The average values for SF WBC/mm3 are provided for most diagnostic categories.
Table 1.
Diagnostic categories of synovial fluids: number of samples, WBC counts, and number of available cell counts.
| Diagnostic Category | No. of Patients | Inflammatory Samples, % | Average WBC/mm3 (range) | No. of Cell Counts Available** |
|---|---|---|---|---|
| Rheumatoid Arthritis | 48 | 94.3 | 22,723 (70–210,000) | 41 |
| Gout | 25 | 84.2 | 22,040 (25–11,950) | 19 |
| Osteoarthritis | 24 | 65.2 | 2,657 (92–21,225) | 23 |
| Pseudogout | 4 | 100.0 | 17,323 (3,470–27,000) | 4 |
| Reiters | 9 | 88.0 | 18,875 (500–70,000) | 8 |
| Psoriatic Arthritis | 3 | 100.0 | −(19,500) | 1 |
| Infectious Arthropathy | 9 | 100.0 | 21,833 (4,250–65,000) | 8 |
| Joint Trauma | 4 | 25.0 | 45,390 (245–134,000) | 4 |
| Systemic Lupus Erythematosus | 5 | 50.0 | 15,570 (37–48,500) | 4 |
| Juvenile Arthritis | 2 | 50.0 | −(238–11,575) | 2 |
| Other/No Confirmed Diagnosis | 11 | 100.0 | 5,907 (500–8,475) | 8 |
| Total | 144 | 122 |
Inflammatory SF have > 2000 WBC/mm3.
Number of samples for each disease with reported WBC counts.
Comparison of amino acid concentrations in SF samples.
Table 2 depicts the SF amino acid concentrations for each diagnosis. The amino acids include glutamate (Glu), aspartate (Asp), glutamate precursor glutamine (Gln), serine (Ser), glycine (Gly), arginine (Arg), citrulline (Ctn), and threonine (Thr). The ranges of the SF amino acid concentrations are quite variable for all diagnoses and in these samples do not segregate to disease. The highest average Glu concentrations are seen for Reiter’s (495 μM), SLE (458 μM), RA (326 μM), gout (362 μM), and infectious arthropathies (378 μM). The diagnostic groups with the lowest average values are OA (266 μM), pseudogout (267 μM), psoriatic arthritis (273 μM), and joint trauma (211 μM). The highest average concentration for Asp values are SLE (67 μM), infectious arthritis (66 μM), Reiter’s (50 μM), psoriatic arthritis (48 μM), RA (38 μM), and gout (36 μM). The lowest values are seen in OA (21 μM) and pseudogout (22 μM). The range of concentrations in all arthritic categories for the EAA Glu and Asp are higher than the other amino acids. Averages and ranges of SF amino acid concentrations derived from 14 joints of 7 cadavers are provided for comparison in the bottom row, Table 2.
Table 2.
Synovial fluid amino acid concentrations by HPLC. Values (μm) are average (range) for each diagnosis.
| Diagnosis | Glu | Asp | Gln* | Ser | Gly | Arg | Ctn | Thr |
|---|---|---|---|---|---|---|---|---|
| RA | 326 (4–608) | 38 (1–107) | 201 (4–721) | 109 (5–226) | 261 (4–791) | 112 (3–384) | 37 (4–88) | 152 (11–292) |
| Gout | 362 (70–663) | 36 (8–86) | 217 (0–539) | 131 (53–295) | 231 (108–408) | 118 (18–405) | 36 (13–128) | 130 (48–264) |
| OA | 266 (0–530) | 21 (2–69) | 206 (26–489) | 72 (28–170) | 232 (110–415) | 110 (25–242) | 34 (11–114) | 114 (78–264) |
| Pseudogout | 267 (139–463) | 22 (6–37) | 154 (4–257) | 96 (92–101) | 211 (179–251) | 67 (39–96) | 23 (15–31) | 81 (72–87) |
| Reiter’s | 495 (268–895) | 50 (19–98) | 94 (8–70) | 168 (86–355) | 310 (131–480) | 138 (64–289) | 35 (9–85) | 151 (72–255) |
| Psoriatic arthritis | 273 (136–459) | 48 (39–56) | 227 (88–459) | 184 (123–250) | 360 (206–558) | 162 (128–223) | 25 (13–35) | 122 (90–162) |
| Infectious arthropathy | 378 (166–556) | 66 (15–264) | 161 (2–480) | 130 (41–176) | 269 (147–460) | 78 (7–156) | 58 (11–228) | 144 (60–348) |
| Joint trauma | 211 (157–287) | 42 (15–65) | 426 (52–617) | 129 (66–169) | 481 (149–1098) | 145 (23–376) | 50 (14–127) | 131 (82–165) |
| SLE | 458 (52–1061) | 67 (4–201) | 148 (5–512) | 165 (33–375) | 468 (149–925) | 209 (67–587) | 27 (9–44) | 91 (60–209) |
| Autopsy samples n = 14** | 6.25 (0.82–22) | 1.39 (0–7.64) | 70.97 (37–116) | 7.59 (2.89–19.4) | 43 (20.94–86.74) | 9 (5.3–13.96) | 2 (0.63–3.83) | 17 (10–30) |
Gln (glutamine) is a precursor to glutamic acid (Glu).
SF obtained from human autopsy specimens without joint disease.
The RA, gout, and OA disease categories had the greatest number of patients for comparison. Mean Glu levels in OA were 81% and 73% of the levels found in RA and gout, respectively. Mean Asp levels in OA were 55% and 61% of the levels found in RA and gout, respectively. The average SF WBC counts/mm3 were RA, 22,723; gout, 20,040; and OA, 2,657 (Tables 1 and 2). These values suggest that the SF WBC count and EAA values are independent. When individual SF WBC counts were plotted against glutamate and aspartate values, there was no consistent correlation to WBC and EAA concentrations (data not shown). SF glutamate and aspartate elevations did not correlate with SF WBC count, peripheral WBC count (when available), red blood cell count, disease duration, or therapy, in single time point determinations (data not shown). Chart review data were insufficient to correlate values to degree of pain experienced by the patient.
Comparison of SF amino acid ratio elevations in patients vs controls.
Figure 1 shows the means and standard errors for the ratios of amino acid concentrations in the joint fluid samples derived from patients with active synovitis divided by the SF concentrations derived from cadaver samples presented in Table 2. The EAA, aspartate and glutamate, were significantly elevated (p < 0.0001) compared to the other amino acids, compared individually or as EAA vs the other amino acids. The average values for the excitatory amino acid concentrations are much higher than the averages for the normal human autopsy models, with a 54-fold increase for glutamate. Aspartate shows a 28-fold increase over control values. Mean SF glutamine level was 3-fold elevated over the cadaver controls and was the closest value of all amino acids. Mean SF serine and glycine ratio elevations were 16 and 6, respectively. Mean SF ratio elevations of amino acids Arg, Ctn, and Thr were 14, 18, and 9, respectively. The mean concentrations of each of the EAA were then compared to those of the other amino acids. The average values of Glu and Asp in arthritic patients were 4-fold and 2-fold higher, respectively, than the combined means of the Ser, Gly, Arg, Ctn, and Thr.
Figure 1.
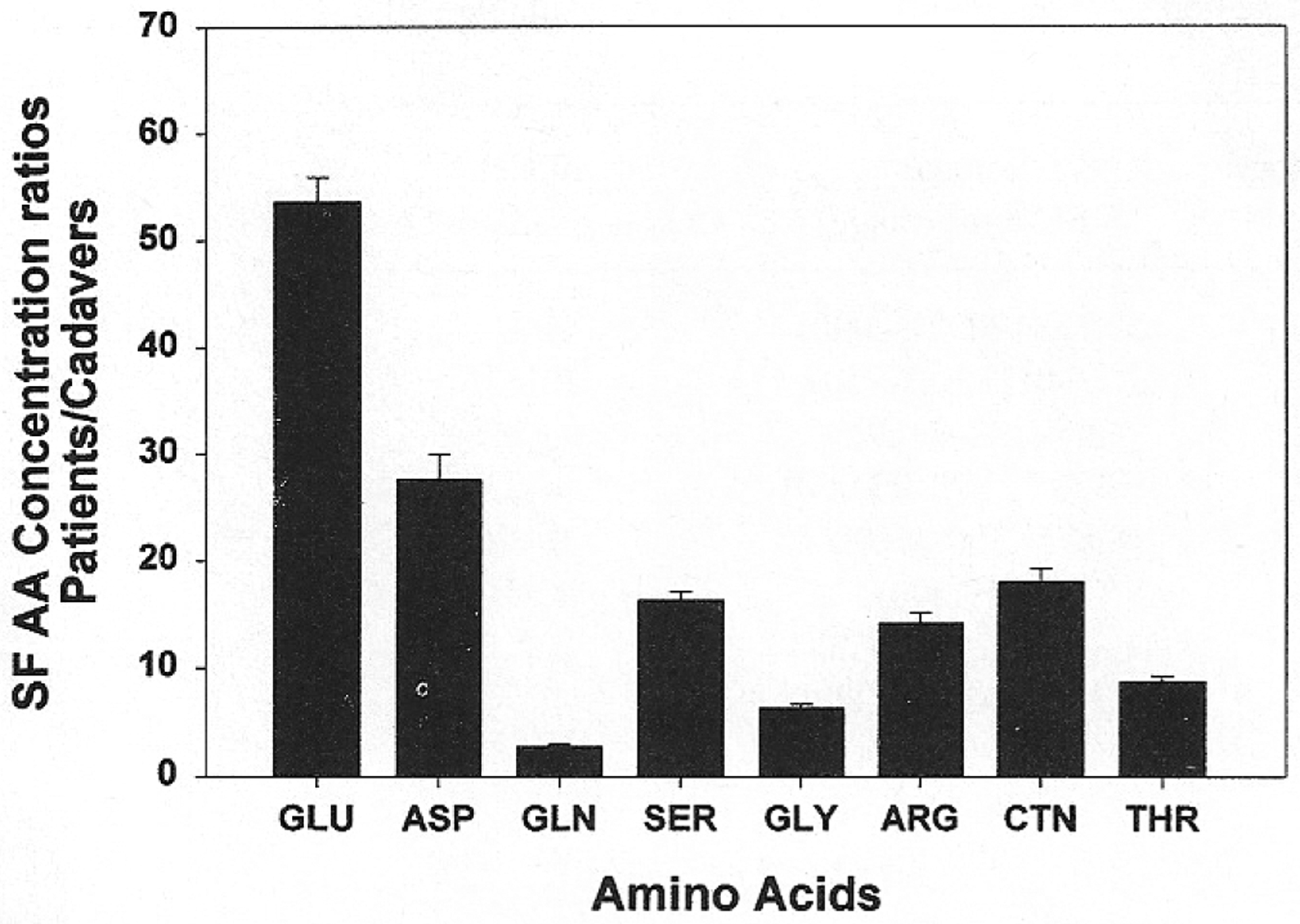
Ratio of amino acid (AA) concentrations (M) of human SF derived from patients with synovitis divided by SF amino acid levels in cadavers. Mean SF AA concentrations from patients with synovitis were divided by the mean SF AA concentrations from 7 cadavers. The EAA aspartate (ASP) and glutamate (GLU) levels are almost 53.65-fold and 27.69-fold higher, respectively, than the levels obtained from cadavers (p < 0.0001). SF increases for other AA: serine (SER) 16.34-fold and glycine (GLY) 6.40-fold. Increases in patient:cadaver mean AA concentrations also include: arginine (ARG) 14.17-fold, citrulline (CTN) 17.98-fold, threonine (THR) 8.71-fold. Glutamine (GLN), a precursor of glutamate, is increased 2.81-fold over cadaver. When corrected for human metabolic standards, glutamate was 4.16-fold and aspartate 2.15-fold increased in SF.
The concentrations of Ser, Arg, and Ctn in the SF were significantly elevated (p < 0.001) compared to Gly, Thr, and Ser. In this study, Arg and Ctn are considered nonspecific standards in SF, although they may be elevated compared to other amino acids, reflecting their involvement in the nitric oxide pathway.
DISCUSSION
This is the first report of amino acid levels in human joint fluids derived from patients with acute synovitis. This study evaluated patient SF amino acid levels from a single time point from several arthritic diseases. Glutamate SF concentrations are not published for normal human joints, and we have not located a repository of such samples. To address this limitation in data interpretation, amino acid values in SF from cadavers are provided as control comparisons for the patient samples. Cadaver selection was based on the time of harvest of the SF (< 24 h) and absence of clinically apparent arthritis.
We observed a greater increase in the patient:cadaver ratio of the SF EAA glutamate and aspartate (4-fold and 2-fold, respectively) compared to increases for other amino acids for all diseases of arthritis studied. Mean SF Glu and Asp concentrations in arthritic patients were 54-fold and 28-fold greater than cadaveric SF controls (Figure 1). The lowest mean SF ratio elevation was observed with glutamine, with a patient:cadaver ratio of 3. Mean patient SF Ser, Gly, Arg, Ctn, and Thr levels exhibited a range of ratio elevations of 6–18-fold.
We analyzed SF samples that were derived from a single time point arthrocentesis during acute illness or exacerbation, when the patients sought medical care for a swollen, painful joint. The SF amino acids were compared to diagnosis and WBC counts. It is difficult to compare the EAA profiles across individual diseases, as the sample sizes for pseudogout, psoriatic arthritis, juvenile arthritis, and SLE are small compared to the RA, OA, and gout groups. All arthritic disease categories showed elevation in both EAA (Table 2).
The majority had elevated EAA regardless of disease or joint fluid cell counts. In the repository SF samples, the reported WBC counts at arthrocentesis did not consistently or predictably correlate with any of the amino acid levels. These data suggest that sources of glutamate other than WBC are involved in arthritic processes. It also suggests that EAA elevations may not be strictly associated with inflammation variables as defined by WBC count, or that EAA concentrations may correlate more closely with other factors, such as pain, or elevated cytokines or other inflammatory mediators.
Thus, these preliminary data suggest that EAA are being concentrated, generated in situ, or deposited in the joints at higher amounts than would be expected, and that are unrelated to WBC generation secondary to inflammatory states. Studies show slightly elevated plasma amino acid levels in patients with RA versus healthy controls, but much lower levels than In the repository SF reported here21,22. Plasma/SF glutamate ratios for the repository samples are under study. Contributory sources for these amino acids include blood cells, mast cells, synoviocytes, or plasma extravasation. It is also possible that the SF EAA reflect degradation products of the inflammatory events. Increases in EAA relative to other amino acids during pathological events have been attributed to cell lysis23. Since glutamate is found in all cells, the sources for this glutamate in a damaged joint are likely varied, including release from blood borne and synovial sources. Alternatively, some of the glutamate and aspartate in human joint cavity may be released from the afferent nerve endings. Regardless of the source, EAA could affect joint nerve activity through specific EAA receptors on the nerves15–17
Figure 2 depicts the roles played by glutamate in normal and pathological conditions. EAA are major neurotransmitters of the central nervous system and are involved in transmission of nociceptive information from the spinal cord level to higher brain centers (A)24. EAA are normally found in high concentration as a transmitter substance in some of the peripheral nerve fibers, including those innervating the joint (B)25–27, and specific glutamate receptors have been localized at nerve endings and in dorsal root ganglia of peripheral nerves15–17. Based on the present data and our experimental model, we propose that under pathological conditions high concentrations of glutamate are released into the joint. Persistent activation of peripheral afferents by glutamate and other algesic agents in the joint, however, produces sensitization of both central and peripheral neuronal afferent fiber endings. Antidromic volleys in primary afferent fibers travel out the peripheral nerve to release neuropeptides and some of the EAA in the joint fluid compartment (C). These EAA are capable of further generating and amplifying pathological nociceptive processes directly in the joint and centrally. They may also contribute indirectly to the central neurogenic events promoting the inflammatory process itself.
Figure 2.
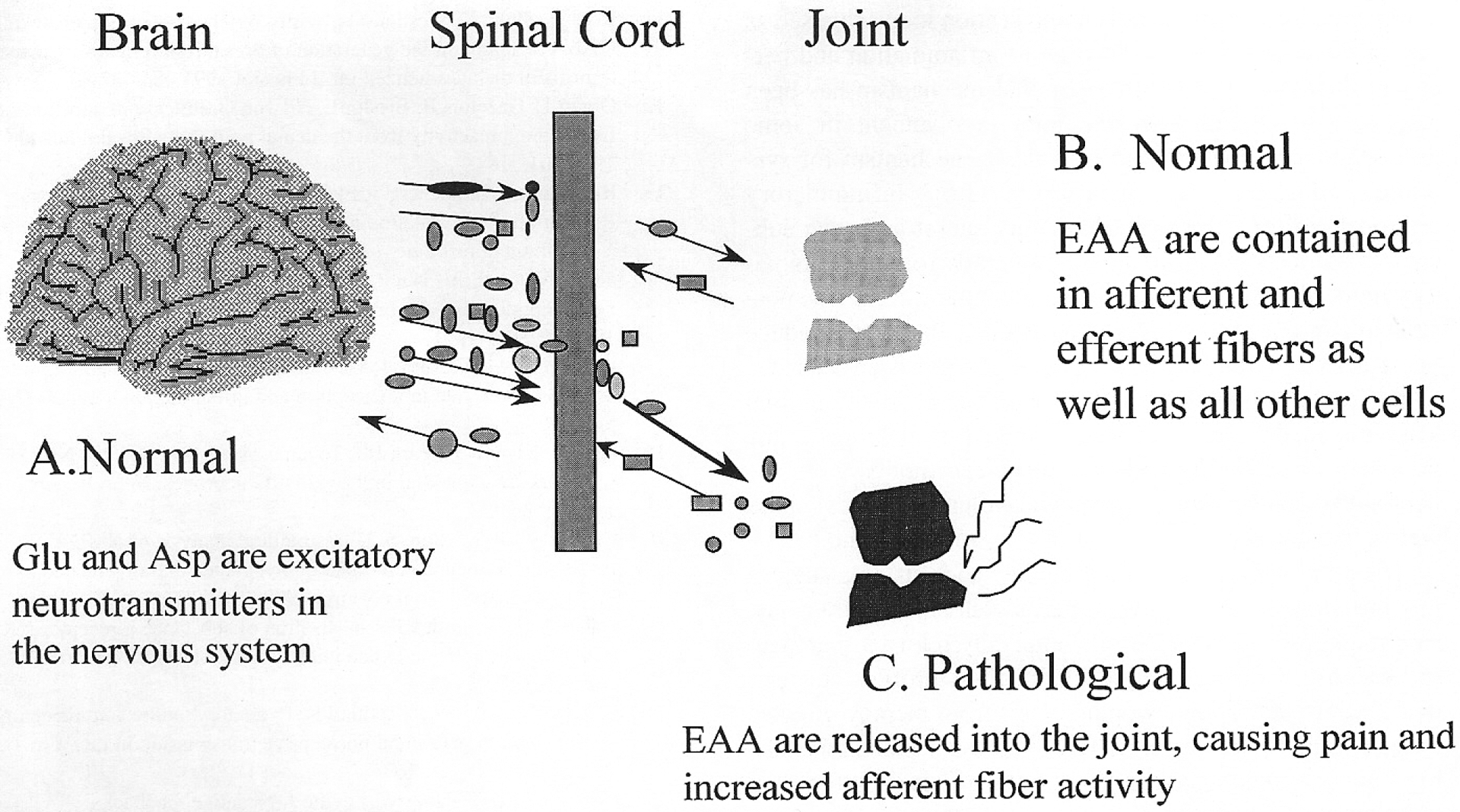
Neurogenic inflammation. A. These EAA are neurotransmitters in the nervous system. B. Neurotransmitters and neuropeptides are contained in afferent and efferent nerve fibers in the joint. C. In the pathological state, EAA are released in excess into the joint fluid and locally incite inflammation and pain. Asp: aspartic acid, Glu: glutamic acid.
Substantial evidence exists in support of peripheral glutamate receptor involvement in the amplification of nociceptive events. In the rat, the involvement of peripheral glutamate receptors in the generation of hyperalgesia and allodynia was revealed in experiments using our acute synovitis model induced by intraarticular injection of kaolin and carrageenan, as well as in experiments in which direct knee joint administration of the EAA glutamate and aspartate induced persistent hyperalgesic nociceptive behavior18. Administration of either NMDA or non-NMDA glutamate receptor antagonists directly into the joint attenuates hyperalgesic nociceptive responses induced by either method. Glutamate antagonist intervention in the periphery has been shown to attenuate secondary hyperalgesia in other models of peripheral experimentally induced nociceptive behaviors28–31. There are related neurogenic contributions to arthritis already described for substance P32, which has been reported to increase in rat arthritic joints13 and to increase severity of arthritis if added to the joint by increasing protein/plasma extravasation33 in experimental models. The neuropeptide substance P has also been shown to potentiate the release of glutamate34 Substance P has been shown to interact with EAA at peripheral nerve endings to enhance and increase the duration of glutamate induced behavioral responses35,36 A case report of a stroke patient who later developed psoriatic arthritis on the nonparalyzed side describes significant elevations in substance P only on the nonparalyzed (arthritic) side37. However, both sides had elevations in joint fluid inflammatory infiltrates, suggesting that increased cell count alone does not account for the inflammatory mediator changes or development of arthritic pathology. Thus, a synergistic cooperation of Glu and SP as well as other neuronally released agents could potentially form a peripheral regenerative element for the amplification loop proposed as part of the neurogenic contribution to inflammation and persistent nociception. A central neuronal mechanism has been suggested to explain why the joint involvement in some arthropathies is bilateral and provides a mechanism for systemic illnesses to manifest with inflammatory arthropathies32,35,36. If the EAA or other neurotransmitter substances are indeed contributing peripherally to the inflammatory induced nociceptive responses in humans, it may be possible to introduce specific receptor blockers that would reduce nerve activity into the joints and quell the persistent increase in nociception associated with inflammation. In the animal synovitis model, inhibiting antidromic nerve firing to the periphery or incoming neuronal input can modify both the neurogenic contribution to peripheral inflammatory events as well as the nociceptive responses of the affected joint7,10,11.
The case reports and animal models and our data suggest that interruption of the aberrant peripheral neuronal transmission might provide an additional clinically relevant therapeutic response. Characterization of the EAA profiles suggests that a variety of patients would benefit from therapy directed to reduction of joint nerve activity to abrogate pain and possibly reverse major pain related behavioral symptoms of inflammation during development of disease pathology.
ACKNOWLEDGMENT
The authors thank Dr. David McAdoo, Director of the Protein Chemistry Laboratory, Department of Anatomy and Neurosciences, University of Texas Medical Branch, for his input to the project. We thank Gregory Robak, research technician at UTMB, for his technical expertise, and Drs. Bruce Baethge and Niti Goel (UTMB) for critical review of the manuscript. UTMB physicians contributing synovial fluid samples to the Gulf Coast Arthritis Registry repository include Bruce Baethge, Jerry Daniels, Niti Goel, and Ulysses Lopez, whose efforts we appreciate. The authors thank Kristin Sawyer for assistance with the manuscript.
Supported by NIH R21 AR048371, P01 NS011255, and NS 32778 Central Control of Arthritis and Arthritic Pain.
REFERENCES
- 1.Nakamura K, Akizuki M, Kimura A, Chino N. A case of polyarthritis developed on the non-paralytic side in a hemiplegic patient. Ryumachi 1994;34:656–61. [PubMed] [Google Scholar]
- 2.Thomason M, Bywater E. Unilateral rheumatoid arthritis following hemiplegia. Ann Rheum Dis 1962;21:370–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Velayos E, Cohen S. The effect of stroke on well established rheumatoid arthritis. Md State Med J 1972;21:38–42. [PubMed] [Google Scholar]
- 4.Glyn J, Clayton M. Sparing effect of hemiplegia on tophaceous gout. Ann Rheum Dis 1976;35:534–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sethi S, Sequeira W. Sparing effect of hemiplegia on scleroderma. Ann Rheum Dis 1990;49:999–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter S. Unilateral Heberden’s nodes in a case of hemiplegia. NY State J Med 1952;52:349–50. [PubMed] [Google Scholar]
- 7.Sluka K, Willis W, Westlund K. The role of dorsal root reflexes in neurogenic inflammation. Pain Forum 1995;4:141–9. [Google Scholar]
- 8.Sluka K, Westlund K. Centrally administered non-NMDA but not NMDA receptor antagonists block peripheral knee joint inflammation. Pain 1993;55:217–25. [DOI] [PubMed] [Google Scholar]
- 9.Sluka K, Jordan H, Willis W, Westlund K. Reduction in joint swelling and hyperalgesia following post-treatment with a non-NMDA glutamate receptor antagonist. Pain 1994;59:95–100. [DOI] [PubMed] [Google Scholar]
- 10.Rees H, Sluka K, Westlund K, Willis W. Do dorsal root reflexes augment peripheral inflammation? Neuroreport 1994;5:821–4. [DOI] [PubMed] [Google Scholar]
- 11.Rees H, Sluka K, Westlund K, Willis W. The role of glutamate and GABA receptors in the generation of dorsal root reflexes by acute arthritis in the anesthetized rat. J Physiol 1995;484:437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olgart L, Gazelius B, Brodin E, Nilsson G. Release of substance P-like immunoreactivity from the dental pulp. Acta Physiol Scand 1977;101:510–2. [DOI] [PubMed] [Google Scholar]
- 13.Bileviciute I, Lundeberg T, Ekblom A, Theodorsson E. Substance P-, neurokinin A-, calcitonin gene-related peptide and neuropeptide Y-like immunoreactivity (-LI) in rat knee joint synovial fluid during acute monoarthritis is not correlated with concentrations of neuropeptide-LI in cerebrospinal fluid and plasma. Neurosci Lett 1994;167:145–8. [DOI] [PubMed] [Google Scholar]
- 14.Lawand N, McNearney T, Westlund K. Amino acid release into the knee joint: key role in nociception and inflammation. Pain 2000;(in press). [DOI] [PubMed] [Google Scholar]
- 15.Sato K, Kiyama H, Park HT, Toyama M. AMPA, KA and NMDA receptors are expressed in the rat DRG neurones. NeuroReport 1993;4:1263–5. [DOI] [PubMed] [Google Scholar]
- 16.Coggeshall R, Carlton S. Ultrastructural analysis of NMDA, AMPA and kainate receptors in unmyelinated and myelinated axons in the periphery. J Comp Neurol 1998;391:78–86. [DOI] [PubMed] [Google Scholar]
- 17.Hanesch U, Schmidt RF. Localization of substance P receptor NK1 in normal and inflamed knee joints of rats [abstract]. Neurosci Abstr 1997;23:1490. [Google Scholar]
- 18.Lawand N, Willis W, Westlund K. Excitatory amino acid receptor involvement in peripheral nociceptive transmission in rats. Eur J Pharmacol 1997;324:169–77. [DOI] [PubMed] [Google Scholar]
- 19.Green P, Miao F, Jänig W, Levine J. Negative feedback neuroendocrine control of the inflammatory response in rats. J Neurosci 1995;15:4678–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Primer on the Rheumatic Diseases. Klippel JH, editor. Atlanta (GA): Arthritis Foundation; 11th ed. 1997:453–64. [Google Scholar]
- 21.Trang L, Furst P, Odeback A-C, Lovgren O. Plasma amino acids in rheumatoid arthritis. Scand J Rheumatol 1985;14:393–402. [DOI] [PubMed] [Google Scholar]
- 22.Partsch G, Tausch G, Eberl R. Plasma amino acid level in rheumatoid arthritis and ankylosing spondylitis and its variation during age. Z Rheumatol 1978;37:105–11. [PubMed] [Google Scholar]
- 23.Caldwell M, Mastrofrancesco B, Shearer D, Bereiter D. Clinical and experimental approaches to dermal and epidermal repair: Normal and chronic wounds. Prog Clin Biol Res 1991;365:205–22. [PubMed] [Google Scholar]
- 24.Willis W, Westlund K. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol 1997;14:2–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westlund K, McNeill DL, Coggeshall RE. Glutamate immunoreactivity in rat dorsal root axons. Neurosci Lett 1989;96:13–7. [DOI] [PubMed] [Google Scholar]
- 26.Westlund K, McNeill DL, Coggeshall RE. Aspartate immunoreactive axons in normal rat L4 dorsal roots. Brain Res 1989;489:347–51. [DOI] [PubMed] [Google Scholar]
- 27.Westlund K, Sun Y, Sluka K, Dougherty P, Willis W. Neuronal changes in acute arthritis in monkeys. II. Increased glutamate immunoreactivity in the medial articular nerve. Brain Res Rev 1992;17:15–27. [DOI] [PubMed] [Google Scholar]
- 28.Davidson E, Coggeshall R, Carlton S. Peripheral NMDA and non-NMDA glutamate receptors contribute to nociceptive behaviors in the rat formalin test. NeuroReport 1997;8:941–6. [DOI] [PubMed] [Google Scholar]
- 29.Zhou S, Bonasera L, Carlton SM. Peripheral administration of NMDA, AMPA or kianate results in pain behaviors in rats. NeuroReport 1996;7:895–900. [DOI] [PubMed] [Google Scholar]
- 30.Prioleau C, Coggeshall R, Carlton S. Peripheral NMDA receptors play a role in capsaicin hyperalgesia [abstract]. Soc Neurosci Abstr 1996;22:1370. [Google Scholar]
- 31.Jackson D, Graff C, Richardson J, Hargreaves K. Glutamate participates in the peripheral modulation of thermal hyperalgesia in rats. Eur J Pharmacol 1995;284:321–5. [DOI] [PubMed] [Google Scholar]
- 32.Levine J, Collier D, Basbaum A, Moskowitz M, Helms C. Hypothesis: the nervous system may contribute to the pathophysiology of rheumatoid arthritis [review]. J Rheumatol 1985;12:406–11. [PubMed] [Google Scholar]
- 33.Levine J, Moskowitz M, Basbaum M. The contribution of neurogenic inflammation in experimental arthritis. J Immunol 1985;135 Suppl 2:843–7. [PubMed] [Google Scholar]
- 34.Kangrga I, Larew J, Randic M. The effects of substance P and calcitonin-gene-related peptide on the efflux of endogenous glutamate and aspartate from the rat spinal dorsal horn in vitro. Neurosci Lett 1990;108:155–60. [DOI] [PubMed] [Google Scholar]
- 35.Rees H, Sluka K, Lu Y, Westlund K, Willis W. Dorsal root reflexes in articular afferents occur bilaterally in a chronic model of arthritis in rats. J Neurophysiol 1996;76:4190–3. [DOI] [PubMed] [Google Scholar]
- 36.Bileviciute I, Lundeberg T, Ekblom A, Theodorsson E. The effect of a single intraperitoneal dose of hrIL-1 alpha on substance P-, neurokinin A-, calcitonin gene-related peptide- and neuropeptide Y-like immunoreactivity in cerebrospinal fluid, plasma and knee joint synovial fluid in the rat. Regul Pept 1994;53:71–6. [DOI] [PubMed] [Google Scholar]
- 37.Veale D, Farrell M, Fitzgerald O. Mechanism of joint sparing in a patient with unilateral psoriatic arthritis and a longstanding hemiplegia. Br J Rheumatol 1993;32:413–6. [DOI] [PubMed] [Google Scholar]