

Abstract
The coordinated differentiation of hematopoietic stem and progenitor cells (HSPCs) into the various mature blood cell types is responsible for sustaining blood and immune system homeostasis. The cell fate decisions underlying this important biological process are made at the level of single cells. Methods to trace the fate of single cells are therefore essential for understanding the hematopoietic system activity in health and disease, and have made a major impact in how we understand and represent hematopoiesis. Here, we discuss the basic methodologies and technical considerations for three important clonal assays: single cell transplantation, lentiviral barcoding, and sleeping beauty barcoding. This Perspective is a synthesis of presentations and discussions from the 2019 International Society for Experimental Hematology (ISEH) Annual Meeting New Investigator Technology Session and the 2019 ISEH Winter Webinar.
Keywords: Hematopoiesis, hematopoietic stem cell, hematopoietic progenitor cell, single cell biology, lineage tracing, HSC transplantation, lentiviral barcoding, sleeping beauty
Introduction
Hematopoietic stem and progenitor cells (HSPCs) are responsible for the sustained production of mature blood cells during homeostasis and in response to hematopoietic stresses1,2. Aberrant HSPC activity is also linked to a spectrum of hematological diseases, from clonal hematopoiesis to leukemias3–6. The classical models of hematopoiesis predict the existence of a homogeneous pool of multipotent and self-renewing hematopoietic stem cells (HSCs), which are equally contributing to the balanced production of all blood cell lineages through progressive lineage differentiation7,8. However, technology advances in the isolation of purified HSPC subsets and the capacity to investigate the behavior of single HSCs has revealed a high level of heterogeneity in their kinetics and patterns of reconstitution1,2,9. This heterogeneity is reflected in variable degrees of self-renewal potential and distinct propensity to differentiate towards the different blood cell lineages. Accumulating evidence suggests that only a fraction of mouse HSCs generate balanced multilineage output. Instead, HSCs with biased and restricted lineage potentials have been identified7,8,10. Since the decision between self-renewal and differentiation are made by individual HSCs, the examination of these processes require analyses at the clonal level.
In this Perspective, we introduce and discuss the technical aspects of three powerful methodologies for single-cell lineage tracing studies in hematology research: single cell transplantation, lentiviral barcoding, and sleeping beauty barcoding. This Perspective represents a synthesis of presentations and discussions from the 2019 International Society for Experimental Hematology (ISEH) Annual Meeting New Investigator Technology Session and the 2019 ISEH Winter Webinar by Drs. Joana Carrelha, Dawn Lin, and Alejo Rodriguez-Fraticelli. The webinar is also available to watch online (https://www.youtube.com/watch?v=ykfPl7hV9zU&feature=youtu.be).
Lineage tracing by single HSC transplantation
In vivo transplantation is a long-standing gold-standard technique for the study of self-renewal and differentiation of HSCs11,12. HSC frequency can be quantified by limiting dilution transplantation assays (LDTA)13–15, which do not require flow cytometry or cell sorting. However, a more detailed analysis of the functional heterogeneity of HSCs has been achieved by sorting and transplanting highly purified single cells15,16. Large-scale studies of the in vivo outputs of single transplanted bone marrow (BM) HSCs15–17 have recently confirmed that the phenotypically-defined HSC pool is substantially heterogeneous, but also demonstrated that there is a specific number of distinct long-term engrafting HSC subtypes. In contrast to all conceivable stochastic variation, only a limited number of sustained lineage-biased and lineage-restricted HSC subtypes are actually observable upon transplantation, in addition to the classically defined multilineage HSC17–24.
Technical considerations for transplantation
Transplantation from adult mouse BM consists of five main steps (Figure 1): (1) BM harvest from donor mice, (2) sorting of HSCs by fluorescence-activated cell sorting (FACS), (3) mixture with unfractionated BM support cells, (4) injection of cells into preconditioned recipient mice, and (5) analysis of donor-derived cells in the recipients. Each of the main steps of this technique can be modified to some extent to fit different model systems and different experimental questions. This technique can be used not only with adult BM HSCs, but also with fetal liver and fetal BM HSCs23.
Figure 1. Experimental steps for transplantation of single murine HSCs from adult bone marrow.
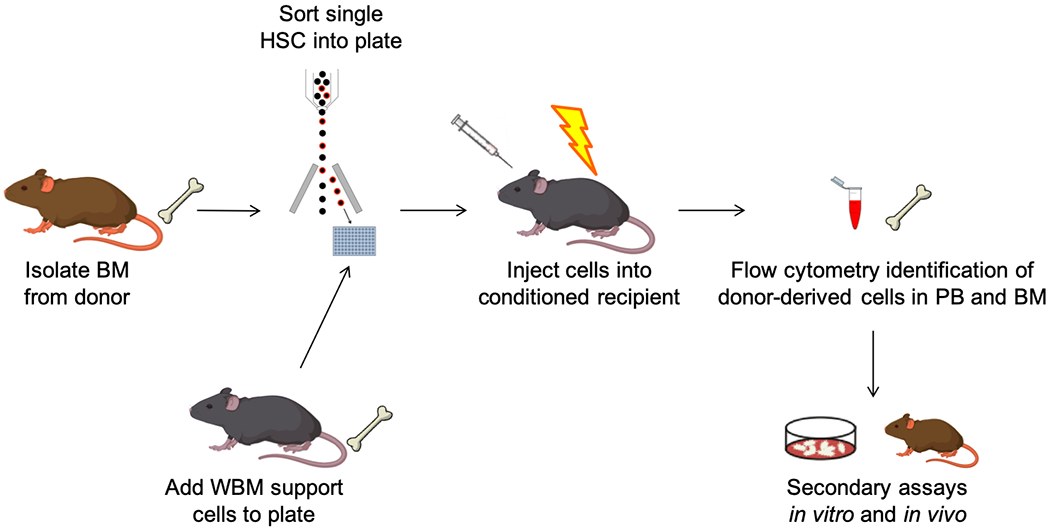
Long bones are harvested from donor mice, and a single cell suspension of BM cells is prepared. BM cells are stained with fluorochrome-labelled antibodies for FACS, and individual phenotypic HSCs are sorted into a cooled collection plate. A specified number of whole BM support cells is added to each well that contains a single sorted HSC, and the contents of each well are then collected into a syringe and injected (typically intravenously) into conditioned recipient mice (typically lethally irradiated). Donor-derived cells can be identified in the recipients through flow cytometry analysis of congenic markers and/or fluorescent reporter signals in serial biopsies (typically peripheral blood sampling or BM aspirations). These donor-derived cells can then be recovered by FACS from relevant tissues, and further tested in a multitude of secondary in vitro and in vivo assays.
Expression of distinct alleles of the pan-hematopoietic marker CD45 in donor and recipient cells13,16, and/or expression of distinct fluorescent reporters17,19 allows quantification by flow cytometry of the contribution of transplanted HSCs to the hematopoietic system of the recipient mouse, at multiple timepoints post-transplantation. The most typical and less invasive approach is the analysis of mature lineages in serial samples of peripheral blood, but donor-derived HSPCs can also be serially analyzed through BM aspirations. Serial sampling allows flexibility of experimental decisions based on the ongoing observation of donor-derived output, before more cell-destructive methods are used for terminal analysis.
Different mouse lines with inducible or constitutive phenotypes can be used as donors and recipients in HSC transplantations. For example, mutant donors can be used to assess the impact of specific mutations on HSC function, Cre reporters can be used as donors to map the contribution of transplanted HSCs to specific lineages, and immune-compromised recipients can be used to improve engraftment. Not many donor and recipient phenotypes have yet been explored in single HSC transplantation studies, but a few fluorescent reporter lines have recently been used, such as Kusabira Orange (KuO)17,18, Vwf-GFP25, Vwf-Tomato/Gata1-GFP19, and Pdzk1ip1-GFP26. However, mixed genetic backgrounds and immunological compatibility issues can lead to quantitatively and qualitatively reduced engraftment, or even to total premature loss of engraftment. Single HSC transplantations are particularly sensitive to this issue, and therefore the effects of new donor/recipient combinations should be carefully assessed.
Preparation and sorting of single HSCs
In order to maximize cell survival and increase the frequency of reconstituted mice in single HSC transplantations assays, processing and sorting of BM samples should be as simplified as possible, and the total time between donor BM harvest and single HSC injection should be minimized. A simple processing and sorting strategy can be very efficient when using a typical phenotypic definition of HSCs (e.g. Lineage- Sca1+ cKit+ CD150+ CD48- CD34-). However, to ensure feasibility of specific experimental setups — for example, when dealing with very rare phenotypic HSC sub-fractions — extra processing steps might be required before single-cell deposition, such as magnetic sorting with microbeads (cKit enrichment or Lineage depletion) or double-sorting with flow cytometry. Pre- or post-sort culture of cells with media of different compositions might also be part of the experimental objectives.27,28 Overall, it must be considered that extra processing steps might affect HSC survival and homing ability. The chosen protocol should always be a reasonable compromise between the experimental objectives, the sorting efficiency and purity, and the quantity and quality of engraftment in recipient mice.
A sorter with an automated cell deposition unit (ACDU) is required for deposition of single cells into U-bottom or V-bottom 96-well tissue culture plates. Most sorters use 0/32/16 as the default yield/purity/phase masks for single-cell deposition, but the less strict mode of 0/32/8 can be equally accurate for low- and mid-range sample concentrations, with the advantage of a significant reduction of the number of discarded target cells. It is possible to confirm single-cell deposition into wells using a brightfield microscope. However, in this technique it is very difficult to discern if lack of engraftment of a particular sorted cell was due to biological or technical factors, such as cell disruption or cell loss within the well or syringe. Single-cell FACS allows the use of index sorting29, meaning that the fluorescence intensity of each marker in the staining panel can be recorded for each individual sorted cell. This information can then be correlated with the functional output observed for each cell.
HSC transplantation
HSCs are most commonly transplanted into recipient mice by intravenous injection, but intraosseous injection can also be performed if HSC homing represents a significant variable within the experimental setting30. Direct deposition into the calvarium can also be performed for the purposes of live imaging31.
Whole-body lethal irradiation of wild-type recipients is the most extensively used method for transplantation of mouse HSCs. Support BM cells must be injected together with the purified HSCs, as a means to provide radioprotective progenitors that ensure survival of the lethally irradiated recipients. A common approach in single HSC transplantations is to use 2−3×105 unfractionated BM cells from a wild-type mouse17–19, but the phenotype and dosage of these support BM cells can be modified according to the experimental purposes. Sub-lethally irradiated W41 mice32 (the W41 mouse strain contains a spontaneous point mutation in the Kit gene that affects hematopoiesis33), have also been used as recipients in single-HSC transplantations27–29. Different methods of preconditioning recipient mice for transplantation can be considered34, and HSC engraftment can also be achieved without preconditioning35, but these methods have not been used, and might not be feasible, for single HSCs.
Single HSC transplantation can require the use of large cohorts of recipient mice in order to obtain robust data. The exact number required depends on the experimental questions and, most importantly, the engraftment frequency obtained with a specific experimental setup. In general, the frequency of long-term engraftment of single adult mouse BM HSCs ranges from 30% to 60%17,19,36.
Reconstitution analysis
Flow cytometry analysis of the output of engrafting HSCs is mostly unambiguous, but especially in single HSC transplantations it is inevitable that some HSCs with extremely low level of contribution might be misidentified as non-engrafting. Appropriate detection thresholds must be set for the analysis of the donor contribution to each lineage of interest. The background fluorescence levels of multiple non-transplanted control mice with the same phenotype as the experimental recipients represents a good measure for setting such a treshold. Identification of donor-derived cells based on positive markers (for example, donor-derived cells are GFP+ in a GFP- recipient) is typically a more sensitive and accurate approach than the opposite (for example, donor-derived cells are GFP- in a GFP+ recipient). Importantly, blood platelets often adhere to other cells during tissue processing. Although it might be impossible to completely avoid this problem, in blood leucocytes it can be reduced by taking steps such as collection of samples into heparin-treated or EDTA-treated tubes, separation of platelets from leucocytes by centrifugation, ammonium chloride incubation, dextran incubation, and increased percentage of serum in the preparation media. When using as HSC donor a fluorescent reporter that labels platelets and also other cell types, it is important to be able to distinguish, for each population, the real donor-derived fluorescent signal of that population from the fluorescent signal of donor-derived platelets attached to non-donor-derived cells. Platelets can be identified in the flow cytometry analysis of other cell types by inclusion of platelet markers like CD41. Furthermore, the intensity of fluorescent reporter signals tends to be lower in platelets, and so limiting gating to cells with high fluorescence intensity can help to exclude platelet contamination from the analysis.
The donor-derived compartments in each recipient of a single HSC encapsulate the activity of that single engrafting HSC, and thus the properties of different HSC subtypes can be characterized. Donor-derived cells can be further tested in situ in particular experimental setups, but most commonly they can be sorted from multiple tissues of the recipient mice and tested with in vitro assays and in vivo serial transplantation assays. Generally, 16 weeks post-transplantation is considered to be the minimum time required to identify long-term HSC engraftment in a primary transplantation setting. However, even longer time, and additionally serial transplantation, might be required to study true long-term HSCs37.
Strengths and weaknesses of single HSC transplantation
Although it is not a high-throughput approach, single HSC transplantation is a powerful method to study functional heterogeneity. It allows quantification and qualification of the in vivo outputs of single HSCs at serial timepoints, and it can be flexibly combined with additional fate mapping techniques and multiple readout methods. This approach has been used to confirm improved purification of engrafting HSCs when using modified phenotypic definitions29 or new reporters38. It has also allowed the detailed description of lineage-biased and lineage-restricted subsets of long-term engrafting HSCs that deviate from the classic definition of multilineage HSC10,17–19. Specific markers for the prospective purification of these lineage-biased and lineage-restricted HSC subsets are yet to be identified, and therefore single HSC transplantation continues to be an important method of HSC isolation based on functional output, and it will continue to play a role in the study of the regulation of different HSC subtypes in normal and stress hematopoiesis. It is important to note that, even though transplantation assays reveal important information about the properties of HSCs, what is observed upon transplantation might not reflect the typical output of HSCs in unperturbed hematopoiesis. To enhance our understanding of the functional heterogeneity and regulation of the HSC compartment, it is essential to combine observations from techniques that assay HSC behavior in both the transplantation setting and the native setting.
Lineage tracing by ex vivo genetic barcoding
Cellular barcoding involves the tagging of individual cells with unique and heritable genetic barcodes to allow interrogation of lineage relationships of large numbers of labelled clones. The technology can be broadly separated into two categories based on differential routes of barcode introduction: exogenous induced (ex vivo) and endogenous induced (in vivo) barcoding which is discussed in the third section of this review. In general, ex vivo barcoding systems introduce DNA barcodes (short stretches of random DNA sequences) into cells via lentiviral or retroviral transduction. Currently, several barcoding libraries/collections have been constructed, each differ in the length of barcode sequences and the size/diversity of available barcodes39–42. To date, several ex vivo barcoding studies in the field of hematopoiesis have highlighted striking fate heterogeneity of a variety of HSPC populations, such as lymphoid-primed multipotent progenitors (LMPPs)41 and common myeloid progenitors (CMPs)43.
A general experimental setup for ex vivo barcoding in hematology research involves the following steps (Figure 2): (1) isolation of HSPC populations of interest; (2) barcode labelling via lenti-viral or retro-viral transduction; (3) transplantation or culturing of barcoded HSPCs to allow development into mature lineages; (4) isolation of mature populations; (5) amplification of barcodes within the isolated populations via PCR; (6) next-generation sequencing of barcodes; (7) barcode data processing and analysis. By comparing the presence or absence of barcodes in the different mature lineages, fate outcomes of barcoded HSPCs (clones) can be established. Furthermore, because the technology is semi-quantitative44,45, the abundance of barcodes within populations can inform relative contribution of these barcoded clones to cell types. In addition, serial sampling of materials from the same mice/culture over time can be performed to interrogate developmental kinetics at a clonal level46,47.
Figure 2. Experimental steps for ex vivo barcoding.
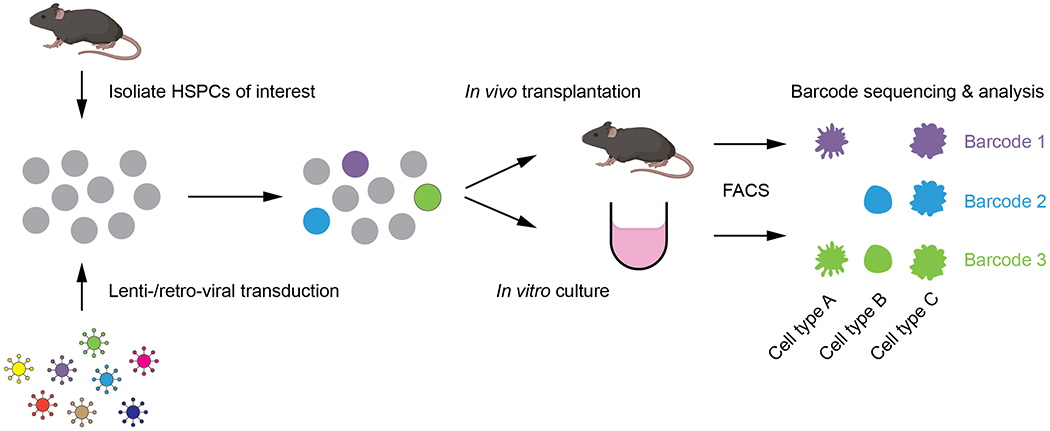
HSPCs are isolated and transduced with a library of lentiviral or retroviral barcodes. These barcoded HSPCs can then be transplanted into recipient mice or seeded in culture to allow development in vivo or in vitro, respectively. Mature progeny cell types are purified using FACS and barcodes within each sorted population are analyzed via PCR amplification and sequencing.
During lentiviral or retroviral transduction, multiple viral particles encoding the same barcode sequences can be integrated into the same cell, especially with a high transduction efficiency. Therefore, to reduce the possibility of multiple integrations, most ex vivo barcoding protocols aim for a low transduction efficiency. In principle, multiple integrations can result in over estimation of the number of clones with the same pattern but will not lead to false interpretation of a clone’s lineage output. In addition, multiple intergration can be detected as it is unlikely that several barcoded clones exhibit highly similar pattern regarding the proportional output to different lineages. In contrast, repeat use of barcodes (i.e. viral particles encoding the same barcode being transduced into different cells) represents a more serious issue, which might lead to false assignment of clonal lineage output. To reduce the possibility of repeat usage, a small percentage of the total library size should be used for individual biological replicates. In addition, it is possible to predetermine this threshold by random sampling different numbers of barcodes from the library and comparing the percentage overlap of barcodes.
Barcode contamination, PCR, and sequencing errors can lead to false identification of barcodes. To increase the accuracy of barcode identification, inclusion of technical replicates in the experimental design is highly recommended. After isolation of mature cell types of interest, each sample can be halved, and PCR amplified separately. This would result in a rough equal splitting of daughter cells with the same barcodes. Therefore, a high correlation of barcode signatures between the technical replicates would be expected. When this is not the case, the robustness of the data should be considered carefully44. If serial sampling is performed from the same mouse/culture over time, it is important to assess whether the barcode distribution from the sampled material is representative of the remaining material. Therefore, it is highly recommended to perform control experiments to determine the amount and frequency of serial sampling in your system. Again, correlation of split controls can be performed for comparison46.
A common way to present barcoding data is to use heatmaps to plot contribution to lineages (i.e. lineage output) of all barcodes detected (Figure 3A). Alternatively, dimension reduction techniques such as t-distributed stochastic neighborhood embedding (t-SNE) and uniform manifold approximation and projection (UMAP) can be used to plot barcodes on a two- or three-dimension map (Figure 3B). Using the relative contribution of barcodes to different lineages as input, t-SNE or UMAP algorithm can determine coordinates for each barcode on each axis, which generally results in barcodes with similar patterns being placed in close proximity to form clusters. To facilitate comprehensive visualization of longitudinal barcoding data, a novel tool called developmental interpolated t-SNE time-lapse movie (DiSNE) was developed recently46 (Figure 3C). Essentially, barcode contributions to all cell types over all time points are used to generate a t-SNE map, which places barcodes with similar patterns in close proximity to form clusters. The proportional output to cell types and clone size are then visualized by plotting individual barcodes as a pie chart on the t-SNE map, where the proportion of each slice corresponds to the proportional output to a cell type and the size of the pie is scaled based on the total number of cells produced by that clone. Finally, linear interpolation is applied between time points to visualize the dynamic changes in both lineage output and size of each clone over time.
Figure 3. Different visualization methods for barcoding data.
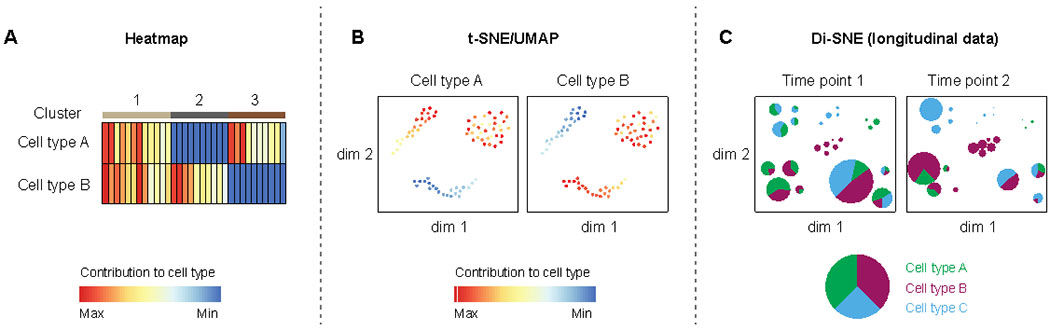
A) Heatmap. B) Dimentionality reduction methods such as t-SNE and UMAP. C) Di-SNE visualization of the dynamic changes in lineage output and clone size over time.
In general, the ultimate goal of a barcoding experiment is to understand the diversity in lineage output of single cells of interest, with a focus on HSPCs in this review. This can be achieved by classification of barcodes based on distinct outcomes (e.g. multi-outcome, oligo-outcome or uni-outcome). Classification can be done by applying thresholds to determine binary outputs of barcodes to each progeny cell type. For example, if the cut-off is set to be 0.01%, any barcodes with less than 0.01% contribution to B cells are defined as a non-B cell generating clone. Another way to classify barcodes is to use hierarchical clustering. This can be performed in conjunction with heatmap visualization for the identification of clusters of barcodes with similar outputs. While this approach captures the complexity of most barcoding data, it might not result in intuitive visualization of some datasets. In a recent study using barcoding to track the development of dendritic cells over time, the use of t-SNE in combination of a DBSCAN clustering method allows better separation of clusters of clones with similar development dynamics46. Collectively, barcode data visualization and analysis remain to be challenging and several analytical approaches should be performed on the same dataset to thoroughly assess the structure of the data and to maximize the extraction of useful biological meanings.
Similar to single cell transplantation assays, a major limitation of ex vivo barcoding is the need to remove HSPCs from their original niche for barcode transduction and sequential transplantation or culturing in a different environment. Therefore, it is important to note that ex vivo barcoding does not allow assessment of clonal fate during hematopoiesis in native conditions. Furthermore, barcode transduction usually involves 6-48 hours of ex vivo culture, which might lead to changes in fate bias of HSPCs of interest. On the other hand, ex vivo barcoding technologies offer multiple advantages. First, ex vivo barcoding is high throughput. Depending on the library size, most protocols enable simultaneous tracking of large numbers of clones (hundreds to millions) in a single biological replicate39–41. Compared to in vivo barcoding systems, most ex vivo barcoding protocols are more straightforward to implement and analyse, and can be ultilized for human studies using xenograft models. Importantly, the system is highly flexible, as different HSPC populations can be easily isolated based on known phenotypic markers using FACS, as opposed to labelling via the generation of in vivo barcoding mice using different gene promoters. Furthermore, purified and barcode-labelled HSPCs can be first expanded in vitro and transplanted into several recipients to compare lineage output of sisters from the same founder clone41.
Several areas can be improved to further advance the application of ex vivo barcoding. First, development of several sub-libraries of barcodes would allow labelling of different populations, followed by transplantation into the same host to assess clonal output in a competitive setting. This could be achieved by introduction of different fluorescent tags, different index sequences and/or different collection of barcodes for each sub-library. Second, integration of DNA barcode detection in single cell RNA sequencing protocols enables simultaneous measurement of cell identity and clonal history. These methods, including CellTagging48 and LARRY49, can be extremely powerful in hematology research for the discovery of early molecular fate determinants in HSPCs.
Transposon-based methods for single-cell lineage tracing in situ
Lentiviral barcoding, as many other procedures modifying cells ex vivo, challenges scientists with the issue of physiological relevance50. The consequences of ex vivo labeling becomes less of a problem when studying transplantation hematopoiesis, a critical therapy that will continue to be increasingly used in the clinic, especially with the current advances in gene therapy and gene editing. However, for most people, blood development and pathogenesis take place in the native setting, i.e. without transplantation. Importantly, native hematopoiesis does not equal to unperturbed hematopoiesis, as most animals in their natural context (including humans) will be exposed to injuries, infections and other perturbations that affect this process51,52. As perturbations also mostly affect hematopoietic progenitors and stem cells in their native context, in situ lineage tracing of hematopoiesis is essential to understand the normal development and regeneration of blood cells.
The labeling of cells in the native setting poses many challenges, across multiple disciplines and tissues53. However, there are some common principles or considerations that are applicable to all these techniques. In order to prospectively trace cells in their native context, investigators have to generate observable and heritable differences among cells in a controllable manner. Unsurprisingly, most techniques rely on genetic mutations (point mutations, transpositions, inversions, deletions), typically triggered by the inducible activity of recombinases (Cre, Flp), nucleases (Cas9) or transposases (Sleeping Beauty). Controlled activation of these enzymes in transgenic animal models induces modifications at the DNA level, which are typically inherited in a stable and long-term manner. Furthermore, all of these alterations can be sequenced, to read out the label information from potentially many thousands of clones at once.
The Sleeping Beauty mouse model
Since PiggyBac (PB) and Sleeping Beauty (SB) transposases were engineered in the early 2000s, they have revolutionized our capacity to induce mutagenesis and deliver transgenes in mammalian cells54,55. Transposon (Tn)-based approaches for gene therapy have only now taken second place due to recent advances in gene editing and lentiviral engineering, but they remain powerful weapons in the arsenal of the molecular biologist56. Since transposable elements can be highly mutagenic, the cut-and-paste transposons like PB and SB are generally preferred for native lineage tracing, and rapid inactivation of the transposase is required after induction. In contrast to the use of transposons in insertional mutagenesis screenings, native lineage tracing only requires one single transposon, which still retains a large diversity of potential integration sites while significantly reducing the mutational capacity of the system57.
Current transposase-based barcoding mouse models consist of three transgenes: (1) a Tet-responsive transactivator (Tet-ON system, such as rtTA), (2) an inducible expression cassette of the SB transposase (SBase), under the control of a Tet-responsive promoter, and (3) a single cognate transposable Tn element, which contains a sequence of variable length, and is flanked by inverted repeats (Figure 4A). In the current iteration of the Sleeping Beauty lineage tracing system, each allele was separately knocked-in into the collagen Col1a1 locus, which allows relatively unbiased transposase expression and transposition in most organs and cell types58. During mouse breeding, the SBase and Tn alleles are maintained separately, in order to avoid possible spontaneous transposition, and bred together only to generate experimental animals.
Figure 4. Lineage tracing with the Sleeping Beauty (SB) mouse model.
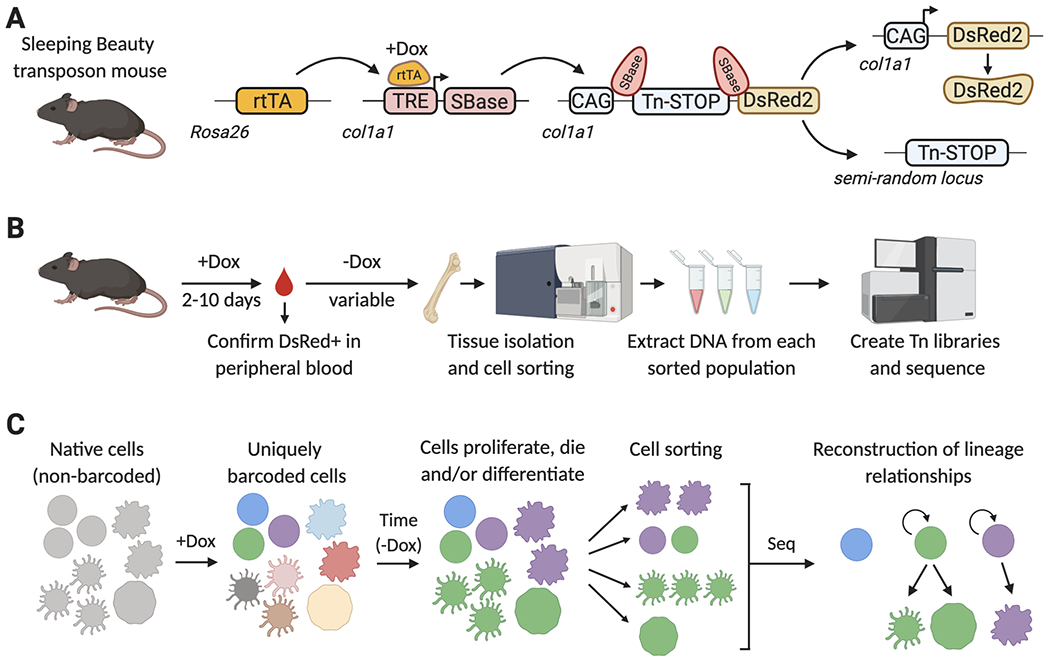
A) SB mouse model. A Dox-inducible transcriptional activator (rtTAM2), an rtTA- and Dox-regulated hyperactive SB transposase (TRE-SBase), and a single-copy SB-dependent transposable element (Tn), are combined in order to allow the inducible mobilization of the Tn element into a semi-random locus in the genome. The Tn element carries a transcriptional STOP cassette, which prevents DsRed2 expression in non-barcoded cells. Upon SBase-mediated Tn excision, DsRed2 is expressed to help avoid processing of non-barcoded cells. B) Summary of the SB-barcoding protocol. Mice are treated with Dox for up to 10 days, and expression of DsRed is confirmed in peripheral blood by flow cytometry. Dox is removed, to prevent further SBase expression and Tn mobilization. During the period of time without Dox, barcoded stem and progenitor cells will divide and differentiate, propagating these barcodes to their progeny. Tissues are then isolated and labeled with fluorescently-tagged antibodies to sort distinct cell populations by FACS. Genomic DNA is isolated from each population, and used to generate Tn-insertion libraries, which specifically amplify the Tn-flanking sequences and add adapters for next-generation sequencing. C) Reconstructing lineages with the SB mouse model. Before Dox addition, each cell in the SB barcoding mouse carries the Tn element in the same col1a1 locus. Upon Dox administration, Tn elements are mobilized and each cell will now have a unique single Tn integration site in a different locus of their genome. Upon Dox withdrawal, this integration site remains stable and establishes a tag or barcode that can be inherited by the cell progeny as HSPCs divide and differentiate into one or multiple cell types. Lists of transposon insertion sites are determined for each FACS-isolated population by next-generation sequencing, and then these are mathematically analyzed to establish lineage relationships between populations.
Transposon-based lineage tracing relies on the semi-random acute mobilization of the single transposable element, and the subsequent use of its random integration sites as heritable and stable cellular labels after the transposase is inactivated58. After transposition, labeled cells of interest are isolated (typically by cell sorting), and integration sites are sequenced and aligned/mapped to the genome in order to generate tables of all detected integrations sites and their abundances (reads, or unique molecular identifiers) from different analyzed populations or timepoints (Figure 4B). Once these tables are obtained, the relationships between cellular progenies and the clonal dynamics of the system of study can be inferred through mathematical analyses.
Unique advantages and challenges of transposon-based single-cell tracing
Given the possibility for integration in approximately any location in the genome, transposase-based methods have a much larger information potential compared to recombinase or lentiviral-barcoding methods59. Although most transposases have mild preferences for integration in particular sequences, they still boast the highest potential diversity of generated barcodes of all methods to date. In practice, the exploration of clonal diversity is more constrained by sequencing depth (number of reads per sample) than by barcode diversity. The large costs of tracing hundreds of thousands of clones with high throughput sequencing is thus an important consideration for using this method. One way to minimize costs is to label and trace a random subsample of all potentially traceable cells (“sublabel”), which can be achieved through titration of the transposase induction time and/or dose60. Then, the absolute numbers of clones can be extrapolated or modelled mathematically from the experimentally sublabeled dataset.
Since the expression of the transposases needs to be reversible (to prevent constant Tn mobilization), they are typically controlled by Tet-regulated promoters. Therefore induction of transposition requires the presence of a Tet-ON reverse transcriptional activator (rtTA) transgene. In the original Sleeping Beauty model, this was achieved by ubiquitous expression through the Rosa26-driven rtTAM2allele. A yet unexploited advantage of Tet-inducibility is the existing availability of conditional or tissue-specific alleles for rtTA expression, such as Rosa26-LoxP-STOP-LoxP-rtTA (SR), which allow transposition induction in specific tissues or even cell types61.
Another advantage of transposon-based lineage tracing is that sequences contained inside the transposon cassette can be exploited to establish a secondary label to serve as a reporter for transposase activity. For instance, in the Sleeping Beauty lineage tracing mouse system, the Tn3 transposon carries a transcriptional STOP cassette inside its terminal repeats58. The left flank of the T3 donor locus contains a CAGGS promoter, while the right flank contains a coding sequence for a red fluorescent protein (DsRed2). As a result, the cells in which transposition occurs, and all of their progeny, are concomitantly labeled with DsRed2 expression. In this way, transposons enable dual tracing systems: (1) the Tn-STOP switch; and (2) the random integration site of the transposon. Furthermore, sorting the DsRed2-positive cells allows researchers to avoid sampling cells in which transposition was ineffective and reveals no valuable lineage information.
An important consideration of the transposase-based methods is their dependency on integration-site sequencing (ISS), a group of still relatively underdeveloped techniques, in spite of their relevance for clonal analysis in the follow-up of lentiviral-delivered gene-therapy patients62. ISS techniques rely on the PCR-based amplification of the integration site, which contains a known sequence on one end, and a completely unknown single piece of genomic DNA on the other (or two, if both ends of the transposon are being sequenced). The attempts to solve this molecular cloning problem in the last 30 years have been diverse and continue to improve in each iteration63–65. However, even in the best-case scenario, one can still only expect to recover 10-20% of single copy barcodes, which is one the lowest sampling efficiencies of sequenced-based barcoding methods. Furthermore, the low sensitivity of these methods typically requires the pretreatment of DNA samples from around 1000 cells. Lower cell numbers can be used in combination with whole-genome DNA amplification, which, however, further amplifies the noise of the barcode signals, especially for the lowest abundant clones. For this and other reasons, the quantitative aspect of transposon-based tracing continues to be one of the major challenges of the method.
Recent findings and future perspectives of in situ single-cell lineage tracing
In spite of their limitations, the unrivaled large diversity and low background of transposon-based tracing achieves something very unique among endogenous barcoding methods: a remarkably low number of false positives. That is, the fraction of cells that get independently labeled with the same particular barcode is close to 0, even for hundreds of thousands of cells. This unique feature has allowed the relatively unbiased retrospective analysis of thousands of lineage trajectories across the entire adult hematopoietic hierarchy in situ without the need for isolation and culture or transplantation of marker-defined progenitor populations, with minimal requirements for error correction. Thanks to these features we were able to observe that most transplantable self-renewing long-term hematopoietic stem cell clones do not contribute extensively to mature blood cell formation at least for the first year of mouse life-span, with the exception of the megakaryocyte lineage58,60. And more recently, we validated the existence of two ontogenies of monocytes, neutrophil-related and dendritic cell-related populations, in situ49.
A major concern of transposon-based lineage tracing methods is their dependence on flow-based cell sorting for isolating prospective populations of interest. While this is not problematic for studying well-defined mature blood cell types, a few issues become evident when analyzing barcodes from hematopoietic stem and progenitor cell populations. First, sequencing transposon integration sites from very rare populations can be challenging and frequently requires genome amplification methods, which may lead to contaminations and misinterpretation of lineage relationships. Second, recent transcriptome-level studies of HSPCs have revealed a continuous nature of the hematopoietic hierarchy, which means that flow cytometry gates cannot recapitulate the heterogeneous landscape of early stem and progenitor cellular states66. To circumvent these limitations, researchers have recently developed mouse models that enable expression of inducible endogenous barcodes, facilitating simultaneous read-out of transcriptomes and lineage tracing information from single cells67,68. The first method, CARLIN (CRISPR-array repair lineage tracing), relies on Dox-inducible Cas9 expression to mutate a battery of guide RNA target sequences that is located in the 3’UTR of an EGFP transgene67. The second method, PolyloxExpress, uses Cre-based recombination of an array of loxP-flanked cassettes, which are located in the 3’UTR of a tdTomato transgene68. In both methods, assignment of lineage barcodes to individual transcriptomic entities avoids contamination with spurious barcodes and provides a more robust quantitative measurement through single-cell counts.
While these techniques allow for the first time to connect cellular stem cell states with lineage fates without the need for ex vivo cell labeling, these methods can still only generate a relatively small diversity of barcodes compared to transposon-based lineage tracing. As a consequence, only small subsamples of cells (carrying the rarest combinations of mutations) can be reliably labeled with unique tags, at least in the current implementations of these methods. It is foreseeable that future transposon-based tracing methods will also implement Tn integration-site capture through single cell sequencing platforms to allow multimodal sequencing with mRNA, protein or chromatin accessibility. In this sense, recently developed self-reporting transposons represent an interesting opportunity that may unlock the multiomics single-cell toolbox for this group of powerful tracing techniques69.
Conclusions
The hierarchical structure of the hematopoietic system, in which a single HSC can generate the whole set of blood cell populations, represents a powerful model for studying cell fate decisions. The methods described above have been developed for tracking cell fate decisions in different biological contexts and represent powerful tools to understand the biological processes controlling homeostasis and disease. Although recent advances in single-cell transplantation assays, barcoding, and transposon-based methods have facilitated this field of research, several aspects of these innovative technologies still require improvement in order to enable the discovery of the molecular determinants for every distinct cell fate of HSPCs at the single-cell level. Future technological advances will most likely address these limitations, and ultimately improve the overall depth of the data generated whilst facilitating the application of those powerful single-cell tools to other hierarchical systems that contribute to homeostasis and disease.
Table 1.
Comparision of clonal methods
| Single cell transplantation | Lentiviral barcoding | Endogenous SB barcoding | |
|---|---|---|---|
| Requirement of ex vivo manipulation | Yes | Yes | No |
| Nº of cells that can be simultaneously tracked in one mouse | 1 | In practice, under 100,000 (due to library production constraints and sequencing/cost limitations). | In practice, under 1M (due to sequencing/cost limitations). |
| Cost (per standard experiment) | Low | Intermediate | High |
| Efficiency | Variable | Variable | Low (up to 50%) |
| Target population for tracing | Only HSCs | Any cell | Any cell |
| Requirement of cell sorting for analysis of results | No. | Depends (if barcodes are expressed or not) | Yes |
| False positives | Extremely rare | Rare (depends on library diversity, target population, and possible contaminations) | Rare (mostly due to contaminations and whole genome amplification) |
| False negatives | Extremely rare. | Rare | Very frequent (due to poorly performing Tn capture techniques) |
| Possible use for investigating human biology | Yes | Yes | No |
Highlights.
This review describes clonal lineage tracing approaches for studying hematopoiesis
We discuss single cell transplantation assays and lentiviral barcoding technologies
We cover sleeping beauty barcoding for studying cell fate in native hematopoiesis
Acknowledgements
We thank the ISEH staff and New Investigator Committee for their support and Dr. Shalin Naik for critical feedback on the manuscript. TCL is supported by a Sir Henry Dale Fellowship from the Wellcome Trust and The Royal Society. ACW is supported by the Leukemia and Lymphoma Society (3385-19) and the NIH (K99HL150218). ARF is supported by the NHLBI (K99HL146983), is a Scholar of the American Society of Hematology and is a Special Fellow of the Leukemia and Lymphoma Society (3391-19). JC is supported by a MRC Molecular Hematology Unit Core award (MC_UU_12009/5).
References
- 1.Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley interdisciplinary reviews Systems biology and medicine. 2010;2(6):640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125(17):2605–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luis TC, Wilkinson AC, Beerman I, Jaiswal S, Shlush LI. Biological implications of clonal hematopoiesis. Exp Hematol. 2019;77:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steensma DP, Ebert BL. Clonal hematopoiesis as a model for premalignant changes during aging. Exp Hematol. 2020;83:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swierczek S, Prchal JT. Clonal hematopoiesis in hematological disorders: Three different scenarios. Exp Hematol. 2020;83:57–65. [DOI] [PubMed] [Google Scholar]
- 6.Horton SJ, Huntly BJ. Recent advances in acute myeloid leukemia stem cell biology. Haematologica. 2012;97(7):966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553(7689):418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas S, Trumpp A, Milsom MD. Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell. 2018;22(5):627–638. [DOI] [PubMed] [Google Scholar]
- 9.Loughran S, Haas S, Wilkinson A, Klein A, Brand M. Lineage commitment of hematopoietic stem cells and progenitors: insights from recent single cell and lineage tracing technologies. Experimental Hematology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto R, Wilkinson AC, Nakauchi H. Changing concepts in hematopoietic stem cells. Science. 2018;362(6417):895–896. [DOI] [PubMed] [Google Scholar]
- 11.Weissman IL, Shizuru JA. The origins of the identification and isolation of hematopoietic stem cells, and their capability to induce donor-specific transplantation tolerance and treat autoimmune diseases. Blood. 2008;112(9):3543–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson AC, Igarashi KJ, Nakauchi H. Haematopoietic stem cell self-renewal in vivo and ex vivo. Nat Rev Genet. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241(4861):58–62. [DOI] [PubMed] [Google Scholar]
- 14.Abramson S, Miller RG, Phillips RA. The identification in adult bone marrow of pluripotent and restricted stem cells of the myeloid and lymphoid systems. The Journal of experimental medicine. 1977;145(6):1567–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith LG, Weissman IL, Heimfeld S. Clonal analysis of hematopoietic stem-cell differentiation in vivo. Proc Natl Acad Sci U S A. 1991;88(7):2788–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273(5272):242–245. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto R, Morita Y, Ooehara J, et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154(5):1112–1126. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto R, Wilkinson AC, Ooehara J, et al. Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment. Cell stem cell. 2018;22(4):600–607 e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrelha J, Meng Y, Kettyle LM, et al. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 2018;554(7690):106–111. [DOI] [PubMed] [Google Scholar]
- 20.Morita Y, Ema H, Nakauchi H. Heterogeneity and hierarchy within the most primitive hematopoietic stem cell compartment. The Journal of experimental medicine. 2010;207(6):1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sieburg HB, Cho RH, Dykstra B, Uchida N, Eaves CJ, Müller-Sieburg CE. The hematopoietic stem compartment consists of a limited number of discrete stem cell subsets. Blood. 2006;107(6):2311–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Copley MR, Beer PA, Eaves CJ. Hematopoietic stem cell heterogeneity takes center stage. Cell stem cell. 2012;10(6):690–697. [DOI] [PubMed] [Google Scholar]
- 23.Benz C, Copley MR, Kent DG, et al. Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell stem cell. 2012;10(3):273–283. [DOI] [PubMed] [Google Scholar]
- 24.Müller-Sieburg CE, Cho RH, Karlsson L, Huang JF, Sieburg HB. Myeloid-biased hematopoietic stem cells have extensive self-renewal capacity but generate diminished lymphoid progeny with impaired IL-7 responsiveness. Blood. 2004;103(11):4111–4118. [DOI] [PubMed] [Google Scholar]
- 25.Sanjuan-Pla A, Macaulay IC, Jensen CT, et al. Platelet-biased stem cells reside at the apex of the haematopoietic stem-cell hierarchy. Nature. 2013;502:232–236. [DOI] [PubMed] [Google Scholar]
- 26.Sawai CM, Babovic S, Upadhaya S, et al. Hematopoietic stem cells are the major source of multilineage hematopoiesis in adult animals. Immunity. 2016;45(3):597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uchida N, Dykstra B, Lyons KJ, Leung FYK, Eaves CJ. Different in vivo repopulating activities of purified hematopoietic stem cells before and after being stimulated to divide in vitro with the same kinetics. Exp Hematol. 2003(31):1338–1347. [DOI] [PubMed] [Google Scholar]
- 28.Dykstra B, Kent D, Bowie M, et al. Long-term propagation of distinct hematopoietic differentiation programs in vivo. Cell stem cell. 2007;1(2):218–229. [DOI] [PubMed] [Google Scholar]
- 29.Wilson NK, Kent DG, Buettner F, et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell stem cell. 2015;16(6):712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhan Y, Zhao Y. Hematopoietic stem cell transplant in mice by intra-femoral injection. Methods Mol Biol. 2008;430:161–169. [DOI] [PubMed] [Google Scholar]
- 31.Turcotte R, Alt C, Runnels JM, et al. Image-guided transplantation of single cells in the bone marrow of live animals. Sci Rep. 2017;7(1):3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geissler EN, McFarland EC, Russell ES. Analysis of Pleiotropism at the Dominant White-Spotting (W) Locus of the House Mouse: A Description of Ten New W Alleles. Genetics. 1981;97(2):337–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma Y, Astle CM, Harrison DE. Heterozygous Kit mutants with little or no apparent anemia exhibit large defects in overall hematopoietic stem cell function. Exp Hematol. 2007;35(2):214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Czechowicz A, Kraft D, Weissman IL, Bhattacharya D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science. 2007;318(5854):1296–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waskow C, Madan V, Bartels S, Costa C, Blasig R, Rodewald HR. Hematopoietic stem cell transplantation without irradiation. Nature methods. 2009;6(4):267–269. [DOI] [PubMed] [Google Scholar]
- 36.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. [DOI] [PubMed] [Google Scholar]
- 37.Benveniste P, Frelin C, Janmohamed S, et al. Intermediate-term hematopoietic stem cells with extended but time-limited reconstitution potential. Cell stem cell. 2010;6(1):48–58. [DOI] [PubMed] [Google Scholar]
- 38.Sawai CM, Babovic S, Upadhaya S, et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity. 2016;45(3):597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerrits A, Dykstra B, Kalmykowa OJ, et al. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood. 2010;115(13):2610–2618. [DOI] [PubMed] [Google Scholar]
- 40.Lu R, Neff NF, Quake SR, Weissman IL. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nature biotechnology. 2011;29(10):928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naik SH, Perie L, Swart E, et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature. 2013;496(7444):229–232. [DOI] [PubMed] [Google Scholar]
- 42.Gerlach C, van Heijst JW, Swart E, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. 2010;207(6):1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perié L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The branching point in erythro-myeloid differentiation. Cell. 2015;163(7):1655–1662. [DOI] [PubMed] [Google Scholar]
- 44.Naik SH, Schumacher TN, Perié L. Cellular barcoding: a technical appraisal. Exp Hematol. 2014;42(8):598–608. [DOI] [PubMed] [Google Scholar]
- 45.Verovskaya E, Broekhuis MJ, Zwart E, et al. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood. 2013;122(4):523–532. [DOI] [PubMed] [Google Scholar]
- 46.Lin DS, Kan A, Gao J, Crampin EJ, Hodgkin PD, Naik SH. DiSNE Movie Visualization and Assessment of Clonal Kinetics Reveal Multiple Trajectories of Dendritic Cell Development. Cell reports. 2018;22(10):2557–2566. [DOI] [PubMed] [Google Scholar]
- 47.Wu C, Li B, Lu R, et al. Clonal tracking of rhesus macaque hematopoiesis highlights a distinct lineage origin for natural killer cells. Cell Stem Cell. 2014;14(4):486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biddy BA, Kong W, Kamimoto K, et al. Single-cell mapping of lineage and identity in direct reprogramming. Nature. 2018;564(7735):219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinreb C, Rodriguez-Fraticelli A, Camargo FD, Klein AM. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. 2020;367(6479). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busch K, Rodewald HR. Unperturbed vs. post-transplantation hematopoiesis: both in vivo but different. Curr Opin Hematol. 2016;23(4):295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hofer T, Busch K, Klapproth K, Rodewald HR. Fate Mapping and Quantitation of Hematopoiesis In Vivo. Annual review of immunology. 2016;34:449–478. [DOI] [PubMed] [Google Scholar]
- 52.Boettcher S, Manz MG. Regulation of Inflammation- and Infection-Driven Hematopoiesis. Trends in immunology. 2017;38(5):345–357. [DOI] [PubMed] [Google Scholar]
- 53.Espinosa-Medina I, Garcia-Marques J, Cepko C, Lee T. High-throughput dense reconstruction of cell lineages. Open Biol. 2019;9(12):190229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91(4):501–510. [DOI] [PubMed] [Google Scholar]
- 55.Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122(3):473–483. [DOI] [PubMed] [Google Scholar]
- 56.Kawakami K, Largaespada DA, Ivics Z. Transposons As Tools for Functional Genomics in Vertebrate Models. Trends Genet. 2017;33(11):784–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moriarity BS, Largaespada DA. Sleeping Beauty transposon insertional mutagenesis based mouse models for cancer gene discovery. Curr Opin Genet Dev. 2015;30:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun J, Ramos A, Chapman B, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514(7522):322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Izsvák Z, Ivics Z. Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther. 2004;9(2):147–156. [DOI] [PubMed] [Google Scholar]
- 60.Rodriguez-Fraticelli AE, Wolock SL, Weinreb CS, et al. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553(7687):212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dow LE, Nasr Z, Saborowski M, et al. Conditional reverse tet-transactivator mouse strains for the efficient induction of TRE-regulated transgenes in mice. PLoS One. 2014;9(4):e95236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paruzynski A, Glimm H, Schmidt M, Kalle C. Analysis of the clonal repertoire of gene-corrected cells in gene therapy. Methods Enzymol. 2012;507:59–87. [DOI] [PubMed] [Google Scholar]
- 63.Kim S, Kim Y, Liang T, Sinsheimer JS, Chow SA. A high-throughput method for cloning and sequencing human immunodeficiency virus type 1 integration sites. J Virol. 2006;80(22):11313–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt M, Schwarzwaelder K, Bartholomae C, et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat Methods. 2007;4(12):1051–1057. [DOI] [PubMed] [Google Scholar]
- 65.Kwon YM, Ricke SC, Mandal RK. Transposon sequencing: methods and expanding applications. Appl Microbiol Biotechnol. 2016;100(1):31–43. [DOI] [PubMed] [Google Scholar]
- 66.Watcham S, Kucinski I, Gottgens B. New insights into hematopoietic differentiation landscapes from single-cell RNA sequencing. Blood. 2019;133(13):1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bowling S, Sritharan D, Osorio FG, et al. An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell. 2020;181(7):1693–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pei W, Shang F, Wang X, et al. Resolving fate and transcriptome of hematopoietic stem cell clones. bioRxiv. 2020:2020.2003.2025.008433. [DOI] [PubMed] [Google Scholar]
- 69.Moudgil A, Wilkinson MN, Chen X, et al. Self-reporting transposons enable simultaneous readout of gene expression and transcription factor binding in single cells. bioRxiv. 2019:538553. [Google Scholar]