

Abstract
Aptamers are nucleic acid-based affinity reagents that are isolated via an in vitro process known as systematic evolution of ligands by exponential enrichment (SELEX). Despite their great potential for a wide range of analytical applications, there are relatively few high-quality small-molecule binding aptamers, especially for “challenging” targets that have low water solubility and/or limited moieties for aptamer recognition. The use of libraries containing chemically modified bases may improve the outcome of some SELEX experiments, but this approach is costly and yields inconsistent results. Here, we demonstrate that a thoughtfully designed SELEX procedure with natural DNA libraries can isolate aptamers with high affinity and specificity for challenging small molecules, including targets for which such selections have previously failed. We first isolate a DNA aptamer with nanomolar affinity and high specificity for (—)-trans-Δ9-tetrahydrocannabinol (THC), a target previously thought to be unsuitable for SELEX with natural DNA libraries. We subsequently isolate aptamers that exhibit high affinity and cross-reactivity to two other challenging targets, synthetic cannabinoids UR-144 and XLR-11, while maintaining excellent specificity against a wide range of non-target interferents. Our findings demonstrate that natural nucleic acid libraries can yield high-quality aptamers for small-molecule targets, and we outline a robust workflow for isolating other such aptamers in future selection efforts.
Graphical Abstract
INTRODUCTION
Aptamers are nucleic acid-based affinity reagents that are isolated from randomized DNA or RNA libraries through an in vitro method termed systematic evolution of ligands by exponential enrichment (SELEX).1,2 They can achieve strong and specific recognition of a wide range of targets and offer many advantages as bioreceptors for molecular sensing and detection relative to antibodies, including high chemical stability, ease and affordability of synthesis, and low batch-to-batch variability.3 Aptamers have been isolated for small-molecule targets such as metabolites, drugs, and environmental toxins.4 However, most small-molecule-binding aptamers isolated to date have insufficient binding affinity or specificity for real-world applications, especially compared to antibodies.5,6 For example, the well-known cocaine-binding aptamer and ATP-binding aptamer have micromolar affinities and undesirable cross-reactivity to other molecules.
Several research groups have pursued strategies for incorporating non-canonical, chemically modified bases into nucleic acid libraries to increase their chemical diversity and thereby improve aptamer performance with respect to their binding properties and metabolic stability.7–9 Although there is strong evidence that modified libraries generally improve the likelihood of isolating protein-binding aptamers with superior binding properties,10 it remains unclear whether the same benefits pertain to the isolation of small-molecule-binding aptamers. This is because, first, there are only a few works on this subject,11–18 and this can be attributed to the requirement for specifically engineered enzymes and modified nucleotides, which are costly and are generally not commercially available. Second, these reports offer conflicting perspectives on whether modified libraries increase the success rate of SELEX. For example, based on moities directly involved in ligand recognition of the human cannabinoid receptor, Mayer and co-workers used a modified DNA library incorporating benzyl-modified deoxyuridine to isolate an aptamer against the cannabinoid (—)-trans-Δ9-tetrahydrocannabinol (THC) following seven unsuccessful SELEX trials with natural DNA and RNA libraries of differing structure and length and various selection conditions. In contrast, the Kuwahara group was unable to isolate aptamers for the negatively charged target glutamic acid, even though they used a library containing bases modified with positively charged arginine residues.16 Furthermore, base-modified aptamers do not consistently exhibit improved binding properties. For example, aptamers isolated from base-modified libraries containing N-(2-(N6-adeninyl)-ethyl))carbamylvinyl deoxyuridine had an order of magnitude greater affinity and specificity for a derivative of camptothecin than those isolated from a natural nucleic acid library.17 On the other hand, ATP-binding aptamers isolated from libraries containing 5-(3-aminopropynyl)-2’-deoxyuridine11 or 5-(3-aminopropyl)uridine12 modifications had similar target-binding affinities to previously isolated unmodified RNA19 and DNA5 aptamers.
The gradual accumulation of knowledge and improvements in the SELEX process,20,21 such as library design and selection strategies, have facilitated the more efficient isolation of high-quality aptamers for small-molecule targets.22–26 This includes aptamers for challenging “low-epitope” targets that are at both ends of the water solubility spectrum, such as steroid hormones22 and glucose.23 These outcomes suggest that, given a properly designed selection strategy, natural DNA and RNA aptamers are capable of effectively binding small-molecule targets regardless of their physicochemical properties. Here, we present findings that further support this notion by isolating aptamers from unmodified libraries that bind to natural and synthetic cannabinoids, which are considered challenging targets because of their low water solubility and limited moieties for aptamer binding. We first isolated a new DNA aptamer that binds THC with an equilibrium dissociation constant (KD) of ~50 nM and high specificity despite the previously described inaccessibility of this target to natural DNA libraries.18 We used this aptamer to develop a colorimetric assay that can detect THC with a limit of detection (LOD) of 250 nM within seconds with no response to structurally similar interferents as well as other illicit drugs. We then isolated two new aptamers that bind cross-reactively to UR-144 and XLR-11, synthetic cannabinoids that exhibit poorer water solubility than THC and have even fewer moieties for binding. After three SELEX trials, we were ultimately able to isolate aptamers with nanomolar affinity and high specificity for these targets. Our results demonstrated the considerable impact that well-chosen selection strategies and conditions have on the outcome of SELEX experiments and should provide greater confidence in the utility of natural nucleic acid libraries for small-molecule targets. Given that unmodified aptamers can be isolated and synthesized at low cost and engineered with greater ease compared to base-modified aptamers, our findings should greatly broaden the applicability of aptamers for sensitive and low-cost small-molecule detection.
EXPERIMENTAL SECTION
SELEX Procedure.
All DNA oligonucleotides were synthesized and HPLC-purified by Integrated DNA Technologies (IDT). Sequences used in this work are listed in Supporting Information (SI), Table S1. DNA was dissolved in PCR grade water before use, and concentrations were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). For SELEX, aptamers were isolated from a library of 73-nt DNA oligonucleotides, comprising a stem-loop structure with a loop of 30 random nucleotides. Conventional SELEX, counter-SELEX, and parallel and serial selection were carried out based on a previously reported library-immobilized SELEX protocol.25,27 Detailed selection conditions (e.g., amount of library strands, the concentration of target or interferent, and concentration of organic co-solvent) for the THC and synthetic cannabinoid SELEX experiments are provided in Tables S2 and S3, respectively. All selections were performed in selection buffer (20 mM Tris-HCl, 0.5 mM MgCl2, 20 mM NaCl, pH 7.4) unless stated otherwise. Ten 250 μL wash volumes of selection buffer were applied before counter-SELEX, and 30 such wash volumes were applied before target elution; for all washes, selection buffer was continually flowed through the column. Aptamer sequence identification for THC and synthetic cannabinoid-binding aptamers was performed by Sanger sequencing and high-throughput sequencing, respectively. Urine and saliva samples were collected from consenting individuals with approval from the Institutional Review Board of Florida International University (FIU IRB: IRB-13–0320). Samples were collected from three males and three females, pooled, and then filtered with a 0.22 μm filter (Millipore). These were then diluted 50% with 2× selection buffer and used for counter-SELEX.
RESULTS AND DISCUSSION
Isolation of a THC-Binding DNA Aptamer.
THC is the main psychoactive component of the plant Cannabis sativa.28 The detection of this compound in complex samples such as plant matter, saliva, and urine is important for a variety of applications, and thus there is a need for bioreceptors for sensor development. THC is a challenging target for SELEX. For one, it has low water solubility, setting a severe limitation on the maximum target concentration that can be used during SELEX. Furthermore, THC has no charge, and very few functional groups capable of forming strong interactions with nucleic acids: bare cyclohexenyl and tetrahydropyran rings, a phenolic group, and an alkyl tail. We were interested in investigating whether the challenges faced by the Mayer group18 represented an inherent limitation of natural libraries for isolating aptamers for THC and, in parallel, determining the influence that selection conditions have on the outcome of SELEX for small-molecule targets.
We used a 73-nt stem-loop structured library containing 30 random nucleotides (SI, Table S1),25 in a library-immobilized SELEX strategy25,27 for aptamer isolation. This selection procedure has successfully yielded nanomolar-affinity aptamers for various small molecules.22–25 This is in contrast to the approaches previously used by the Mayer group,18 in which the target was immobilized on microbeads and the aptamers were free in solution. We believe that our approach using the target in its native state potentially increases the likelihood of finding aptamers because the entirety of the molecule can freely interact with the oligonucleotides. Additionally, 2.6% methanol was included in the selection buffer as a co-solvent to completely dissolve THC. In total, we performed 11 rounds of selection (Supporting Information, Table S2). In the first eight rounds, we used high quantities of target to ensure enrichment of all strands binding to THC. We then increased the selection stringency by reducing the amount of target in rounds 9–11 by two fold to isolate high-affinity aptamers. Cannabis is typically consumed as dried plant material or crude extracts but may also be blended with other herbs such as peppermint, damiana, lemon balm, and thyme for smoking. To remove non-specific aptamers that bind to endogenous compounds in these nontarget plants, we performed counter-SELEX29 from rounds 2–11 against methanol extracts of these herbs. From rounds 3–11, we also performed counter-SELEX against pharmaceutical and illicit drugs (cocaine, clonazepam, methamphetamine, amphetamine, pentylone, α-pyrrolidinopentiophenone (α-PVP), and ibuprofen) and common drug adulterants (procaine, acetaminophen, and pseudoephedrine). This was done to enrich highly specific aptamers for THC. The amount of counter-target used in each round was gradually increased to enhance the stringency of counter-SELEX. Finally, in round 11, we included 50% urine and 50% saliva in the counter-SELEX process to remove sequences that bind interferents in these biomatrices (SI, Table S2).
After eight rounds of selection, we used a gel elution assay30 to evaluate the target-binding affinity of the enriched pool and found that the KD of the pool toward THC was 3.7 μM and 27% of the library eluted with 100 μM target (SI, Figure S1A). The fraction of target-eluted library increased in round 9 to 52%, 61% in round 10, and 59% in round 11 (SI, Figure S2A), while the KD of these pools did not significantly differ from the round 8 pool (SI, Figures S1B,C and S2B), indicating that high-affinity aptamers had been sufficiently enriched. We also used the same gel elution assay to characterize the specificity of the round 11 pool (SI, Figure S2C). No meaningful elution was observed with 200 μM ibuprofen or clonazepam; 500 μM cocaine, methcathinone, procaine, pseudoephedrine, acetaminophen, amphetamine, pentylone, or α-PVP; or 0.16 mg/mL peppermint, damiana, lemon balm, or thyme extract relative to the low levels of elution seen in a buffer-only control. These results demonstrated that the enriched aptamer pool had high affinity and specificity for THC. Notably, the pool was also cross-reactive to (−)-11-nor-9-carboxy-Δ9-tetrahydrocannabinol (THC-COOH), the major metabolite of THC.28 This is actually advantageous as it enables the evaluation of marijuana consumption by detecting THC-COOH in urine. Simultaneous detection of THC and its metabolites also increases the assay sensitivity and detection window in saliva.
Aptamer Sequencing and Characterization.
The results above indicated that a majority of the oligonucleotides in the round 11 pool bound to THC. We therefore cloned this pool and performed Sanger sequencing of 44 clones. As expected, the pool had very low diversity as 30 of these clones (68%) shared the same consensus sequence (Figure 1A and SI, Table S4). We truncated the 15- and 12-nt primer-binding sites from the 5’ and 3’ ends of this consensus sequence to produce the 47-nt aptamer termed THC1.2. During sequencing analysis, we noticed that this aptamer has a 31-nt rather than a 30-nt binding domain; this is most likely due to a nucleotide insertion in the random domain during SELEX. Like the aptamer isolated by the Mayer group,18 THC1.2 is G-rich, having 60% G-content in the putative binding domain. Based solely on Watson–Crick base pairing rules, THC1.2 has a 10-bp stem and a 27-nt loop (SI, Figure S3). We used circular dichroism to determine the actual secondary structure of the aptamer. The aptamer’s circular dichroism spectrum consisted of a negative peak at 240 nm and a broad positive peak from 260 to 280 nm (SI, Figure S4), which could correspond to a structure comprising a mixture of double-stranded B-form DNA and a parallel G-quadruplex.31,32 Upon addition of THC, the shoulder of the positive peak decreased, and new negative peaks formed at 205 and ~290 nm (SI, Figure S4), indicating a B- to Z-DNA transition.31
Figure 1.
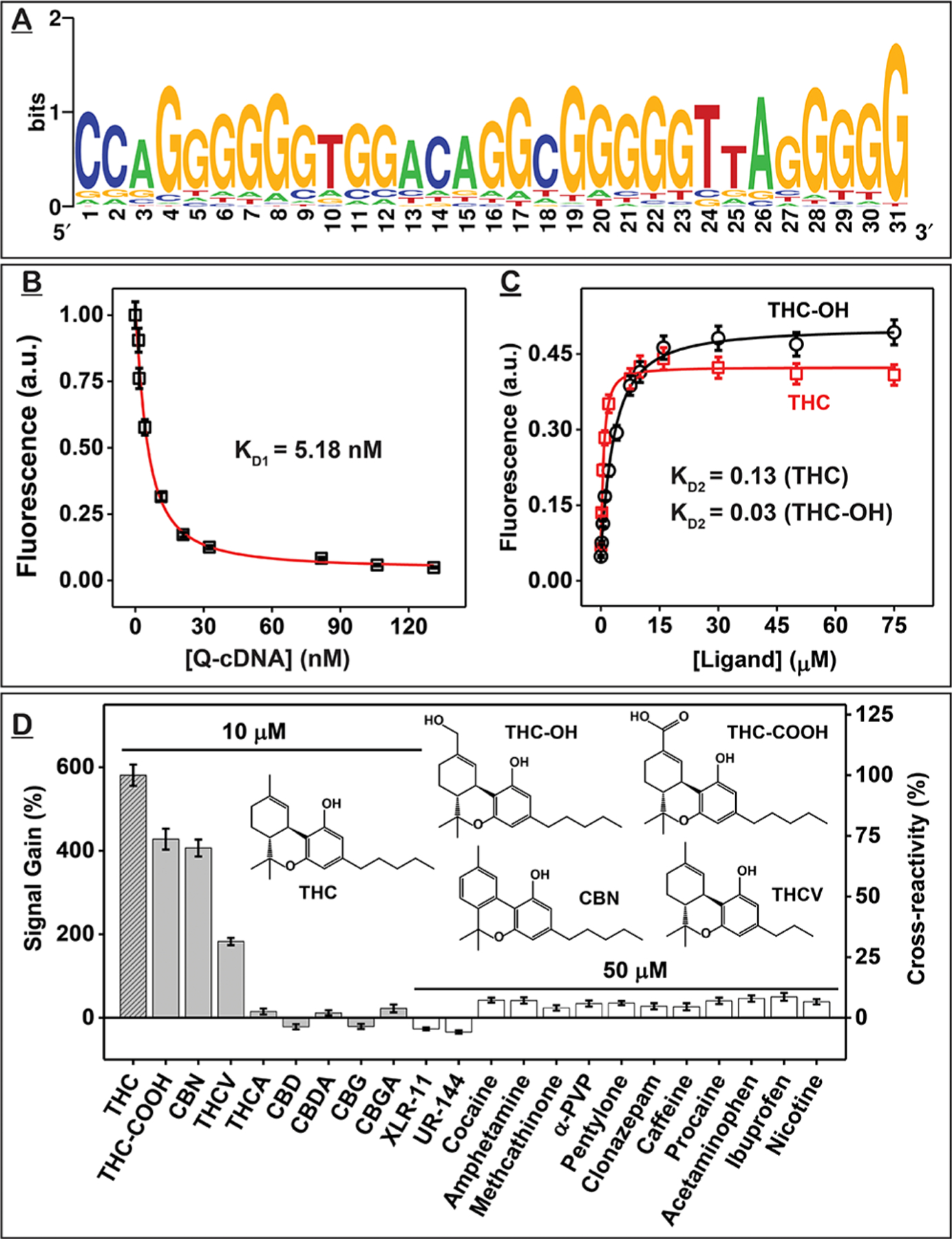
Identification and characterization of THC-binding aptamer THC1.2. (A) Sequence logo for the 44 clones obtained from the round 11 pool showing the nucleotide diversity at each position of the random domain. A larger font size represents higher frequency. (B–D) Strand-displacement fluorescence assay for determining the affinity and specificity of THC1.2. (B) KD1 was determined by titrating different concentrations of Iowa Black RQ-labeled complementary DNA strand (Q-cDNA) into Cy5-labeled THC1.2 (F-THC1.2) and measuring fluorescence quenching at 668 nm. (C) KD2 was determined from fluorescence recovery at 668 nm of F-THC1.2–Q-cDNA complexes combined with varying concentrations of THC or THC-OH. (D) Signal gains produced by cannabinoids and interferents and their cross-reactivity relative to THC. Error bars represent the standard deviation of measurements from three individual experiments.
We then used isothermal titration calorimetry (ITC), a gold standard method for measuring the affinity between aptamers and their targets,33 to characterize THC1.2. The standard protocol for ITC entails titrating a relatively large quantity of small-molecule ligand into a lower concentrations of receptor. Cannabinoids such as THC typically have very low water solubility, whereas the aptamer has high solubility. Therefore, we performed a “reverse” titration by titrating higher concentrations of THC1.2 into lower concentrations of cannabinoid targets (SI, Table S5). Using a single-site binding model, ITC showed that THC1.2 binds to THC with a KD of 61 ± 25 nM. We determined that the aptamer also binds two major metabolites of THC, 11-hydroxy-THC (THC-OH) and THC-COOH, with KDs of 556 ± 64 and 180 ± 36 nM, respectively (SI, Figure S5). Based on our ITC data, binding of THC1.2 to THC, THC-OH, or THC-COOH is solely enthalpy-driven (SI, Table S6). This is observed for most ligand–receptor interactions.
We noticed that the binding stoichiometries obtained from these ITC experiments did not equal 1, which is possibly due to self-association of THC1.2 (a G-rich aptamer) at high concentrations. We therefore used a previously reported strand-displacement fluorescence assay25 to verify that the binding affinities of THC1.2 determined via ITC were accurate. We first determined KD1, the affinity of the Cy5-labeled aptamer (F-THC1.2) for an Iowa Black RQ-labeled complementary DNA strand (Q-cDNA), and then measured KD2, which reflects the affinity of the aptamer-cDNA complex for THC, THC-OH, or THC-COOH. The KD of THC1.2 for each ligand was then calculated based on KD1/KD2. We measured KD1 by titrating different concentrations of Q-cDNA into a solution containing a fixed concentration of F-THC1.2. When F-THC1.2 is free in solution, it emits strong fluorescence (SI, Figure S6A); hybridization with Q-cDNA brings the quencher into close proximity to the fluorophore, greatly decreasing fluorescence (SI, Figure S6B). Based on the titration curve, we obtained a KD1 of 5.18 nM (Figure 1B). We then titrated the various concentrations of the targets into solutions of F-THC1.2-Q-cDNA complexes. Target binding to the aptamer induces dissociation of Q-cDNA, resulting in recovery of fluorescence (SI, Figure S6C). We measured KD2s of 0.13, 0.03, and 0.04 for THC, THC-OH, and THC-COOH, respectively, which in turn yielded KD measurements of 40, 173, and 130 nM (Figure 1C and SI, Figure S7). These results confirm that THC1.2 binds tightly to THC and its metabolites with nanomolar affinity. Notably, our aptamer achieves affinity that is at least three-fold better than the previously reported benzyl-modified aptamer, C11.41 (KD = 113 nM) and also two-fold than its naphthalene-modified version (KD = 88 nM).18
We used the same strand-displacement fluorescence assay to characterize the specificity of THC1.2. We first assessed the aptamer’s cross-reactivity to other major cannabinoids (SI, Figure S8), including THC-COOH, tetrahydrocannabinolic acid (THCA), tetrahydrocannabivarin (THCV), cannabigerolic acid (CBGA), cannabidiolic acid (CBDA), cannabigerol (CBG), cannabidiol (CBD), and cannabinol (CBN) at a concentration of 10 μM. THC1.2 was cross-reactive to THC-COOH, CBN, and THCV but did not bind to other cannabinoids (Figure 1D). Structure—cross-reactivity relationships showed that adding substituents to the aromatic ring (e.g., THCA) or opening of the ether group (e.g., CBD, CBG) completely impaired aptamer recognition, which implies that these functional groups are involved in aptamer binding. This suggests a different binding mechanism from C11.41, which tolerates alterations to the aromatic ring. For THC1.2, shortening of the alkyl tail (e.g., THCV) resulted in a great reduction in affinity, implying that the tail is involved in binding. On the other hand, modification of the cyclohexene ring (i.e., CBN) only moderately reduced binding affinity, indicating less involvement of this region.
We also challenged THC1.2 with another 15 interferents in the strand-displacement fluorescence assay, including common illicit drugs (XLR-11, UR-144, cocaine, methamphetamine, amphetamine, methcathinone, α-PVP, and pentylone), pharmaceuticals (clonazepam, pseudoephedrine, acetaminophen, and ibuprofen), cutting agents (caffeine and procaine), and nicotine. All interferents showed less than 10% cross-reactivity at a 5-fold higher concentration relative to 10 μM THC (Figure 1D). Notably, the synthetic cannabinoids XLR-11 and UR-144, which also bind to human cannabinoid receptors CB1 and CB2,34 do not bind to THC1.2, indicating a different recognition mechanism for this aptamer compared with these receptors. These results clearly demonstrate that aptamers with strong affinity and excellent specificity for targets as challenging as THC can be successfully isolated from unmodified DNA libraries with well-designed SELEX conditions. Using the strand-displacement fluorescence assay, we observed that both THC and THC-COOH can be easily detected at a concentration of 200 nM (SI, Figure S9). Given that the concentration of THC in oral fluids is usually above 300 nM within 2 h of consumption, and THC-COOH can be detected in urine at 200 nM many hours after consumption, these results suggest that THC1.2 could be used for on-site detection of recent cannabis use in both urine and saliva samples.28
Detection of THC Using a Dye-Displacement Assay.
After confirming the high affinity and specificity of THC1.2, we developed a dye-displacement assay for rapid colorimetric detection of THC and THC-COOH. We found that the DNA-binding dye 3,3-di(3-sulfopropyl)−4,5,4,5-dibenzo-9-ethylthia-carbocyanine (ETC)35 can bind to THC1.2 as a monomer with absorption maxima at 575 nm. Binding of THC to the aptamer–dye complex displaced the dye from the aptamer, resulting in J-aggregates that absorb at 660 nm. Based on the difference between the area under the curve of the monomer (500–620 nm) and aggregate peak (620–680 nm) with and without target (Figure 2A), we were able to quantify THC concentrations in the range of 0.25–20 μM spectroscopically (Figure 2B).
Figure 2.
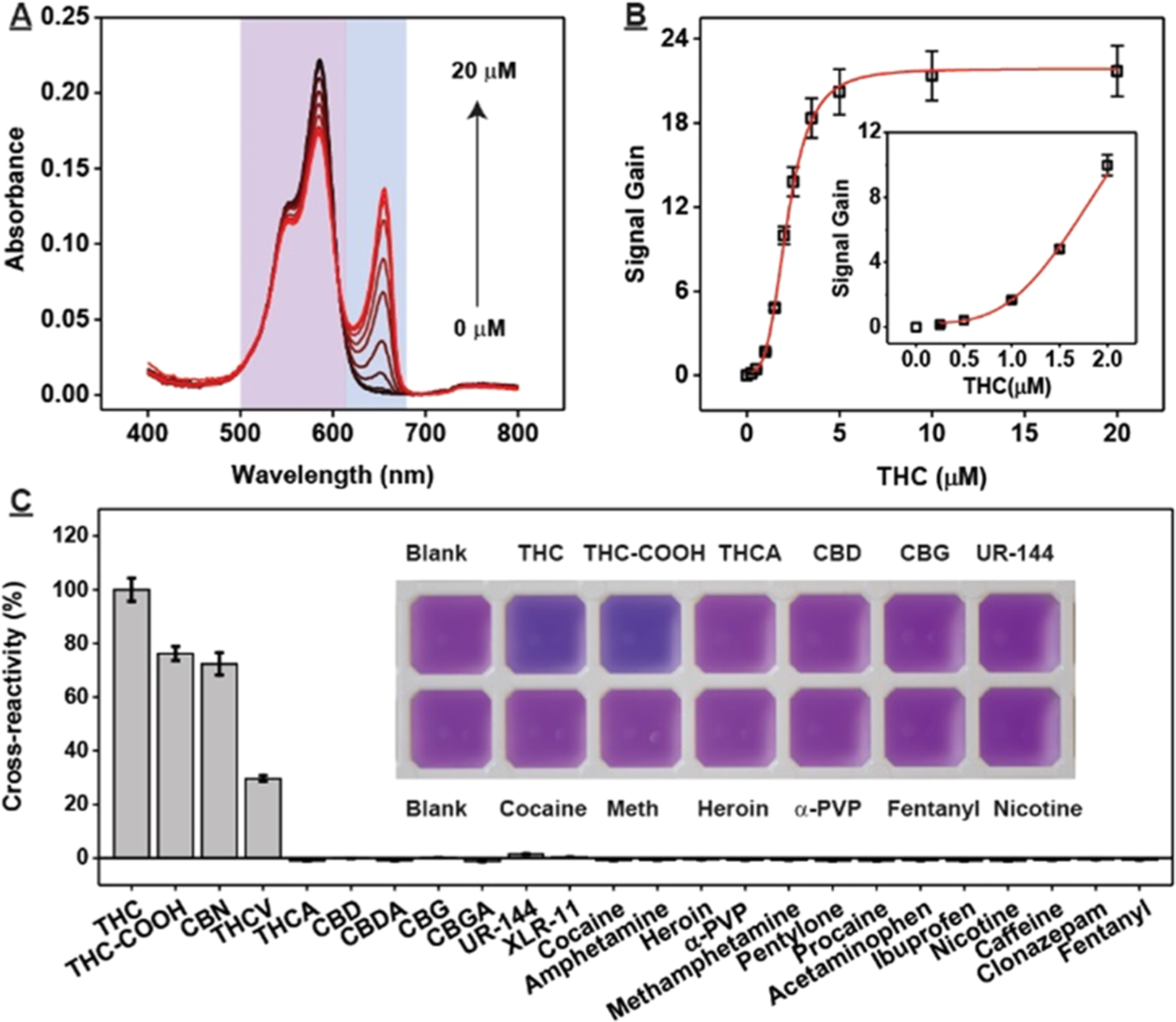
Dye-displacement assay for colorimetric detection of THC. (A) Spectra of ETC at various concentrations of THC (0, 0.25, 0.5, 1, 1.5, 2, 2.5, 3.5, 5, 10, and 20 μM) and (B) corresponding calibration curves. Inset shows response at low concentrations of target. (C) Cross-reactivity of 5 μM THC-COOH, CBN and THCA; 25 μM CBD, CBDA, CBG, CBGA, UR-144, and XLR-11; and other small-molecule drugs at a concentration of 100 μM. Cross-reactivity was calculated relative to the signal gain produced by 5 μM THC. Inset shows photographs of the assay for selected specificity tests. Error bars represent the standard deviation of measurements from three individual experiments. Abbreviation: α-PVP = α-pyrrolidinopentiophenone, Meth = methamphetamine.
This dye-displacement assay also displayed high specificity against structurally similar interferents including THCA, CBD, and CBG at 5-fold higher concentrations relative to THC, and other drugs and adulterants like cocaine, amphetamine, fentanyl, heroin, procaine, nicotine, and caffeine at 20-fold higher concentrations (Figure 2C). Importantly, THC and THC-COOH can be detected at concentrations as low as 3 μM via a purple-to-blue color change visible to the naked eye, with no such change from any tested interferents (Figure 2C). Given the high sensitivity and specificity of this assay as well as its ease of use and second-scale turnaround time, we believe that it would be highly amenable for on-site THC and THC-COOH detection.
Isolation of DNA Aptamers that Bind to both UR-144 and XLR-11.
To further probe the capabilities of natural DNA libraries, we isolated an aptamer binding to UR-144 and XLR-11, two widely abused synthetic cannabinoids.36 As newly emerging designer drugs, there are presently no affinity reagents that recognize these targets. Structurally, UR-144 and XLR-11 are similar; both consist of an indole ring, ketone, pentyl tail, and tetramethylcyclopropyl ring. To our knowledge, no aptamer has been isolated for any synthetic cannabinoids.
We used the same library design and library-immobilized SELEX technique described above with our recently reported parallel-and-serial selection strategy24 (inspired by the toggle-SELEX strategy)37 to isolate an aptamer capable of binding both UR-144 and XLR-11. First, we performed SELEX in parallel to individually enrich binders for each target. These parallel pools were then combined for serial selection, and the pool was challenged with each target consecutively to isolate aptamers capable of binding both targets. Overall, we needed to perform three independent SELEX experiments using various selection conditions and strategies to isolate aptamers with high affinity and specificity for both targets.
In the first approach, six rounds of parallel selection were performed with UR-144 and XLR-11 (SI, Figure S10, RD 1.1–RD 6.1). Since our targets have extremely low water solubility but high solubility in DMSO (>100 mM), we included 5% DMSO and 0.05% (v/v) Tween 20 to increase their solubility in the selection buffer. In the first round, we employed low selection stringency with a large library (1 nmol) and relatively high amount of target (375 nmol) to prevent loss of potential aptamers. As SELEX progressed, the quantity of library and target were gradually reduced to enrich high-affinity aptamers. Additionally, to enhance aptamer specificity, counter-SELEX was performed from the second round onward against plant extracts (leaves of damiana, lemon balm, mint, and thyme), commonly used drugs (nicotine, caffeine, cocaine, methamphetamine, α-PVP, and THC), and structurally similar compounds (serotonin, tryptophan, tocopherol, albendazole, and granisetron). Unfortunately, no specific target elution was observed after six rounds of parallel selection. Given that the concentration of Tween 20 in the buffer was above the critical micelle concentration (0.007% v/v),38 we hypothesized that target molecules may have been trapped in micelles, preventing their interaction with library strands. This surfactant may have also increased non-specific elution of library strands, reducing selection efficiency.
For the second approach, we removed Tween 20 from the selection buffer and performed a similar parallel SELEX strategy with minor modifications (SI, Figure S10, RD 1.2–RD 8.2). After four rounds of parallel selection, we observed considerable target elution in both pools. This confirmed that the presence of Tween 20 precluded the enrichment of aptamers binding to our targets. We combined these two pools (SI, Figure S10, RD 4.2) and characterized their target-binding affinity using the gel elution assay. This combined pool had high affinity for XLR-11 (KD = 49 μM) (SI, Figure S11A), high cross-reactivity to UR-144 (86%), and no cross-reactivity to most interferents (SI, Figure S11B). However, it still showed moderate cross-reactivity to THC (18%) and significant cross-reactivity to the plant extracts, and so we used more stringent counter-SELEX conditions in subsequent rounds. From the fifth round, we performed serial selection with the combined pool (SI, Figure S10, RD 5.2–RD 8.2) to rapidly isolate aptamers that recognizes both targets. Specifically, we performed selection with UR-144 in rounds 5.2 and 7.2 and XLR-11 in rounds 6.2 and 8.2. We used the gel elution assay on the round 8.2 pool to measure an affinity of 51 μM for XLR-11, with 107% cross-reactivity to UR-144 (SI, Figure S12A). However, the pool exhibited far stronger cross-reactivity to THC (129%) than for the selection targets themselves.
To remedy this specificity problem, we pursued a third approach in which we restarted serial selection from the round 4 combined pool (SI, Figure S10, RD 5.3–RD 8.3) with more stringent counter-SELEX and 4-fold higher concentrations of THC. The number of washes during counter-SELEX was increased from 3 to 8 in rounds 5 and 6 and 16 in rounds 7 and 8 to remove interferent binders. In addition, for rounds 6–8, we used a pre-treatment strategy in which we incubated the library with THC overnight prior to bead immobilization. We anticipated that THC-bound strands would be unable to duplex with the cDNA employed for the immobilization and thus would be excluded from the SELEX process. We performed a gel elution assay to evaluate the affinity and specificity of the round 8 (RD 8.3) pool, which bound to both XLR-11 (KD = 40 μM) and UR-144 (with 82% cross-reactivity); but even with extremely stringent counter-SELEX against THC, the pool still bound to this interferent with even greater cross-reactivity (114%) (SI, Figure S12B).
We hypothesized that THC was being retained in the column even after extensive washing with buffer due to its high hydrophobicity and viscosity, leading to inadvertent enrichment of THC-binding sequences. To address this, we ceased performing counter-SELEX against THC in subsequent rounds of selection (SI, Figure S10, RD 9.3–RD 17.3). We also used XLR-11 as the sole target from this point forward since the round 8 pool cross-reacted strongly to both targets. Over the next several rounds of selection, XLR-11 elution and affinity of the pool improved while retaining high cross-reactivity to UR-144 (SI, Figure S13A–C). Importantly, cross-reactivity to THC greatly decreased (SI, Figure S13D), confirming our hypothesis regarding THC contamination. Pool elution did not further improve by round 17, and we therefore characterized the target affinity and specificity of this pool. We observed a high XLR-11-binding affinity (KD = 6.2 μM), with a high cross-reactivity to UR-144 (101%) and high specificity against all interferents, including THC (15% cross-reactivity) (SI, Figure S12C).
Identification and Characterization of XA1 and XA2.
Due to the low target-elution percentage of the final round’s pool (~35%), we performed high-throughput sequencing of the round 16 and 17 pools to identify aptamer sequences. The round 16 and 17 pools respectively contained 90,341 and 93,680 unique sequences, out of 681,673 and 526,186 total reads. Cutadapt was used to remove primer-binding sites from the sequences for subsequent clustering .39 Two highly abundant sequences were identified (Figure 3A). The most popular sequence, XA1, composed 40% of the round 16 pool and was further enriched to 50% of the round 17 pool. The other sequence (XA2) comprised 38% of the round 16 pool but was reduced to 23% abundance in the round 17 pool.
Figure 3.
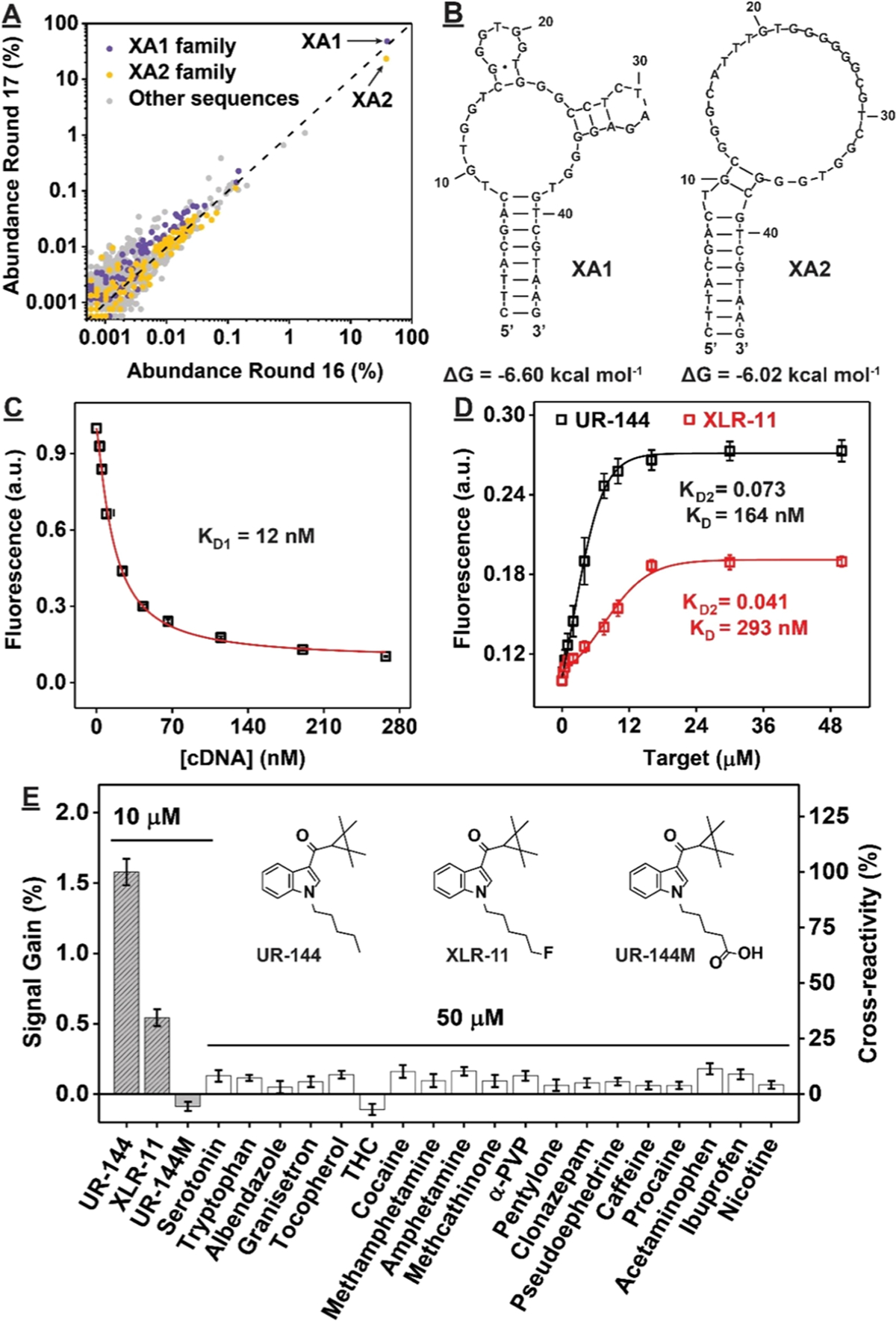
Identification and characterization of synthetic cannabinoid aptamers XA1 and XA2. (A) Population of sequences in the XA1 family (purple), XA2 family (yellow), and other sequences (gray) in the round 16 and 17 pools based on high-throughput sequencing. (B) Secondary structures of XA1 and XA2, and their free energy as predicted by Mfold.41 (C, D) Affinity and (E) specificity of XA1 as determined by strand-displacement fluorescence assay. Bar plot shows signal gains produced by different compounds and their cross-reactivity relative to UR-144. Inset shows structures of UR-144, XLR-11, and the metabolite UR-144 pentanoic acid (UR-144M).
We removed the primer binding sites of these sequences by truncating the 15- and 12-nt from the 5’ and 3’ ends of XA1 and XA2 to form 46-nt aptamers with 8-bp stems (Figure 3B) and analyzed their structure via circular dichroism. The spectrum of XA1 included a positive peak at 215 nm, a negative peak spanning from 240 to 260 nm, and a broad positive band at 265–300 nm with a peak at ~280 nm. This indicated that the aptamer contains an antiparallel G-quadruplex, based on the 210 and 255 nm peaks, and B- form duplex DNA, based on the 265–300 nm band.31,32 Upon the addition of XLR-11 or UR-144, a slight shift was observed for the negative band from 255 to 250 nm (SI, Figure S14A) as well as an increase in the positive 210 nm peak, which suggests that conformational changes primarily occur in the G-quadruplex region upon target binding. The spectrum for XA2 included a positive peak at 215 nm, a negative band consisting of peaks at 234 and 255 nm joined by a 240 nm shoulder, and a broad positive band spanning 265–315 nm, centered at 290 nm. This indicated the presence of an antiparallel G-quadruplex, based on the peaks at 210, 255, and 290 nm peaks, and B-form duplex DNA, based on the 265–315 nm band.31,32 Upon the addition of XLR-11 or UR-144, a slight shift was observed for the negative band from 255 to 254 nm and the positive band at 290–288 nm, with an increase in intensity, (SI, Figure S14B). This suggests that the targets induce formation of B-form duplex DNA while still retaining the G-quadruplex domain.
We then characterized the affinity of XA1 and XA2 for XLR-11 and UR-144 using ITC (SI, Table S5). XA1 bound tightly to XLR-11 and UR-144, with a KD of 310 ± 70 and 127 ± 32 nM, respectively (SI, Figure S15A,B). XA2 also bound strongly to both targets, albeit with slightly lower affinities, with KDs of 394 ± 93 and 170 ± 44 nM for XLR-11 and UR-144, respectively (SI, Figure S15C,D). The binding of XA1 to XLR-11 or UR-144 is solely enthalpy-driven, with a decrease in entropy. However, the binding of XA2 to XLR-11 and UR-144 is different in that it is both enthalpy- and entropy-driven, which highlighted the hydrophobic interaction between XA2 and the targets (SI, Table S6). We subsequently used the above-described strand-displacement fluorescence assay to further characterize the affinity and specificity of XA1. The results were consistent with the ITC data, with KDs of 293 and 164 nM for XLR-11 and UR-144, respectively (Figure 3C,D). Due to the high affinity of the aptamer, we could easily detect XLR-11 or UR-144 at concentrations as low as 200 nM (SI, Figure S16). This LOD is similar to or higher than the KD of the aptamer due to competition between the cDNA and the target for binding to the aptamer.30 Although XA1 binds UR- 144 with submicromolar affinity, it generated no response to the putative metabolite UR-144 N-pentanoic acid40 (Figure 3E, UR-144M). This means that the addition of just a single carboxylate group to the alkyl tail of the target impairs binding, highlighting the excellent specificity of our aptamer. XA1 showed minimal cross-reactivity to all other interferents tested, even at a 5-fold higher concentration than the targets.
CONCLUSIONS
Many aptamers have been isolated for a variety of small-molecule targets, but a majority of them are unsuitable for real-world applications because of their poor binding affinities and specificities. This has fueled speculation that natural DNA or RNA aptamers may be inadequate as bioreceptors, and that chemically modified bases might be required to improve their performance. However, several recent studies have suggested that well-designed SELEX experiments with natural DNA libraries may indeed be sufficient for isolating high-quality small-molecule binding aptamers, even for “challenging” targets. Here, we have formally confirmed this notion by isolating aptamers binding to cannabinoids including THC, UR-144, and XLR-11 from natural DNA libraries. These targets are considered challenging because they are highly hydrophobic and contain very few functional groups to mediate recognition by aptamers. Indeed, a previous report suggested that it was not possible to isolate THC-binding aptamers from natural nucleic acid libraries.18 However, we were able to isolate THC aptamers from a natural DNA library in a single SELEX experiment by employing alternative selection conditions and strategies. Importantly, our aptamer has three-fold better affinity for THC compared to a recently-isolated base-modified aptamer18 and exhibits high specificity against many structurally similar interferents. Both aptamers are G-rich and form a parallel G-quadruplex structure, but their sequences are different. They most likely have different binding mechanisms as the aptamer reported by the Mayer group requires the benzyl-modified deoxyuridine base to bind to THC and has a KD of 117 nM. In contrast, our aptamer contains only natural nucleotides and binds THC with a KD of 61 nM (ITC) or 40 nM (strand-displacement fluorescence assay). Changing the benzyl-modified uridine to a naphthyl-modified uridine further improved the THC-binding affinity of the Mayer group’s aptamer (KD = 88 nM), but our natural DNA aptamer still exhibits slightly better affinity. One possible reason is that we employed a different selection strategy and conditions than the Mayer group.18 We also isolated new DNA aptamers that bind to the synthetic cannabinoids XLR-11 and UR-144 with high affinity and specificity. Over the course of three independent SELEX experiments, we determined that selection strategies and conditions profoundly affect the success or failure of a selection as well as the quality of the isolated aptamers. In our screening process, we adjusted conditions such as buffer components and selection stringency as well as the use of screening strategies like counter-SELEX and parallel-and serial-selection. This is in keeping with previous reports—for example, Stojanovic and co-workers demonstrated that by switching their library design from an N8 three-way junction42 to an N30 stem-loop and applying a more stringent selection strategy,22 they could isolate aptamers with greatly improved affinity and specificity for steroid hormones. We have also previously observed substantial improvements in aptamer target cross-reactivity and specificity by changing library design and selection strategies.24,30 For in vivo studies, our DNA aptamer would encounter stability issues due to its susceptibility to digestion by endogenous nucleases. However, we do not foresee any issues for in vitro detection of cannabinoids in plant extracts or biosamples such as saliva, urine, and serum given that assays typically require relatively miniscule sample exposure times.
Through this effort, we have learned several important pieces of information for isolating high-quality aptamers for small molecules. Prior to initiating SELEX, it is crucial to identify the application of the aptamer, as this will help establish the necessary target specificity, intended analytical matrix, and potential interferents. The selection conditions should ideally mirror those of the chosen matrix since the structure (and thus the function) of aptamers are sensitive to physicochemical conditions. Another important consideration is the solubility of the target molecule. For targets with low water solubility, organic co-solvents should be included in the selection buffer to ensure complete target dissolution. For our cannabinoid targets, it was crucial to include small amounts of methanol or DMSO in our selection buffers. In terms of the selection strategy, if highly target-specific aptamers are desired, it is important to perform counter-SELEX against interferents likely to be present in the intended sample matrix. We generally try to include as many counter-targets as we feasibly can—for example, our counter-SELEX regime for the THC aptamer selection included five illicit drugs, five adulterants, four different plant extracts, and human saliva and urine. However, if cross-reactive aptamers for a family of structurally similar small molecules are desired, we suggest a parallel-and-serial SELEX strategy like the one used in our synthetic cannabinoid selection. However, this should still be supplemented with a well-designed counter-SELEX procedure. With regard to the design of the library, we have generally had successful outcomes using the library-immobilized SELEX process with the stem-loop structured 30N DNA library designed by Stojanovic and co-workers.25 This approach eliminates the challenges associated with immobilizing small-molecule targets and allows the aptamer to interact with the native target, which, we believe, promotes the isolation of high-affinity aptamers. Furthermore, examination of the of myriads of SELEX experiments and aptamers isolated for small molecules over the past few decades have revealed that libraries containing between 30 and 50 randomized nucleotides are sufficient for isolating high-quality aptamers.43
It is crucial to constantly monitor the selection process using a simple and reliable assay such as our gel elution assay.30 We recommend extensive evaluation of the affinity and specificity of enriched pools once an initial uptick in target elution is observed. Early pool binding affinity (typically Kd 100–1000 μM can be observed after ~8 rounds) and levels of target elution can be rather low, but they are important indicators of whether the selection conditions and strategies are allowing target-binding aptamers to be enriched, as well as how to fine-tune these conditions if needed. For instance, data from our gel elution assay helped us make sound judgments on when to decrease target concentrations to promote the enrichment of high-affinity aptamers. As another example, we were able to determine based on the lack of XLR-11/UR-144 pool binding affinity in our first SELEX trial that Tween 20 should be eliminated from the selection buffer. The specificity and cross-reactivity of the pool also reveals whether the employed selection strategies are appropriately designed. If the level of cross-reactivity is excessive or insufficient, it is advisable to restart SELEX with an adjusted selection strategy. For example, in our second SELEX trial for XLR-11/UR-144, we identified unwanted cross-reactivity to THC that prompted us to reexamine our selection strategies—first by increasing the concentration of THC and then eventually (and counterintuitively) removing it over the course of our third SELEX trial. Once the pool has the desired binding profile, SELEX can be continued until the percentage of oligonucleotides eluted by the target saturates. At this point, we recommend performing one last thorough examination of the binding affinity and specificity of the enriched pool. We choose to sequence pools once they have KD <100 μM and relatively low cross-reactivity to the counter-targets (similar elution as buffer). Sanger sequencing is useful when target elution in the final pool is high (>40%), but when target elution is lower, high-throughput sequencing and bioinformatics might be required. Based on these determinations, we have formulated an algorithm that should serve as a useful guide for those aiming to isolate high-quality aptamers for small molecules using SELEX (SI, Figure S17). Given the cost and complexity associated with working with chemically modified nucleotides and engineered polymerases, we believe that this workflow will create opportunities for the more efficient and cost-effective development of high-quality aptamers for small-molecule targets in a range of applications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute of Justice, Grant Office of Justice Programs, U.S. Department of Justice award 2015-R2-CX-0034, and the National Institutes of Health, National Institute on Drug Abuse R21DA045334-01A1.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c04592.
Materials, high-throughput sequencing; gel elution assay; isothermal titration calorimetry(ITC); fluorophore-quencher assay; dye displacement assay for detection of THC; DNA sequences used in this work; SELEX conditions for isolating a THC-binding aptamer; experimental conditions for our three UR-144 and XLR-11 SELEX experiments; sequences of the random region and counts for clones from the round 11 THC SELEX pool; experimental parameters for ITC experiments and experimental results of ITC; THC affinity of the pools from rounds 8, 9, and 10 as determined by a gel elution assay; characterization of SELEX THC pools using a gel elution assay; secondary structure of THC1.2 and free energy; circular dichroism spectra of THC1.2 in the absence and presence of THC; characterization of the ligand-binding affinity of THC1.2 using ITC; schematic of the strand-displacement fluorescence assay used for determining the THC-binding affinity of THC1.2; KD of THC1.2 for THC-COOH based on a strand-displacement fluorescence assay; chemical structures of different cannabinoids, with substituents relative to THC; calibration curve of THC1.2 for THC and THC-COOH based on the strand-displacement fluorescence assay; three SELEX approaches for isolating an aptamer that binds UR-144 and XLR-11; characterization of the combined round 4.2 pool after four rounds of parallel selection for XLR-11; characterization of pool affinity and specificity for XLR-11 and UR-144 using a gel elution assay; selection progress for isolating an XLR-11/UR-144-binding aptamer; circular dichroism spectra of XLR-11 binding aptamers XA1 and XA2 in the absence and presence of 10 μM XLR-11 or UR-144; characterization of target-binding affinity of XA1 and XA2 using ITC; calibration curve of XA1 for XLR-11 and UR-144 for the strand-displacement fluorescence assay; and an algorithm to guide SELEX isolation of high-quality aptamers for small molecules (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c04592
The authors declare no competing financial interest.
Contributor Information
Haixiang Yu, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States.
Yingping Luo, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States; State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, Nanjing 211198, P. R. China.
Boyang Yu, State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, Nanjing 211198, P. R. China.
Yi Xiao, Department of Chemistry and Biochemistry, Florida International University, Miami, Florida 33199, United States;.
REFERENCES
- (1).Tuerk C; Gold L Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (2).Ellington AD; Szostak JW Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- (3).Zhou J; Rossi J Nat. Rev. Drug Discovery 2017, 16, 181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dunn MR; Jimenez RM; Chaput JC Nat. Rev. Chem 2017, 1, 0076. [Google Scholar]
- (5).Huizenga DE; Szostak JW Biochemistry 1995, 34, 656–665. [DOI] [PubMed] [Google Scholar]
- (6).Stojanovic MN; de Prada P; Landry DW J. Am. Chem. Soc 2001, 123, 4928–4931. [DOI] [PubMed] [Google Scholar]
- (7).Rohloff JC; Gelinas AD; Jarvis TC; Ochsner UA; Schneider DJ; Gold L; Janjic N Mol. Ther. -Nucleic Acids 2014, 3, No. e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sefah K; Yang Z; Bradley KM; Hoshika S; Jimenez E; Zhang L; Zhu G; Shanker S; Yu F; Turek D; Weihong T; Benner SA Proc. Natl. Acad. Sci. U. S. A 2014, 111, 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hili R; Niu J; Liu DR J.Am. Chem. Soc 2013, 135, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gold L; Ayers D; Bertino J; Bock C; Bock A; Brody EN; Carter J; Dalby AB; Eaton BE; Fitzwater T; Flather D; Forbes A; Foreman T; Fowler C; Gawande B; Goss M; Gunn M; Gupta S; Halladay D; Heil J; Heilig J; Hicke B; Husar G; Janjic N; Jarvis T; Jennings S; Katilius E; Keeney TR; Kim N; Koch TH; Kraemer S; Kroiss L; Le N; Levine D; Lindsey W; Lollo B; Mayfield W; Mehan M; Mehler R; Nelson SK; Nelson M; Nieuwlandt D; Nikrad M; Ochsner U; Ostroff RM; Otis M; Parker T; Pietrasiewicz S; Resnicow DI; Rohloff J; Sanders G; Sattin S; Schneider D; Singer B; Stanton M; Sterkel A; Stewart A; Stratford S; Vaught JD; Vrkljan M; Walker JJ; Watrobka M; Waugh S; Weiss A; Wilcox SK; Wolfson A; Wolk SK; Zhang C; Zichi D PLoS One 2010, 5, e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Battersby TR; Ang DN; Burgstaller P; Jurczyk SC; Bowser MT; Buchanan DD; Kennedy RT; Benner SA J. Am. Chem. Soc 1999, 121, 9781–9789. [DOI] [PubMed] [Google Scholar]
- (12).Vaish NK; Larralde R; Fraley AW; Szostak JW; McLaughlin LW Biochemistry 2003, 42, 8842–8851. [DOI] [PubMed] [Google Scholar]
- (13).Ito Y; Suzuki A; Kawazoe N; Imanishi Y Bioconjugate Chem 2001, 12, 850–854. [DOI] [PubMed] [Google Scholar]
- (14).Masud MM; Kuwahara M; Ozaki H; Sawai H Bioorg. Med. Chem 2004, 12, 1111–1120. [DOI] [PubMed] [Google Scholar]
- (15).Shoji A; Kuwahara M; Ozaki H; Sawai HJ Am. Chem. Soc 2007, 129, 1456–1464. [DOI] [PubMed] [Google Scholar]
- (16).Ohsawa K; Kasamatsu T; Nagashima J-I; Hanawa K; Kuwahara M; Ozaki H; Sawai H Anal. Sci 2008, 24, 167–172. [DOI] [PubMed] [Google Scholar]
- (17).Imaizumi Y; Kasahara Y; Fujita H; Kitadume S; Ozaki H; Endoh T; Kuwahara M; Sugimoto NJ Am. Chem. Soc 2013, 135, 9412–9419. [DOI] [PubMed] [Google Scholar]
- (18).Rosenthal M; Pfeiffer F; Mayer G Angew. Chem. Int. Ed 2019, 58, 10752–10755. [DOI] [PubMed] [Google Scholar]
- (19).Sassanfar M; Szostak JW Nature 1993, 364, 550–553. [DOI] [PubMed] [Google Scholar]
- (20).Ruscito A; McConnell EM; Koudrina A; Velu R; Mattice C; Hunt V; McKeague M; DeRosa MC Curr. Protoc. Chem. Biol 2017, 9, 233–268. [DOI] [PubMed] [Google Scholar]
- (21).Yang K-A; Pei R; Stojanovic MN Methods 2016, 106, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yang K-A; Chun H; Zhang Y; Pecic S; Nakatsuka N; Andrews AM; Worgall TS; Stojanovic MN ACS Chem. Biol 2017, 12, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nakatsuka N; Yang K-A; Abendroth JM; Cheung KM; Xu X; Yang H; Zhao C; Zhu B; Rim YS; Yang Y; Weiss PS; Stojanovic MN; Andrews AM Science 2018, 362, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yang W; Yu H; Alkhamis O; Liu Y; Canoura J; Fu F; Xiao Y Nucleic Acids Res 2019, 47, No. e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Yang K-A; Barbu M; Halim M; Pallavi P; Kim B; Kolpashchikov DM; Pecic S; Taylor S; Worgall TS; Stojanovic MN Nat. Chem 2014, 6, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Han Y; Diao D; Lu Z; Li X; Guo Q; Huo Y; Xu Q; Li Y; Cao S; Wang J; Wang Y; Zhao J; Li Z; He M; Luo Z; Luo X Anal. Chem 2017, 89, 5270–5277. [DOI] [PubMed] [Google Scholar]
- (27).Nutiu R; Li Y Angew. Chem. Int. Ed 2005, 44, 1061–1065. [DOI] [PubMed] [Google Scholar]
- (28).Huestis MA Chem. Biodiversity 2007, 4, 1770–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jenison RD; Gill SC; Pardi A; Polisky B Science 1994, 263, 1425–1429. [DOI] [PubMed] [Google Scholar]
- (30).Yu H; Yang W; Alkhamis O; Canoura J; Yang K-A; Xiao Y Nucleic Acids Res 2018, 46, No. e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kypr J; Kejnovska I; Renciuk D; Vorlfckova M Nucleic Acids Res. 2009, 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).del Villar-Guerra R; Trent JO; Chaires JB Angew. Chem. Int. Ed 2018, 57, 7171–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Slavkovic S; Johnson PE Aptamers 2018, 2, 45–51. [Google Scholar]
- (34).Frost JM; Dart MJ; Tietje KR; Garrison TR; Grayson GK; Daza AV; El-Kouhen OF; Yao BB; Hsieh GC; Pai M; Zhu CZ; Chandran P; Meyer MD J. Med. Chem 2010, 53, 295–315. [DOI] [PubMed] [Google Scholar]
- (35).Yang Q; Xiang J; Yang S; Li Q; Zhou Q; Guan A; Zhang X; Zhang H; Tang Y; Xu G Chem. Commun 2010, 38, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Langer N; Lindigkeit R; Schiebel H-M; Ernst L; Beuerle T Drug Test. Anal 2014, 6, 59–71. [DOI] [PubMed] [Google Scholar]
- (37).White R; Rusconi C; Scardino E; Wolberg A; Lawson J; Hoffman M; Sullenger B Mol. Ther 2001, 4, 567–573. [DOI] [PubMed] [Google Scholar]
- (38).Helenius A; McCaslin DR; Fries E; Tanford C Methods Enzymol. 1979, 56, 734–749. [DOI] [PubMed] [Google Scholar]
- (39).Martin M EMBnet J. 2011, 17, 10–12. [Google Scholar]
- (40).Wintermeyer A; Moller I; Thevis M; Jubner M; Beike J; Rothschild MA; Bender K Anal. Bioanal. Chem 2010, 398, 2141–2153. [DOI] [PubMed] [Google Scholar]
- (41).Zuker M Nucleic Acids Res. 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yang K-A; Pei R; Stefanovic D; Stojanovic MN J. Am. Chem. Soc 2012, 134, 1642–1647. [DOI] [PubMed] [Google Scholar]
- (43).McKeague M; McConnell EM; Cruz-Toledo J; Bernard ED; Pach A; Mastronardi E; Zhang X; Beking M; Francis T; Giamberardino A; Cabecinha A; Ruscito A; Aranda-Rodriguez R; Dumontier M; DeRosa MC J. Mol. Evol 2015, 81, 150–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.